
T Cell Bispecific Antibodies: An Antibody-Based Delivery System for Inducing Antitumor Immunity


Abstract
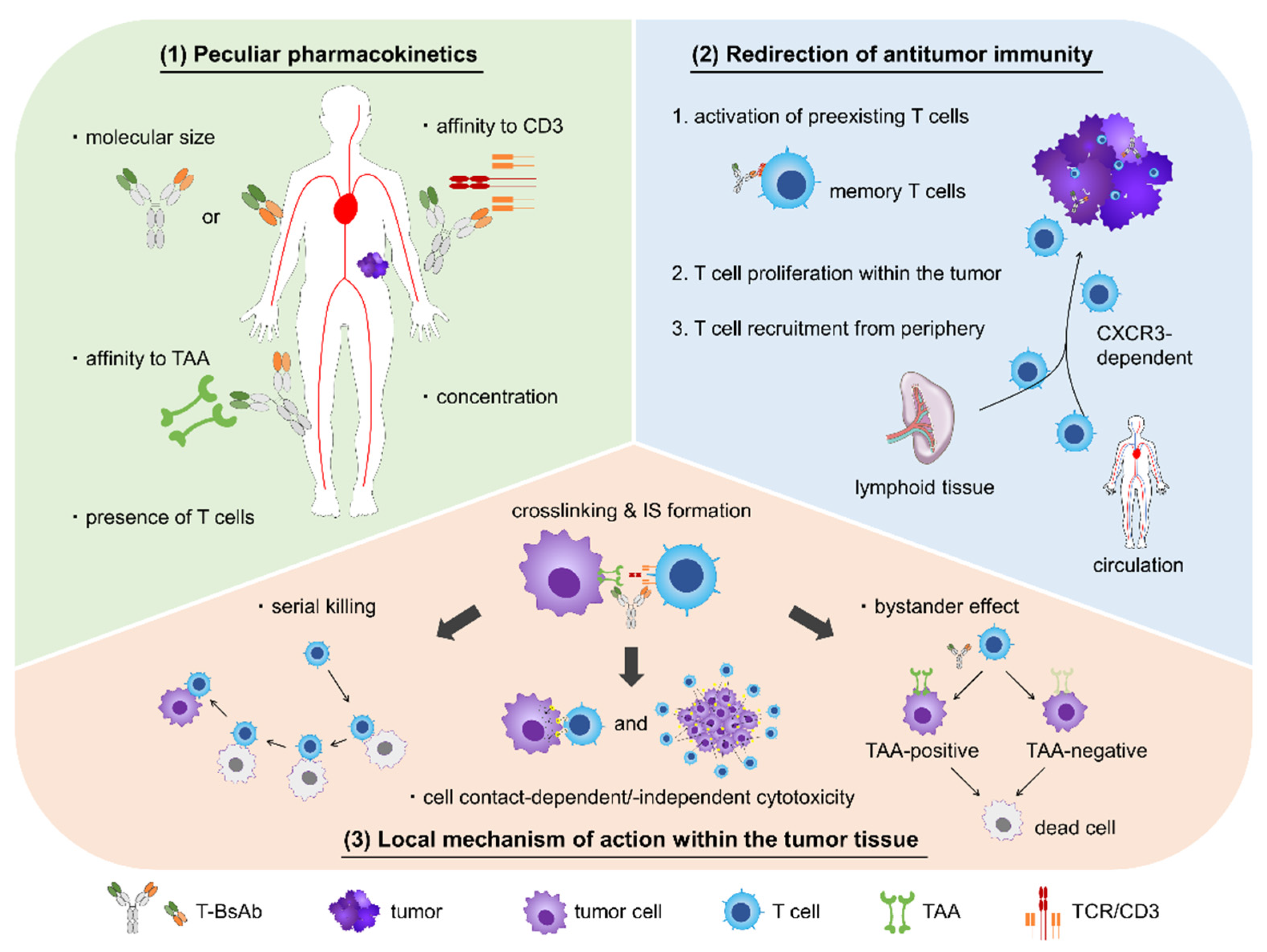
:1. Introduction
2. Pharmacology of T-BsAbs
2.1. Unique PK
2.2. Redirection of Antitumor Immunity
2.3. Local Mechanism of Action within Tumor Tissues
3. T-BsAbs in Development for Solid Tumors
3.1. Preclinical Research

{kind=link}
| Strategy | Executive Summary | Reference |
|---|---|---|
| Antigen Selection | • HER2, EGFR, EpCAM, and CEA are common antigens that have been targeted by T-BsAbs. | [97,111,116,120] |
| • Aimed at mitigating on-target toxicity to normal tissues, more tumor-specific antigens, such as mutant proteins and complexes of a glycan and a protein, have been exploited as a T-BsAb target. | [110,122,123] | |
| Format Selection | • Asymmetrical conformation consisting of an anti-TAA moiety and an anti-CD3 moiety provides an advantage in the purification of the targeted heterodimer. | [98] |
| • Although multivalency of T-BsAbs for a TAA is beneficial for its potency and specificity, one for a CD3 has pros and cons. | [102,104] | |
| • A suitable domain order of variant fragments brings about efficient crosslinking of two antigens and T cell-mediated cytotoxicity. | [106,114] | |
| Combination therapy | • T-BsAb therapy in combination with CPI therapy has synergistic effects against multiple types of tumor, including hematological and solid tumors. | [57,70,97] |
| • Inducing costimulatory signals with an agonist, CD28 or 4-1BB ligand, strengthens therapeutic efficacy of T-BsAbs. | [80,136] | |
| Utilization of oncolytic virusis | • Tumor-specific viruses can be applied as a delivery tool for T-BsAbs | [137,138] |
| • Pretreatment with oncolytic virus promotes T cell infiltration by inducing immune reactions within tumor tissues, which results in enhanced antitumor activity of subsequent T-BsAb therapy. | [141] |
3.2. Clinical Research
4. Future Perspectives, Challenges, and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Schulze, H.J.; Cribier, B.; Requena, L.; Reifenberger, J.; Ferrándiz, C.; Garcia Diez, A.; Tebbs, V.; McRae, S. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: Results from a randomized vehicle-controlled phase III study in Europe. Br. J. Dermatol. 2005, 152, 939–947. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laetsch, T.W.; Myers, G.D.; Baruchel, A.; Dietz, A.C.; Pulsipher, M.A.; Bittencourt, H.; Buechner, J.; De Moerloose, B.; Davis, K.L.; Nemecek, E.; et al. Patient-reported quality of life after tisagenlecleucel infusion in children and young adults with relapsed or refractory B-cell acute lymphoblastic leukaemia: A global, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 1710–1718. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Kümpers, C.; Jokic, M.; Haase, O.; Offermann, A.; Vogel, W.; Grätz, V.; Langan, E.A.; Perner, S.; Terheyden, P. Immune Cell Infiltration of the Primary Tumor, Not PD-L1 Status, Is Associated With Improved Response to Checkpoint Inhibition in Metastatic Melanoma. Front. Med. 2019, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.-Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin. Cancer Res. 2019, 25, 5002–5014. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Tian, Y.; Li, Y.; Zhang, W.; Cai, W.; Liu, Y.; Ren, Y.; Liang, Z.; Zhou, P.; Zhang, Y.; et al. In vivo therapeutic effects of affinity-improved-TCR engineered T-cells on HBV-related hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e001748. [Google Scholar] [CrossRef]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]
- Trabolsi, A.; Arumov, A.; Schatz, J.H. T Cell-Activating Bispecific Antibodies in Cancer Therapy. J. Immunol. 2019, 203, 585–592. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Renner, C.; Held, G.; Ohnesorge, S.; Bauer, S.; Gerlach, K.; Pfitzenmeier, J.P.; Pfreundschuh, M. Role of naive and memory T cells in tumor cell lysis mediated by bi-specific antibodies. Immunobiology 1997, 197, 122–132. [Google Scholar] [CrossRef]
- Huo, Y.; Sheng, Z.; Lu, D.R.; Ellwanger, D.C.; Li, C.M.; Homann, O.; Wang, S.; Yin, H.; Ren, R. Blinatumomab-induced T cell activation at single cell transcriptome resolution. BMC Genom. 2021, 22, 145. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Keizer, R.J.; Huitema, A.D.; Schellens, J.H.; Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharm. 2010, 49, 493–507. [Google Scholar] [CrossRef]
- Unverdorben, F.; Richter, F.; Hutt, M.; Seifert, O.; Malinge, P.; Fischer, N.; Kontermann, R.E. Pharmacokinetic properties of IgG and various Fc fusion proteins in mice. MAbs 2016, 8, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Yasunaga, M.; Manabe, S.; Tsuji, A.; Furuta, M.; Ogata, K.; Koga, Y.; Saga, T.; Matsumura, Y. Development of Antibody-Drug Conjugates Using DDS and Molecular Imaging. Bioengineering 2017, 4, 78. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Krippendorff, B.F.; Shah, D.K. Influence of Molecular size on the clearance of antibody fragments. Pharm. Res. 2017, 34, 2131–2141. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: An emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef]
- Einsele, H.; Borghaei, H.; Orlowski, R.Z.; Subklewe, M.; Roboz, G.J.; Zugmaier, G.; Kufer, P.; Iskander, K.; Kantarjian, H.M. The BiTE (bispecific T-cell engager) platform: Development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer 2020, 126, 3192–3201. [Google Scholar] [CrossRef]
- Huang, L.; Shah, K.; Barat, B.; Lam, C.K.; Gorlatov, S.; Ciccarone, V.; Tamura, J.; Moore, P.A.; Diedrich, G. Multispecific, Multivalent Antibody-Based Molecules Engineered on the DART® and TRIDENT(TM) Platforms. Curr. Protoc. Immunol. 2020, 129, e95. [Google Scholar] [CrossRef]
- Bühler, P.; Molnar, E.; Dopfer, E.P.; Wolf, P.; Gierschner, D.; Wetterauer, U.; Schamel, W.W.; Elsässer-Beile, U. Target-dependent T-cell activation by coligation with a PSMA x CD3 diabody induces lysis of prostate cancer cells. J. Immunother. 2009, 32, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Asano, R.; Kawaguchi, H.; Watanabe, Y.; Nakanishi, T.; Umetsu, M.; Hayashi, H.; Katayose, Y.; Unno, M.; Kudo, T.; Kumagai, I. Diabody-based recombinant formats of humanized IgG-like bispecific antibody with effective retargeting of lymphocytes to tumor cells. J. Immunother. 2008, 31, 752–761. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Hummel, H.D.; Kufer, P.; Grüllich, C.; Seggewiss-Bernhardt, R.; Deschler-Baier, B.; Chatterjee, M.; Goebeler, M.E.; Miller, K.; de Santis, M.; Loidl, W.; et al. Pasotuxizumab, a BiTE® immune therapy for castration-resistant prostate cancer: Phase I, dose-escalation study findings. Immunotherapy 2021, 13, 125–141. [Google Scholar] [CrossRef]
- Pishvaian, M.; Morse, M.A.; McDevitt, J.; Norton, J.D.; Ren, S.; Robbie, G.J.; Ryan, P.C.; Soukharev, S.; Bao, H.; Denlinger, C.S. Phase 1 Dose Escalation Study of MEDI-565, a Bispecific T-Cell Engager that Targets Human Carcinoembryonic Antigen, in Patients With Advanced Gastrointestinal Adenocarcinomas. Clin. Colorectal. Cancer 2016, 15, 345–351. [Google Scholar] [CrossRef] [PubMed]
- von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’Brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.A.; Shah, K.; Yang, Y.; Alderson, R.; Roberts, P.; Long, V.; Liu, D.; Li, J.C.; Burke, S.; Ciccarone, V.; et al. Development of MGD007, a gpA33 x CD3-Bispecific DART Protein for T-Cell Immunotherapy of Metastatic Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root, A.R.; Cao, W.; Li, B.; LaPan, P.; Meade, C.; Sanford, J.; Jin, M.; O’Sullivan, C.; Cummins, E.; Lambert, M.; et al. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies 2016, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Deegen, P.; Thomas, O.; Nolan-Stevaux, O.; Li, S.; Wahl, J.; Bogner, P.; Aeffner, F.; Friedrich, M.; Liao, M.Z.; Matthes, K.; et al. The PSMA Targeting Half-Life Extended BiTE® Therapy AMG 160 Has Potent Antitumor Activity in Preclinical Models of Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Karle, A.; Meissburger, B.; Höfig, I.; Stork, R.; Kontermann, R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007, 282, 12650–12660. [Google Scholar] [CrossRef] [Green Version]
- Suurs, F.V.; Lorenczewski, G.; Bailis, J.M.; Stienen, S.; Friedrich, M.; de Vries, E.G.E.; de Groot, D.J.A.; Lub-de Hooge, M.N. Abstract 2769: PET imaging shows dose-dependent pharmacokinetics of a 89Zr-labeled mesothelin/CD3 half-life extended bispecific T-cell engager molecule in a syngeneic mouse model. Cancer Res. 2020, 80, 2769. [Google Scholar] [CrossRef]
- Chao, D.T.; Ma, X.; Li, O.; Park, H.; Law, D. Functional characterization of N297A, a murine surrogate for low-Fc binding anti-human CD3 antibodies. Immunol. Investig. 2009, 38, 76–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Alegre, M.L.; Varga, S.S.; Rothermel, A.L.; Collins, A.M.; Pulito, V.L.; Hanna, L.S.; Dolan, K.P.; Parren, P.W.; Bluestone, J.A.; et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol. 2000, 200, 16–26. [Google Scholar] [CrossRef]
- Schlothauer, T.; Herter, S.; Koller, C.F.; Grau-Richards, S.; Steinhart, V.; Spick, C.; Kubbies, M.; Klein, C.; Umaña, P.; Mössner, E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 2016, 29, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakahara, H.; Endo, K.; Koizumi, M.; Nakashima, T.; Kunimatsu, M.; Watanabe, Y.; Kawamura, Y.; Nakamura, T.; Tanaka, H.; Kotoura, Y.; et al. Relationship between in vitro binding activity and in vivo tumor accumulation of radiolabeled monoclonal antibodies. J. Nucl. Med. 1988, 29, 235–240. [Google Scholar] [PubMed]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [Green Version]
- Tsumura, R.; Manabe, S.; Takashima, H.; Koga, Y.; Yasunaga, M.; Matsumura, Y. Influence of the dissociation rate constant on the intra-tumor distribution of antibody-drug conjugate against tissue factor. J. Control. Release 2018, 284, 49–56. [Google Scholar] [CrossRef]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K.; et al. Relative Target Affinities of T-Cell-Dependent Bispecific Antibodies Determine Biodistribution in a Solid Tumor Mouse Model. Mol. Cancer Ther. 2018, 17, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- List, T.; Neri, D. Biodistribution studies with tumor-targeting bispecific antibodies reveal selective accumulation at the tumor site. MAbs 2012, 4, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; Perera, R.; Grimm, H.P.; Sam, J.; Colombetti, S.; Fauti, T.; Fahrni, L.; Schaller, T.; Freimoser-Grundschober, A.; Zielonka, J.; et al. In Vivo Fluorescence Imaging of the Activity of CEA TCB, a Novel T-Cell Bispecific Antibody, Reveals Highly Specific Tumor Targeting and Fast Induction of T-Cell-Mediated Tumor Killing. Clin. Cancer Res. 2016, 22, 4417–4427. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, T.; Sano, Y.; Komatsu, S.I.; Kamata-Sakurai, M.; Kaneko, A.; Kinoshita, Y.; Shiraiwa, H.; Azuma, Y.; Tsunenari, T.; Kayukawa, Y.; et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Cooke, K.; Estrada, J.; Zhan, J.; Werner, J.; Caenepeel, S.; Giffin, M.; Bailis, J.M.; Coxon, A.; Hughes, P.E.; Canon, J. Abstract 4558: Antitumor activity of AMG757, a half-life extended (HLE) bispecific T-cell engager (BiTE®) immune therapy targeting DLL3, in human PDX and orthotopic mouse models of small cell lung cancer (SCLC). Cancer Res. 2020, 80, 4558. [Google Scholar] [CrossRef]
- Mathur, D.; Root, A.R.; Bugaj-Gaweda, B.; Bisulco, S.; Tan, X.; Fang, W.; Kearney, J.C.; Lucas, J.; Guffroy, M.; Golas, J.; et al. A Novel GUCY2C-CD3 T-Cell Engaging Bispecific Construct (PF-07062119) for the Treatment of Gastrointestinal Cancers. Clin. Cancer Res. 2020, 26, 2188–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Hoseini, S.S.; Xu, H.; Ponomarev, V.; Cheung, N.K. Silencing Fc Domains in T cell-Engaging Bispecific Antibodies Improves T-cell Trafficking and Antitumor Potency. Cancer Immunol. Res. 2019, 7, 2013–2024. [Google Scholar] [CrossRef]
- Haber, L.; Olson, K.; Babb, R.; Kelly, M.; Crawford, A.; Retter, M.; DiLillo, D.; Ullman, E.; Finney, J.; Canova, L.; et al. Abstract 4555: Selection of CD3 affinity allows generation of T-cell redirecting bispecific antibodies with unique pharmacokinetic and biodistribution properties. Cancer Res. 2020, 80, 4555. [Google Scholar] [CrossRef]
- Holland, C.J.; Crean, R.M.; Pentier, J.M.; de Wet, B.; Lloyd, A.; Srikannathasan, V.; Lissin, N.; Lloyd, K.A.; Blicher, T.H.; Conroy, P.J.; et al. Specificity of bispecific T cell receptors and antibodies targeting peptide-HLA. J. Clin. Investig. 2020, 130, 2673–2688. [Google Scholar] [CrossRef] [Green Version]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef]
- Kim, A.; Han, C.J.; Driver, I.; Olow, A.; Sewell, A.K.; Zhang, Z.; Ouyang, W.; Egen, J.G.; Yu, X. LILRB1 Blockade Enhances Bispecific T Cell Engager Antibody-Induced Tumor Cell Killing by Effector CD8(+) T Cells. J. Immunol. 2019, 203, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- Meermeier, E.W.; Welsh, S.J.; Sharik, M.E.; Du, M.T.; Garbitt, V.M.; Riggs, D.L.; Shi, C.X.; Stein, C.K.; Bergsagel, M.; Chau, B.; et al. Tumor burden limits bispecific antibody efficacy through T cell exhaustion averted by concurrent cytotoxic therapy. Blood Cancer Discov. 2021, 2, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Benonisson, H.; Altıntaş, I.; Sluijter, M.; Verploegen, S.; Labrijn, A.F.; Schuurhuis, D.H.; Houtkamp, M.A.; Verbeek, J.S.; Schuurman, J.; van Hall, T. CD3-Bispecific Antibody Therapy Turns Solid Tumors into Inflammatory Sites but Does Not Install Protective Memory. Mol. Cancer Ther. 2019, 18, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.R.; Root, A.; Fisher, T.; Norberg, R.; David, J.; Clark, T.; Cohen, J.; May, C.; Giddabasappa, A. Molecular imaging reveals biodistribution of P-cadherin LP-DART bispecific and trafficking of adoptively transferred T cells in mouse xenograft model. Oncotarget 2020, 11, 1344–1357. [Google Scholar] [CrossRef] [Green Version]
- Hipp, S.; Tai, Y.T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2017, 31, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Zuch de Zafra, C.L.; Fajardo, F.; Zhong, W.; Bernett, M.J.; Muchhal, U.S.; Moore, G.L.; Stevens, J.; Case, R.; Pearson, J.T.; Liu, S.; et al. Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-cell–recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin. Cancer Res. 2019, 25, 3921–3933. [Google Scholar] [CrossRef] [Green Version]
- Chiu, D.; Tavaré, R.; Haber, L.; Aina, O.H.; Vazzana, K.; Ram, P.; Danton, M.; Finney, J.; Jalal, S.; Krueger, P.; et al. A PSMA-Targeting CD3 Bispecific Antibody Induces Antitumor Responses that Are Enhanced by 4-1BB Costimulation. Cancer Immunol. Res. 2020, 8, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Sam, J.; Colombetti, S.; Fauti, T.; Roller, A.; Biehl, M.; Fahrni, L.; Nicolini, V.; Perro, M.; Nayak, T.; Bommer, E.; et al. Combination of T-Cell Bispecific Antibodies With PD-L1 Checkpoint Inhibition Elicits Superior Anti-Tumor Activity. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ybarra, R.; Mak, J.; Herault, A.; De Almeida, P.; Arrazate, A.; Ziai, J.; Totpal, K.; Junttila, M.R.; Walsh, K.B.; et al. IFNγ-induced Chemokines Are Required for CXCR3-mediated T-Cell Recruitment and Antitumor Efficacy of Anti-HER2/CD3 Bispecific Antibody. Clin. Cancer Res. 2018, 24, 6447–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franciszkiewicz, K.; Boissonnas, A.; Boutet, M.; Combadière, C.; Mami-Chouaib, F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012, 72, 6325–6332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppaluri, R.; Sheehan, K.C.; Wang, L.; Bui, J.D.; Brotman, J.J.; Lu, B.; Gerard, C.; Hancock, W.W.; Schreiber, R.D. Prolongation of cardiac and islet allograft survival by a blocking hamster anti-mouse CXCR3 monoclonal antibody. Transplantation 2008, 86, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremasco, F.; Menietti, E.; Speziale, D.; Sam, J.; Sammicheli, S.; Richard, M.; Varol, A.; Klein, C.; Umana, P.; Bacac, M.; et al. Cross-linking of T cell to B cell lymphoma by the T cell bispecific antibody CD20-TCB induces IFNγ/CXCL10-dependent peripheral T cell recruitment in humanized murine model. PLoS ONE 2021, 16, e0241091. [Google Scholar] [CrossRef]
- Hoffmann, P.; Hofmeister, R.; Brischwein, K.; Brandl, C.; Crommer, S.; Bargou, R.; Itin, C.; Prang, N.; Baeuerle, P.A. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int. J. Cancer 2005, 115, 98–104. [Google Scholar] [CrossRef]
- Bromley, S.K.; Burack, W.R.; Johnson, K.G.; Somersalo, K.; Sims, T.N.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse. Annu. Rev. Immunol. 2001, 19, 375–396. [Google Scholar] [CrossRef]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Roda-Navarro, P.; Álvarez-Vallina, L. Understanding the Spatial Topology of Artificial Immunological Synapses Assembled in T Cell-Redirecting Strategies: A Major Issue in Cancer Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 370. [Google Scholar] [CrossRef]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Rossi, E.A.; Rossi, D.L.; Cardillo, T.M.; Chang, C.-H.; Goldenberg, D.M. Redirected T-Cell Killing of Solid Cancers Targeted with an Anti-CD3/Trop-2–Bispecific Antibody Is Enhanced in Combination with Interferon-α. Mol. Cancer Ther. 2014, 13, 2341–2351. [Google Scholar] [CrossRef] [Green Version]
- Kamakura, D.; Asano, R.; Kawai, H.; Yasunaga, M. Mechanism of action of a T cell-dependent bispecific antibody as a breakthrough immunotherapy against refractory colorectal cancer with an oncogenic mutation. Cancer Immunol. Immunother. 2021, 70, 177–188. [Google Scholar] [CrossRef]
- Choudhuri, K.; Wiseman, D.; Brown, M.H.; Gould, K.; van der Merwe, P.A. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature 2005, 436, 578–582. [Google Scholar] [CrossRef] [PubMed]
- James, J.R.; Vale, R.D. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature 2012, 487, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Bluemel, C.; Hausmann, S.; Fluhr, P.; Sriskandarajah, M.; Stallcup, W.B.; Baeuerle, P.A.; Kufer, P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010, 59, 1197–1209. [Google Scholar] [CrossRef]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuellner, U.; Klupsch, K.; Buller, F.; Attinger-Toller, I.; Santimaria, R.; Zbinden, I.; Henne, P.; Grabulovski, D.; Bertschinger, J.; Brack, S. Bispecific CD3/HER2 Targeting FynomAb Induces Redirected T Cell-Mediated Cytolysis with High Potency and Enhanced Tumor Selectivity. Antibodies 2015, 4, 426–440. [Google Scholar] [CrossRef] [Green Version]
- Santich, B.H.; Park, J.A.; Tran, H.; Guo, H.F.; Huse, M.; Cheung, N.V. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Renner, C.; Held, G.; Ohnesorge, S.; Bauer, S.; Gerlach, K.; Pfitzenmeier, J.P.; Pfreundschuh, M. Role of perforin, granzymes and the proliferative state of the target cells in apoptosis and necrosis mediated by bispecific-antibody-activated cytotoxic T cells. Cancer Immunol. Immunother. 1997, 44, 70–76. [Google Scholar] [CrossRef]
- Gruen, M.; Bommert, K.; Bargou, R.C. T-cell-mediated lysis of B cells induced by a CD19xCD3 bispecific single-chain antibody is perforin dependent and death receptor independent. Cancer Immunol. Immunother. 2004, 53, 625–632. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.J.; Iwasa, S.; Sugimoto, N.; Ryu, M.H.; Sakai, D.; Chung, H.C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef]
- Ross, S.L.; Sherman, M.; McElroy, P.L.; Lofgren, J.A.; Moody, G.; Baeuerle, P.A.; Coxon, A.; Arvedson, T. Bispecific T cell engager (BiTE®) antibody constructs can mediate bystander tumor cell killing. PLoS ONE 2017, 12, e0183390. [Google Scholar] [CrossRef] [PubMed]
- Voynov, V.; Adam, P.J.; Nixon, A.E.; Scheer, J.M. Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors. Antibodies 2020, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Junttila, T.T.; Li, J.; Johnston, J.; Hristopoulos, M.; Clark, R.; Ellerman, D.; Wang, B.E.; Li, Y.; Mathieu, M.; Li, G.; et al. Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells. Cancer Res. 2014, 74, 5561–5571. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Zhang, J.; Yan, Y.; Yao, X.; Fang, L.; Xiong, H.; Liu, Y.; Chu, Q.; Zhou, P.; Wu, K. A novel asymmetrical anti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2-positive tumor cells. J. Exp. Clin. Cancer Res. 2019, 38, 355. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Albaitero, A.; Xu, H.; Guo, H.; Wang, L.; Wu, Z.; Tran, H.; Chandarlapaty, S.; Scaltriti, M.; Janjigian, Y.; de Stanchina, E.; et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody. Oncoimmunology 2017, 6, e1267891. [Google Scholar] [CrossRef] [Green Version]
- Park, J.A.; Cheung, N.V. GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J. Hematol. Oncol. 2020, 13, 172. [Google Scholar] [CrossRef]
- Ahmed, M.; Cheng, M.; Cheung, I.Y.; Cheung, N.K. Human derived dimerization tag enhances tumor killing potency of a T-cell engaging bispecific antibody. Oncoimmunology 2015, 4, e989776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaga, D.; Ellerman, D.; Lombana, T.N.; Vij, R.; Li, J.; Hristopoulos, M.; Clark, R.; Johnston, J.; Shelton, A.; Mai, E.; et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Asano, R.; Ikoma, K.; Sone, Y.; Kawaguchi, H.; Taki, S.; Hayashi, H.; Nakanishi, T.; Umetsu, M.; Katayose, Y.; Unno, M.; et al. Highly enhanced cytotoxicity of a dimeric bispecific diabody, the hEx3 tetrabody. J. Biol. Chem. 2010, 285, 20844–20849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reusch, U.; Harrington, K.H.; Gudgeon, C.J.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.H.; Zhukovsky, E.A.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 Tetravalent Bispecific Tandem Diabodies (TandAbs) for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeau, M.R.; Miller, R.E.; Bader, R.; Gottschalk, R.; Daugherty, M.; Sewell, T.; Misher, L.; Parr, L.; DeFrancesco, M.; Bienvenue, D.; et al. Abstract 1786: APVO436, a bispecific anti-CD123 x anti-CD3 ADAPTIR™ molecule for redirected T-cell cytotoxicity, induces potent T-cell activation, proliferation and cytotoxicity with limited cytokine release. Cancer Res. 2018, 78, 1786. [Google Scholar] [CrossRef]
- Asano, R.; Shimomura, I.; Konno, S.; Ito, A.; Masakari, Y.; Orimo, R.; Taki, S.; Arai, K.; Ogata, H.; Okada, M.; et al. Rearranging the domain order of a diabody-based IgG-like bispecific antibody enhances its antitumor activity and improves its degradation resistance and pharmacokinetics. MAbs 2014, 6, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Cheng, M.; Guo, H.; Chen, Y.; Huse, M.; Cheung, N.-K.V. Retargeting T Cells to GD2 Pentasaccharide on Human Tumors Using Bispecific Humanized Antibody. Cancer Immunol. Res. 2015, 3, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.; Clark, G.; Wong, S.; Levin, W.; Ullrich, A.; McGuire, W. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Press, M.F.; Cordon-Cardo, C.; Slamon, D.J. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene 1990, 5, 953–962. [Google Scholar]
- Rius Ruiz, I.; Vicario, R.; Morancho, B.; Morales, C.B.; Arenas, E.J.; Herter, S.; Freimoser-Grundschober, A.; Somandin, J.; Sam, J.; Ast, O.; et al. p95HER2-T cell bispecific antibody for breast cancer treatment. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Reusch, U.; Sundaram, M.; Davol, P.A.; Olson, S.D.; Davis, J.B.; Demel, K.; Nissim, J.; Rathore, R.; Liu, P.Y.; Lum, L.G. Anti-CD3 × Anti-Epidermal Growth Factor Receptor (EGFR) Bispecific Antibody Redirects T-Cell Cytolytic Activity to EGFR-Positive Cancers In vitro and in an Animal Model. Clin. Cancer Res. 2006, 12, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Gedeon, P.C.; Schaller, T.H.; Chitneni, S.K.; Choi, B.D.; Kuan, C.-T.; Suryadevara, C.M.; Snyder, D.J.; Schmittling, R.J.; Szafranski, S.E.; Cui, X.; et al. A Rationally Designed Fully Human EGFRvIII:CD3-Targeted Bispecific Antibody Redirects Human T Cells to Treat Patient-derived Intracerebral Malignant Glioma. Clin. Cancer Res. 2018, 24, 3611–3631. [Google Scholar] [CrossRef] [Green Version]
- Harwood, S.L.; Alvarez-Cienfuegos, A.; Nuñez-Prado, N.; Compte, M.; Hernández-Pérez, S.; Merino, N.; Bonet, J.; Navarro, R.; Van Bergen En Henegouwen, P.M.P.; Lykkemark, S.; et al. ATTACK, a novel bispecific T cell-recruiting antibody with trivalent EGFR binding and monovalent CD3 binding for cancer immunotherapy. Oncoimmunology 2017, 7, e1377874. [Google Scholar] [CrossRef] [PubMed]
- Asano, R.; Kuroki, Y.; Honma, S.; Akabane, M.; Watanabe, S.; Mayuzumi, S.; Hiyamuta, S.; Kumagai, I.; Sode, K. Comprehensive study of domain rearrangements of single-chain bispecific antibodies to determine the best combination of configurations and microbial host cells. MAbs 2018, 10, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.C.; Wabnitz, G.H.; Samstag, Y.; Moldenhauer, G.; Ludwig, T. Functional analysis of bispecific antibody (EpCAMxCD3)-mediated T-lymphocyte and cancer cell interaction by single-cell force spectroscopy. Int. J. Cancer 2011, 128, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Dorado, J.; Baeuerle, P.A.; Heeschen, C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin. Cancer Res. 2012, 18, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Shi, B.; Gao, H.; Jiang, H.; Kong, J.; Yan, J.; Pan, X.; Li, K.; Zhang, P.; Yao, M.; et al. An EpCAM/CD3 bispecific antibody efficiently eliminates hepatocellular carcinoma cells with limited galectin-1 expression. Cancer Immunol. Immunother. 2014, 63, 121–132. [Google Scholar] [CrossRef]
- Osada, T.; Patel, S.P.; Hammond, S.A.; Osada, K.; Morse, M.A.; Lyerly, H.K. CEA/CD3-bispecific T cell-engaging (BiTE) antibody-mediated T lymphocyte cytotoxicity maximized by inhibition of both PD1 and PD-L1. Cancer Immunol. Immunother. 2015, 64, 677–688. [Google Scholar] [CrossRef]
- Bacac, M.; Klein, C.; Umana, P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology 2016, 5, e1203498. [Google Scholar] [CrossRef] [Green Version]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Gao, W.; Zhang, Y.F.; Ho, M. Glypicans as Cancer Therapeutic Targets. Trends Cancer 2018, 4, 741–754. [Google Scholar] [CrossRef]
- Lund, M.E.; Howard, C.B.; Thurecht, K.J.; Campbell, D.H.; Mahler, S.M.; Walsh, B.J. A bispecific T cell engager targeting Glypican-1 redirects T cell cytolytic activity to kill prostate cancer cells. BMC Cancer 2020, 20, 1214. [Google Scholar] [CrossRef]
- Crawford, A.; Haber, L.; Kelly, M.P.; Vazzana, K.; Canova, L.; Ram, P.; Pawashe, A.; Finney, J.; Jalal, S.; Chiu, D.; et al. A Mucin 16 bispecific T cell-engaging antibody for the treatment of ovarian cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Shang, T.; Ma, P.; Sun, X.; Zhao, J.; Sun, X.; Zhang, M. Bispecific anti-CD3 x anti-B7-H3 antibody mediates T cell cytotoxic ability to human melanoma in vitro and in vivo. Investig. New Drugs 2019, 37, 1036–1043. [Google Scholar] [CrossRef]
- Iizuka, A.; Nonomura, C.; Ashizawa, T.; Kondou, R.; Ohshima, K.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; Maruyama, K.; et al. A T-cell-engaging B7-H4/CD3-bispecific Fab-scFv Antibody Targets Human Breast Cancer. Clin. Cancer Res. 2019, 25, 2925–2934. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.R.; Zhang, T.; Gacerez, A.T.; Coupet, T.A.; DeMars, L.R.; Sentman, C.L. B7H6-Specific Bispecific T Cell Engagers Lead to Tumor Elimination and Host Antitumor Immunity. J. Immunol. 2015, 194, 5305–5311. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.A.; Johnston, J.; Li, J.; Mandikian, D.; Hristopoulos, M.; Clark, R.; Nickles, D.; Liang, W.-C.; Hötzel, K.; Dunlap, D.; et al. Anti-LYPD1/CD3 T-Cell-Dependent Bispecific Antibody for the Treatment of Ovarian Cancer. Mol. Cancer Ther. 2021, 20, 716–725. [Google Scholar] [CrossRef]
- Kodama, T.; Kochi, Y.; Nakai, W.; Mizuno, H.; Baba, T.; Habu, K.; Sawada, N.; Tsunoda, H.; Shima, T.; Miyawaki, K.; et al. Anti-GPRC5D/CD3 Bispecific T-Cell-Redirecting Antibody for the Treatment of Multiple Myeloma. Mol. Cancer Ther. 2019, 18, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- de Jong, G.; Bartels, L.; Kedde, M.; Verdegaal, E.M.E.; Gillissen, M.A.; Levie, S.E.; Cercel, M.G.; van Hal-van Veen, S.E.; Fatmawati, C.; van de Berg, D.; et al. Melanoma cells can be eliminated by sialylated CD43 × CD3 bispecific T cell engager formats in vitro and in vivo. Cancer Immunol. Immunother. 2021, 70, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Köhnke, T.; Krupka, C.; Tischer, J.; Knösel, T.; Subklewe, M. Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J. Hematol. Oncol. 2015, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Feucht, J.; Kayser, S.; Gorodezki, D.; Hamieh, M.; Döring, M.; Blaeschke, F.; Schlegel, P.; Bösmüller, H.; Quintanilla-Fend, L.; Ebinger, M.; et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget 2016, 7, 76902–76919. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Ubukawa, K.; Fujishima, M.; Takahashi, N. Correlation between increased immune checkpoint molecule expression and refractoriness to blinatumomab evaluated by longitudinal T cell analysis. Int. J. Hematol. 2021, 113, 600–605. [Google Scholar] [CrossRef]
- Schreiner, J.; Thommen, D.S.; Herzig, P.; Bacac, M.; Klein, C.; Roller, A.; Belousov, A.; Levitsky, V.; Savic, S.; Moersig, W.; et al. Expression of inhibitory receptors on intratumoral T cells modulates the activity of a T cell-bispecific antibody targeting folate receptor. Oncoimmunology 2016, 5, e1062969. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Zhao, P.; Zhang, Z.; Zhang, J.; Zhang, Z.; Hua, Y.; Han, B.; Li, N.; Zhao, X.; Hou, L. TIM-3 blockade combined with bispecific antibody MT110 enhances the anti-tumor effect of γδ T cells. Cancer Immunol. Immunother. 2020, 69, 2571–2587. [Google Scholar] [CrossRef]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Hüsser, T.; et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, C.A.; Guedan, S.; Rojas, L.A.; Moreno, R.; Arias-Badia, M.; de Sostoa, J.; June, C.H.; Alemany, R. Oncolytic Adenoviral Delivery of an EGFR-Targeting T-cell Engager Improves Antitumor Efficacy. Cancer Res. 2017, 77, 2052–2063. [Google Scholar] [CrossRef] [Green Version]
- de Sostoa, J.; Fajardo, C.A.; Moreno, R.; Ramos, M.D.; Farrera-Sal, M.; Alemany, R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J. Immunother. Cancer 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijs, P.R.A.; Verhagen, J.H.E.; van Eijck, C.H.J.; van den Hoogen, B.G. Oncolytic viruses: From bench to bedside with a focus on safety. Hum. Vaccines Immunother. 2015, 11, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Groeneveldt, C.; Kinderman, P.; van den Wollenberg, D.J.M.; van den Oever, R.L.; Middelburg, J.; Mustafa, D.A.M.; Hoeben, R.C.; van der Burg, S.H.; van Hall, T.; van Montfoort, N. Preconditioning of the tumor microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Burges, A.; Wimberger, P.; Kümper, C.; Gorbounova, V.; Sommer, H.; Schmalfeldt, B.; Pfisterer, J.; Lichinitser, M.; Makhson, A.; Moiseyenko, V.; et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: A phase I/II study. Clin. Cancer Res. 2007, 13, 3899–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, P.; Kluge, M.; Jäger, M.; Burges, A.; Volovat, C.; Heiss, M.M.; Hess, J.; Wimberger, P.; Brandt, B.; Lindhofer, H. Pharmacokinetics, immunogenicity and bioactivity of the therapeutic antibody catumaxomab intraperitoneally administered to cancer patients. Br. J. Clin. Pharmacol. 2010, 69, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, M.; Passlick, B.; Friccius-Quecke, H.; Jäger, M.; Lindhofer, H.; Kanniess, F.; Wiewrodt, R.; Thiel, E.; Buhl, R.; Schmittel, A. Treatment of non-small cell lung cancer patients with the trifunctional monoclonal antibody catumaxomab (anti-EpCAM x anti-CD3): A phase I study. Cancer Immunol. Immunother. 2007, 56, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K.; Pfisterer, J.; Wimberger, P.; Burchardi, N.; Kurzeder, C.; du Bois, A.; Loibl, S.; Sehouli, J.; Huober, J.; Schmalfeldt, B.; et al. Intraperitoneal treatment with the trifunctional bispecific antibody Catumaxomab in patients with platinum-resistant epithelial ovarian cancer: A phase IIa study of the AGO Study Group. Gynecol. Oncol. 2011, 123, 27–32. [Google Scholar] [CrossRef]
- Atanackovic, D.; Reinhard, H.; Meyer, S.; Spöck, S.; Grob, T.; Luetkens, T.; Yousef, S.; Cao, Y.; Hildebrandt, Y.; Templin, J.; et al. The trifunctional antibody catumaxomab amplifies and shapes tumor-specific immunity when applied to gastric cancer patients in the adjuvant setting. Hum. Vaccines Immunother. 2013, 9, 2533–2542. [Google Scholar] [CrossRef]
- Sehouli, J.; Reinthaller, A.; Marth, C.; Reimer, D.; Reimer, T.; Stummvoll, W.; Angleitner-Boubenizek, L.; Brandt, B.; Chekerov, R. Intra- and postoperative catumaxomab in patients with epithelial ovarian cancer: Safety and two-year efficacy results from a multicentre, single-arm, phase II study. Br. J. Cancer 2014, 111, 1519–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokemeyer, C.; Stein, A.; Ridwelski, K.; Atanackovic, D.; Arnold, D.; Wöll, E.; Ulrich, A.; Fischer, R.; Krüger, C.; Schuhmacher, C. A phase II study of catumaxomab administered intra- and postoperatively as part of a multimodal approach in primarily resectable gastric cancer. Gastric Cancer 2015, 18, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Borlak, J.; Länger, F.; Spanel, R.; Schöndorfer, G.; Dittrich, C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcγ receptors. Oncotarget 2016, 7, 28059–28074. [Google Scholar] [CrossRef]
- Kiewe, P.; Hasmüller, S.; Kahlert, S.; Heinrigs, M.; Rack, B.; Marmé, A.; Korfel, A.; Jäger, M.; Lindhofer, H.; Sommer, H.; et al. Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 3085–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35, 3002. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Trinklein, N.D.; Pham, D.; Schellenberger, U.; Buelow, B.; Boudreau, A.; Choudhry, P.; Clarke, S.C.; Dang, K.; Harris, K.E.; Iyer, S.; et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. MAbs 2019, 11, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Piskol, R.; Ybarra, R.; Chen, Y.J.; Li, J.; Slaga, D.; Hristopoulos, M.; Clark, R.; Modrusan, Z.; Totpal, K.; et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef]
- Braig, F.; Brandt, A.; Goebeler, M.; Tony, H.P.; Kurze, A.K.; Nollau, P.; Bumm, T.; Böttcher, S.; Bargou, R.C.; Binder, M. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 2017, 129, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Aldoss, I.; Qu, C.; Crawford, J.C.; Gu, Z.; Allen, E.K.; Zamora, A.E.; Alexander, T.B.; Wang, J.; Goto, H.; et al. Tumor-intrinsic and -extrinsic determinants of response to blinatumomab in adults with B-ALL. Blood 2021, 137, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Exposito, R.; Semiannikova, M.; Griffiths, B.; Khan, K.; Barber, L.J.; Woolston, A.; Spain, G.; von Loga, K.; Challoner, B.; Patel, R.; et al. CEA expression heterogeneity and plasticity confer resistance to the CEA-targeting bispecific immunotherapy antibody cibisatamab (CEA-TCB) in patient-derived colorectal cancer organoids. J. Immunother. Cancer 2019, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.Q.; Grantham, A.; Landry, C.; Granda, B.; Chopra, R.; Chakravarthy, S.; Deutsch, S.; Vogel, M.; Russo, K.; Seiss, K.; et al. A CRISPR Screen Reveals Resistance Mechanisms to CD3-Bispecific Antibody Therapy. Cancer Immunol. Res. 2021, 9, 34–49. [Google Scholar] [CrossRef]

| Cancer Type | Name | Target TAA | Format | Phase | Identifier |
|---|---|---|---|---|---|
| Solid tumor | HPN536 | mesothelin | TriTAC 1 | 1/2 | NCT03872206 |
| ERY974 | GPC3 | IgG-like BsAb | 1 | NCT02748837 | |
| JNJ-63898081 | PSMA | DuoBody (IgG-like BsAb) | 1 | NCT03926013 | |
| PF-06671008 | CDH3 (P-cadherin) | DART-Fc 2 | 1 | NCT02659631 | |
| MGD009/orlotamab | B7-H3 | DART-Fc | 1 | NCT02628535 | |
| M701 | EpCAM | Fab/scFv-Fc BsAb | 1 | NCT04501744 | |
| M802 | HER2 | Fab/scFv-Fc BsAb | 1 | NCT04501770 | |
| BTRC4017A/RG6194 | HER2 | IgG-like BsAb | 1 | NCT03448042 | |
| GEM3PSCA | PSCA | ATAC 3 | 1 | NCT03927573 | |
| AMV564 | CD33 | bivalent BiTE | 1 | NCT04128423 | |
| GEN1044 | 5T4 | DuoBody (IgG-like BsAb) | 1/2 | NCT04424641 | |
| Glioblastoma | AMG596 | EGFRvIII | BiTE | 1 | NCT03296696 |
| Neuroblastoma | Hu3F8-BsAb | GD2 | Bivalent Fab/scFv-Fc BsAb | 1/2 | NCT03860207 |
| Small cell lung cancer | AMG757 | DLL3 | BiTE-Fc | 1 | NCT03319940 |
| NSCLC | RO6958688/RG7802/ cibisatamab | CEA | 2 + 1 Fab-Fc BsAb | 1/2 | NCT03866239 |
| Breast cancer | GBR1302/ISB1302 | HER2 | Fab/scFv-Fc BsAb | 1/2 | NCT03983395 |
| NET and GIST | XmAb18087/tidutamab | SSTR2 | XmAb (Fab/scFv-Fc BsAb) | 1 | NCT03411915 |
| Gastric cancer | AMG199 | MUC17 | BiTE-Fc | 1 | NCT04117958 |
| AMG910 | CLDN18.2 | BiTE-Fc | 1 | NCT04260191 | |
| catumaxomab | EpCAM | IgG-like BsAb | 3 | NCT04222114 | |
| Gastrointestinal cancer | MEDI-565/AMG211/ MT-111 | CEA | BiTE | 1 | NCT01284231 |
| PF-07062119 | GUCY2C | DART-Fc | 1 | NCT04171141 | |
| Colorectal cancer | MGD007 | gpA33 | DART-Fc | 1 | NCT02248805 |
| Prostate cancer | AMG212/MT-112/ BAY2010112/ pasotuxizumab | PSMA | BiTE | 1 | NCT01723475 |
| HPN424 | PSMA | TriTAC | 1 | NCT03577028 | |
| AMG160 | PSMA | BiTE-Fc | 1 | NCT03792841 | |
| AMG509 | STEAP1 | Fab/scFv-Fc BsAb | 1 | NCT04221542 | |
| ES414/APVO414/MOR209 | PSMA | ADAPTIRTM | 1 | NCT02262910 | |
| CCW702/ABBV-154 | PSMA | bispecific antibody-small molecule conjugates | 1 | NCT04077021 | |
| CC-1 | PSMA | Bivalent Fab/scFv-Fc BsAb | 1 | NCT04104607 | |
| ovarian cancer | REGN4018 | MUC16 | IgG-like BsAb | 1/2 | NCT03564340 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamakura, D.; Asano, R.; Yasunaga, M. T Cell Bispecific Antibodies: An Antibody-Based Delivery System for Inducing Antitumor Immunity. Pharmaceuticals 2021, 14, 1172. https://doi.org/10.3390/ph14111172
Kamakura D, Asano R, Yasunaga M. T Cell Bispecific Antibodies: An Antibody-Based Delivery System for Inducing Antitumor Immunity. Pharmaceuticals. 2021; 14(11):1172. https://doi.org/10.3390/ph14111172
Chicago/Turabian StyleKamakura, Daisuke, Ryutaro Asano, and Masahiro Yasunaga. 2021. "T Cell Bispecific Antibodies: An Antibody-Based Delivery System for Inducing Antitumor Immunity" Pharmaceuticals 14, no. 11: 1172. https://doi.org/10.3390/ph14111172