1. Introduction
DNA sequencing of the ITS1, 5.8S and ITS2 region identified the ascomycete
Hypocrea virens (Chaverri, Samuels and Steward) as the teleomorph of
Trichoderma virens (Miller, Giddens and Foster) Arx [
1,
2,
3]. By further analyzing the newly identified
H. virens isolate and comparing its ITS barcode with four different reference strains of the anamorph
T. virens, both types were found to be indistinguishable [
1].
T. virens is well known under its common name
Gliocladium virens Mill. and is also referred to as
Gliocladium flavofuscum Mill. or
Trichoderma flavofuscum Mill. The genus
Hypocrea/Trichoderma consists of about 200 species described by molecular data [
4]. In molecular studies for the reconstruction of phylogenetic relationships based on RNA polymerase II subunit b (rpb2) and translation elongation factor 1 alpha (tef1) [
5] as well as on ITS sequences of the rDNA repeat [
6],
T. virens shows a distinct grouping in the resulting phylogenetic trees. Tree positions are supported by high bootstrap values and are localized between sections
Longibrachiatum/Pachybasium and within section
Pachybasium B, respectively, with
T. crassum Bisset being the closest relative in both cases. From these data, it can be concluded that
T. virens is a well-defined species that can certainly be identified by molecular taxonomy approaches.
Trichoderma spp. have attracted growing interest due to their economic value and are widely used in several branches of the industry. A key aspect is the production of diverse extracellular enzymes. Enzyme preparations and living strains of
Trichoderma are products that are commonly used in agricultural biotechnology. Living strains are utilized for bioremediation of contaminated agricultural land and
T. asperellum Samuels H15 has demonstrated that it is able to degrade polycyclic aromatic hydrocarbons (PAHs) in soils [
7],
T. viride Pers. strain NFCCI-2745 has the capability of decomposing phenolic pollutants in soil [
8] and the
T. viride strain FRP3 has the ability to biodegrade glyphosate and to be potentially useful in areas where this herbicide is extensively used [
9]. In agricultural crop production and plant protection, living strains, mainly of
T. harzianum Rifai,
T. virens,
T. atroviride Karst,
T. koningii Oudem,
T. hamatum Bonord and
T. asperellum, are placed close to the root system in greenhouse and field crops. Strains of these species have been identified as avirulent plant symbionts that enter root cells and have been characterized as root endophytes that form intracellular structures [
10] which are assumed to be responsible for substrate exchange and molecular crosstalk. Penetration of the root system by
Trichoderma frequently causes elevated resistance levels in their host plants via salicylate and jasmonate induced pathways [
11]. Further positive effects of the root connected hyphal network include a delay in drought symptoms, enhanced root growth and nutrient uptake. Therefore they are often used as so-called biofertilizers or biostimulants and are available on the market as formulated spore-inoculum products. In natural ecosystems,
Trichoderma species have been recognized as common and persistent members of the soil microbiome and rhizosphere microbial communities [
12].
Mycoparasitism is a common feature among
Trichoderma spp. but is only rarely reported in other families of the fungi [
13,
14]. The antagonistic mode of primarily
T. virens,
T. harzianum and
T. asperellum against other fungi and particularly soil-borne plant pathogenic fungi is exploited by the development of biocontrol agents and biological control strategies in integrated plant protection.
Trichoderma biocontrol products are well established on the market and available as spore-inocula. Numerous reports exist about inhibition of prominent fungal plant pathogens by
Trichoderma spp., among these,
Rhizoctonia solani Kühn,
Fusarium solani Mart. and
Sclerotinia sclerotiorium de Bary [
15];
Fusarium culmorum Sm. and
Fusarium graminearum Schwabe [
16]; and
Alternaria altanata Kreissl.,
Botrytis cinerea Pers. and
Fusarium spp. [
17]. The mode of action in terms of direct interaction is based on a mycotrophic lifestyle including the production of hydrolytic cell wall degrading enzymes like chitinase and ß-1-3 glucanase [
18] as well as biocidal substances [
19].
Trichoderma spp. actively attack other fungi by coiling around their hyphae releasing the above-mentioned substances and feeding on the parasitized mycelia, which often leads to de-vitalization of the prey fungus. This sometimes also causes negative impact in the commercial production of culinary fungi like
Agaricus bisporus Lange and
Pleurotus ostreatus Krumm. There are even isolates that are effective against members of the Oomycota through direct interaction [
20] or by triggering defense reactions in their host plants [
21].
The estimation of root colonization efficiencies of different
Trichoderma strains, their ability to induce systemic resistance in host plants and their capability of inhibiting plant pathogenic fungi in the rhizosphere under field conditions requires precise re-identification to track and trace strains experimentally released to the root system. In this context, diagnostic molecular markers are a prerequisite for studying plant-microbe interaction, the persistence of released strains in the rhizosphere and their impact on rhizosphere microbial communities. Despite the fact that three complete genome sequences (
T. reesei Simmons,
T. virens and
T. atroviride) are available on the ISTH webpage (
www.isth.info/links.php, International Subcommission on
Trichoderma and
Hypocrea Taxonomy) genomic data have not yet been intensively exploited for the development of locus-specific molecular markers. Reports about this marker type are sparse in
Trichoderma spp. and comprise solely the development of sequence characterized amplified regions (SCAR markers) obtained from sequence data of cloned random amplified polymorphic DNA (RAPD) fragments [
22,
23]. The vast majority of studies on discriminating the strains of
Trichoderma species are based on anonymous markers like (universally primed) UP-PCR, RAPDs or restriction fragment length polymorphisms (RFLPs) [
24,
25,
26]. The anonymous approach also holds true for
Trichoderma microsatellite studies, where ISSRs (inter simple sequence repeat) and RAMS (random amplified microsatellites) have been used so far [
27,
28]. To our knowledge, no sequence-tagged microsatellite site (STMS) markers based on PCR amplification of unique SSR loci [
29] are currently available for
Trichoderma spp. Therefore, the invaluable genomic information of
T. virens was exploited to develop a set of SSR markers that is capable of identifying strains in root tissue, rhizosphere soil and environmental samples. Moreover, markers were tested for their potential use in real-time PCR applications to estimate and quantify root colonization efficiencies of certain strains and combinations of strains. Furthermore, transferability of the markers to other
Trichoderma species, in particular to
Trichoderma harzianum, was evaluated.
2. Experimental Section
2.1. Screening of Databases and Primer Design
The
T. virens genome sequence was accessed via the ISTH website by clicking on the links button and going to the genome portal of the Department of Energy Joint Genome Institute [
30]. In total, 93 scaffolds ranging from 4.407 bp (scaffold 7) to 3,456,807 bp (scaffold 6) were available and downloaded, of which scaffold 7 was by far the smallest. The majority of the scaffolds were larger than 20 kb and 43 were larger than 50 kb. Roughly 9 scaffolds were at least 1.7 Mb in length and covered half of the genome. The total genome size was 39 Mb containing 12,427 structurally and functionally annotated genes. Further information is available on the homepage of the U.S. Department of Energy’s Joint Genome Institute. All scaffolds (in FASTA format) were loaded into the BioEdit Sequence Editor (Ibis Biosciences: Carlsbad, CA, USA) by using the select/slide mode. This made it possible to search for all scaffolds in one go. Screening the genome for microsatellite motifs was based on published data about existing motifs that were detected in
T. virens by ISSR and RAMS analysis [
31,
32]. Microsatellite loci sufficiently flanked by unique sequences were selected for primer design using the Primer3 software [
33] with parameter settings that included the GC-clamp option and an allowance for primer annealing at approximately 60 °C. The primer sequences obtained were searched in the genome of
T. virens to avoid multiple annealing sites. After that, about half of the developed primers (
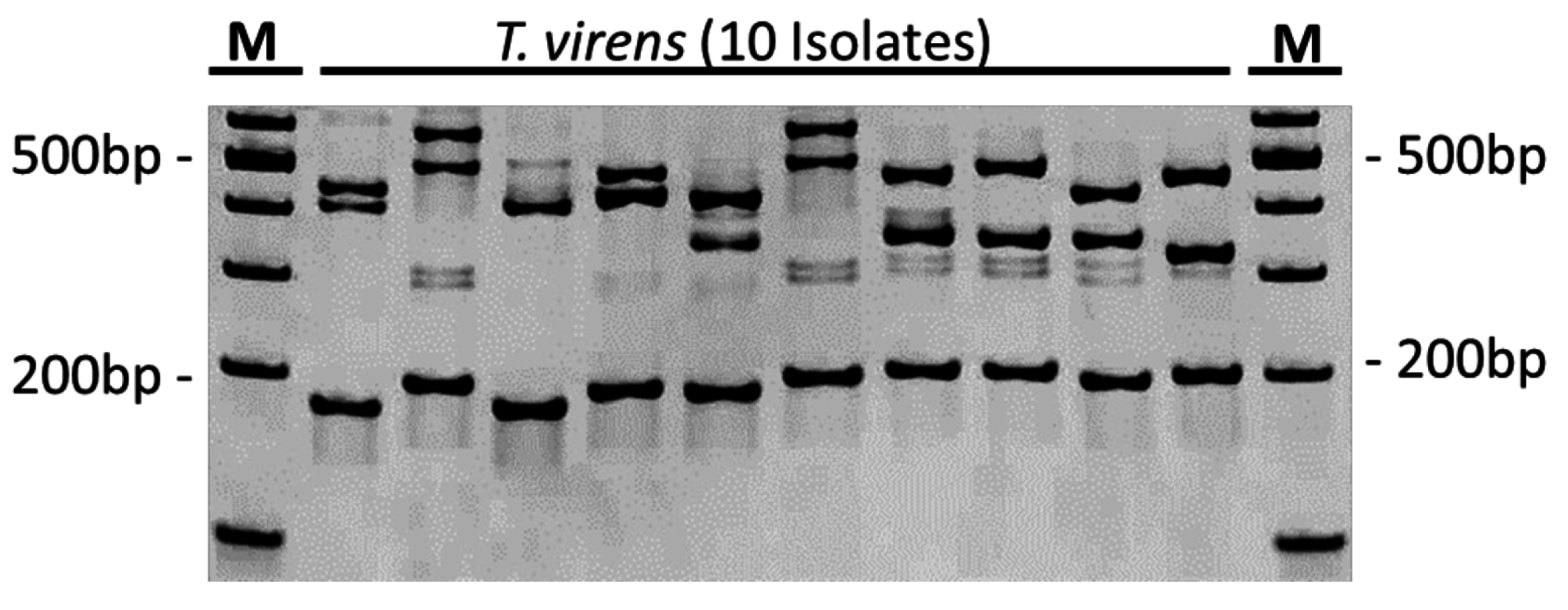
Table 1) displayed clear single-band PCR amplification products on agarose gels, while in some cases stuttering bands became visible on PAA gels when the same primers were used.
2.2. Fungal Strains
In order to evaluate the variability at the selected microsatellite loci, a set of
T. virens isolates of geographically diverse origin (
Table 2) was obtained from the CBS-KWAS (Centraalbureau Schimmelcultures–Koninklijke Nederlandse Academie van Wetenschappen) Fungal Biodiversity Centre (Utrecht, The Netherlands). The cultures were grown according to the supplier’s recommendations and the DNA was isolated from liquid cultures as described below. The molecular fingerprints of the CBS isolates obtained were compared to a local strain of
T. virens, which was isolated from maize roots in August 2012. Briefly, the roots of a healthy
Zea mays L. plant from a maize field in Central Germany (silty loam loess chernozem, pH 7.2–7.5, 2.5%–3.0% humus, 80 m a.s.l., 51°80′ N, 11°73′ E, 511 mm average annual precipitation, 9.7 °C average annual temperature) were obtained by carefully removing residual soil and selecting fine roots (1–2 mm in diameter). The roots were thoroughly washed with sterile water and cleaned with a soft brush to remove any remaining soil. The cleaned roots were submerged in 3% Na-hypochlorite for 5 min, rinsed with sterile water, cut into pieces (approximately 1.5 cm long) and placed on a Petri dish containing potato dextrose agar (PDA, 1.5%, pH 5.8). Outgrowing mycelia were transferred to fresh plates and grown at room temperature until sporulation. A 20 µL spore suspension was taken, diluted 100-fold and 10–100 µL were plated on fresh PDA. One colony originating from a single spore was selected and the mycelium cultivated further. Agar blocks (approximately 5 mm × 5 mm × 5 mm) were excised from actively growing mycelium and stored at −80 °C in a solution containing 50% glycerol, 25% culture medium and 25% distilled water. A liquid culture was prepared and DNA isolated as described below. The ITS region of the nuclear rDNA was amplified using the primers ITS1F [
34] and ITS4 [
35] and the resulting PCR fragment directly sequenced following the Sanger method. An online homology search was carried out in the NCBI GenBank nucleotide database by means of the algorithm BLASTn and results confirmed the identity of the isolate, which was then designated as
Trichoderma virens strain Maize9B.
In order to test for cross-species amplification the primers derived from the T. virens genome were applied to genomic DNA of a commercially available T. harzianum isolate, notably strain T22 (Koppert B.V.: Berkel en Rodenrijs, The Netherlands).
2.3. Culture Conditions
Conserved fungal cultures were retrieved from −80 °C stocks by thawing the tubes on ice, removing one agar block, rinsing it with sterile water to wash off residual glycerol and placing it on a Petri dish containing potato dextrose agar (1.5%, pH 5.8). Plates were incubated at 20 °C in a 12/12 h day/night cycle. Outgrowing mycelia from the agar blocks were either left until sporulation to harvest spores for root inoculation or—after 4 to 5 days of growth—small agar blocks were taken from the actively growing front of the mycelium and placed in Erlenmeyer flasks containing 50 mL potato dextrose bouillon (pH 5.8). Cultures were incubated on a rotary shaker (120 rpm) under the same conditions as above and mycelia for DNA isolation were harvested after 2 to 3 days by filtration.
2.4. Plant Material and Root Inoculation
All root inoculation experiments were performed with tomato plants Solanum lycopersicum L. cv. MOBIL (Breeding Company: ZKI, Zöldségtermesztési Kutató Intézet, Kecskemét, Hungary). This variety is a robust cultivar frequently used in Hungary for field production of tomatoes.
Well sporulated plates of T. virens were covered with a layer of 25 mM MgSO4 solution, left to rest for 20 min and then gently stirred with a glass rod to obtain a dark green spore suspension which was transferred to fresh tubes. Spore concentrations were determined using an Abbe-Zeiss counting cell chamber and spore concentrations adjusted to 5 × 107 spores/mL by either diluting or centrifuging the suspension.
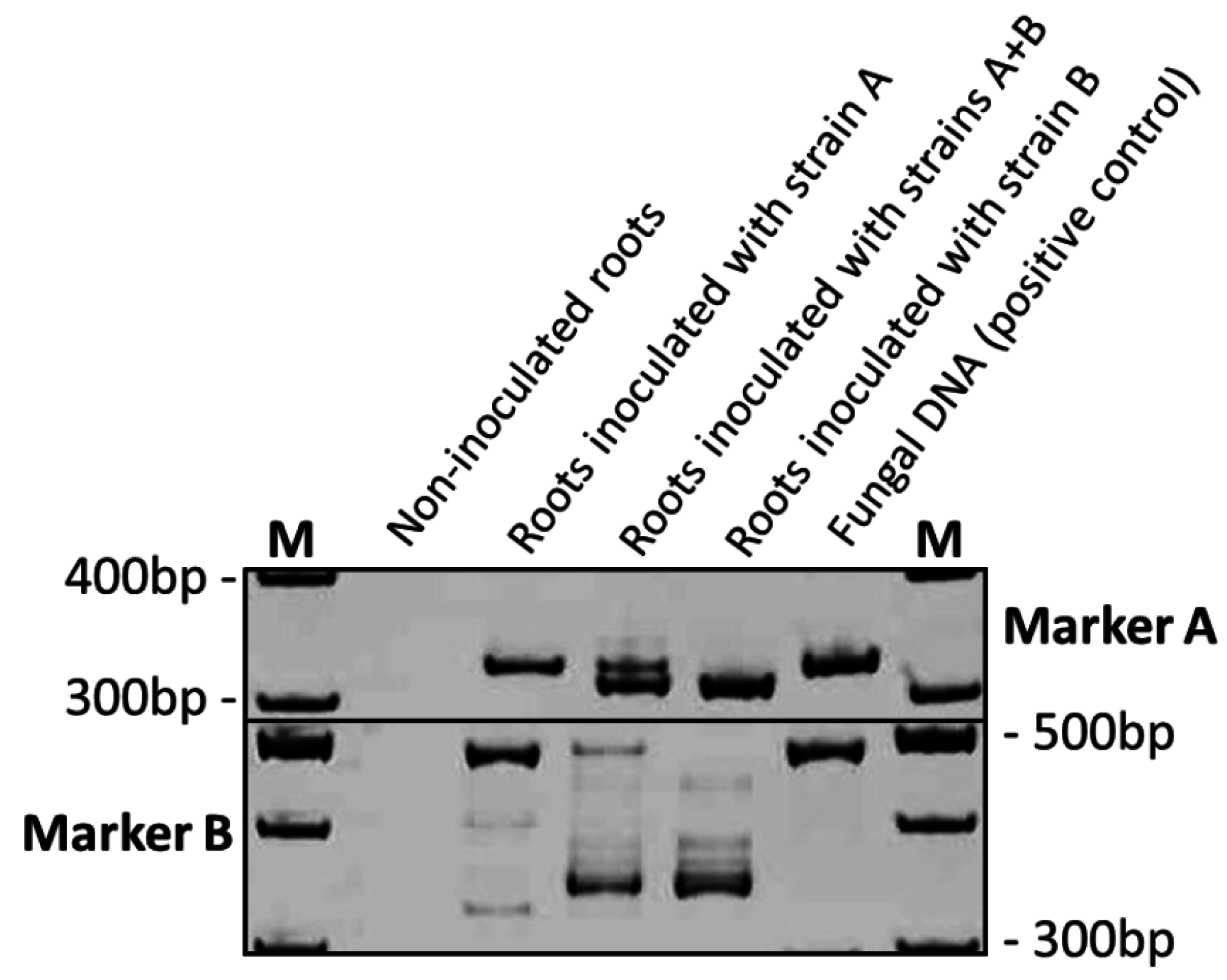
Tomato seeds were germinated in Fruhstorfer Soil Type P (Hawita-Group: Vechta, Germany) and seedlings (10 to 12 cm in height) were transferred to 2.5 L planting pots containing 2 kg (dry weight) of local natural farmland topsoil as described above for the maize field in Central Germany. Before transplanting, the soil was pre-wetted, a planting hole opened in the pots, and 2.5 × 104 spores/g substrate pipetted into the holes (1 mL of the stock solution). Seedlings were carefully removed from the germination soil and transplanted into the prepared planting cavity. In the case of co-inoculation with two different T. virens strains half of the amount of each isolate was pipetted into the same planting cavity to reach the same spore concentration as single inoculations (2 × 0.5 mL stock solution from each isolate). Inoculated seedlings were grown under semi-controlled greenhouse conditions. Pots were watered on demand from the trivet, temperatures ranged from 18 to 26 °C with a day/night cycle of 10/14 h at an illumination intensity of 20,000 Lux. Roots were harvested 42 days after inoculation and the DNA was immediately isolated as described below.
Table 1.
PCR primer sequences for unique microsatellite loci in T. virens. Repeat numbers in primer names refer to the T. virens reference genome. “Fragment Sizes” were determined in isolate Maize9B; “Range” refers to the other strains analyzed in this study and “Reference Sequence” to the virtual fragment size in the reference genome. “Scaffold No.” marks the super contig from which loci were retrieved and the T. harzianum column shows which markers were transferable to T. harzianum. Primers for multiplex PCR are in bold.
Table 1.
PCR primer sequences for unique microsatellite loci in T. virens. Repeat numbers in primer names refer to the T. virens reference genome. “Fragment Sizes” were determined in isolate Maize9B; “Range” refers to the other strains analyzed in this study and “Reference Sequence” to the virtual fragment size in the reference genome. “Scaffold No.” marks the super contig from which loci were retrieved and the T. harzianum column shows which markers were transferable to T. harzianum. Primers for multiplex PCR are in bold.
| Primer Name | Primer Sequence (5′-3′) | Locus No. | Tm | Fragment Sizes in bp: Maize9B, (Range), Reference Sequence | NCBI Accession No. | Alleles | Scaffold No. | T. harzianum |
|---|
| TvCTT56f | CTTGATGACAAGCCAAAAGG | | 58.4 | 289 (283–439) 456 | KM010303 | 8 | 64 | − |
| TvCTT56r | GAAGAGAGGACATAGGGTCTGG | L1 | 59.2 | | | | | |
| TvCAT32f | GTGTAGCAGCCCAACAGTCC | | 60.7 | 409(364–457) 481 | KM010304 | 8 | 89 | − |
| TvCAT32r | CAGGTGTCGTGACAGATTCG | L2 | 60.3 | | | | | |
| TvCTTT29f | GGAAGATAGCACGATGAAGTCG | | 61.1 | 350 (291–410) 402 | KM010305 | 9 | 81 | − |
| TvCTTT29r | AACCGTGGAAGTAGGTGTCG | L3 | 60.0 | | | | | |
| TvCTTTT27f | TCATCCACCCTGCTAACTCG | | 61.2 | 420 (378–563) 482 | KM010306 | 9 | 81 | − |
| TvCTTTT27r | CGCTGCGTCATCCTAAACC | L4 | 61.7 | | | | | |
| TvAAC21f | CACCATTCCATTATTACGCGACG | | 60.4 | 234 (210–269) 268 | KM010307 | 7 | 2 | + |
| TvAAC21r | CTGCACTCCCTCCCAATGC | L5 | 60.8 | | | | | |
| TvCAG13f | CCCAGGAAACCCTCAGAACG | | 60.3 | 180(161–180) 206 | KM010308 | 7 | 92 | − |
| TvCAG13r | TCTTTGCAGTTTCCAAGTCGG | L6 | 59.1 | | | | | |
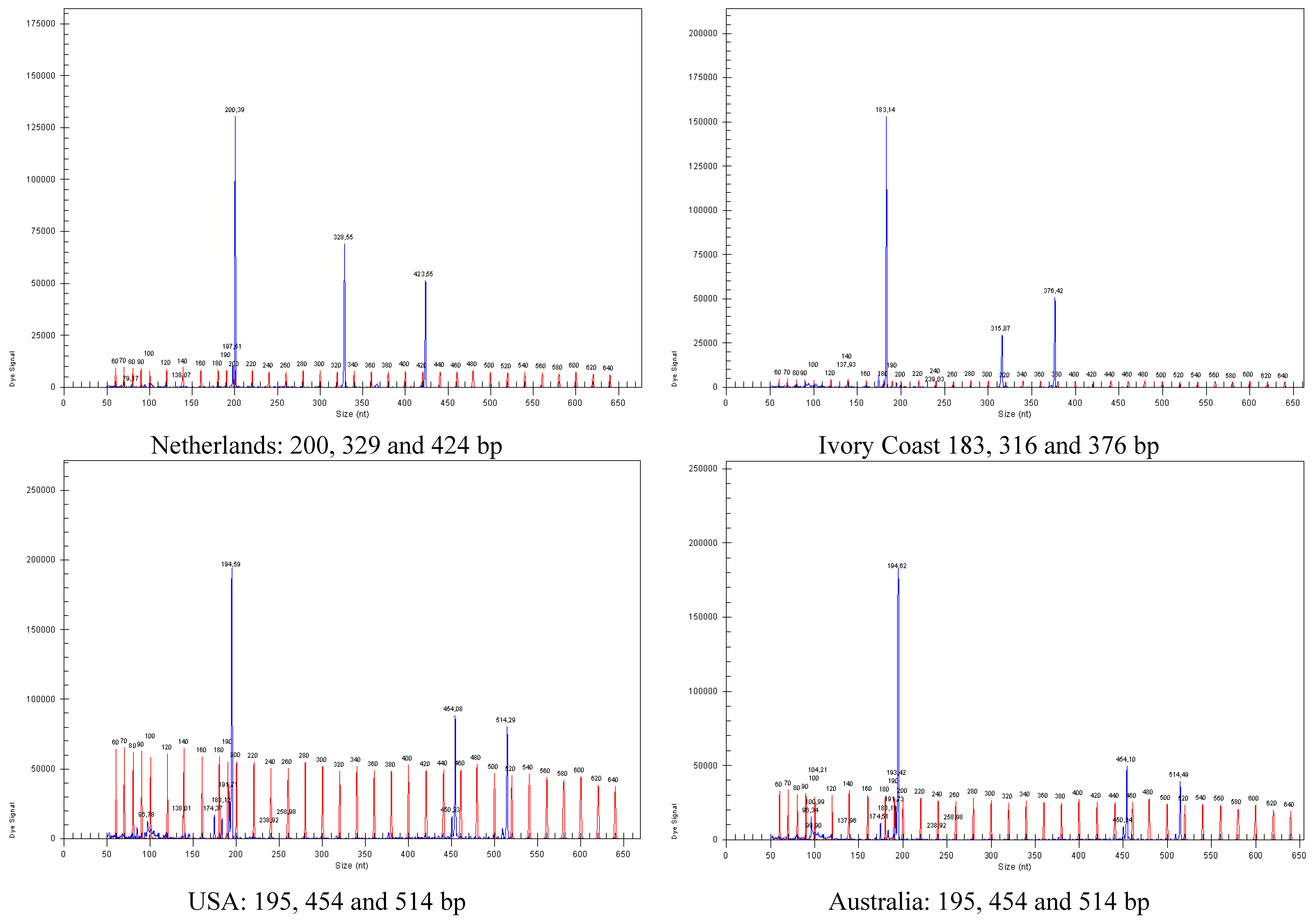
| TvGAAA34f | GGGGTGCTGAATAGCTAACG | | 59.7 | 325 (315–491) 423 | KM010310 | 6 | 3 | + |
| TvGAAA34r | TGCCGTCTTGTCTTATTTTCG | L7 | 60.3 | | | | | |
| TvTGTC18f | GTGGTGAGGACTTGCTTGG | | 59.3 | 425 (393–469) 483 | KM010311 | 7 | 2 | + |
| TvTGTC18r | TCTGCCTGTCAGTTGTTTGC | L8 | 60.0 | | | | | |
| TvGAT18f | GGGATCTGATTTGGCCTACC | | 60.7 | 371 (333–423) 387 | KM010312 | 8 | 3 | + |
| TvGAT18r | ACTTCCCCCATCCAATAACG | L9 | 60.9 | | | | | |
| TvCA39f | GCATCTGCACCTGATATATTCC | | 58.6 | 256 (236–271) 306 | KM010313 | 8 | 6 | − |
| TvCA39r | CCTTGTACGATCTCCAGAACC | L10 | 58.7 | | | | | |
| TvGTT23f | GCATCAAAGCGTGCTGTTGG | | 60.8 | 216 (206–237) 279 | KM010309 | 4 | 87 | − |
| TvGTT23r | GCAAACACAAGCTGACAATGC | L11 | 60.6 | | | | | |
| TvAG29f | TGTGCCCACTGAGATTTCG | | 60.8 | 449 (423–462) 470 | KM010314 | 8 | 93 | − |
| TvAG29r | TCAGCATGAGATTACACATACCG | L12 | 60.0 | | | | | |
Table 2.
CBS numbers of the utilized T. virens strains with additional information retrieved from the CBS catalogue.
Table 2.
CBS numbers of the utilized T. virens strains with additional information retrieved from the CBS catalogue.
| Isolate Origin | CBS Number | Habitat | Year of Isolation |
|---|
| Ivory Coast | CBS 123790 | soil | Unknown |
| Italy | CBS 116947 | sandy soil, Pinus pinea | 1982 |
| Iran | CBS 111249 | soil | 2001 |
| Guadeloupe | CBS 100946 | rain forest soil | 1998 |
| Papua New Guinea | CBS 350.96 | coastal region soil | 1995 |
| The Netherlands | CBS 609.95 | compost | 1995 |
| Australia (Perth) | CBS 497.84 | sandy soil, lettuce | 1984 |
| Moldova | CBS 512.66 | soil | 1966 |
| USA (Maryland) | CBS 430.54 | soil | 1954 |
| Germany | - | maize roots | 2012 |
2.5. DNA Extraction
2.5.1. Mycelia
Fungal DNA of approximately 100 mg fresh mycelium from liquid culture was extracted by using the peqGOLD Fungal DNA Mini Kit (VWR Peqlab: Erlangen, Germany). Mycelia were filtered, briefly dried between tissue paper and mechanically homogenized in 2 mL tubes containing 700 µL peqGOLD lysis buffer in a FastPrep24 instrument (MP Biomedicals: Heidelberg, Germany) for 3 × 30 s at a speed of 6 m/s with 1.0 mm silica spheres and an additional 0.25 inch ceramic bead. After each homogenization cycle samples were cooled on ice for 1 min. DNA was further extracted following the manufacturer’s instructions. The DNA was tested on 1% agarose gels and concentrations were determined using the NanoDrop ND-1000 photometer (VWR Peqlab).
2.5.2. Roots
Tomato roots were carefully removed from the planting pots, thoroughly cleaned with water and a soft brush until no residual soil adhered, briefly dried between paper towels, and cut into small pieces. Approximately 100 mg of fine roots were placed in 2 mL tubes containing 1.0 mm silica spheres and a single 0.25 inch ceramic bead (MP Biomedicals) and 700 µL peqGOLD lysis buffer, homogenized for 30 s at a speed of 6 m/s three times (FastPrep 24 device, MP Biomedicals). After each cycle, samples were cooled on ice for 1 min. DNA was extracted with the peqGOLD Fungal DNA Kit (VWR Peqlab) following the manufacturer’s instructions. DNA quality was determined as described above.
2.5.3. Leaves
DNA from tomato leaves was extracted with the Plant Fast DNA Spin Kit (MP Biomedicals: Heidelberg, Germany), following the supplier’s instructions. Tissue disruption with the FastPrep 24 device was performed similarly to that of root tissue. Leaf DNA was used to ensure that the T. virens SSR primers did not show cross-reactions with tomato DNA, which could cause confusion when examining results from in-root detection of fungal endophytes.
2.6. PCR Conditions
2.6.1. Touchdown PCR
PCR was carried out in 20 µL volumes containing 10 ng of extracted DNA, 0.5 µM of each primer and 2× Phusion High-Fidelity PCR Master Mix (Thermo Scientific: Schwerte, Germany). Microsatellite amplification was performed in a thermal cycler (Labcycler, SensoQuest: Göttingen, Germany) with the following thermal profile: initial denaturation at 96 °C for 3 min, 9 cycles consisting of 95 °C for 20 s, 64 °C for 25 s (−0.5 °C per cycle), 72 °C for 30 s, followed by 31 cycles consisting of 95 °C for 20 s, 60 °C for 25 s, and 72 °C for 30 s. A final elongation step at 72 °C for 5 min completed the protocol.
2.6.2. Multiplex PCR
For multiplex PCR the primer pairs TvCTTTT27, TvGAAA34 and TvCAG13 (0.5 µM each) were used in a single PCR. PCR conditions and thermal profile conformed to touchdown PCR conditions.
2.6.3. In-Root Detection of Fungal DNA
In-root detection of endophytic T. virens strains was performed with primer pairs TvGAAA34 and TvCTT56, respectively. PCR conditions and thermal profile were identical to touchdown PCR, except that 50 ng of total root DNA was used for specific amplification of the fungal DNA fraction.
2.6.4. Real-Time PCR
Real-time PCR was performed using a PikoReal 96 real-time PCR system (Biozym: Hessisch Oldendorf, Germany). PCR reactions consisted of 0.5 µM forward and reverse primers (TvGAAA34 and TvCTT56, respectively), 2× Phusion High-Fidelity PCR Master Mix, 1× SybrGreen I Nucleic Acid Stain (Lonza: Basel, Switzerland), and a dilution series of DNA with a total volume of 20 µL. The following thermal profile was used: initial denaturation at 96 °C for 3 min, 37 cycles consisting of 95 °C for 20 s, 60 °C for 25 s, and 72 °C for 30 s. After amplification, a melting curve was created by increasing the temperature from 60 °C to 95 °C in 0.2 °C steps. The results were analyzed using the PikoRealTM 2.2 software.
2.7. Electrophoresis
2.7.1. Agarose Gels
PCR products from microsatellite amplification were checked on 3% agarose gels containing 2.5 µL Roti-GelStain/50 mL 1× TAE buffer (Carl Roth: Karlsruhe, Germany). Electrophoresis was performed at 6 V/cm for approximately 45 min. Then the gels were documented under UV light (Genius Bio Imaging System, Syngene: Cambridge, UK).
2.7.2. Polyacrylamide (PAA) Gels
5% Mini-Protean TBE precast PAA gels (Bio-Rad: Munich, Germany) were used to analyze small molecular weight differences caused by variable microsatellite repeats. Electrophoresis was conducted in a 1× TBE buffer at 10 V/cm for 90–120 min depending on fragment sizes using the Mini-Protean Tetra Cell System (Bio-Rad). Gels were stained with ethidium bromide (5 µL ethidium bromide (10 mg/mL)/100 mL 1× TBE buffer) for 15 min on a rotary shaker. After washing with water, the gels were de-stained with double-distilled water for 15 min on an orbital shaker. Finally, the gels were photographed under UV light (Genius Bio Imaging System, Syngene: Cambridge, UK), the images were imported into the phoretix grabber software version 3.01 (Phoretix International: Newcastle upon Tyne, UK), and fragments sizes were calculated by the software according to molecular weight standards co-separated with the SSR fragments.
2.7.3. Capillary Gel Electrophoresis
The CEQ 8000 capillary sequencer device was used (Beckman Coulter: Pasadena, CA, USA) for fragment analyses of multiplex PCR. Multiplex PCR was performed with Cy5-labeled forward primers and unlabeled reverse primers. PCR products were diluted 100–200 fold and then mixed with a 30 µL sample loading solution (Beckman Coulter) and 0.5–1 µL of 10 fold diluted 20 bp DNA standard ladder (CEQ DNA Size Standard Kit—600, Beckman Coulter). A drop of mineral oil was placed on the sample and the method “Frag-4” was started (capillary temperature 50 °C, sample denaturation at 90 °C for 120 s, injection with 2.0 kV for 30 s, and separation with 4.8 kV for 60 min). Fragment sizes were determined using the CEQ Main Software, Version 9.0.25.
2.8. Re-Sequencing
Extracted DNA of
T. virens strain Maize9B was amplified using all 12 microsatellite primer pairs (
Table 1). PCR products were excised from agarose gels and purified using the Wizard SV Gel and PCR Clean-Up Kit (Promega: Madison, WI, USA). The concentration of the purified fragments was determined using the NanoDrop ND-1000 photometer (VWR Peqlab). Fragments were ligated into the pJET 1.2 blunt cloning vector (CloneJET PCR Cloning Kit, Life Technologies: Paisley, UK) and transformed in competent
E. coli JM109 cells. One positive clone of each microsatellite amplification product was sequenced in forward and reverse orientation using a 30 pmol primer in 10 mM Tris/HCL following the Sanger method. Sequence alignments were carried out using the Vector NTI Advance 11.5 software (Life Technologies: Carlsbad, CA, USA). Complete microsatellite sequences of 12 distinct loci were submitted to NCBI (GenBank accession numbers: KM010303 to KM010314).
2.9. Statistical Analysis
To establish the relationships and to assess the genetic diversity of the analyzed
T. virens isolates, a 0/1 matrix was prepared from the absence and presence of bands at distinct molecular weights through microsatellite amplification. The resulting matrix (
Supplementary Material 1) was then used to calculate D
ST, Nei’s standard genetic distance [
36]. The molecular variance (AMOVA) was analyzed using GenAIEx 6.5 software [
37,
38]. Haploid diversity by population and haploid diversity by locus were determined and values for numbers of different alleles (Na), numbers of effective alleles (Ne), Shannon’s information index (I), diversity (h) and unbiased diversity (uh) were calculated. For AMOVA, isolates were split into 2 populations (the first five isolates originating from the Eurasian continent and the second five isolates originating from the Americas, Australia, the Pacific and Africa). Phi
PT (modified F
ST) was calculated among and within populations by applying 999 permutations to estimate the total molecular variance and molecular variance by locus. The probability (P) of Phi
PT was based on standard permutations across the full data set (
Supplementary Material 2).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




