Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitylation as a Novel Pharmaceutical Target for Cystic Fibrosis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
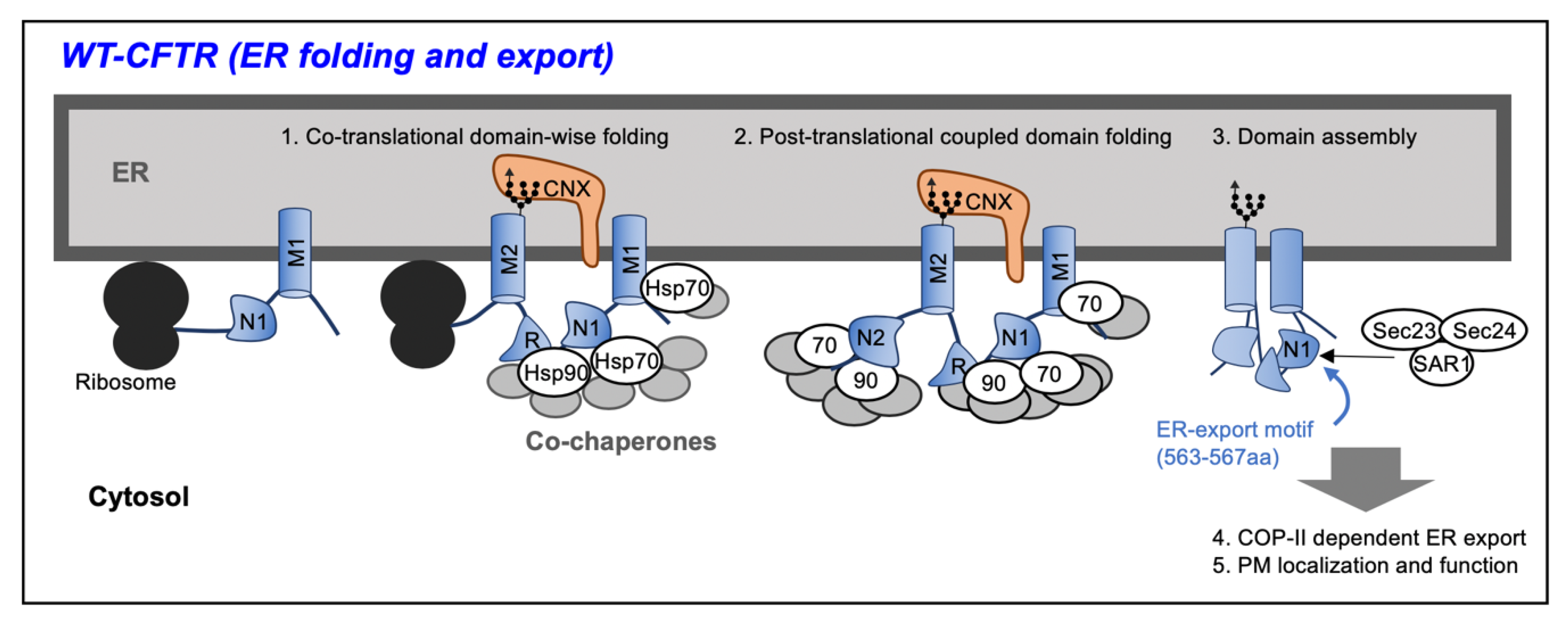
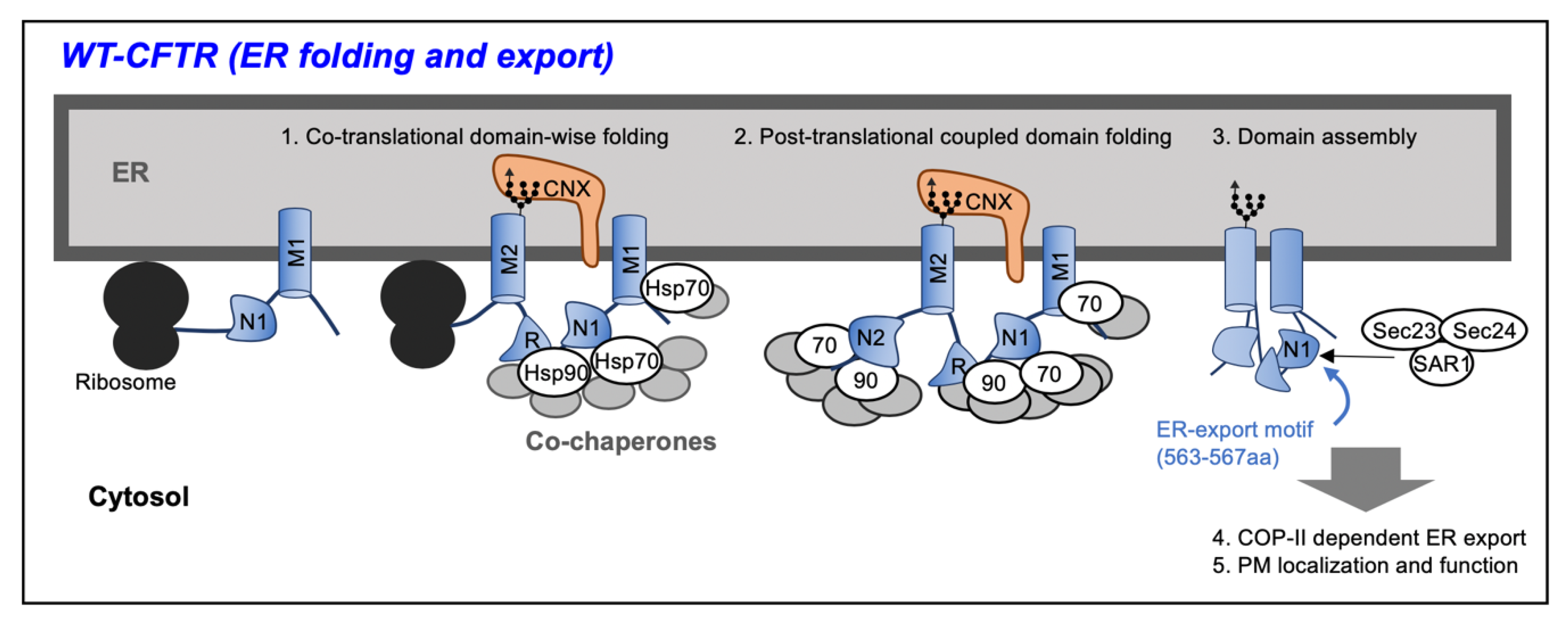
2. Biosynthesis of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Protein
3. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Endoplasmic Reticulum Quality Control (ERQC)
4. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Plasma Membrane Quality Control (PMQC)
5. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators
6. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitylation as a Cystic Fibrosis (CF) Drugs Target
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miquéias, L.-P. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.L.; Omura, S.; Kopito, R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995, 83, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Barrière, H.; Bagdány, M.; Rabeh, W.M.; Du, K.; Höhfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Lukacs, G.L. Fixing cystic fibrosis by correcting CFTR domain assembly. J. Cell Biol. 2012, 199, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Lukacs, G.L. Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell 2009, 20, 1903–1915. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Sharma, M.; Lukacs, G.L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. [Google Scholar] [CrossRef]
- He, L.; Aleksandrov, A.A.; Serohijos, A.W.; Hegedus, T.; Aleksandrov, L.A.; Cui, L.; Dokholyan, N.V.; Riordan, J.R. Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J. Biol. Chem. 2008, 283, 26383–26390. [Google Scholar] [CrossRef] [Green Version]
- Pind, S.; Riordan, J.R.; Williams, D.B. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1994, 269, 12784–12788. [Google Scholar]
- Okiyoneda, T.; Harada, K.; Takeya, M.; Yamahira, K.; Wada, I.; Shuto, T.; Suico, M.A.; Hashimoto, Y.; Kai, H. Delta F508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol. Biol. Cell 2004, 15, 563–574. [Google Scholar] [CrossRef]
- Loo, M.A.; Jensen, T.J.; Cui, L.; Hou, Y.; Chang, X.B.; Riordan, J.R. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998, 17, 6879–6887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. [Google Scholar] [CrossRef]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Qu, B.H.; Thomas, P.J. Alteration of the cystic fibrosis transmembrane conductance regulator folding pathway. J. Biol. Chem. 1996, 271, 7261–7264. [Google Scholar] [CrossRef] [Green Version]
- Rabeh, W.M.; Bossard, F.; Xu, H.; Okiyoneda, T.; Bagdany, M.; Mulvihill, C.M.; Du, K.; di Bernardo, S.; Liu, Y.; Konermann, L.; et al. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 2012, 148, 150–163. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Yoon, J.S.; Shishido, H.; Yang, Z.; Rooney, L.A.; Barral, J.M.; Skach, W.R. Protein folding. Translational tuning optimizes nascent protein folding in cells. Science 2015, 348, 444–448. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Oliver, K.; Apaja, P.M.; Perdomo, D.; Bidaud-Meynard, A.; Lin, S.T.; Guo, J.; Icyuz, M.; Sorscher, E.J.; Hartman, J.L.; et al. Ribosomal Stalk Protein Silencing Partially Corrects the ΔF508-CFTR Functional Expression Defect. PLoS Biol. 2016, 14, e1002462. [Google Scholar] [CrossRef]
- Wang, X.; Matteson, J.; An, Y.; Moyer, B.; Yoo, J.S.; Bannykh, S.; Wilson, I.A.; Riordan, J.R.; Balch, W.E. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J. Cell Biol. 2004, 167, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.B.; Cui, L.; Hou, Y.X.; Jensen, T.J.; Aleksandrov, A.A.; Mengos, A.; Riordan, J.R. Removal of multiple arginine-framed trafficking signals overcomes misprocessing of delta F508 CFTR present in most patients with cystic fibrosis. Mol. Cell 1999, 4, 137–142. [Google Scholar] [CrossRef]
- Kim Chiaw, P.; Huan, L.J.; Gagnon, S.; Ly, D.; Sweezey, N.; Rotin, D.; Deber, C.M.; Bear, C.E. Functional rescue of DeltaF508-CFTR by peptides designed to mimic sorting motifs. Chem. Biol. 2009, 16, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Niibori, A.; Harada, K.; Kohno, T.; Michalak, M.; Duszyk, M.; Wada, I.; Ikawa, M.; Shuto, T.; Suico, M.A.; et al. Role of calnexin in the ER quality control and productive folding of CFTR; differential effect of calnexin knockout on wild-type and DeltaF508 CFTR. Biochim. Biophys. Acta 2008, 1783, 1585–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Janich, S.; Cohn, J.A.; Wilson, J.M. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. USA 1993, 90, 9480–9484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.J.; Loo, M.A.; Pind, S.; Williams, D.B.; Goldberg, A.L.; Riordan, J.R. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995, 83, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Heath-Engel, H.; Zhang, D.; Nguyen, N.; Thomas, D.Y.; Hanrahan, J.W.; Shore, G.C. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell 2008, 133, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef]
- Sato, S.; Ward, C.L.; Kopito, R.R. Cotranslational ubiquitination of cystic fibrosis transmembrane conductance regulator in vitro. J. Biol. Chem. 1998, 273, 7189–7192. [Google Scholar] [CrossRef] [Green Version]
- Younger, J.M.; Chen, L.; Ren, H.Y.; Rosser, M.F.; Turnbull, E.L.; Fan, C.Y.; Patterson, C.; Cyr, D.M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006, 126, 571–582. [Google Scholar] [CrossRef] [Green Version]
- El Khouri, E.; Le Pavec, G.; Toledano, M.B.; Delaunay-Moisan, A. RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2013, 288, 31177–31191. [Google Scholar] [CrossRef] [Green Version]
- Morito, D.; Hirao, K.; Oda, Y.; Hosokawa, N.; Tokunaga, F.; Cyr, D.M.; Tanaka, K.; Iwai, K.; Nagata, K. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol. Biol. Cell 2008, 19, 1328–1336. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Chiba, T.; Tokunaga, F.; Kawasaki, H.; Iwai, K.; Suzuki, T.; Ito, Y.; Matsuoka, K.; Yoshida, M.; Tanaka, K.; et al. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002, 418, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Osterhaus, S.R.; Parekh, K.R.; Jacobi, A.M.; Behlke, M.A.; McCray, P.B. SYVN1, NEDD8, and FBXO2 Proteins Regulate ΔF508 Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitin-mediated Proteasomal Degradation. J. Biol. Chem. 2016, 291, 25489–25504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassink, G.C.; Zhao, B.; Sompallae, R.; Altun, M.; Gastaldello, S.; Zinin, N.V.; Masucci, M.G.; Lindsten, K. The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 2009, 10, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahner, A.; Gong, X.; Schmidt, B.Z.; Peters, K.W.; Rabeh, W.M.; Thibodeau, P.H.; Lukacs, G.L.; Frizzell, R.A. Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell 2013, 24, 74–84. [Google Scholar] [CrossRef]
- Gong, X.; Ahner, A.; Roldan, A.; Lukacs, G.L.; Thibodeau, P.H.; Frizzell, R.A. Non-native Conformers of Cystic Fibrosis Transmembrane Conductance Regulator NBD1 Are Recognized by Hsp27 and Conjugated to SUMO-2 for Degradation. J. Biol. Chem. 2016, 291, 2004–2017. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Liao, Y.; Ahner, A.; Larsen, M.B.; Wang, X.; Bertrand, C.A.; Frizzell, R.A. Different SUMO paralogues determine the fate of wild-type and mutant CFTRs: Biogenesis versus degradation. Mol. Biol. Cell 2019, 30, 4–16. [Google Scholar] [CrossRef]
- Kälin, N.; Claass, A.; Sommer, M.; Puchelle, E.; Tümmler, B. DeltaF508 CFTR protein expression in tissues from patients with cystic fibrosis. J. Clin. Investig. 1999, 103, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Glozman, R.; Okiyoneda, T.; Mulvihill, C.M.; Rini, J.M.; Barriere, H.; Lukacs, G.L. N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol. 2009, 184, 847–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Benharouga, M.; Hu, W.; Lukacs, G.L. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J. Biol. Chem. 2001, 276, 8942–8950. [Google Scholar] [CrossRef] [Green Version]
- Benharouga, M.; Haardt, M.; Kartner, N.; Lukacs, G.L. COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments. J. Cell Biol. 2001, 153, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentzsch, M.; Chang, X.B.; Cui, L.; Wu, Y.; Ozols, V.V.; Choudhury, A.; Pagano, R.E.; Riordan, J.R. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Bagdany, M.; Veit, G.; Fukuda, R.; Avramescu, R.G.; Okiyoneda, T.; Baaklini, I.; Singh, J.; Sovak, G.; Xu, H.; Apaja, P.M.; et al. Chaperones rescue the energetic landscape of mutant CFTR at single molecule and in cell. Nat. Commun. 2017, 8, 398. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Apaja, P.M.; Lukacs, G.L. Protein quality control at the plasma membrane. Curr. Opin. Cell Biol. 2011, 23, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Veit, G.; Sakai, R.; Aki, M.; Fujihara, T.; Higashi, M.; Susuki-Miyata, S.; Miyata, M.; Fukuda, N.; Yoshida, A.; et al. Chaperone-Independent Peripheral Quality Control of CFTR by RFFL E3 Ligase. Dev. Cell 2018, 44, 694–708.e7. [Google Scholar] [CrossRef] [Green Version]
- McDonald, E.R.; El-Deiry, W.S. Suppression of caspase-8- and -10-associated RING proteins results in sensitization to death ligands and inhibition of tumor cell growth. Proc. Natl. Acad. Sci. USA 2004, 101, 6170–6175. [Google Scholar] [CrossRef] [Green Version]
- Araki, K.; Kawamura, M.; Suzuki, T.; Matsuda, N.; Kanbe, D.; Ishii, K.; Ichikawa, T.; Kumanishi, T.; Chiba, T.; Tanaka, K.; et al. A palmitoylated RING finger ubiquitin ligase and its homologue in the brain membranes. J. Neurochem. 2003, 86, 749–762. [Google Scholar] [CrossRef]
- Sakai, R.; Fukuda, R.; Unida, S.; Aki, M.; Ono, Y.; Endo, A.; Kusumi, S.; Koga, D.; Fukushima, T.; Komada, M.; et al. The integral function of the endocytic recycling compartment is regulated by RFFL-mediated ubiquitylation of Rab11 effectors. J. Cell Sci. 2019, 132, jcs228007. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Guggino, W. Ubiquitination and degradation of CFTR by the E3 ubiquitin ligase MARCH2 through its association with adaptor proteins CAL and STX6. PLoS ONE 2013, 8, e68001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.; Cihil, K.; Stolz, D.B.; Pilewski, J.M.; Stanton, B.A.; Swiatecka-Urban, A. c-Cbl facilitates endocytosis and lysosomal degradation of cystic fibrosis transmembrane conductance regulator in human airway epithelial cells. J. Biol. Chem. 2010, 285, 27008–27018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caohuy, H.; Jozwik, C.; Pollard, H.B. Rescue of DeltaF508-CFTR by the SGK1/Nedd4-2 signaling pathway. J. Biol. Chem. 2009, 284, 25241–25253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Rab, A.; Tang, L.; Bebok, Z.; Rowe, S.M.; Bartoszewski, R.; Collawn, J.F. ΔF508 CFTR surface stability is regulated by DAB2 and CHIP-mediated ubiquitination in post-endocytic compartments. PLoS ONE 2015, 10, e0123131. [Google Scholar] [CrossRef] [Green Version]
- Bomberger, J.M.; Barnaby, R.L.; Stanton, B.A. The deubiquitinating enzyme USP10 regulates the post-endocytic sorting of cystic fibrosis transmembrane conductance regulator in airway epithelial cells. J. Biol. Chem. 2009, 284, 18778–18789. [Google Scholar] [CrossRef] [Green Version]
- Bomberger, J.M.; Ye, S.; Maceachran, D.P.; Koeppen, K.; Barnaby, R.L.; O’Toole, G.A.; Stanton, B.A. A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog. 2011, 7, e1001325. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.; Brouillette, C.G.; Forman-Kay, J.D. Direct Binding of the Corrector VX-809 to Human CFTR NBD1: Evidence of an Allosteric Coupling between the Binding Site and the NBD1:CL4 Interface. Mol. Pharmacol. 2017, 92, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharmacol. 2013, 86, 612–619. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Nakatsukasa, K.; Huyer, G.; Michaelis, S.; Brodsky, J.L. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 2008, 132, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Grove, D.E.; Rosser, M.F.; Ren, H.Y.; Naren, A.P.; Cyr, D.M. Mechanisms for rescue of correctable folding defects in CFTRDelta F508. Mol. Biol. Cell 2009, 20, 4059–4069. [Google Scholar] [CrossRef] [Green Version]
- Tomati, V.; Sondo, E.; Armirotti, A.; Caci, E.; Pesce, E.; Marini, M.; Gianotti, A.; Jeon, Y.J.; Cilli, M.; Pistorio, A.; et al. Genetic Inhibition of the Ubiquitin Ligase Rnf5 Attenuates Phenotypes Associated to F508del Cystic Fibrosis Mutation. Sci. Rep. 2015, 5, 12138. [Google Scholar] [CrossRef]
- Sondo, E.; Falchi, F.; Caci, E.; Ferrera, L.; Giacomini, E.; Pesce, E.; Tomati, V.; Mandrup Bertozzi, S.; Goldoni, L.; Armirotti, A.; et al. Pharmacological Inhibition of the Ubiquitin Ligase RNF5 Rescues F508del-CFTR in Cystic Fibrosis Airway Epithelia. Cell Chem. Biol. 2018, 25, 891–905. [Google Scholar] [CrossRef]
- Min, J.N.; Whaley, R.A.; Sharpless, N.E.; Lockyer, P.; Portbury, A.L.; Patterson, C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell. Biol. 2008, 28, 4018–4025. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.U.; Moulin, M.; Coumailleau, F.; Wong, W.W.; Miasari, M.; Carter, H.; Silke, J.; Cohen-Tannoudji, M.; Vince, J.E.; Vaux, D.L. CARP2 deficiency does not alter induction of NF-kappaB by TNFalpha. Curr. Biol. 2009, 19, R7–R9. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, R.; Okiyoneda, T. Peripheral Protein Quality Control as a Novel Drug Target for CFTR Stabilizer. Front. Pharmacol. 2018, 9, 1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuda, R.; Okiyoneda, T. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitylation as a Novel Pharmaceutical Target for Cystic Fibrosis. Pharmaceuticals 2020, 13, 75. https://doi.org/10.3390/ph13040075
Fukuda R, Okiyoneda T. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitylation as a Novel Pharmaceutical Target for Cystic Fibrosis. Pharmaceuticals. 2020; 13(4):75. https://doi.org/10.3390/ph13040075
Chicago/Turabian StyleFukuda, Ryosuke, and Tsukasa Okiyoneda. 2020. "Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitylation as a Novel Pharmaceutical Target for Cystic Fibrosis" Pharmaceuticals 13, no. 4: 75. https://doi.org/10.3390/ph13040075
APA StyleFukuda, R., & Okiyoneda, T. (2020). Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitylation as a Novel Pharmaceutical Target for Cystic Fibrosis. Pharmaceuticals, 13(4), 75. https://doi.org/10.3390/ph13040075