
Anticancer Drug-Induced Cardiotoxicity: Insights and Pharmacogenetics

 , ,
, ,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. Mode of Cardiotoxicity Induction
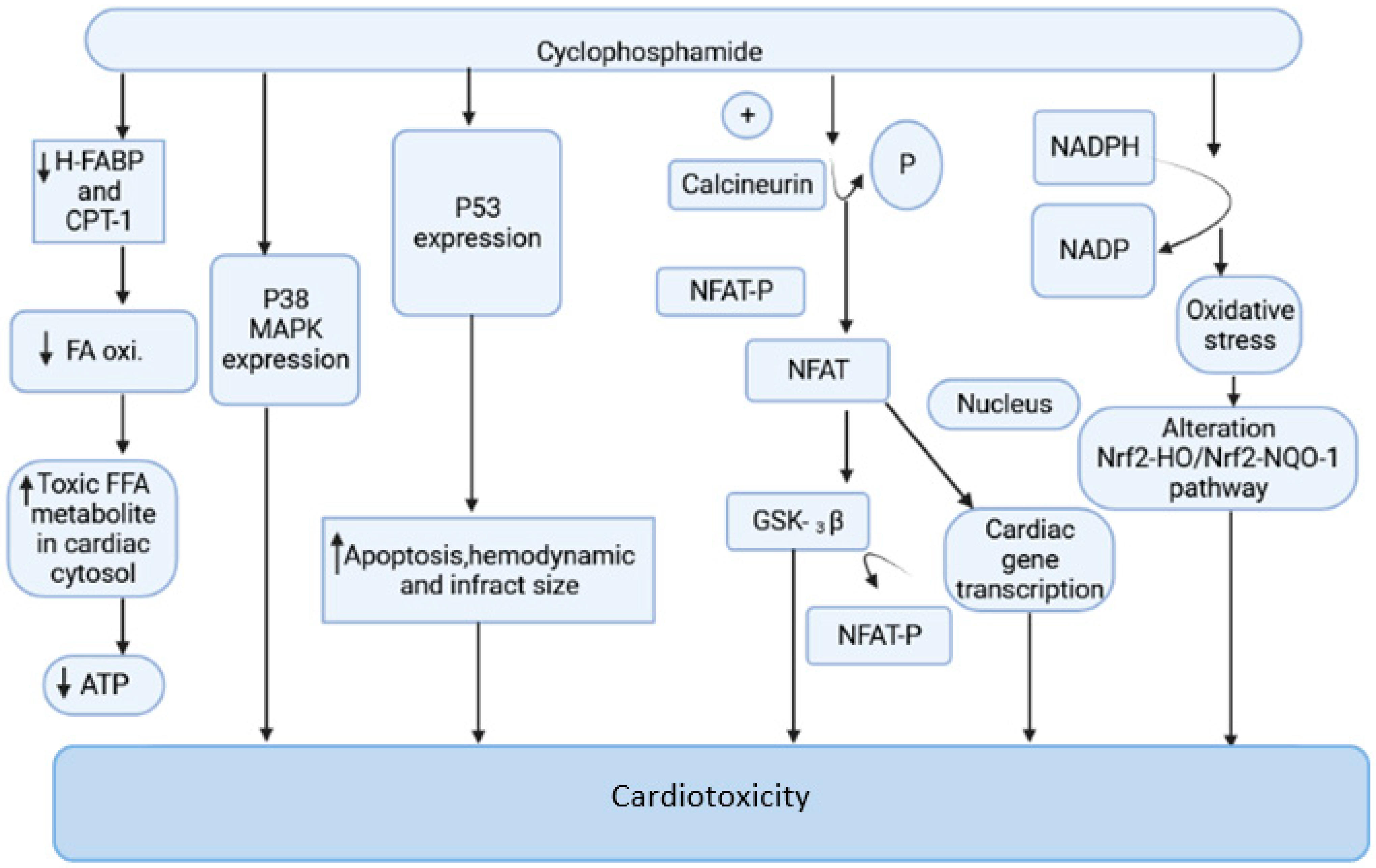
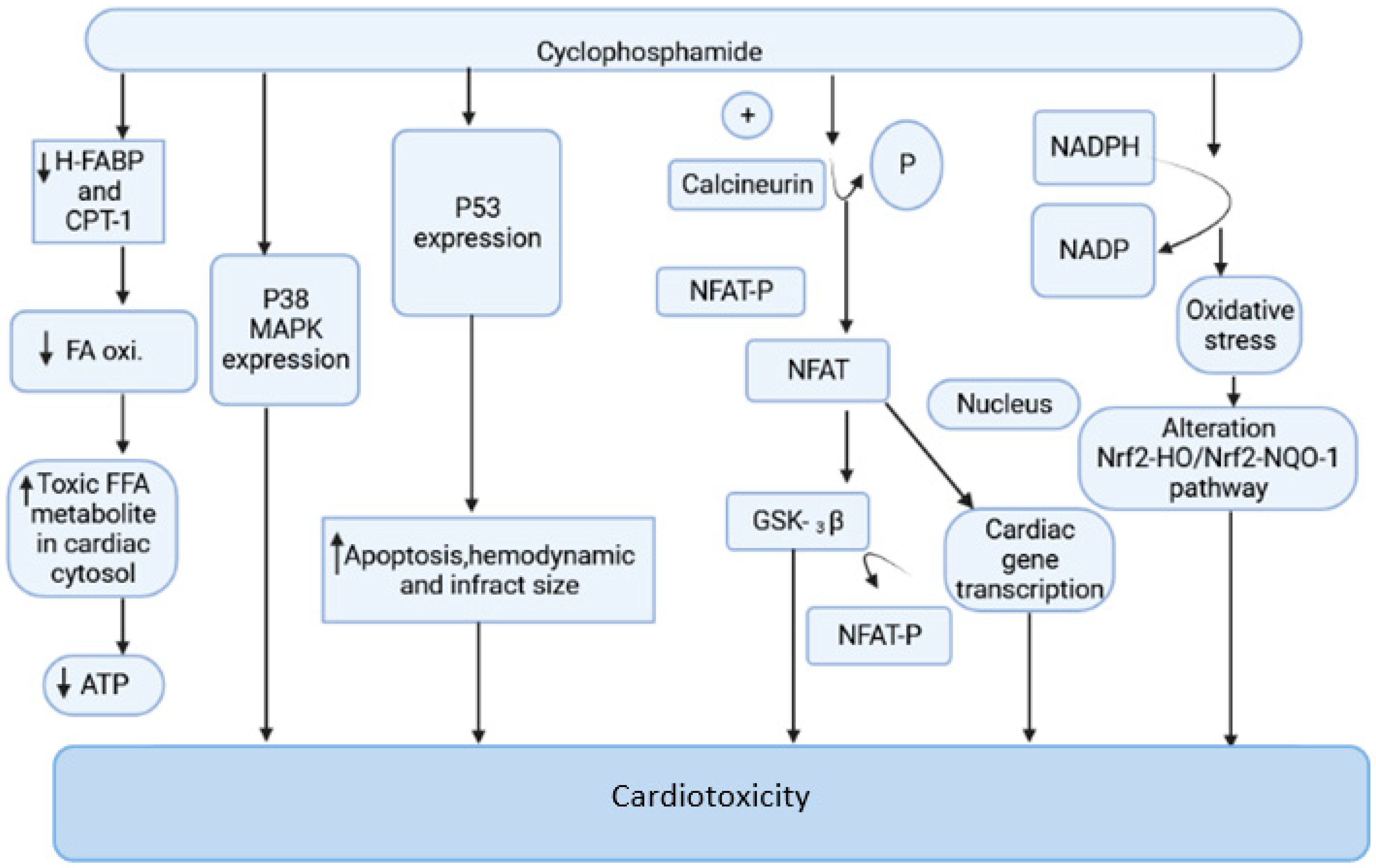
2.1. Cyclophosphamide-Induced Carditoxicity
2.1.1. Mitochondrial-Dependent ROS Production
2.1.2. Oxidative Stress Produced by NADPH
2.1.3. Oxidative Stress and Nrf2 Expression
2.1.4. Endoplasmic Reticulum Stress Associated with CP
2.1.5. Cyclophosphamide and Nitric Oxide
2.2. Doxorubicin-Induced Cardiotoxicity
2.2.1. Mechanism of Doxorubicin-Induced Cardiotoxicity
Oxidative Stress
- A.
- Altered mitochondrial functions
- B.
- Fe–Dox complex
- C.
- Role of NADPH in the generation of ROS
- D.
- Generation of reactive oxygen species by nitric oxide
- E.
- Generation of oxidative stress by nrf2
Apoptosis
Necrosis
Pyroptosis
Autophagy
Fibrosis
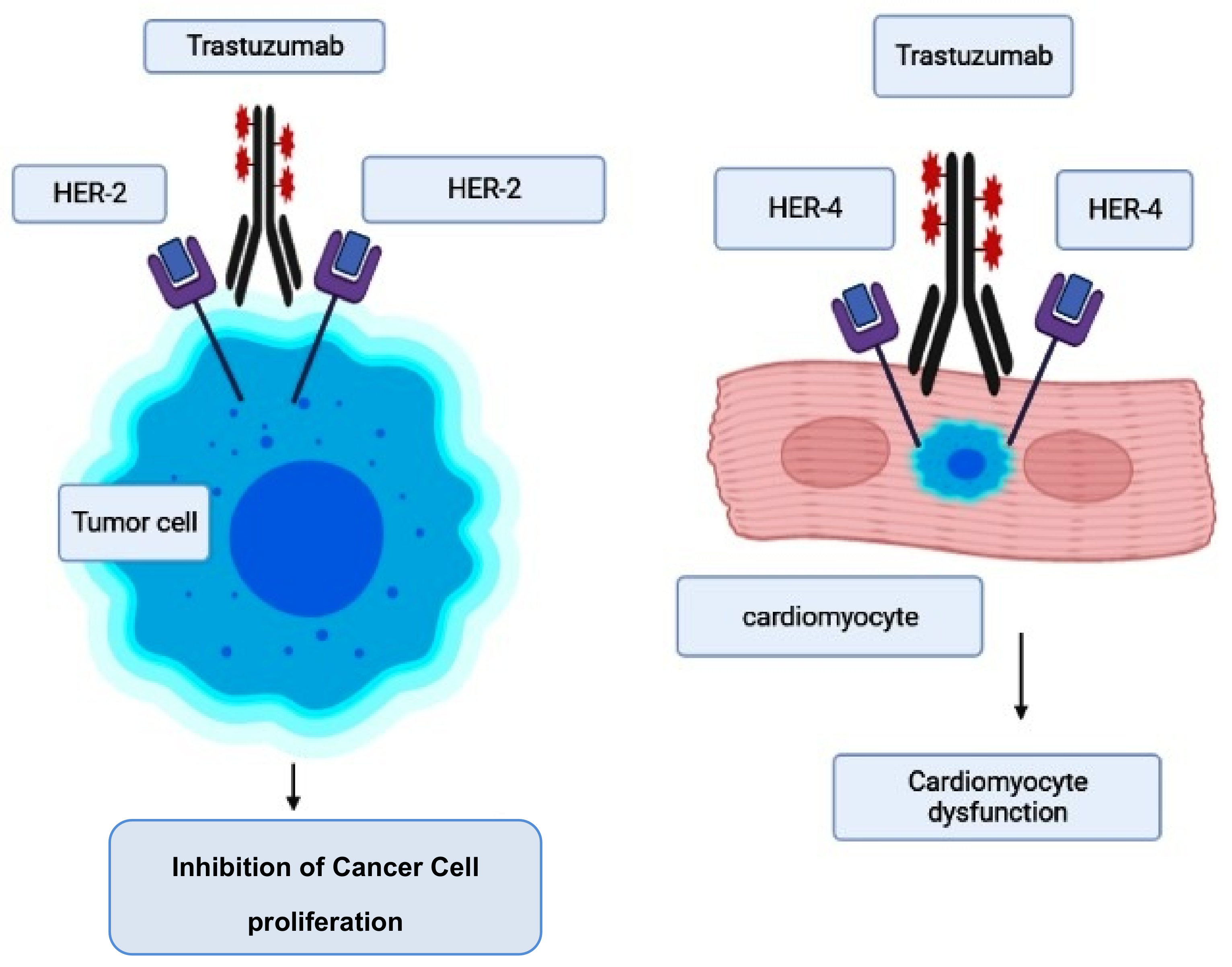
2.3. Trastuzumab-Induced Cardiotoxicity
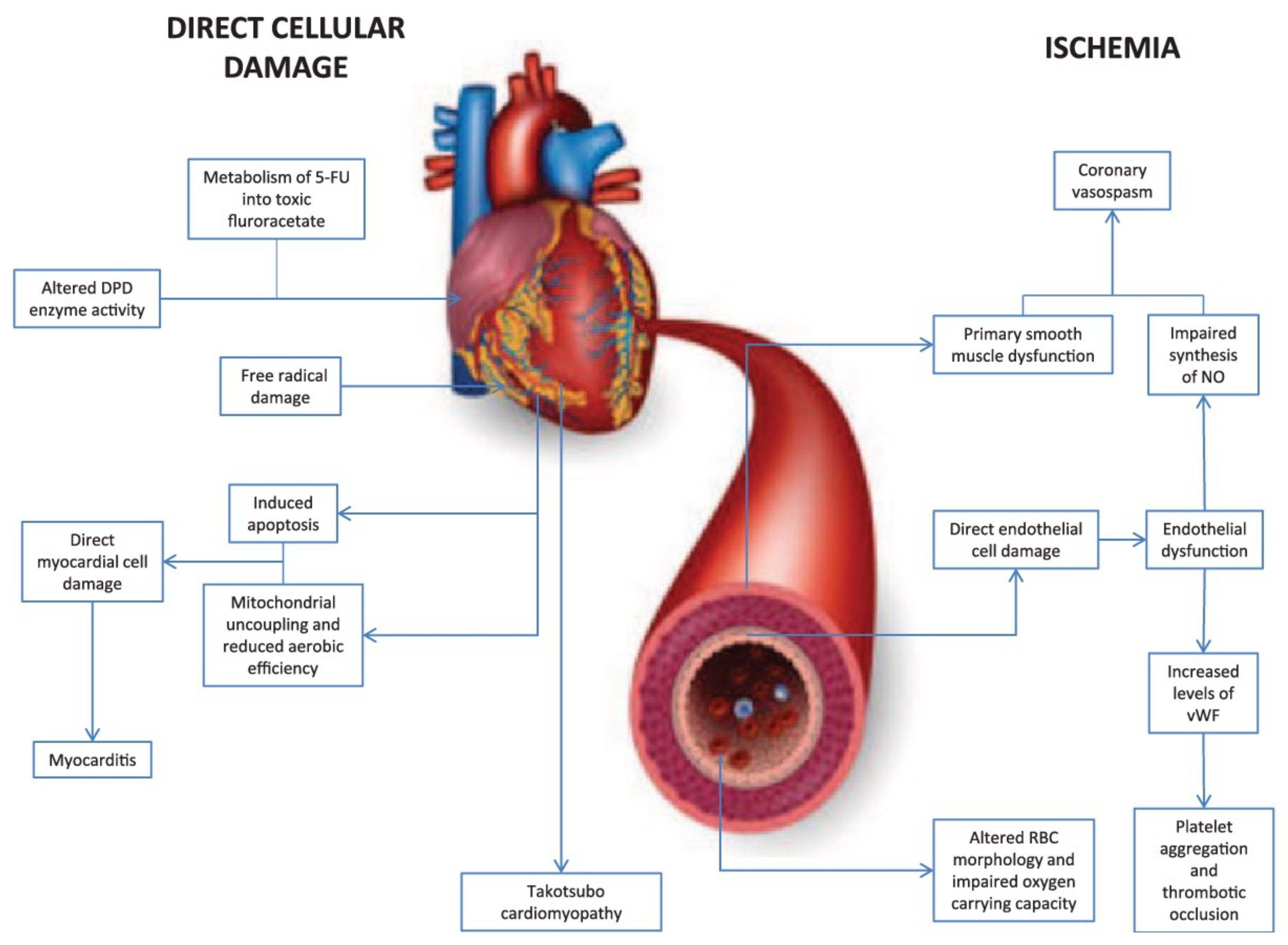
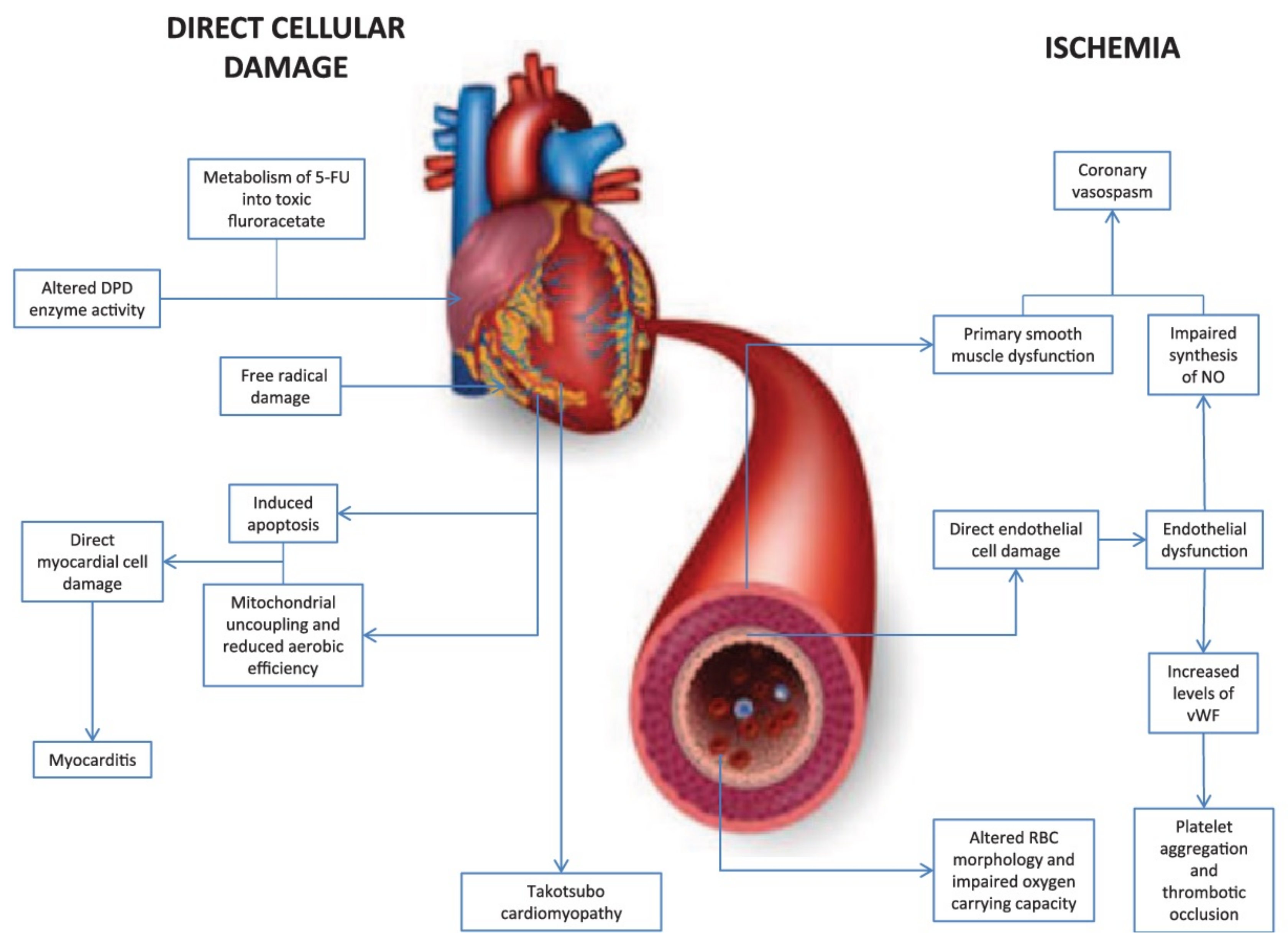
2.4. Fluorouracil-Induced Cardiotoxicity
2.5. Cisplatin-Induced Cardiotoxicity
2.6. Immunotherapy-Induced Cardiotoxicity
2.6.1. Immune Checkpoint Inhibitors and Cardiac Complications
2.6.2. CDK4/6 Inhibitors
2.6.3. VEGF Inhibitors
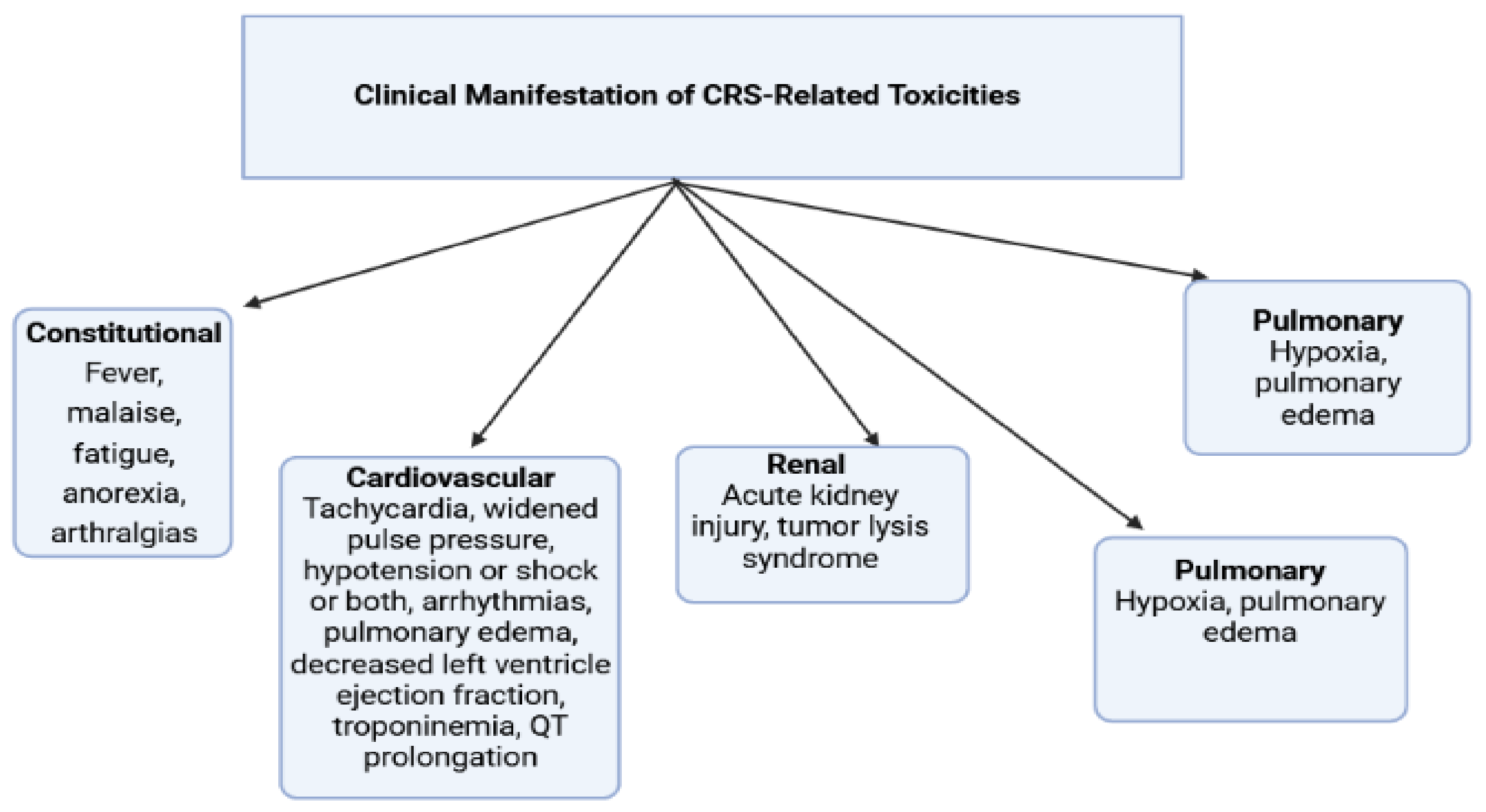
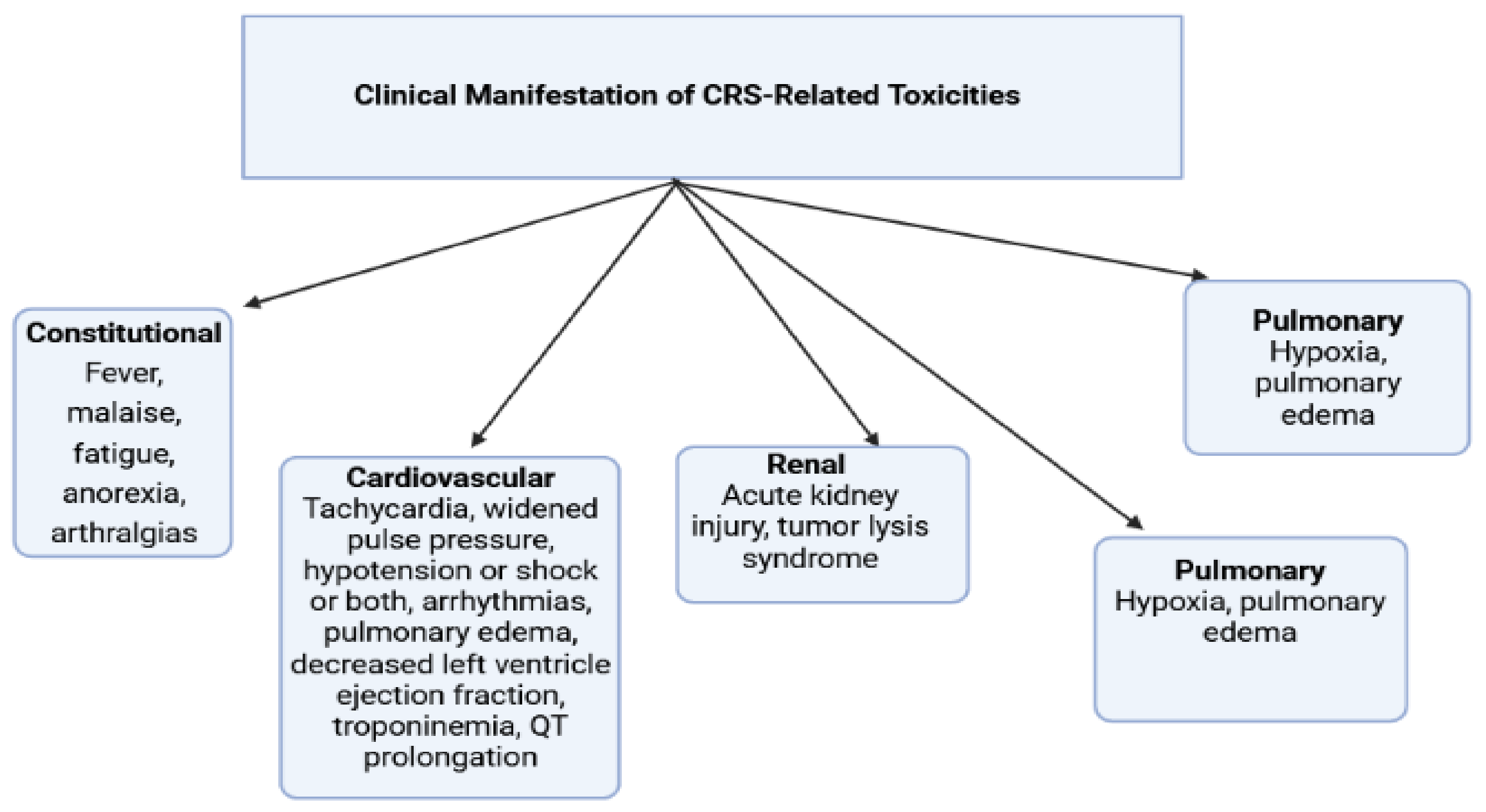
2.6.4. Chimeric Antigen Receptor (CAR) T Cell Therapy
3. Genes’ Susceptibility to Cardiotoxicity Induced by Chemotherapeutic Agents
3.1. ErbB2 Gene
3.2. NOX2
3.3. CBR Gene
3.4. TTN Gene
3.5. G Protein-Coupled Receptor 35 (GPR35)
3.6. CELF4, RARG, SLC28A3, UGT1A6
4. Radiation Therapy-Induced Cardiotoxicity
5. Role of Herbs as Antioxidants in the Inhibition of Anticancer Drug-Induced Cardiac Toxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Animals Used | Method and Intervention | Major Findings |
|---|---|---|---|
| Bhatt et al. [12] | Wistar rats | Rats were exposed to CP toxicity with the dose of (200 mg/kg, i.p.) on day 1 of the treatment protocol. The animals were treated with 100 mg/kg of mangiferin for 10 days. | The treatment with mangiferin restored serum biomarker enzymes, antioxidant levels, lipid profile, electrocardiographic parameters, and histological score and mortality. |
| Ayza et al. [16] | Sprague Dawley rats | Rats were exposed to CP toxicity with the dose of (200 mg/kg, i.p.) on day 1 of the treatment protocol. The animals were treated with 100 mg/kg of mangiferin for 10 days. | The crude extract and ethyl acetate and aqueous fractions of Croton macrostachyus exhibited in vitro free radical scavenging activities in DPPH free radical scavenging assay. The treatment also restored serum biomarker enzymes, lipid profile, and histological score. |
| Qin et al. [32] | Wistar rats | Rats were exposed to CP toxicity with the dose of (07 mg/kg, i.p.) on day 6 of the treatment protocol. The animals were treated with 5, 15 and 45 mg/kg of resveratrol for 10 days. | Resveratrol treatment reported synergistic antineoplastic activity with cisplatin to A549 adenocarcinoma cells. Resveratrol treatment in a dose-dependent manner restored blood pressure, heart rate, serum biomarker enzymes, tissue antioxidant level, and histopathology of myocardial cell against CP-induced cardiotoxicity. |
| Bahadır et al. [103] | Wistar rats | Rats were exposed to cisplatin toxicity with the dose of 5 mg/kg/week for two weeks. The animals were treated with curcumin (200 mg/kg) and beta-carotene (100 mg/kg) | Curcumin and beta-carotene reported significant improvement in tissue antioxidant levels, tumor necrosis factor-α, interleukin-1β, and interleukin-6 against cisplatin-induced cardiotoxicity. |
| El-Hawwary et al. [106] | Wistar rats | Rats were exposed to cisplatin toxicity with the dose (2 mg/kg/day) for 1 week. The animals were treated with ginger (500 mg/kg) for 12 days. | Ginger treatment reported significant restoration of cardiac histology ultrastructure and a decrease in P53 and TNF-α immune expressions and creatinine kinase and lactate dehydrogenase levels against cisplatin-induced cardiotoxicity. |
| Ahmed et al. [157] | Wistar rats | Rats were exposed to cisplatin toxicity with the dose of doxorubicin (25 mg/kg i.p.) on 7th day. The animals were treated with methyl gallate (150 and 300 mg/kg) for 7 days. | Methyl gallate treatment restored ECG recording, serum biomarkers, and tissue antioxidant and lipid profile levels against doxorubicin-induced cardiotoxicity. |
| Birari et al. [158] | Wistar rats | Rats were intoxicated with a dose of (6 mg/kg, i.p.) with doxorubicin on alternate days (cumulative dose 30 mg/kg). The rats were treated with aloin as aqueous solution (1, 5, 25 and 125 mg/kg, p.o., once a day) | Aloin treatment restored ECG tracings and tissue antioxidant levels and reduced the levels of proinflammatory cytokines TNF-α, IL-1β, and IL-6 against doxorubicin-induced cardiotoxicity. |
| Hu X et al. [159] | C57BL/6 mice | Mice were administered with doxorubicin with a dose of (15 mg/kg, i.p.). Mice were treated with asiatic acid (10 mg/kg and 30 mg/kg) two weeks before doxorubicin treatment | Asiatic acid treatment restored echocardiographic and tissue antioxidant level. Asiatic acid reduced oxidative stress and apoptosis induced by doxorubicin by AKT signaling pathway. |
| Meng et al. [160] | C57BL/6 mice | Mice were administered with doxorubicin with a dose of (15 mg/kg, i.p.). Mice were treated with geniposide (25 mg/kg and 50 mg/kg) for 10 days, which was started three days before doxorubicin treatment. | Geniposide witnessed cardio-protection against doxorubicin-induced cardiotoxicity by the activation of AMP-activated protein kinase α. |
| Zhang et al. [161] | C57BL/6 mice | Mice were treated with doxorubicin with a dose of (20 mg/kg, i.p.). Mice were treated with oroxylin A for 10 days, which was started five days before doxorubicin treatment | Oroxylin A treatment restored oxidative damage and reduced inflammation accumulation and myocardial apoptosis in vivo and in vitro. Oroxylin A showed protection by activation of sirtuin 1 signaling pathway via the cAMP/protein kinase A. |
6. Proposed Future Hopes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaulin, A.M.; Abashina, O.E.; Duplyakov, D.V. Pathophysiological mechanisms of cardiotoxicity in chemotherapeutic agents. Russ. Open Med. J. 2020, 9, 1–9. [Google Scholar] [CrossRef]
- Moudgil, R.; Yeh, E.T. Mechanisms of cardiotoxicity of cancer chemotherapeutic agents: Cardiomyopathy and beyond. Can. J. Cardiol. 2016, 32, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Chen, H. Cardiotoxicity of anticancer therapeutics. Front. Cardiovasc. Med. 2018, 7, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Grieve, D.J.; Davidson, S.M. New insights into cardiotoxicity caused by chemotherapeutic agents: Editorial. Br. J. Pharmacol. 2017, 174, 3675–3676. [Google Scholar] [CrossRef] [Green Version]
- Jain, D.; Aronow, W. Cardiotoxicity of cancer chemotherapy in clinical practice. Hosp. Pract. 2019, 47, 6–15. [Google Scholar] [CrossRef]
- Rhea, I.B.; Oliveira, G.H. Cardiotoxicity of novel targeted chemotherapeutic agents. Curr. Treat. Options Cardiovasc. Med. 2018, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, M.W.; Hamo, C.E.; Cardinale, D.; Ky, B.; Nohria, A.; Baer, L.; Skopicki, H.; Lenihan, D.J.; Gheorghiade, M.; Lyon, A.R.; et al. Cancer therapy–related cardiac dysfunction and heart failure. Circ. Heart Fail. 2016, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, I.E.; Taveras Alam, S.; Hernandez, G.A.; Sancassani, R. Cancer therapy-related cardiac dysfunction: An overview for the clinician. Clin. Med. Insights Cardiol. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, J.; Cautela, J.; Ederhy, S.; Damaj, G.L.; Salem, J.E.; Barlesi, F.; Farnault, L.; Charbonnier, A.; Mirabel, M.; Champiat, S.; et al. Cardiovascular toxicity related to cancer treatment: A pragmatic approach to the American and European cardio-oncology guidelines. J. Am. Heart Assoc. 2020, 9, e018403. [Google Scholar] [CrossRef]
- Bansal, N.; Adams, M.J.; Ganatra, S.; Colan, S.D.; Aggarwal, S.; Steiner, R.; Amdani, S.; Lipshultz, E.R.; Lipshultz, S.E. Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardio-Oncology 2019, 5, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, L.; Sebastian, B.; Joshi, V. Mangiferin protects rat myocardial tissue against cyclophosphamide-induced cardiotoxicity. J. Ayurveda Integr. Med. 2017, 8, 62–67. [Google Scholar] [CrossRef]
- Ayza, M.A.; Zewdie, K.A.; Tesfaye, B.A.; Wondafrash, D.Z.; Berhe, A.H. The role of antioxidants in ameliorating cyclophosphamide-induced cardiotoxicity. Oxid. Med. Cell Longev. 2020, 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Iqubal, M.K.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Ali, S.M.; Ali, J.; Haque, S.E. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: Old drug with a new vision. Life Sci. 2019, 218, 112–131. [Google Scholar] [CrossRef] [PubMed]
- Jeelani, R.; Khan, S.N.; Shaeib, F.; Kohan-Ghadr, H.-R.; Aldhaheri, S.R.; Najafi, T.; Thakur, M.; Morris, R.; Abu-Soud, H.M. Cyclophosphamide and acrolein induced oxidative stress leading to deterioration of metaphase II mouse oocyte quality. Free Radic. Biol. Med. 2017, 110, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ayza, M.A.; Balasubramanian, R.; Berhe, A.H. Cardioprotective effect of Croton macrostachyus stem bark extract and solvent fractions on cyclophosphamide-induced cardiotoxicity in rats. Evid. Based Complement. Altern. Med. 2020, 2020, 8467406. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Nickel, A.G.; Maack, C. Mitochondrial energetics and calcium coupling in the heart. J. Physiol. 2017, 595, 3753–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, D.; Zhao, Y.; O’Neill, K.M.; Edgar, K.S.; Dunne, P.D.; Kearney, A.M.; Grieve, D.J.; McDermott, B.J. Signalling mechanisms underlying doxorubicin and Nox2 NADPH oxidase-induced cardiomyopathy: Involvement of mitofusin-2. Br. J. Pharmacol. 2017, 174, 3677–3695. [Google Scholar] [CrossRef] [Green Version]
- Varghese, M.V.; Abhilash, M.; Paul, M.S.; Alex, M.; Nair, R.H. Omega-3 fatty acid protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Cardiovasc. Toxicol. 2017, 17, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Hamm, N.C.; Stammers, A.N.; Susser, S.E.; Hlynsky, M.W.; Kimber, D.E.; Kehler, D.S.; Duhamel, T.A. Regulation of cardiac sarco (endo) plasmic reticulum calcium-ATPases (SERCA2a) in response to exercise. In Regulation of Ca2+-ATPases, V-ATPases and F-ATPases; Springer: Cham, Switzerland, 2016; pp. 187–206. [Google Scholar]
- Fabris, S.; MacLean, D.A. Doxorubicin chemotherapy affects intracellular and interstitial nitric oxide concentrations in skeletal muscle. Cell Biol. Toxicol. 2016, 32, 121–131. [Google Scholar] [CrossRef]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: A scientific statement from the American Heart Association. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef] [PubMed]
- Kurauchi, K.; Nishikawa, T.; Miyahara, E.; Okamoto, Y.; Kawano, Y. Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res. Notes 2017, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Komolafe, O.A.; Arayombo, B.E.; Abiodun, A.A.; Saka, O.S.; Abijo, A.Z.; Ojo, S.K.; Fakunle, O.O. Immunohistochemical and histological evaluations of cyclophosphamide-induced acute cardiotoxicity in wistar rats: The role of turmeric extract (curcuma). Morphologie 2020, 104, 133–142. [Google Scholar] [CrossRef]
- Elrashidy, R.A.; Hasan, R.A. Cilostazol preconditioning alleviates cyclophosphamide-induced cardiotoxicity in male rats: Mechanistic insights into SIRT1 signaling pathway. Life Sci. 2021, 266, 118822. [Google Scholar] [CrossRef] [PubMed]
- El-Agamy, D.S.; Elkablawy, M.A.; Abo-Haded, H.M. Modulation of cyclophosphamide-induced cardiotoxicity by methyl palmitate. Cancer Chemother. Pharmacol. 2017, 79, 399–409. [Google Scholar] [CrossRef]
- Omran, M.M.; Mansour, H.H.; Hasan, H.F. Metformin and/or low dose radiation reduces cardiotoxicity and apoptosis induced by cyclophosphamide through SIRT-1/SOD and BAX/Bcl-2 pathways in rats. Mol. Biol. Rep. 2020, 47, 5115–5126. [Google Scholar]
- Refaie, M.M.; Shehata, S.; El-Hussieny, M.; Abdelraheem, W.M.; Bayoumi, A.M. Role of ATP-sensitive potassium channel (K ATP) and eNOS in mediating the protective effect of nicorandil in cyclophosphamide-induced cardiotoxicity. Cardiovasc. Toxicol. 2020, 20, 71–81. [Google Scholar] [CrossRef]
- Liu, W.; Zhai, X.; Wang, W.; Zheng, B.; Zhang, Z.; Fan, X.; Chen, Y.; Wang, J. Aldehyde dehydrogenase 2 activation ameliorates cyclophosphamide-induced acute cardiotoxicity via detoxification of toxic aldehydes and suppression of cardiac cell death. J. Mol. Cell. Cardiol. 2018, 121, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Gunes, S.; Sahinturk, V.; Karasati, P.; Sahin, I.K.; Ayhanci, A. Cardioprotective effect of selenium against cyclophosphamide-induced cardiotoxicity in rats. Biol. Trace Elem. Res. 2017, 177, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Sandamali, J.A.; Hewawasam, R.P.; Fernando, M.A.; Jayatilaka, K.A.; Madurawe, R.D.; Sathananthan, P.P.; Ekanayake, U.; Horadugoda, J. Anthracycline-induced cardiotoxicity in breast cancer patients from Southern Sri Lanka: An echocardiographic analysis. BioMed Res. Int. 2020, 2020, 1847159. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Chang, F.; Wang, Z.; Jiang, W. Correlation of circulating pro-angiogenic miRNAs with cardiotoxicity induced by epirubicin/cyclophosphamide followed by docetaxel in patients with breast cancer. Cancer Biomark. 2018, 23, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Avci, H.; Epikmen, E.T.; İpek, E.; Tunca, R.; Birincioglu, S.S.; Akşit, H.; Sekkin, S.; Akkoç, A.N.; Boyacioglu, M. Protective effects of silymarin and curcumin on cyclophosphamide-induced cardiotoxicity. Exp. Toxicol. Pathol. 2017, 69, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Syed, M.A.; Ali, J.; Alam, M.M.; Ahmad, S.; Haque, S.E. Nerolidol attenuates cyclophosphamide-induced cardiac inflammation, apoptosis and fibrosis in Swiss Albino mice. Eur. J. Pharmacol. 2019, 863, 172666. [Google Scholar] [CrossRef] [PubMed]
- Abushouk, A.I.; Ismail, A.; Salem, A.M.; Afifi, A.M.; Abdel-Daim, M.M. Cardioprotective mechanisms of phytochemicals against doxorubicin-induced cardiotoxicity. Biomed. Pharmacother. 2017, 90, 935–946. [Google Scholar] [CrossRef]
- Gazia, M.A.; El-Magd, M.A. Ameliorative effect of cardamom aqueous extract on doxorubicin-induced cardiotoxicity in rats. Cells Tissues Organs 2018, 206, 62–72. [Google Scholar]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663. [Google Scholar] [CrossRef] [Green Version]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into doxorubicin-induced cardiotoxicity: Molecular mechanisms, preventive strategies, and early monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Renu, K.; Abilash, V.G.; Pichiach, B.T.P.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Podyacheva, E.Y.; Kushnareva, E.A.; Karpov, A.A.; Toropova, Y.G. Analysis of models of doxorubicin-induced cardiomyopathy in rats and mice. A modern view from the perspective of the pathophysiologist and the clinician. Front. Pharmacol. 2021, 12, 1398. [Google Scholar] [CrossRef] [PubMed]
- Singal, P.; Li, T.; Kumar, D.; Danelisen, I.; Iliskovic, N. Adriamycin-induced heart failure: Mechanisms and modulation. Mol. Cell. Biochem. 2000, 207, 77–86. [Google Scholar] [CrossRef]
- Mobaraki, M.; Faraji, A.; Zare, M.; Dolati, P.; Ataei, M.; Manshadi, H.D. Molecular mechanisms of cardiotoxicity: A review on major side-effect of doxorubicin. Indian J. Pharm. Sci. 2017, 79, 335–344. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Shabalala, S.; Muller, C.J.; Louw, J.; Johnson, R. Polyphenols, autophagy and doxorubicin-induced cardiotoxicity. Life Sci. 2017, 180, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Nickel, B.E.; Edel, A.L.; Fandrich, R.R.; Ravandi, A.; Kardami, E. Oxidized phospholipids in Doxorubicin-induced cardiotoxicity. Chem. Biol. Interact. 2019, 303, 35–39. [Google Scholar] [CrossRef]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Al Qahtani, A.A.; Osman, A.M.; Damanhouri, Z.A.; Al-Kreathy, H.M.; Al-Malky, H.S.; Ramadan, W.S.; Alharthi, S.E.; Kamel, F.O. Cardioprotective effect of marine Astaxanthin on doxorubicin-induced cardiotoxicity in normal rats. J. Pharm. Res. Int. 2019, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Qi, Y.; Xu, L.; Tao, X.; Han, X.; Yin, L.; Peng, J. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2018, 15, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Corremans, R.; Adão, R.; De Keulenaer, G.W.; Leite-Moreira, A.F.; Brás-Silva, C. Update on pathophysiology and preventive strategies of anthracycline-induced cardiotoxicity. Clin. Exp. Pharmacol. Physiol. 2019, 46, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.Z.; Zhang, L.; Wu, Z.X.; Shan, T.T.; Xiong, C. Berberine ameliorates doxorubicin-induced cardiotoxicity via a SIRT1/p66Shc-mediated pathway. Oxid. Med. Cell. Longev. 2019, 2019, 339. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Yi, M.; Huang, Y.P. Oxymatrine ameliorates doxorubicin-induced cardiotoxicity in rats. Cell. Physiol. Biochem. 2017, 43, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Alkuraishy, H.M.; Al-Gareeb, A.I.; Al-hussaniy, H.A. Doxorubicin-induced cardiotoxicity: Molecular mechanism and protection by conventional drugs and natural products. Int. J. Clin. Oncol. Cancer Res. 2017, 2, 31–44. [Google Scholar]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhang, X.; Song, P.; Yuan, Y.P.; Kong, C.Y.; Wu, H.M.; Xu, S.C.; Ma, Z.G.; Tang, Q.Z. Meteorin-like protein attenuates doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1 pathway. Redox Biol. 2020, 37, 101747. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, C.; Kong, C.Y.; Song, P.; Wu, H.M.; Xu, S.C.; Yuan, Y.P.; Deng, W.; Ma, Z.G.; Tang, Q.Z. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020, 27, 540–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, V.Y.; Wang, J.J. Pharmacogenetics of chemotherapy-induced cardiotoxicity. Curr. Oncol. Rep. 2018, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhong, T.; Ma, Y.; Wan, X.; Qin, A.; Yao, B.; Zou, H.; Song, Y.; Yin, D. Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci. 2020, 242, 2–9. [Google Scholar] [CrossRef]
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The cardioprotective effect of metformin in doxorubicin-induced cardiotoxicity: The role of autophagy. Molecules 2018, 23, 1184. [Google Scholar] [CrossRef] [Green Version]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 1–5. [Google Scholar] [CrossRef]
- Ruggeri, C.; Gioffré, S.; Achilli, F.; Colombo, G.I.; D’Alessandra, Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: An overview of preclinical models and cancer patients. Heart Fail. Rev. 2018, 23, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Timm, K.N.; Perera, C.; Ball, V.; Henry, J.A.; Miller, J.J.; Kerr, M.; West, J.A.; Sharma, E.; Broxholme, J.; Logan, A.; et al. Early detection of doxorubicin-induced cardiotoxicity in rats by its cardiac metabolic signature assessed with hyperpolarized MRI. Commun. Biol. 2020, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef]
- Osataphan, N.; Phrommintikul, A.; Chattipakorn, S.C.; Chattipakorn, N. Effects of doxorubicin-induced cardiotoxicity on cardiac mitochondrial dynamics and mitochondrial function: Insights for future interventions. J. Cell. Mol. Med. 2020, 24, 6534–6557. [Google Scholar] [CrossRef]
- Mancilla, T.R.; Iskra, B.; Aune, G.J. Doxorubicin-induced cardiomyopathy in children. Compr. Physiol. 2019, 9, 905. [Google Scholar] [CrossRef]
- Gallo, S.; Spilinga, M.; Albano, R.; Ferrauto, G.; Di Gregorio, E.; Casanova, E.; Balmativola, D.; Bonzano, A.; Boccaccio, C.; Sapino, A.; et al. Activation of the MET receptor attenuates doxorubicin-induced cardiotoxicity in vivo and in vitro. Br. J. Pharmacol. 2020, 177, 3107–3122. [Google Scholar] [CrossRef]
- Lee, Y.; Kwon, I.; Jang, Y.; Cosio-Lima, L.; Barrington, P. Endurance exercise attenuates doxorubicin-induced cardiotoxicity. Med. Sci. Sports Exerc. 2020, 52, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.M.; Li, C.B.; Liu, Q.L.; Li, P.; Yang, H. Ginsenoside Rg1 prevents doxorubicin-induced cardiotoxicity through the inhibition of autophagy and endoplasmic reticulum stress in mice. Int. J. Mol. Sci. 2018, 19, 3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmohammadi, F.; Rezaee, R.; Karimi, G. Natural compounds against doxorubicin-induced cardiotoxicity: A review on the involvement of Nrf2/ARE signaling pathway. Phytother. Res. 2021, 35, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Boliang, W.; Xiaoxi, T.; Guoqiang, F.; Jianbo, X.; Gang, W. Cardamonin protects against doxorubicin-induced cardiotoxicity in mice by restraining oxidative stress and inflammation associated with Nrf2 signaling. Biomed. Pharmacother. 2020, 122, 109547. [Google Scholar] [CrossRef]
- Wu, Z.Z.; Rao, M.; Xu, S.; Hu, H.Y.; Tang, Q.Z. Coumestrol ameliorates doxorubicin-induced cardiotoxicity via activating AMPKα. Free Radic. Res. 2020, 54, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Guida, F.; Paparo, L.; Trinchese, G.; Aitoro, R.; Avagliano, C.; Fiordelisi, A.; Napolitano, F.; Mercurio, V.; Sala, V.; et al. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur. J. Heart Fail. 2019, 21, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Deng, H.; Zhang, J.; Zhang, Y.; Zhang, X.; Cui, G. Mitochondria-targeting small molecules effectively prevent cardiotoxicity induced by doxorubicin. Molecules 2018, 23, 1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Shayesteh, M.R.; Mortezaee, K.; Farhood, B.; Haghi-Aminjan, H. The role of melatonin on doxorubicin-induced cardiotoxicity: A systematic review. Life Sci. 2020, 241, 117173. [Google Scholar] [CrossRef]
- Yu, J.; Wang, C.; Kong, Q.; Wu, X.; Lu, J.J.; Chen, X. Recent progress in doxorubicin-induced cardiotoxicity and protective potential of natural products. Phytomedicine 2018, 40, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, P.; Kyriakidis, M.; Perpinia, A.; Karavidas, A.; Zimeras, S.; Mamalis, N.; Kouvela, M.; Charpidou, A. The role of Metoprolol and Enalapril in the prevention of doxorubicin-induced cardiotoxicity in lymphoma patients. Anticancer Res. 2019, 39, 5703–5707. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Wei, L.; Zhang, N.; Wei, W.Y.; Hu, C.; Deng, W.; Tang, Q.Z. Levosimendan protects against doxorubicin-induced cardiotoxicity by regulating the PTEN/Akt pathway. BioMed Res. Int. 2020, 2020, 1–11. [Google Scholar]
- Ni, C.; Ma, P.; Wang, R.; Lou, X.; Liu, X.; Qin, Y.; Xue, R.; Blasig, I.; Erben, U.; Qin, Z. Doxorubicin-induced cardiotoxicity involves IFNγ-mediated metabolic reprogramming in cardiomyocytes. J. Pathol. 2019, 247, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, H.; Xiang, D.; Guo, W. Pharmaceutical measures to prevent doxorubicin-induced cardiotoxicity. Mini Rev. Med. Chem. 2017, 17, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Anthony, F.Y.; Flynn, J.R.; Moskowitz, C.S.; Scott, J.M.; Oeffinger, K.C.; Dang, C.T.; Liu, J.E.; Jones, L.W.; Steingart, R.M. Long-term cardiopulmonary consequences of treatment-induced cardiotoxicity in survivors of ERBB2-positive breast cancer. JAMA Cardiol. 2020, 5, 309–317. [Google Scholar]
- Barish, R.; Gates, E.; Barac, A. Trastuzumab-induced cardiomyopathy. Cardiol. Clin. 2019, 37, 407–418. [Google Scholar] [CrossRef]
- Kitani, T.; Ong, S.G.; Lam, C.K.; Rhee, J.W.; Zhang, J.Z.; Oikonomopoulos, A.; Ma, N.; Tian, L.; Lee, J.; Telli, M.L.; et al. Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation 2019, 139, 2451–2465. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Kim, K.H.; Kim, H.Y.; Park, H.; Cho, J.Y.; Hong, Y.J.; Park, H.W.; Kim, J.H.; Ahn, Y.; Jeong, M.H.; et al. Impacts of non-recovery of trastuzumab-induced cardiomyopathy on clinical outcomes in patients with breast cancer. Clin. Res. Cardiol. 2019, 108, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Koulaouzidis, G.; Yung, A.E.; Yung, D.E.; Skonieczna-Żydecka, K.; Marlicz, W.; Koulaouzidis, A.; Charisopoulou, D. Conventional cardiac risk factors associated with trastuzumab-induced cardiotoxicity in breast cancer: Systematic review and meta-analysis. Curr. Probl. Cancer. 2021, 6, 100723. [Google Scholar] [CrossRef] [PubMed]
- Florido, R.; Smith, K.L.; Cuomo, K.K.; Russell, S.D. Cardiotoxicity from human epidermal growth factor receptor-2 (HER 2) targeted therapies. J. Am. Heart Assoc. 2017, 6, 1–14. [Google Scholar] [CrossRef]
- Pecoraro, M.; Pinto, A.; Popolo, A. Trastuzumab-induced cardiotoxicity and role of mitochondrial connexin43 in the adaptive response. Toxicol. In Vitro 2020, 46, 1–8. [Google Scholar]
- Tahir, H.; Bardia, N.; Bath, K.; Ahmed, Y.; Rafique, M.; Omar, B.; Malozzi, C. Trastuzumab-Induced cardiomyopathy and intermittent left bundle branch block. Cardiol. Res. 2019, 10, 230. [Google Scholar] [CrossRef] [Green Version]
- Jawa, Z.; Perez, R.M.; Garlie, L.; Singh, M.; Qamar, R.; Khandheria, B.K.; Jahangir, A.; Shi, Y. Risk factors of trastuzumab-induced cardiotoxicity in breast cancer: A meta-analysis. Medicine 2016, 95, 1–7. [Google Scholar] [CrossRef]
- Cho, D.H.; Lim, I.R.; Kim, J.H.; Kim, M.N.; Kim, Y.H.; Park, K.H.; Park, S.M.; Shim, W.J. Protective effects of statin and angiotensin receptor blocker in a rat model of doxorubicin-and trastuzumab-induced cardiomyopathy. J. Am. Soc. Echocardiogr. 2020, 33, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, A.; Rodolico, A.; Galdieri, A.; Russo, M.; Campi, G.; Franco, R.; Bruno, D.; Aran, L.; Carannante, A.; Attanasio, U.; et al. Heart failure and cancer: Mechanisms of old and new cardiotoxic drugs in cancer patients. Card. Fail. Rev. 2019, 5, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Saneeymehri, S.S.; Markey, K.R.; Mahipal, A. Paradoxical effect of capecitabine in 5-fluorouracil-induced cardiotoxicity: A case vignette and literature review. J. Oncol. Pharm. Pract. 2016, 22, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Shiga, T.; Hiraide, M. Cardiotoxicities of 5-Fluorouracil and other fluoropyrimidines. Curr. Treat. Options Oncol. 2020, 21, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alter, P.; Herzum, M.; Soufi, M.; Schaefer, J.R.; Maisch, B. Cardiotoxicity of 5-fluorouracil. Cardiovasc. Hematol. Agents Med. Chem. 2006, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sara, J.D.; Kaur, J.; Khodadadi, R.; Rehman, M.; Lobo, R.; Chakrabarti, S.; Herrmann, J.; Lerman, A.; Grothey, A. 5-fluorouracil and cardiotoxicity: A review. Ther. Adv. Med. Oncol. 2018, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Charkviani, M.; Murvelashvili, N.; Barrera, F.; Sharma, A.; Eldin, R.S.; Nabil, N.U. Rare presentation of cardiotoxicity related to 5-Fluorouracil. Case Rep. Oncol. Med. 2020, 2020, 214. [Google Scholar] [CrossRef]
- Dyhl-Polk, A.; Vaage-Nilsen, M.; Schou, M.; Vistisen, K.K.; Lund, C.M.; Kümler, T.; Appel, J.M.; Nielsen, D.L. Incidence and risk markers of 5-fluorouracil and capecitabine cardiotoxicity in patients with colorectal cancer. Acta Oncol. 2020, 59, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Nohria, A. The clinical conundrum of managing 5-fluorouracil–induced vasospasm in colorectal carcinoma. Cancer 2019, 125, 4346–4349. [Google Scholar] [CrossRef] [PubMed]
- Mishra, T.; Shokr, M.; Ahmed, A.; Afonso, L. Acute reversible left ventricular systolic dysfunction associated with 5-fluorouracil therapy: A rare and increasingly recognised cardiotoxicity of a commonly used drug. BMJ Case Rep. CP 2019, 12, e230499. [Google Scholar] [CrossRef]
- Muhammad, R.N.; Sallam, N.; El-Abhar, H.S. Activated ROCK/Akt/eNOS and ET-1/ERK pathways in 5-fluorouracil-induced cardiotoxicity: Modulation by simvastatin. Sci. Rep. 2020, 10, 1–4. [Google Scholar] [CrossRef]
- Bayrak, S.; Aktaş, S.; Altun, Z.; Çakir, Y.; Tütüncü, M.; Kum Özşengezer, S.; Yilmaz, O.; Olgun, N. Antioxidant effect of acetyl-l-carnitine against cisplatin-induced cardiotoxicity. J. Int. Med. Res. 2020, 48, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gunturk, E.E.; Yucel, B.; Gunturk, I.; Yazici, C.E.; Yay, A.R.; Kose, K. The effects of N-acetylcysteine on cisplatin induced cardiotoxicity. Bratislavskelekarskelisty 2019, 120, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, B.; Zhao, B.; Mei, D.; Gu, Q.; Tian, Z. Cisplatin-induced cardiotoxicity with midrange ejection fraction: A case report and review of the literature. Medicine 2018, 97, e13807. [Google Scholar] [CrossRef]
- Bahadır, A.; Ceyhan, A.; Gergin, Ö.Ö.; Yalçın, B.; Ülger, M.; Özyazgan, T.M.; Yay, A. Protective effects of curcumin and beta-carotene on cisplatin-induced cardiotoxicity: An experimental rat model. Anatol. J. Cardiol. 2018, 19, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Afsar, T.; Razak, S.; Almajwal, A.; Shabbir, M.; Khan, M.R. Evaluating the protective potency of Acacia hydaspica R. Parker on histological and biochemical changes induced by Cisplatin in the cardiac tissue of rats. BMC Complement. Altern. Med. 2019, 19, 1–2. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Bakhaat, G.A.; Tammam, H.G.; Mohamed, R.M.; El-Naggar, S.A. Cardioprotective effect of green tea extract and vitamin E on Cisplatin-induced cardiotoxicity in mice: Toxicological, histological and immunohistochemical studies. Biomed. Pharmacother. 2019, 113, 108731. [Google Scholar] [CrossRef]
- El-Hawwary, A.A.; Omar, N.M. The influence of ginger administration on cisplatin-induced cardiotoxicity in rat: Light and electron microscopic study. Actahistochemica 2019, 121, 553–562. [Google Scholar] [CrossRef]
- Darby, S.C.; Cutter, D.J.; Boerma, M.; Constine, L.S.; Fajardo, L.F.; Kodama, K.; Mabuchi, K.; Marks, L.B.; Mettler, F.A.; Pierce, L.J.; et al. Radiation-related heart disease: Current knowledge and future prospects. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varricchi, G.; Galdiero, M.R.; Marone, G.; Criscuolo, G.; Triassi, M.; Bonaduce, D.; Marone, G.; Tocchetti, C.G. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open 2017, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dal’bo, N.; Patel, R.; Parikh, R.; Shah, S.P.; Guha, A.; Dani, S.S.; Ganatra, S. Cardiotoxicity of contemporary anticancer immunotherapy. Curr. Treat. Options Cardiovasc. Med. 2020, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ganatra, S.; Parikh, R.; Neilan, T.G. Cardiotoxicity of immune therapy. Cardiol. Clin. 2019, 37, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Upadhrasta, S.; Elias, H.; Patel, K.; Zheng, L. Managing cardiotoxicity associated with immune checkpoint inhibitors. Chronic Dis. Transl. Med. 2019, 5, 6–14. [Google Scholar] [CrossRef]
- Lobenwein, D.; Kocher, F.; Dobner, S.; Gollmann-Tepeköylü, C.; Holfeld, J. Cardiotoxic mechanisms of cancer immunotherapy–A systematic review. Int. J. Cardiol. 2021, 323, 179–187. [Google Scholar] [CrossRef]
- Brüstle, K.; Heidecker, B. Checkpoint inhibitor induced cardiotoxicity: Managing the drawbacks of our newest agents against cancer. Oncotarget 2017, 8, 106165–106166. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.B.; Youn, J.C.; Youn, H.J. Cardiovascular complications of novel anti-cancer immunotherapy: Old problems from new agents? Korean Circ. J. 2020, 50, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer. 2016, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Varricchi, G.; Marone, G.; Mercurio, V.; Galdiero, M.R.; Bonaduce, D.; Tocchetti, C.G. Immune checkpoint inhibitors and cardiac toxicity: An emerging issue. Curr. Med. Chem. 2018, 25, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Zarifa, A.; Albittar, A.; Kim, P.Y.; Hassan, S.; Palaskas, N.; Iliescu, C.; Durand, J.B.; Lopez-Mattei, J. Cardiac toxicities of anticancer treatments: Chemotherapy, targeted therapy and immunotherapy. Curr. Opin. Cardiol. 2019, 34, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Stein-Merlob, A.F.; Rothberg, M.V.; Holman, P.; Yang, E.H. Immunotherapy-associated cardiotoxicity of immune checkpoint inhibitors and chimeric antigen receptor T cell therapy: Diagnostic and management challenges and strategies. Curr. Cardiol. Rep. 2021, 23, 1. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Chen, D.H.; Guha, A.; Mackenzie, S.; Walker, J.M.; Roddie, C. CAR T cell therapy–related cardiovascular outcomes and management: Systemic disease or direct cardiotoxicity? Cardio Oncol. 2020, 2, 97–109. [Google Scholar]
- O’leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; Roberts, T.M.; Kim, H.; Mcallister, S.S.; Jean, J. CDK4/6 inhibition triggers anti-tumor immunity. Nature 2017, 548, 471–475. [Google Scholar]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 inhibition in cancer: Beyond cell cycle arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Scott, S.C.; Lee, S.S.; Abraham, J. Mechanisms of therapeutic CDK4/6 inhibition in breast cancer. Semin. Oncol. 2017, 44, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Martel, S.; Maurer, C.; Lambertini, M.; Pondé, N.; De Azambuja, E. Breast cancer treatment-induced cardiotoxicity. Expert Opin. Drug Saf. 2017, 16, 1021–1038. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Occhipinti, G.; Romagnoli, E.; Miccini, F.; Scoccia, L.; Giulietti, M.; Principato, G.; Saladino, T.; Piva, F.; Battelli, N. Different cardiotoxicity of palbociclib and ribociclib in breast cancer: Gene expression and pharmacological data analyses, biological basis, and therapeutic implications. BioDrugs 2019, 33, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to anti-angiogenic therapy in cancer—Alterations to anti-VEGF pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [Green Version]
- Lyons, T.G.; Ku, G.Y. Systemic therapy for esophagogastric cancer: Targeted therapies. Chin. Clin. Oncol. 2017, 6, 48–55. [Google Scholar] [CrossRef]
- Cohen, A.C.; Roane, B.M.; Leath, C.A. Novel therapeutics for recurrent cervical cancer: Moving towards personalized therapy. Drugs 2020, 80, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Lu, Y. New developments in molecular targeted therapy of ovarian cancer. Discov. Med. 2018, 26, 219–229. [Google Scholar]
- Lal Goel, H.; Mercurio, A.M. VEGF as a potential target in lung cancer. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar]
- Zhang, H.; Liu, J.; Chen, Q.; Mi, P. Ligand-installed anti-VEGF genomic nanocarriers for effective gene therapy of primary and metastatic tumors. J. Control. Release 2020, 320, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Alvi, R.M.; Frigault, M.J.; Fradley, M.G.; Jain, M.D.; Mahmood, S.S.; Awadalla, M.; Lee, D.H.; Zlotoff, D.A.; Zhang, L.; Drobni, Z.D.; et al. Cardiovascular events among adults treated with chimeric antigen receptor T-cells (CAR-T). J. Am. Coll. Cardiol. 2019, 74, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Ganatra, S.; Carver, J.R.; Hayek, S.S.; Ky, B.; Leja, M.J.; Lenihan, D.J.; Lenneman, C.; Mousavi, N.; Park, J.H.; Perales, M.A.; et al. Chimeric antigen receptor T-cell therapy for cancer and heart: JACC council perspectives. J. Am. Coll. Cardiol. 2019, 74, 3153–3163. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, G.; Yang, T.; Guan, M.; An, N.; Yang, F.; Dai, Q.; Zhong, C.; Luo, C.; Gao, Y.; et al. Possible susceptibility genes for intervention against chemotherapy-induced cardiotoxicity. Oxid. Med. Cell Longev. 2020, 2020, 1–30. [Google Scholar] [CrossRef]
- Shimauchi, T.; Numaga-Tomita, T.; Ito, T.; Nishimura, A.; Matsukane, R.; Oda, S.; Hoka, S.; Ide, T.; Koitabashi, N.; Uchida, K.; et al. TRPC3-Nox2 complex mediates doxorubicin-induced myocardial atrophy. JCI Insight 2017, 2, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, E.; Gibbins, J.M.; Holbrook, L.M.; Palomo, I. NADPH oxidase 2 (NOX2): A key target of oxidative stress-mediated platelet activation and thrombosis. Trends Cardiovasc. Med. 2018, 28, 429–434. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative stress and cellular response to doxorubicin: A common factor in the complex milieu of anthracyclinecardiotoxicity. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Morgan, R.A.; Beck, K.R.; Nixon, M.; Homer, N.Z.; Crawford, A.A.; Melchers, D.; Houtman, R.; Meijer, O.C.; Stomby, A.; Anderson, A.J.; et al. Carbonyl reductase 1 catalyzes 20β-reduction of glucocorticoids, modulating receptor activation and metabolic complications of obesity. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fornaro, A.; Olivotto, I.; Rigacci, L.; Ciaccheri, M.; Tomberli, B.; Ferrantini, C.; Coppini, R.; Girolami, F.; Mazzarotto, F.; Chiostri, M.; et al. Comparison of long-term outcome in anthracycline-related versus idiopathic dilated cardiomyopathy: A single centre experience. Eur. J. Heart Fail. 2018, 20, 898–906. [Google Scholar] [CrossRef]
- Breysse, D.H.; Boone, R.M.; Long, C.M.; Merrill, M.E.; Schaupp, C.M.; White, C.C.; Kavanagh, T.J.; Schmidt, E.E.; Merrill, G.F. Carbonyl reductase 1 plays a significant role in converting doxorubicin to cardiotoxicdoxorubicinol in mouse liver, but the majority of the doxorubicinol-forming activity remains unidentified. Drug Metab. Dispos. 2020, 48, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, V.; Pirozzi, F.; Lazzarini, E.; Marone, G.; Rizzo, P.; Agnetti, G.; Tocchetti, C.G.; Ghigo, A.; Ameri, P. Models of heart failure based on the cardiotoxicity of anticancer drugs. J. Card. Fail. 2016, 22, 449–458. [Google Scholar] [CrossRef]
- Turcotte, M.; Allard, D.; Mittal, D.; Bareche, Y.; Buisseret, L.; José, V.; Pommey, S.; Delisle, V.; Loi, S.; Joensuu, H.; et al. CD73 promotes resistance to HER2/ErbB2 antibody therapy. Cancer Res. 2017, 77, 5652–5663. [Google Scholar] [CrossRef] [Green Version]
- Dokmanovic, M.; King, K.E.; Mohan, N.; Endo, Y.; Wu, W.J. Cardiotoxicity of ErbB2-targeted therapies and its impact on drug development, a spotlight on trastuzumab. Expert Opin. Drug Metab. Toxicol. 2017, 13, 755–766. [Google Scholar] [CrossRef]
- Belmonte, F.; Das, S.; Sysa-Shah, P.; Sivakumaran, V.; Stanley, B.; Guo, X.; Paolocci, N.; Aon, M.A.; Nagane, M.; Kuppusamy, P.; et al. ErbB2 overexpression upregulates antioxidant enzymes, reduces basal levels of reactive oxygen species, and protects against doxorubicin cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, 1271–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentassuglia, L.; Timolati, F.; Seifriz, F.; Abudukadier, K.; Suter, T.M.; Zuppinger, C. Inhibition of ErbB2/neuregulinsignalling augments paclitaxel-induced cardiotoxicity in adult ventricular myocytes. Exp. Cell Res. 2007, 313, 1588–1601. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Khalifa, H.A.; Ahmed, A.A. Allicin ameliorates doxorubicin-induced cardiotoxicity in rats via suppression of oxidative stress, inflammation and apoptosis. Cancer Chemother. Pharmacol. 2017, 80, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Linschoten, M.; Teske, A.J.; Baas, A.F.; Vink, A.; Dooijes, D.; Baars, H.F.; Asselbergs, F.W. Truncating Titin (TTN) variants in chemotherapy-induced cardiomyopathy. J. Card. Fail. 2017, 23, 476–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.Q.; Zhu, S.Y.; He, Y.; Yu, K.D. Association between a tri-allelic polymorphism in the estrogen metabolism oxidoreductase NRH: Quinone oxidoreductase 2 gene and risk of breast cancer by molecular subtype. Front. Genet. 2021, 12, 343. [Google Scholar]
- Ronkainen, V.P.; Tuomainen, T.; Huusko, J.; Laidinen, S.; Malinen, M.; Palvimo, J.J.; Ylä-Herttuala, S.; Vuolteenaho, O.; Tavi, P. Hypoxia-inducible factor 1-induced G protein-coupled receptor 35 expression is an early marker of progressive cardiac remodelling. Cardiovasc. Res. 2014, 101, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, S.; Pathak, A.; Franck, D.; Latorzeff, I.; Jimenez, G.; Fondard, O.; Lapeyre, M.; Colombier, D.; Bruguiere, E.; Lairez, O.; et al. Early detection and prediction of cardiotoxicity after radiation therapy for breast cancer: The BACCARAT prospective cohort study. Radiat. Oncol. 2016, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vošmik, M.; Hodek, M.; Buka, D.; Sýkorová, P.; Grepl, J.; Paluska, P.; Paulíková, S.; Sirák, I. Cardiotoxicity of radiation therapy in esophageal cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Mudd, T.W., Jr.; Khalid, M.; Guddati, A.K. Cardiotoxicity of chemotherapy and targeted agents. Am. J. Cancer Res. 2021, 11, 1132. [Google Scholar]
- Bojan, A.; Torok-Vistai, T.; Parvu, A. Assessment and management of cardiotoxicity in hematologic malignancies. Dis. Markers 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Loap, P.; Kirov, K.; Kirova, Y. Cardiotoxicity in breast cancer patients treated with radiation therapy: From evidences to controversies. Crit. Rev. Oncol. Hematol. 2020, 31, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Shoukat, S.; Zheng, D.; Yusuf, S.W. Cardiotoxicity related to radiation therapy. Cardiol. Clin. 2019, 37, 449–458. [Google Scholar] [CrossRef]
- Boero, I.J.; Paravati, A.J.; Triplett, D.P.; Hwang, L.; Matsuno, R.K.; Gillespie, E.F.; Yashar, C.M.; Moiseenko, V.; Einck, J.P.; Mell, L.K.; et al. Modern radiation therapy and cardiac outcomes in breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.Z.; Satyam, S.M.; Shetty, P.; D’Souza, M.R. Methyl gallate attenuates doxorubicin-induced cardiotoxicity in rats by suppressing oxidative stress. Scientifica 2021, 2021, 6694340. [Google Scholar] [CrossRef] [PubMed]
- Birari, L.; Wagh, S.; Patil, K.R.; Mahajan, U.B.; Unger, B.; Belemkar, S.; Goyal, S.N.; Ojha, S.; Patil, C.R. Aloin alleviates doxorubicin-induced cardiotoxicity in rats by abrogating oxidative stress and pro-inflammatory cytokines. Cancer Chemother. Pharmacol. 2020, 86, 419–426. [Google Scholar] [CrossRef]
- Hu, X.; Li, B.; Li, L.; Li, B.; Luo, J.; Shen, B. Asiatic acid protects against doxorubicin-induced cardiotoxicity in mice. Oxid. Med. Cell. Longev. 2020, 2020, 5347204. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.Y.; Yuan, Y.P.; Zhang, X.; Kong, C.Y.; Song, P.; Ma, Z.G.; Tang, Q.Z. Protection against doxorubicin-induced cytotoxicity by geniposide involves AMPKα signaling pathway. Oxid. Med. Cell. Longev. 2019, 2019, 7901735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.B.; Zheng, Y.F.; Wu, Y.G. Protective effects of oroxylin a against doxorubicin-induced cardiotoxicity via the activation of Sirt1 in mice. Oxid. Med. Cell. Longev. 2021, 2021, 6610543. [Google Scholar] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2015, 15, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, K.D. Dietary polyphenols: Antioxidants or not? Arch. Biochem. Biophys. 2016, 595, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandu, M.; Bîrsă, L.M.; Bahrin, L.G. Flavonoids–small molecules, high hopes. Acta Chem. Iasi 2017, 25, 6–23. [Google Scholar] [CrossRef] [Green Version]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The immunomodulatory and anti-inflammatory role of polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, D.L.; Nohria, A. Prevention of chemotherapy induced cardiomyopathy. Curr. Heart Fail. Rep. 2017, 14, 398–403. [Google Scholar] [CrossRef]

| Drugs Causing Ischemia or Thromboembolism | Cisplatin, Thalidomide, Fluorouracil, Capecitabine, Paclitaxel, Docetaxel, Trastuzumab, Anthracyclines/Anthraquinones, Cyclophosphamide |
|---|---|
| Drugs that Cause Hypertension | bevacizumab, cisplatin, sunitinib, sorafenib |
| Tamponade and Endomyocardial Fibrosis | busulfan |
| Autonomic Neuropathy | vincristine |
| Bradyarrhythmias | paclitaxel |
| Myocarditis with Hemorrhage (rare) | cyclophosphamide (high-dose therapy) |
| Pulmonary Fibrosis | bleomycin, methotrexate, busulfan, cyclophosphamide |
| Raynaud’s Phenomenon | vinblastine, bleomycin |
| Torsades de Pointes or QT Prolongation | arsenic trioxide |
| Title | Drugs under Trial | Phase | Justification of the Study |
|---|---|---|---|
| Anticancer Drug-Induced Cardiac Toxicity in High-Risk Patients (NCT00292526) | Enalapril | Phase 4 | The patients who received chemotherapy experienced an elevation of troponin I responsible for development of left ventricular dysfunction and altered cardiovascular functions. The activation of renin–angiotensin system is responsible for development of several myocardial dysfunctions and resulted in chemotherapy-induced cardiotoxicity (CTIC). This study found the effect of ACE inhibitors in the prevention of CTIC in high-risk cancer patients. |
| Protective Effects of the Nutritional Supplement Sulforaphane on Doxorubicin-Associated Cardiac Dysfunction (NCT03934905) | Sulforaphane as supplement | Phase 2 | Sulforaphane (SFN) is responsible for activation of transcription factor Nrf2 and induces defense mechanisms in normal cells. SFN was shown to inhibit carcinogenesis and metastases and increase the sensitivity of cancer cells to doxorubicin. |
| Cardiotoxicity Prevention in Breast Cancer Patients Treated with Anthracyclines and/or Trastuzumab (NCT02236806) | Bisoprolol and ramipril | Phase 3 | This study showed a protective effect of beta blockers and ACE inhibitors against breast cancer patients treated with anthracyclines with or without trastuzumab. |
| STOP-CA (Statins TO Prevent the Cardiotoxicity from Anthracyclines) (NCT02943590) | Statins | Phase 2 | This study demonstrated the protective effect of atorvastatin, a drug used in the treatment of hyperlipidemia against doxorubicin-induced cardiac damage. |
| Carvedilol Effect in Preventing Chemotherapy-Induced Cardiotoxicity (NCT01724450) | Carvedilol | Phase 3 | This research found the preventive effect of carvedilol against chemotherapy-induced cardiotoxicity in breast cancer patients. |
| Prevention of Chemotherapy-Induced Cardiotoxicity in Children with Bone Tumors and Acute Myeloid Leukemia (NCT03389724) | Captopril | Phase 3 | This study found the protective effect of captopril against chemotherapy-induced cardiotoxicity in children with bone tumors and acute myeloid leukemia. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adhikari, A.; Asdaq, S.M.B.; Al Hawaj, M.A.; Chakraborty, M.; Thapa, G.; Bhuyan, N.R.; Imran, M.; Alshammari, M.K.; Alshehri, M.M.; Harshan, A.A.; et al. Anticancer Drug-Induced Cardiotoxicity: Insights and Pharmacogenetics. Pharmaceuticals 2021, 14, 970. https://doi.org/10.3390/ph14100970
Adhikari A, Asdaq SMB, Al Hawaj MA, Chakraborty M, Thapa G, Bhuyan NR, Imran M, Alshammari MK, Alshehri MM, Harshan AA, et al. Anticancer Drug-Induced Cardiotoxicity: Insights and Pharmacogenetics. Pharmaceuticals. 2021; 14(10):970. https://doi.org/10.3390/ph14100970
Chicago/Turabian StyleAdhikari, Archana, Syed Mohammed Basheeruddin Asdaq, Maitham A. Al Hawaj, Manodeep Chakraborty, Gayatri Thapa, Nihar Ranjan Bhuyan, Mohd. Imran, Mohammed Kanan Alshammari, Mohammed M. Alshehri, Aishah Ali Harshan, and et al. 2021. "Anticancer Drug-Induced Cardiotoxicity: Insights and Pharmacogenetics" Pharmaceuticals 14, no. 10: 970. https://doi.org/10.3390/ph14100970
APA StyleAdhikari, A., Asdaq, S. M. B., Al Hawaj, M. A., Chakraborty, M., Thapa, G., Bhuyan, N. R., Imran, M., Alshammari, M. K., Alshehri, M. M., Harshan, A. A., Alanazi, A., Alhazmi, B. D., & Sreeharsha, N. (2021). Anticancer Drug-Induced Cardiotoxicity: Insights and Pharmacogenetics. Pharmaceuticals, 14(10), 970. https://doi.org/10.3390/ph14100970