Patterns of Spontaneous Nucleotide Substitutions in Grape Processed Pseudogenes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic Sequence
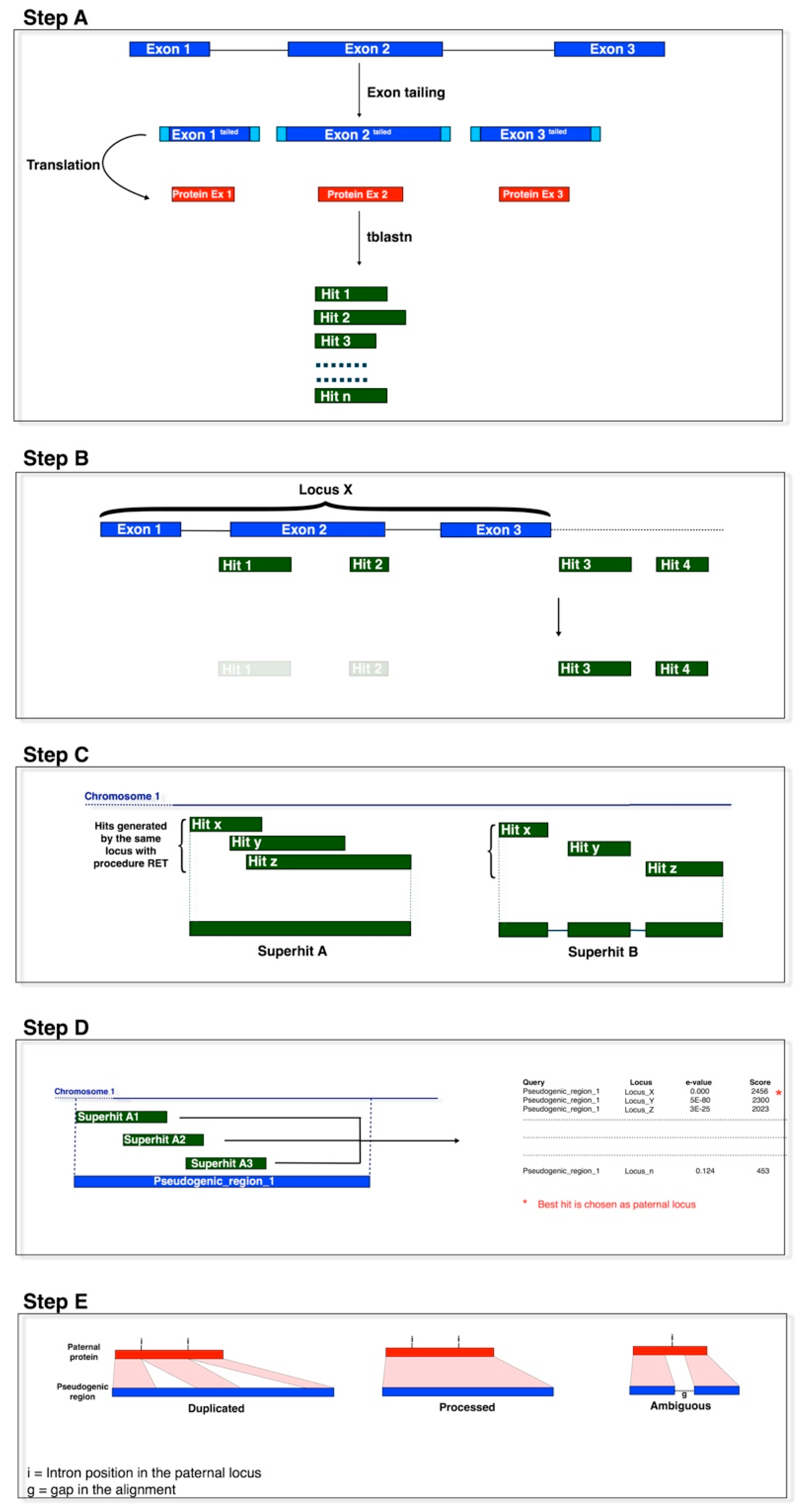
2.2. Pseudogene Identification
2.3. Ka/Ks Rate Determination
2.4. Analysis of Nucleotide Substitutions
2.4.1. The Non-Synonymous Dataset
2.4.2. The Parsimony-Based Dataset
3. Results
3.1. Identification of Primary Processed Pseudogenes
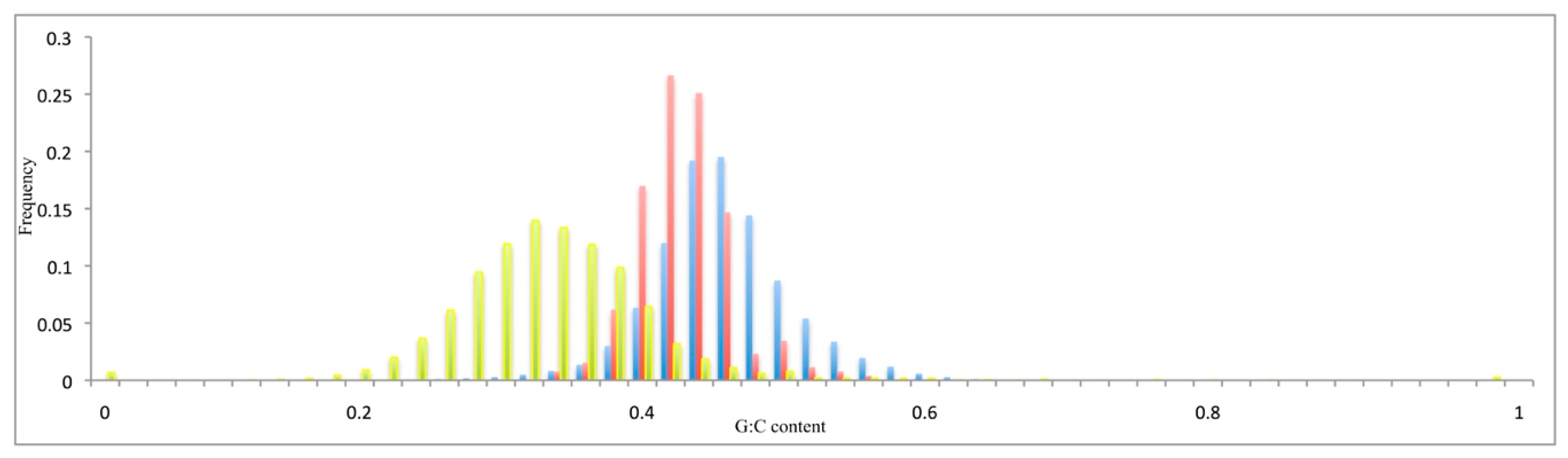
3.2. Compositional Features of Functional Genes and Pseudogenes
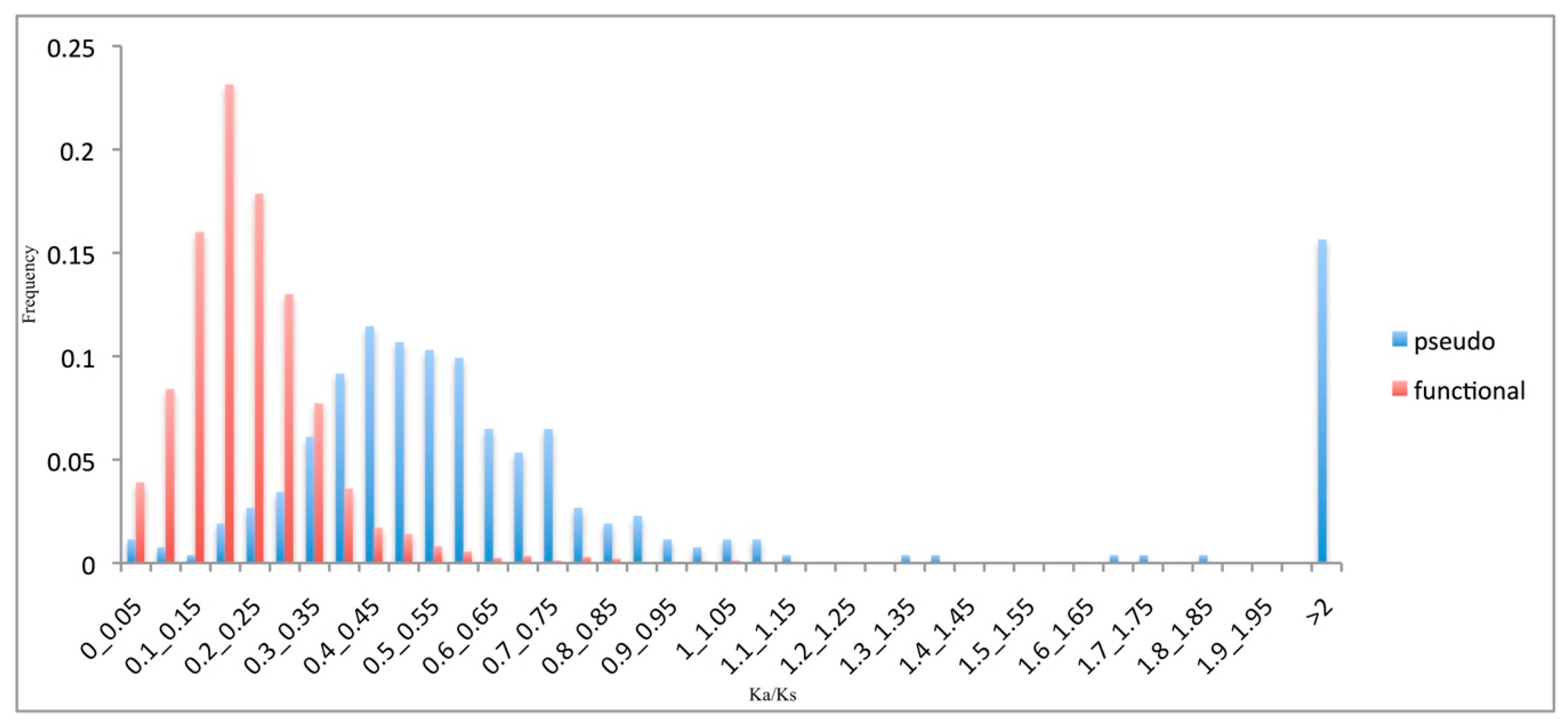
3.3. Pseudogene Evolutionary Rates
3.4. Transitions Are More Freequent Than Transversions
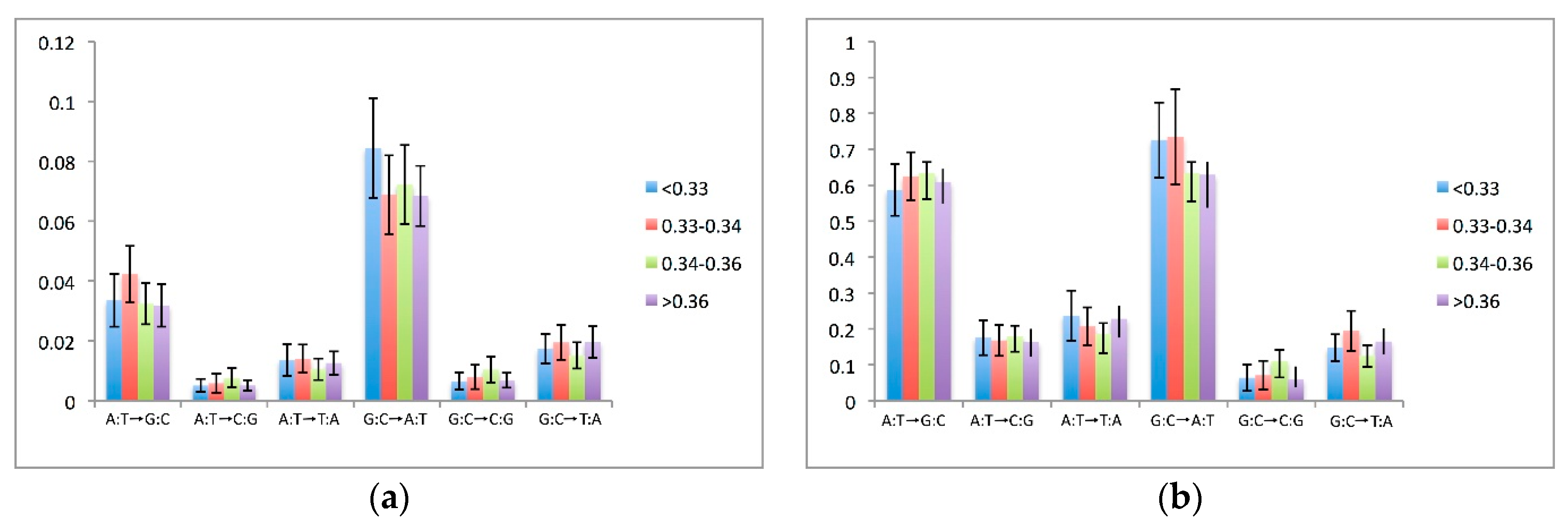
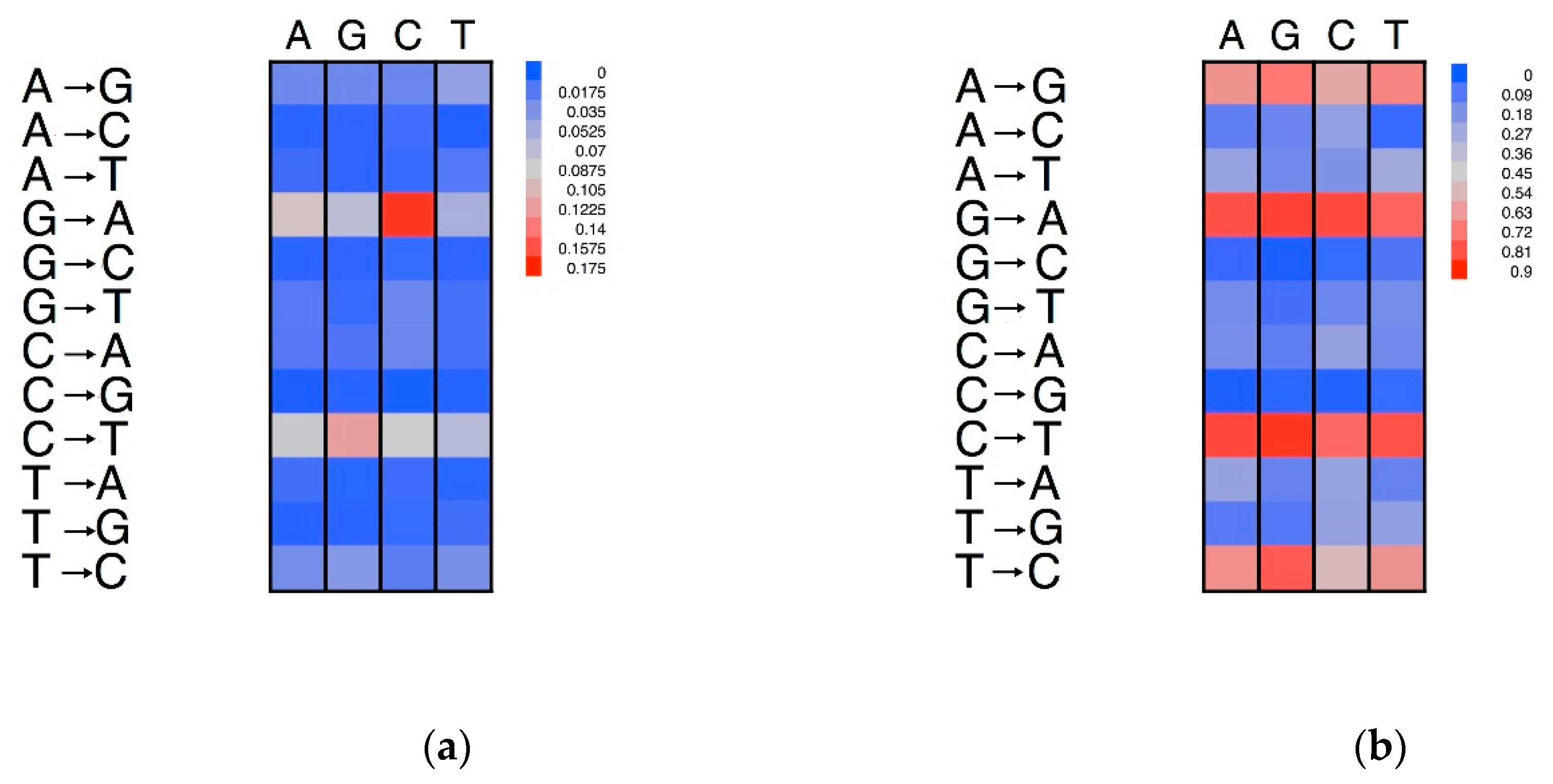
3.5. Effects of Compositional Background on Nucleotide Substitution
3.6. Neigbouring Effects on Nucleotide Substitutions
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fitch, W.M. Evidence suggesting a non-random character to nucleotide replacements in naturally occurring mutations. J. Mol. Biol. 1967, 26, 499–507. [Google Scholar] [CrossRef]
- Kimura, M. Estimation of evolutionary distances between homologous nucleotide sequences. Proc. Natl Acad. Sci. USA 1981, 78, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino-acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Morton, B.R.; Dar, V.-U.-N.; Wright, S.I. Analysis of site frequency spectra from Arabidopsis with context-dependent corrections for ancestral misinference. Plant. Physiol. 2009, 149, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Morton, B.R.; Bi, I.V.; McMullen, M.D.; Gaut, B.S. Variation in mutation dynamics across the maize genome as a function of regional and flanking base composition. Genetics 2006, 172, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gerstein, M. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 2003, 31, 5338–5348. [Google Scholar] [CrossRef] [PubMed]
- Gojobori, T.; Li, W.H.; Graur, D. Patterns of nucleotide substitution in pseudogenes and functional genes. J. Mol. Evol. 1982, 18, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.A.; Hartl, D.L. Patterns of nucleotide substitution in Drosophila and mammalian genomes. Proc. Natl. Acad. Sci. USA 1999, 96, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Benovoy, D.; Drouin, G. Processed pseudogenes, processed genes, and spontaneous mutations in the Arabidopsis genome. J. Mol. Evol. 2006, 62, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Vanin, E.F. Processed pseudogenes. Characteristics and evolution. Biochim. Biophys. Acta 1984, 782, 231–241. [Google Scholar] [CrossRef]
- Esnault, C.; Maestre, J.; Heidmann, T. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 2000, 24, 363–367. [Google Scholar] [PubMed]
- Tutar, Y. Pseudogenes. Comp. Funct. Genom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-Z.; Zheng, L.-L.; Qu, L.-H.; Ayala, F.J.; Lun, Z.-R. Pseudogenes are not pseudo any more. RNA Biol. 2012, 9, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Ophir, R.; Graur, D. Patterns and rates of indel evolution in processed pseudogenes from humans and murids. Gene 1997, 205, 191–202. [Google Scholar] [CrossRef]
- Mitchell, A.; Graur, D. Inferring the pattern of spontaneous mutation from the pattern of substitution in unitary pseudogenes of Mycobacterium leprae and a comparison of mutation patterns among distantly related organisms. J. Mol. Evol. 2005, 61, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schneeberger, K.; Clark, R.M.; Lanz, C.; Warthmann, N.; Weigel, D. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008, 18, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, O.; Aury, J.-M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Camiolo, S.; Porceddu, A. gff2sequence, a new user friendly tool for the generation of genomic sequences. BioData Min. 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Camiolo, S.; Porceddu, A. Identification of Pseudogenes in Brachipodium dystachion. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1667, pp. 1–15. [Google Scholar]
- Zheng, D.; Gerstein, M.B. A computational approach for identifying pseudogenes in the ENCODE regions. Genome Biol. 2006, 7 (Suppl. 1), S13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Carriero, N.; Zheng, D.; Karro, J.; Harrison, P.M.; Gerstein, M. PseudoPipe: An automated pseudogene identification pipeline. Bioinformatics 2006, 22, 1437–1439. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Khelifi, A.; Adel, K.; Duret, L.; Laurent, D.; Mouchiroud, D.; Dominique, M. HOPPSIGEN: A database of human and mouse processed pseudogenes. Nucleic Acids Res. 2005, 33, D59–D66. [Google Scholar] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 1993, 36, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Thibaud-Nissen, F.; Ouyang, S.; Buell, C.R. Identification and characterization of pseudogenes in the rice gene complement. BMC Genom. 2009, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Porceddu, A.; Camiolo, S. Spatial analyses of mono, di and trinucleotide trends in plant genes. PLoS ONE 2011, 6, e22855. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Keller, I.; Bensasson, D.; Nichols, R.A. Transition-transversion bias is not universal: A counter example from grasshopper pseudogenes. PLoS Genet. 2007, 3, e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.D.; Frankish, A.; Hunt, T.; Harrow, J.; Gerstein, M. Identification and analysis of unitary pseudogenes: Historic and contemporary gene losses in humans and other primates. Genome Biol. 2010, 11, R26. [Google Scholar] [CrossRef] [PubMed]
- Costantini, M.; Clay, O.; Auletta, F.; Bernardi, G. An isochore map of human chromosomes. Genome Res. 2006, 16, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.F.; D’Addabbo, P.; Alkan, C.; Bergamini, C.; Catacchio, C.R.; Anaclerio, F.; Chiatante, G.; Marra, A.; Giannuzzi, G.; Perniola, R.; et al. Inter-varietal structural variation in grapevine genomes. Plant. J. 2016, 88, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Mercenaro, L.; Nieddu, G.; Porceddu, A.; Pezzotti, M.; Camiolo, S. Sequence polymorphisms and structural variations among four grapevine (Vitis vinifera L.) cultivars representing Sardinian agriculture. Front. Plant. Sci. 2017, 8, 1279. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide | Total | Unchanged | Deletions | Substitutions a | ||||
|---|---|---|---|---|---|---|---|---|
| Type | Group | (n) | (n (%Total)) | (n (%Total)) | Total | Transitions (n (%Substitutions)) | Transversions (n (%Substitutions)) | Transitions: Transversions Ratio |
| A | All | 13,546 | 12,655 (93.42) | 198 (1.46) | 693 (5.12) | 421 (60.75) | 272 (39.25) | 1.55 |
| G | All | 11,928 | 10,476 (87.83) | 172 (1.44) | 1280 (10.73) | 994 (77.65) | 286 (22.34) | 3.48 |
| CpG | 429 | 343 (79.95) | 6 (1.39) | 80 (18.65) | 63 (78.75) | 17 (21.25) | 3.71 | |
| Non-CpG | 11,499 | 10,133 (88.12) | 166 (1.44) | 1200 (10.44) | 931 (77.58) | 269 (22.42) | 3.46 | |
| C | All | 8618 | 7516 (87.21) | 102 (1.18) | 1000 (11.60) | 786 (78.60) | 214 (21.40) | 3.67 |
| CpG | 429 | 336 (78.32) | 6 (1.39) | 87 (20.28) | 81 (93.10) | 6 (6.90) | 13.50 | |
| Non-CpG | 8189 | 7180 (87.68) | 96 (1.34) | 913 (11.15) | 705 (77.21) | 208 (22.78) | 3.39 | |
| T | All | 13,835 | 12,955 (93.64) | 158 (1.14) | 722 (5.22) | 446 (61.77) | 276 (38.23) | 1.62 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porceddu, A.; Camiolo, S. Patterns of Spontaneous Nucleotide Substitutions in Grape Processed Pseudogenes. Diversity 2017, 9, 45. https://doi.org/10.3390/d9040045
Porceddu A, Camiolo S. Patterns of Spontaneous Nucleotide Substitutions in Grape Processed Pseudogenes. Diversity. 2017; 9(4):45. https://doi.org/10.3390/d9040045
Chicago/Turabian StylePorceddu, Andrea, and Salvatore Camiolo. 2017. "Patterns of Spontaneous Nucleotide Substitutions in Grape Processed Pseudogenes" Diversity 9, no. 4: 45. https://doi.org/10.3390/d9040045
APA StylePorceddu, A., & Camiolo, S. (2017). Patterns of Spontaneous Nucleotide Substitutions in Grape Processed Pseudogenes. Diversity, 9(4), 45. https://doi.org/10.3390/d9040045