The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions



Abstract
:1. Introduction
2. Exploiting the Blood-Brain Barrier and the Immune System against Glioblastoma: The Wrong Notion of the Immune Desert in Glioblastoma
2.1. Factors Related to BBB
2.2. Factors Related to Glioblastoma Immunosuppressive Microenvironment
2.3. Factors Related to Tumor Heterogeneity and Genetic Signature of Glioblastoma Subtypes
2.4. Extrinsic Factors Related to the Microenvironment Modulating Effect of Previous Glioblastoma Treatments
3. Immunotherapy in the Treatment of Glioblastoma
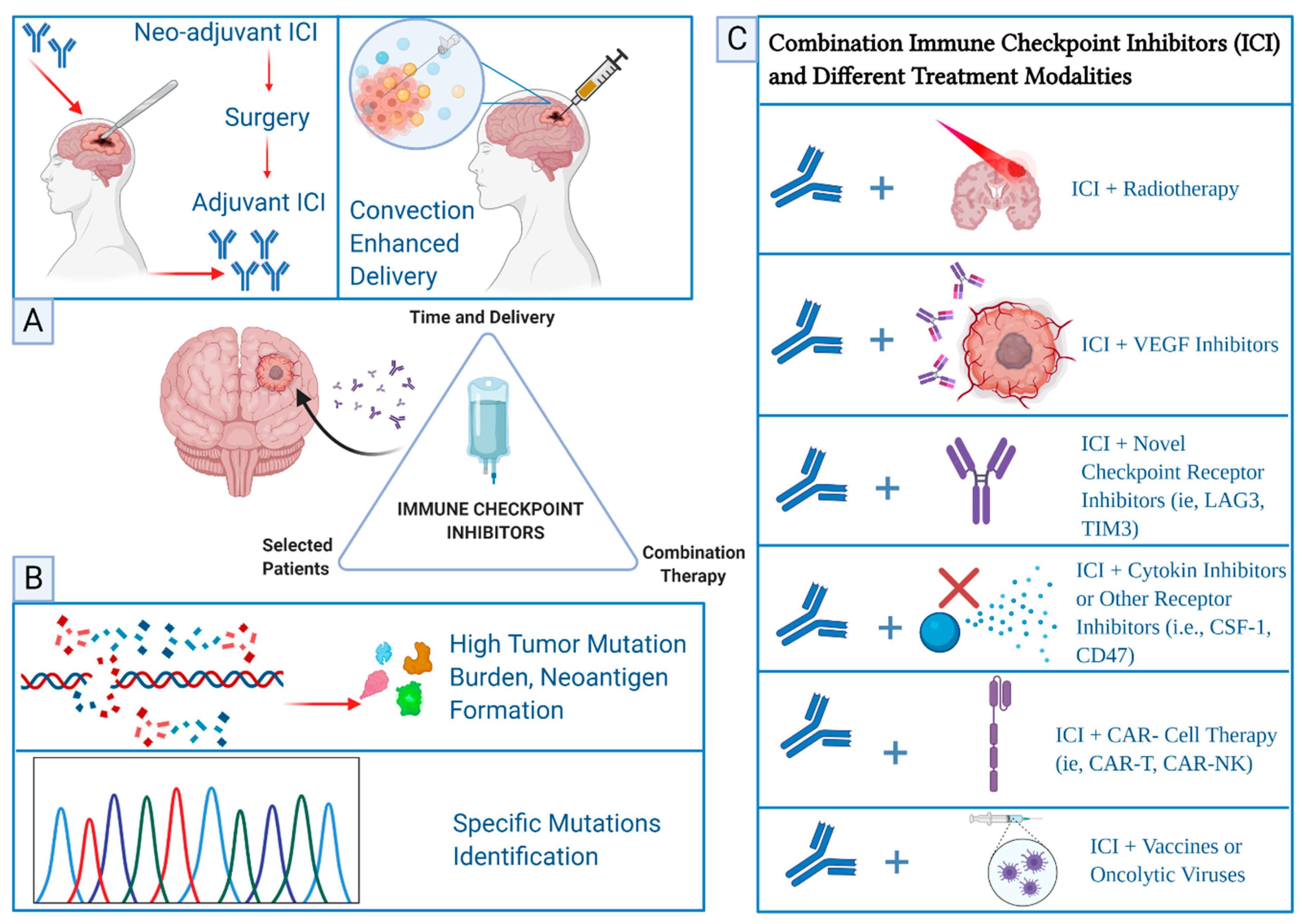
3.1. Immune Checkpoint Inhibitors in Glioblastoma
3.2. Adoptive Cell Therapy in Glioblastoma
3.3. Therapeutic Vaccine Therapy in Glioblastoma
3.4. Oncolytic Virus Therapy
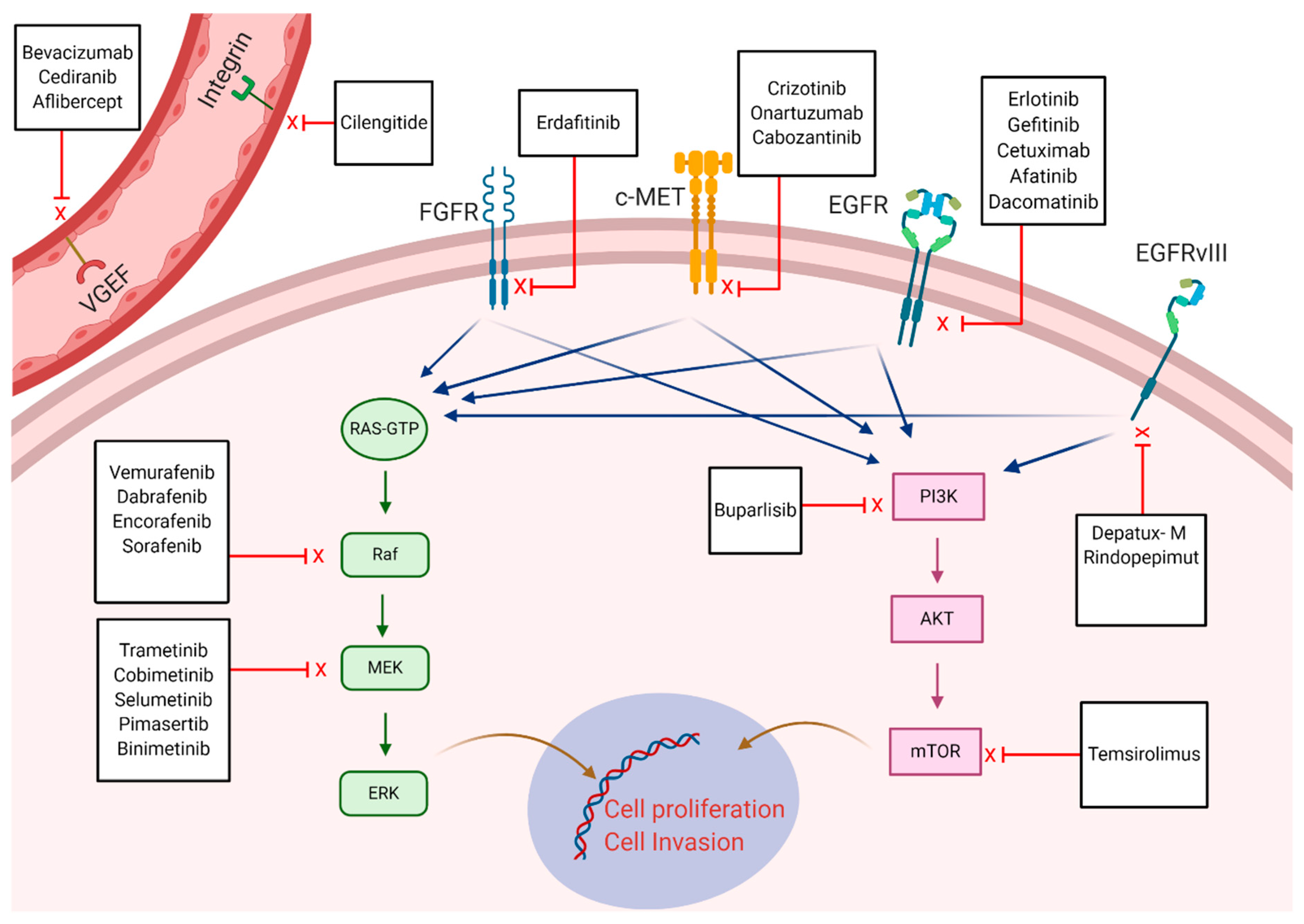
4. Targeted Therapies in Glioblastoma
5. Future Perspective of Glioblastoma Research
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109, Erratum in 2007, 114, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro-Oncology 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. cbtrus statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20 (Suppl. 4), iv1–iv86. [Google Scholar] [CrossRef] [Green Version]
- Sminia, P.; Westerman, B.A. Blood-brain barrier crossing and breakthroughs in glioblastoma therapy. Br. J. Clin. Pharmacol. 2016, 81, 1018–1020. [Google Scholar] [CrossRef] [Green Version]
- Parker, N.R.; Ekhong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular heterogeneity in glioblastoma: Potential clinical implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Bent, M.J.V.D.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs. maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316, Erratum in 2018, 319, 1824. [Google Scholar] [CrossRef] [Green Version]
- Fabian, D.; Eibl, M.D.P.G.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.K.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the addition of Tumor-Treating Fields (TTF): A review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and bevacizumab in progressive glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, J.-F.; Denizot, F.; Luciani, M.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nat. Cell Biol. 1987, 328, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Ardolino, L.; Joshua, A. Immune checkpoint inhibitors in malignancy. Aust. Prescr. 2019, 42, 62–67. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; Von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Galsky, M.D.; E Rosenberg, J.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76, Erratum in 2017, 390, 848. [Google Scholar] [CrossRef] [Green Version]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.; Hahn, N.M.; De Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Seock-Ah IMpassion130 Trial Investigators; Wright, G.S.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 inhibitors: From bench to bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gautier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; Steinmetz, R.N.; Riddell, S.R.; et al. High rate of durable complete remission in follicular lymphoma after CD19 CAR-T cell immunotherapy. Blood 2019, 134, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Hambardzumyan, D. Immune microenvironment in glioblastoma subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the brain: Approaches to enhance brain drug delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar] [CrossRef]
- Dhermain, F.G.; Hau, P.; Lanfermann, H.; Jacobs, A.H.; van den Bent, M.J. Advanced MRI and PET imaging for assessment of treatment response in patients with gliomas. Lancet Neurol. 2010, 9, 906–920. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellisdavies, G.C.R.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [Green Version]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [Green Version]
- van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Platt, M.P.; Agalliu, D.; Cutforth, T. Hello from the other side: How autoantibodies circumvent the blood-brain barrier in autoimmune encephalitis. Front. Immunol. 2017, 8, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majerova, P.; Michalicova, A.; Cente, M.; Hanes, J.; Vegh, J.; Kittel, Á.; Kosikova, N.; Cigankova, V.; Mihaljevic, S.; Jadhav, S.; et al. Trafficking of immune cells across the blood-brain barrier is modulated by neurofibrillary pathology in tauopathies. PLoS ONE 2019, 14, e0217216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, L.; Coll, C.; Erthal, L.C.S.; de la Torre, C.; Serrano, D.R.; Martínez-Máñez, R.; Santos-Martínez, M.J.O.; Ruiz-Hernández, E. Drug delivery nanosystems for the localized treatment of glioblastoma multiforme. Materials 2018, 11, 779. [Google Scholar] [CrossRef] [Green Version]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850, Erratum in 2020, 11, 701. [Google Scholar] [CrossRef]
- Erel-Akbaba, G.; Carvalho, L.A.; Tian, T.; Zinter, M.; Akbaba, H.; Obeid, P.J.; Chiocca, E.A.; Weissleder, R.; Kantarci, A.G.; Tannous, B.A. Radiation-Induced Targeted Nanoparticle-Based Gene Delivery for Brain Tumor Therapy. ACS Nano 2019, 13, 4028–4040. [Google Scholar] [CrossRef]
- Shi, M.; Sanche, L. Convection-enhanced delivery in malignant gliomas: A review of toxicity and efficacy. J. Oncol. 2019, 2019, 9342796. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Ii, J.E.H.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.; Nair, S.; et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Toki, M.I.; Kumar, D.; Ahmed, F.S.; Rimm, D.L.; Xu, M.L. Benign lymph node microenvironment is associated with response to immunotherapy. Precis. Clin. Med. 2020, 3, 44–53. [Google Scholar] [CrossRef]
- Kipnis, J. Multifaceted interactions between adaptive immunity and the central nervous system. Science 2016, 353, 766–771. [Google Scholar] [CrossRef] [Green Version]
- Gomez, G.G.; Kruse, C.A. Mechanisms of malignant glioma immune resistance and sources of immunosuppression. Gene Ther. Mol. Biol. 2006, 10, 133–146. [Google Scholar]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341, Erratum in 2016, 533, 278. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; DeChant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468, Erratum in 2019, 25, 529. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Wiendl, H.; Mitsdoerffer, M.; Hofmeister, V.; Wischhusen, J.; Bornemann, A.; Meyermann, R.; Weiss, E.H.; Melms, A.; Weller, M. A functional role of HLA-G expression in human gliomas: An alternative strategy of immune escape. J. Immunol. 2002, 168, 4772–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, D.; Balça-Silva, J.; Da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/astrocytes-glioblastoma crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, S.; Strommer, K.; Frei, K.; Siepl, C.; De Tribolet, N.; Heid, I.; Fontana, A. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J. Immunol. 1989, 143, 3222–3229. [Google Scholar] [PubMed]
- Huettner, C.; Czub, S.; Kerkau, S.; Roggendorf, W.; Tonn, J.-C. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer. Res. 1997, 17, 3217–3224. [Google Scholar] [PubMed]
- Schartner, J.M.; Hagar, A.R.; Van Handel, M.; Zhang, L.; Nadkarni, N.; Badie, B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 2005, 51, 279–285. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, T.; Li, G.; Nagpal, S. History and current state of immunotherapy in glioma and brain metastasis. Ther. Adv. Med. Oncol. 2017, 9, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wöhrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Garber, S.T.; Hashimoto, Y.; Weathers, S.-P.; Xiu, J.; Gatalica, Z.; Verhaak, R.G.W.; Zhou, S.; Fuller, G.N.; Khasraw, M.; De Groot, J.; et al. Immune checkpoint blockade as a potential therapeutic target: Surveying CNS malignancies. Neuro-Oncology 2016, 18, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301, Erratum in 2015, 21, 662. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Li, G.; Wang, Z.; Zhang, C.; Liu, X.; Cai, J.; Wang, Z.; Hu, H.; Wu, F.; Bao, Z.; Liu, Y.; et al. Molecular and clinical characterization of TIM-3 in glioma through 1,024 samples. OncoImmunology 2017, 6, e1328339. [Google Scholar] [CrossRef]
- Chang, N.; Ahn, S.H.; Kong, D.-S.; Lee, H.W.; Nam, D.-H. The role of STAT3 in glioblastoma progression through dual influences on tumor cells and the immune microenvironment. Mol. Cell. Endocrinol. 2017, 451, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Didenko, V.V.; Ngo, H.N.; Minchew, C.; Baskin, D.S. Apoptosis of T lymphocytes invading glioblastomas multiforme: A possible tumor defense mechanism. J. Neurosurg. 2002, 96, 580–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068, Erratum in 2013, 494, 506. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6, Erratum in 2018, 33, 152. [Google Scholar] [CrossRef] [Green Version]
- Herting, C.J.; Chen, Z.; Pitter, K.L.; Szulzewsky, F.; Kaffes, I.; Kaluzova, M.; Park, J.C.; Cimino, P.J.; Brennan, C.; Wang, B.; et al. Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia 2017, 65, 1914–1926. [Google Scholar] [CrossRef]
- Cantanhede, I.; Oliveira, J.R.M. PDGF family expression in glioblastoma multiforme: Data compilation from ivy glioblastoma atlas project database. Sci. Rep. 2017, 7, 15271. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [Green Version]
- Engler, J.R.; Robinson, A.E.; Smirnov, I.; Hodgson, J.G.; Berger, M.S.; Gupta, N.; James, C.D.; Molinaro, A.; Phillips, J.J. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS ONE 2012, 7, e43339. [Google Scholar] [CrossRef] [Green Version]
- Luoto, S.; Hermelo, I.; Vuorinen, E.M.; Hannus, P.; Kesseli, J.; Nykter, M.; Granberg, K.J. Computational characterization of suppressive immune microenvironments in glioblastoma. Cancer Res. 2018, 78, 5574–5585. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wöhrer, A.; Marosi, C.; Hegi, M.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro-Oncology 2017, 19, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; A Crane, C.; Parney, I.F.; Barry, J.J.; Cachola, K.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.D.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Sa, J.K.; Choi, S.W.; Zhao, J.; Lee, Y.; Zhang, J.; Kong, D.; Choi, J.W.; Seol, H.J.; Lee, J.; Iavarone, A.; et al. Hypermutagenesis in untreated adult gliomas due to inherited mismatch mutations. Int. J. Cancer 2019, 144, 3023–3030. [Google Scholar] [CrossRef]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; De Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro-Oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, I.-H.; Cho, H.J.; Park, C.-K.; Jung, Y.-S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.-S.; et al. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Mahlokozera, T.; Vellimana, A.K.; Li, T.; Mao, D.D.; Zohny, Z.S.; Kim, D.H.; Tran, D.D.; Marcus, D.S.; Fouke, S.J.; Campian, J.L.; et al. Biological and therapeutic implications of multisector sequencing in newly diagnosed glioblastoma. Neuro-Oncology 2018, 20, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of hypermutated glioblastoma: A patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016, 6, 1230–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; De Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karachi, A.; Dastmalchi, F.; A Mitchell, D.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncology 2018, 20, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Chitadze, G.; Flüh, C.; Quabius, E.S.; Freitag-Wolf, S.; Peters, C.; Lettau, M.; Bhat, J.; Wesch, D.; Oberg, H.-H.; Luecke, S.; et al. In-depth immunophenotyping of patients with glioblastoma multiforme: Impact of steroid treatment. OncoImmunology 2017, 6, e1358839. [Google Scholar] [CrossRef]
- Grossman, S.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S.; NABTT CNS Consortium. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef] [Green Version]
- Petrelli, F.; Signorelli, D.; Ghidini, M.; Ghidini, A.; Pizzutilo, E.G.; Ruggieri, L.; Cabiddu, M.; Borgonovo, K.; Dognini, G.; Brighenti, M.; et al. Association of steroids use with survival in patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis. Cancers 2020, 12, 546. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Tanaka, T.; Ohara, K.; Miyake, K.; Morimoto, Y.; Yamamoto, Y.; Kanai, R.; Akasaki, Y.; Murayama, Y.; Tamiya, T.; et al. Persistent restoration to the immunosupportive tumor microenvironment in glioblastoma by bevacizumab. Cancer Sci. 2019, 110, 499–508. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Morimoto, Y.; Kuranari, Y.; Yamamoto, Y.; Takei, J.; Murayama, Y.; Yoshida, K.; Sasaki, H. Alterations of the tumor microenvironment in glioblastoma following radiation and temozolomide with or without bevacizumab. Ann. Transl. Med. 2020, 8, 297. [Google Scholar] [CrossRef]
- Zhang, C.; Fan, Y.; Che, X.; Zhang, M.; Li, Z.; Li, C.; Wang, S.; Wen, T.; Hou, K.; Shao, X.; et al. Anti-PD-1 Therapy Response Predicted by the Combination of Exosomal PD-L1 and CD28. Front. Oncol. 2020, 10, 760. [Google Scholar] [CrossRef]
- Zappasodi, R.; Wolchok, J.; Merghoub, T. Strategies for predicting response to checkpoint inhibitors. Curr. Hematol. Malign Rep. 2018, 13, 383–395, Erratum in 2019, 14, 62. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.-Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2016, 18, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, M.; Pistillo, M.P.; Carosio, R.; Morabito, A.; Banelli, B. Immune checkpoints and innovative therapies in glioblastoma. Front. Oncol. 2018, 8, 464. [Google Scholar] [CrossRef] [PubMed]
- Hanihara, M.; Kawataki, T.; Oh-Oka, K.; Mitsuka, K.; Nakao, A.; Kinouchi, H. Synergistic antitumor effect with indoleamine 2, 3-dioxygenase inhibition and temozolomide in a murine glioma model. J. Neurosurg. 2016, 124, 1594–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020, 6, 1003. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb. Phase-3 CheckMate-498 Study Did Not Meet Primary Endpoint of Overall Survival with Opdivo nivolumab Plus Radiation in Patients with Newly Diagnosed MGMT Unmethylated Glioblastoma Multiforme. 9 May 2019. Available online: https://news.bms.com/news/corporate-financial/2019/Bristol-Myers-Squibb-Announces-Phase-3-CheckMate--498-Study-Did-Not-Meet-Primary-Endpoint-of-Overall-Survival-with-Opdivo-nivolumab-Plus-Radiation-in-Patients-with-Newly-Diagnosed-MGMT-Unmethylated-Glioblastoma-Multiforme/default.aspx (accessed on 1 October 2020).
- Reardon, D.A.; Nayak, L.; Peters, K.B.; Clarke, J.L.; Jordan, J.T.; De Groot, J.F.; Nghiemphu, P.L.; Kaley, T.J.; Colman, H.; Gaffey, S.C.; et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J. Clin. Oncol. 2018, 36 (Suppl. 15), 2006. [Google Scholar] [CrossRef]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9 (Suppl. 1), S3. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125554lbl.pdf (accessed on 13 November 2020).
- Available online: https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf (accessed on 10 September 2020).
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367, eaax0182. [Google Scholar] [CrossRef]
- Versluis, J.M.; Long, G.V.; Blank, C.U. Learning from clinical trials of neoadjuvant checkpoint blockade. Nat. Med. 2020, 26, 475–484. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.; Hugo, W.; Lee, A.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; de Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Yang, Z.; Wei, S.; Deng, Y.; Wang, Z.; Liu, L. Clinical significance of tumour mutation burden in immunotherapy across multiple cancer types: An individual meta-analysis. Jpn. J. Clin. Oncol. 2020, 50, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.; Murphy, A.; Le, D.T.; Diaz, L.A., Jr. Mismatch repair deficiency and response to immune checkpoint blockade. Oncologist 2016, 21, 1200–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya-Matsuoka, C.; Metrus, N.R.; Shaw, K.R.; Penas-Prado, M.; Weathers, S.-P.; Loghin, M.E.; Alfaro-Munoz, K.; Yuan, Y.; O’Brien, B.J.; Harrison, R.A.; et al. The natural course of hypermutator gliomas. J. Clin. Oncol. 2018, 36 (Suppl. 15), 2014. [Google Scholar]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination therapy with Anti-PD-1, Anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Weenink, B.; French, P.J.; Smitt, P.S.; Debets, R.; Geurts, M. Immunotherapy in glioblastoma: Current shortcomings and future perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef] [Green Version]
- Gholamin, S.; Mitra, S.S.; Feroze, A.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Osuchowska, I.; Maciejewski, R.; Roliński, J.; Grajkowska, W.A.; Grochowski, C. Micro RNA Molecules as Modulators of Treatment Resistance, Immune Checkpoints Controllers and Sensitive Biomarkers in Glioblastoma Multiforme. Int. J. Mol. Sci. 2020, 21, 1507. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Riddell, S.R.; Schumacher, T.N. Adoptive cellular therapy: A race to the finish line. Sci. Transl. Med. 2015, 7, 280ps7. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Dietrich, J.; Martinez-Lage, M.; Leick, M.; Choi, B.D.; DeFilipp, Z.; Chen, Y.-B.; Abramson, J.; Crombie, J.; Armand, P.; et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood 2019, 134, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.R.; Wright, C.; et al. Bioactivity and safety of IL13Rα2-Redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor T-Cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK cells for the treatment of glioblastoma: Turning innate effectors into precision tools for cancer immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Schlom, J.; Hodge, J.W.; Palena, C.; Tsang, K.-Y.; Jochems, C.; Greiner, J.W.; Farsaci, B.; Madan, R.A.; Heery, C.R.; Gulley, J.L. Therapeutic cancer vaccines. Adv. Cancer Res. 2014, 121, 67–124. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477, Erratum in 2014, 157, 753. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncology 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; A Goldlust, S.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for patients with relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a double-blind randomized phase II trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Platten, M.; Schilling, D.; Bunse, L.; Wick, A.; Bunse, T.; Riehl, D.; Green, E.; Sanghvi, K.; Karapanagiotou-Schenkel, I.; Harting, I.; et al. ATIM-33. NOA-16: A first-in-man multicenter phase i clinical trial of the german neurooncology working group evaluating a mutation-specific peptide vaccine targeting idh1r132h in patients with newly diagnosed malignant astrocytomas. Neuro-Oncology 2018, 20 (Suppl. 6), vi8–vi9. [Google Scholar] [CrossRef] [Green Version]
- Ampie, L.; Choy, W.; Lamano, J.B.; Fakurnejad, S.; Bloch, O.; Parsa, A.T. Heat shock protein vaccines against glioblastoma: From bench to bedside. J. Neurooncol. 2015, 123, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Crane, C.A.; Han, S.J.; Ahn, B.; Oehlke, J.; Kivett, V.; Fedoroff, A.; Butowski, N.; Chang, S.M.; Clarke, J.; Berger, M.S.; et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin. Cancer Res. 2013, 19, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-Oncology 2014, 16, 274–279. [Google Scholar] [CrossRef]
- Ji, N.; Zhang, Y.; Liu, Y.; Xie, J.; Wang, Y.; Hao, S.; Gao, Z. Heat shock protein peptide complex-96 vaccination for newly diagnosed glioblastoma: A phase I, single-arm trial. JCI Insight 2018, 3, e99145. [Google Scholar] [CrossRef] [Green Version]
- Bregy, A.; Wong, T.M.; Shah, A.H.; Goldberg, J.M.; Komotar, R.J. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat. Rev. 2013, 39, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142, Erratum in 2018, 16, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef]
- Johanns, T.M.; Miller, C.A.; Liu, C.J.; Perrin, R.J.; Bender, D.; Kobayashi, D.K.; Campian, J.L.; Chicoine, M.R.; Dacey, R.G.; Huang, J.; et al. Detection of neoantigen-specific T cells following a personalized vaccine in a patient with glioblastoma. OncoImmunology 2019, 8, e1561106. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245, Erratum in 2019, 566, E13. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.; Essand, M. Virus-based immunotherapy of glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Iorgulescu, J.B.; Reardon, D.A.; Chiocca, E.A.; Wu, C.J. Immunotherapy for glioblastoma: Going viral. Nat. Med. 2018, 24, 1094–1096. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Paget, M.; Tshilenge, K.-T.; Saha, D.; Antoszczyk, S.; Baars, A.; Frost, T.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Restriction of replication of oncolytic herpes simplex virus with a deletion of γ34.5 in glioblastoma stem-like cells. J. Virol. 2018, 92, e00246:1–e00246:18. [Google Scholar] [CrossRef] [Green Version]
- Ji, N.; Weng, D.; Liu, C.; Gu, Z.; Chen, S.; Guo, Y.; Fan, Z.; Wang, X.; Chen, J.; Zhao, Y.; et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget 2016, 7, 4369–4378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloughesy, T.F.; Landolfi, J.; Hogan, D.J.; Bloomfield, S.; Carter, B.; Chen, C.C.; Elder, J.B.; Kalkanis, S.N.; Kesari, S.; Lai, A.; et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 2016, 8, 341ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.A.; Sabel, M.C.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.M.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.-F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef] [Green Version]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.A.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-Oncology 2010, 12, 95–103. [Google Scholar] [CrossRef]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-Oncology 2015, 17, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Sepúlveda-Sánchez, J.M.; Vaz, M. Ángeles; Balañá, C.; Gil-Gil, M.; Reynés, G.; Gallego, Óscar; Martínez-García, M.; Vicente, E.; Quindós, M.; Luque, R.; et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro-Oncology 2017, 19, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Lassman, A.B.; Bent, M.J.V.D.; Gan, H.K.; A Reardon, D.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro-Oncology 2019, 21, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bent, M.J.V.D.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2020, 22, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, Y.; Kim, S.-I.; Park, C.-K.; Paek, S.H.; Lee, S.-T.; Park, S.-H. C-MET overexpression and amplification in gliomas. Int. J. Clin. Exp. Pathol. 2015, 8, 14932–14938. [Google Scholar]
- Wen, P.Y.; Schiff, D.; Cloughesy, T.F.; Raizer, J.J.; Laterra, J.; Smitt, M.; Wolf, M.; Oliner, K.S.; Anderson, A.; Zhu, M.; et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro-Oncology 2011, 13, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, double-blind, placebo-controlled, multicenter phase II study of onartuzumab plus bevacizumab versus placebo plus bevacizumab in patients with recurrent glioblastoma: Efficacy, safety, and hepatocyte growth factor and O6-Methylguanine-DNA methyltransferase biomarker analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: An open-label, multicenter, multi-arm, phase II trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-Oncology 2015, 17, 1261–1269. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; Bent, M.J.V.D.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.-F.; Wick, A.; et al. Phase II study of radiotherapy and temsirolimus versus radiochemotherapy with temozolomide in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [Green Version]
- Behling, F.; Barrantes-Freer, A.; Skardelly, M.; Nieser, M.; Christians, A.; Stockhammer, F.; Rohde, V.; Tatagiba, M.; Hartmann, C.; Stadelmann, C.; et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn. Pathol. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, A.; Chavez, L.; Sharma, T.; Ryzhova, M.; Schrimpf, D.; Stichel, D.; Capper, D.; Sturm, D.; Kool, M.; Habel, A.; et al. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol. 2018, 28, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanns, T.M.; Ferguson, C.J.; Grierson, P.M.; Dahiya, S.; Ansstas, G. Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with braf-mutated high-grade glioma. J. Natl. Compr. Cancer Netw. 2018, 16, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnirsky, M.; Feun, L.G.; Gultekin, S.H.; De La Fuente, M.I. Prolonged complete response with combined dabrafenib and trametinib after BRAF inhibitor failure in BRAF-mutant glioblastoma. JCO Precis Oncol. 2020, 4, PO.19.00272. [Google Scholar] [CrossRef]
- Kaley, T.; Touat, M.; Subbiah, V.; Hollebecque, A.; Rodon, J.; Lockhart, A.C.; Keedy, V.; Bielle, F.; Hofheinz, R.-D.; Joly, F.; et al. BRAF Inhibition in BRAFV600-mutant gliomas: Results from the VE-BASKET study. J. Clin. Oncol. 2018, 36, 3477–3484. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; Bent, M.V.D.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Li, A.; Yi, M.; Qin, S.; Chu, Q.; Luo, S.; Wu, K. Prospects for combining immune checkpoint blockade with PARP inhibition. J. Hematol. Oncol. 2019, 12, 98. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.-F.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of temozolomide sensitivity by PARP inhibitors in mismatch repair deficient glioblastoma is independent of base excision repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro-Oncology 2020, noaa104, Epub ahead of print. [Google Scholar] [CrossRef]
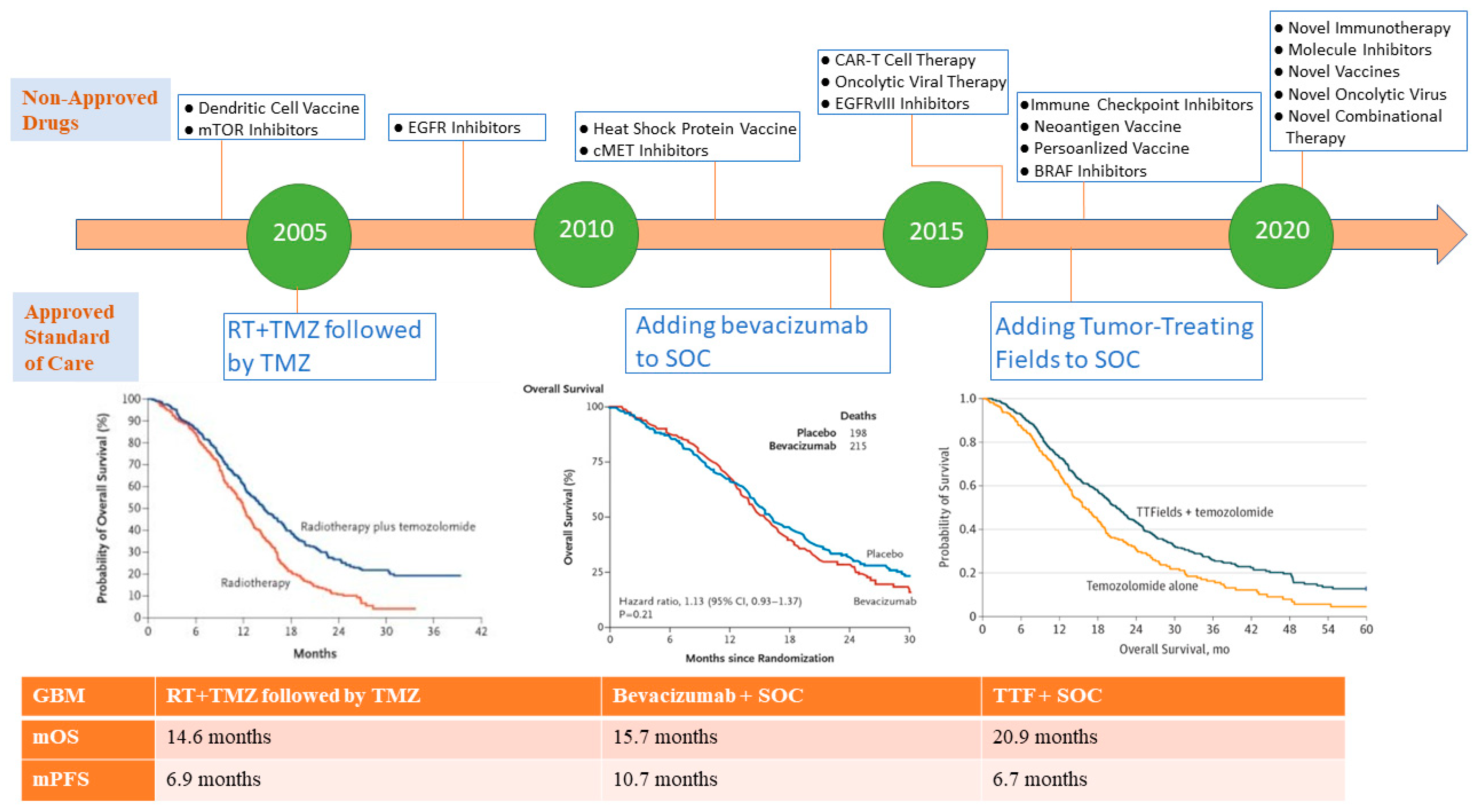
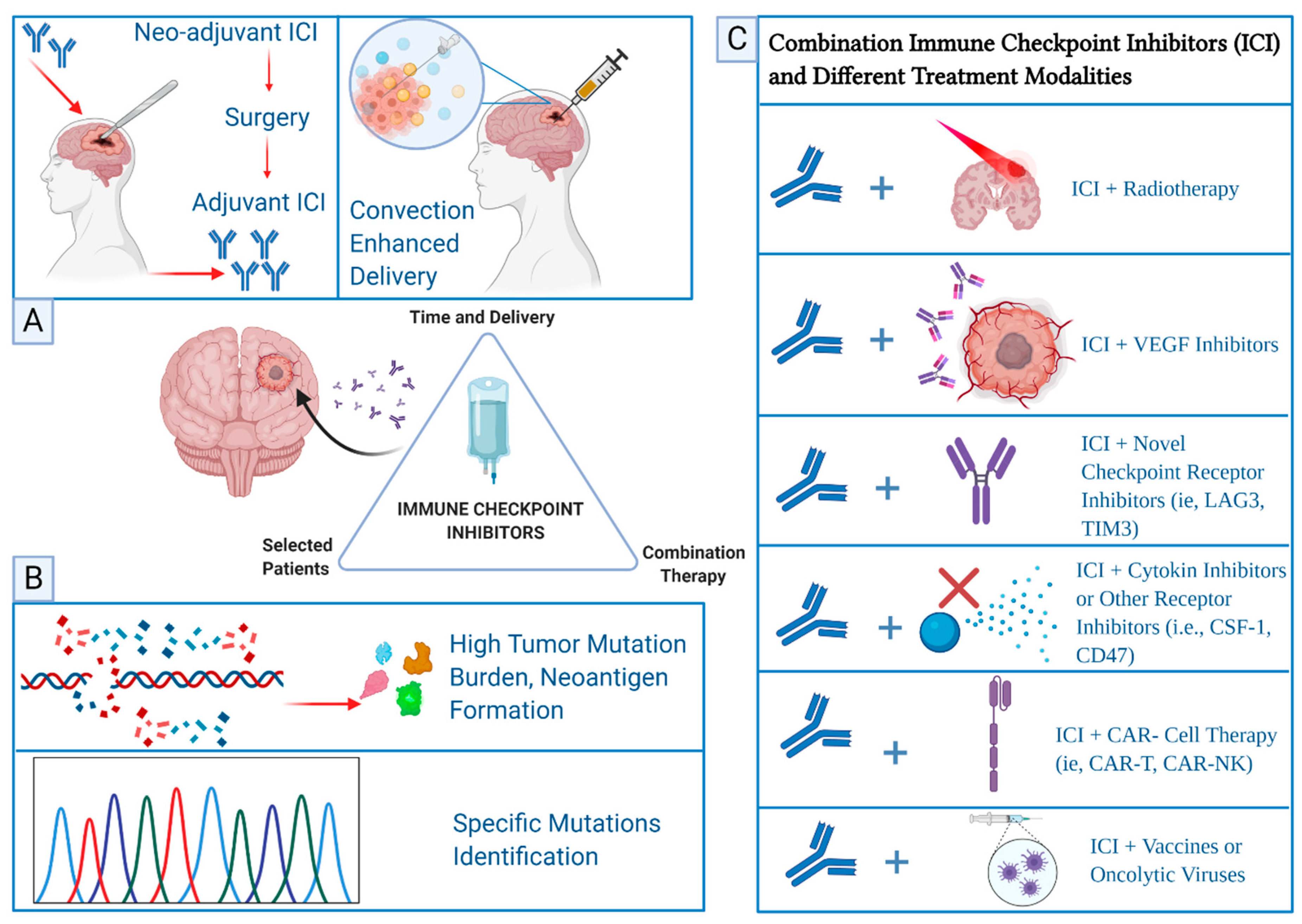
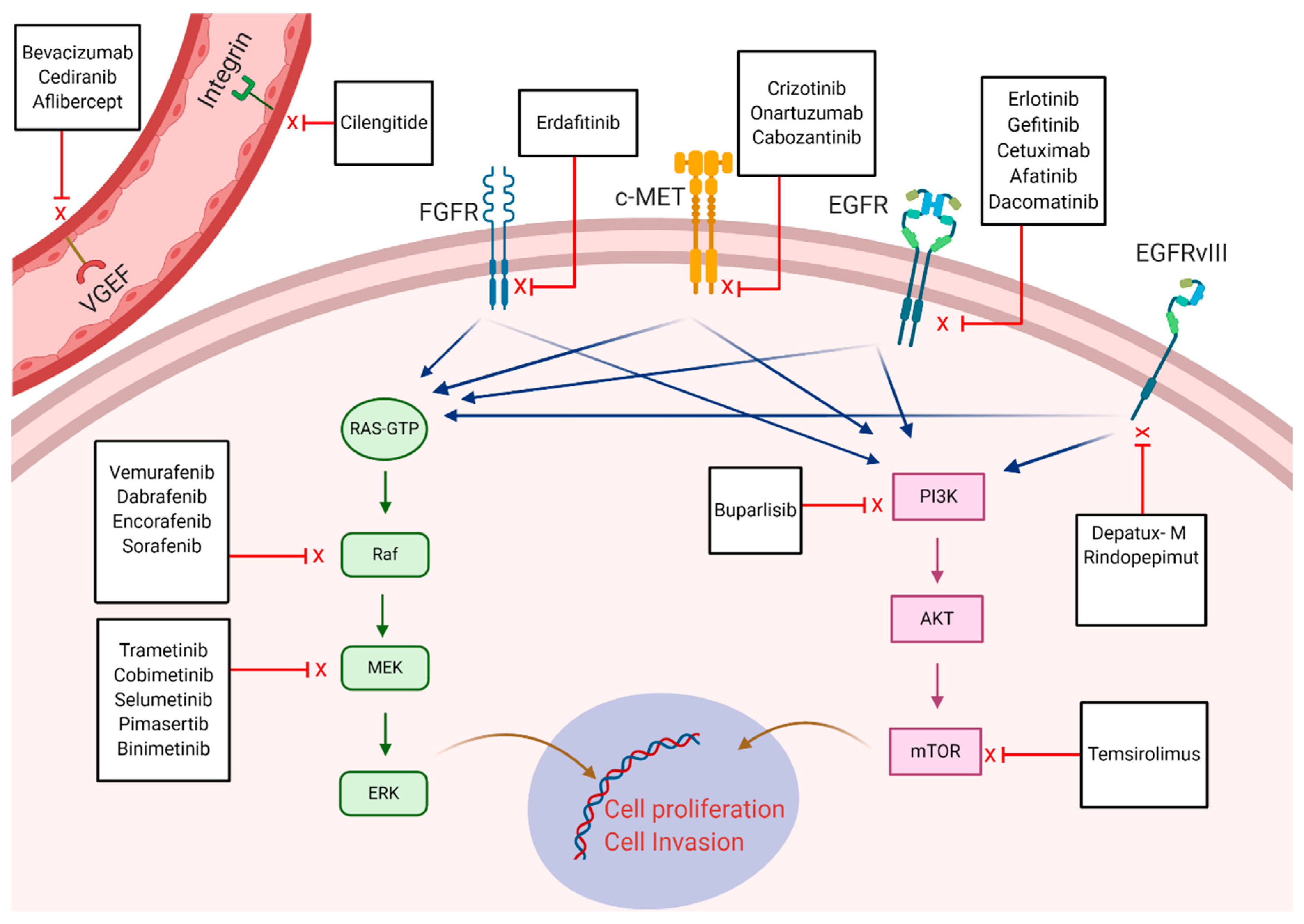

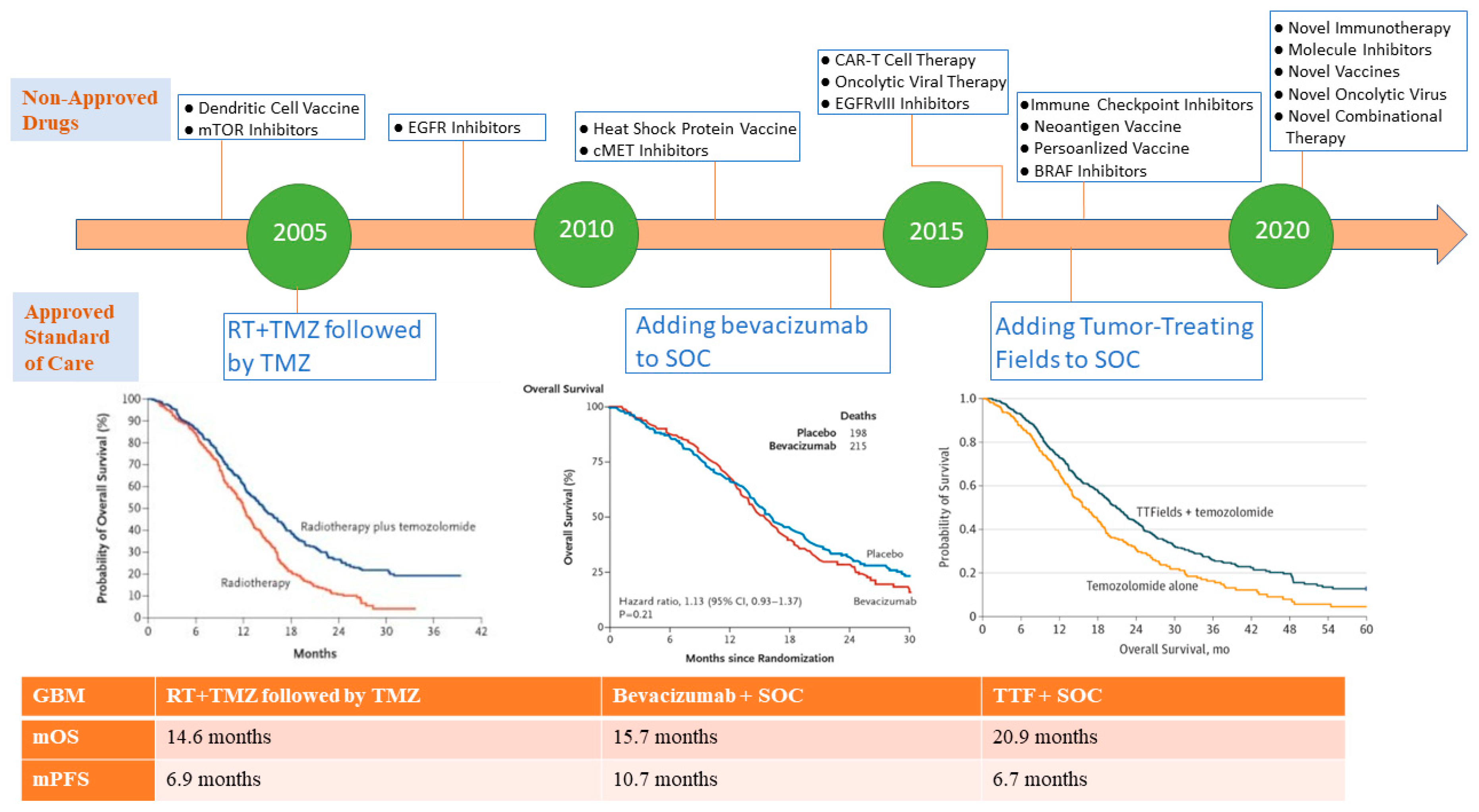{kind=link}
{kind=link}
{kind=link}
| Factors Related to Glioblastoma | Possible Solutions | Reference |
|---|---|---|
| Restricted drug delivery due to blood brain barrier | Convection enhanced delivery (CED) and nano particles. Disruption of blood brain barrier with radiation therapy combined with immune checkpoint inhibitors Adoptive cell therapy | [44,45] [121] [127,128] |
| Effector T-cell suppression and peripheral T-cell entrapment | Use of immune checkpoint inhibitors, therapeutic vaccines or adoptive cell transfer therapy | [52,53,54,55] |
| Increased expression of checkpoint receptors | Use of combination checkpoint inhibitors targeting several checkpoint receptors | [64,65,66,67,68,69,70,121] |
| Suppression of natural killer cells | Use of checkpoint inhibitors Natural killer cell adoptive transfer | [131] |
| Abundance of tumor associated macrophages (TAM) (protumorigenic phenotype) | Boosting host immunity through exploiting plasticity in TAM to express inflammatory phenotype M1 | [73] |
| Increased secretion of TGF-β | Use of checkpoint inhibitors with TGF-β inhibitors | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khaddour, K.; Johanns, T.M.; Ansstas, G. The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions. Pharmaceuticals 2020, 13, 389. https://doi.org/10.3390/ph13110389
Khaddour K, Johanns TM, Ansstas G. The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions. Pharmaceuticals. 2020; 13(11):389. https://doi.org/10.3390/ph13110389
Chicago/Turabian StyleKhaddour, Karam, Tanner M. Johanns, and George Ansstas. 2020. "The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions" Pharmaceuticals 13, no. 11: 389. https://doi.org/10.3390/ph13110389
APA StyleKhaddour, K., Johanns, T. M., & Ansstas, G. (2020). The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions. Pharmaceuticals, 13(11), 389. https://doi.org/10.3390/ph13110389