
Research and Clinical Landscape of Bispecific Antibodies for the Treatment of Solid Malignancies

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
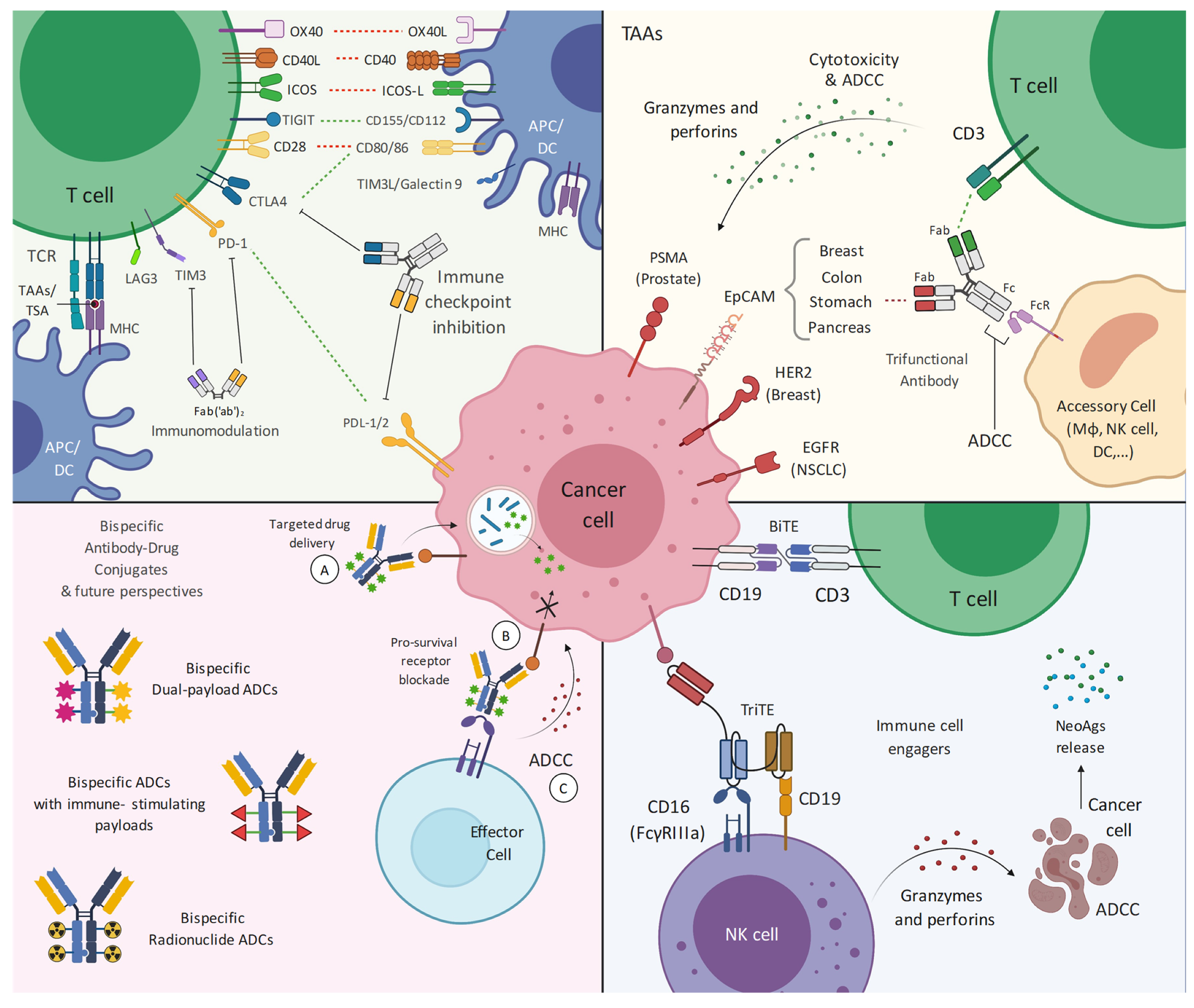
2. Bispecific Antibodies Design and Development
2.1. Structural Classification of Bispecific Antibodies
2.1.1. Fc-Free Bispecific Antibodies
2.1.2. Fc-Bearing Bispecific Antibodies
2.2. Mechanistic Classification of Bispecific Antibodies
2.2.1. Tumor-Associated Antigens
2.2.2. Antibody Drug Conjugates
2.2.3. Immune Cell Engagers
2.2.4. Immune Checkpoint Inhibitors/Blockers
3. State-of-the Art of Bispecific Antibodies in Solid Tumors
4. TAA-Bispecific Targeting
5. Bispecific-Antibody Drug Conjugates
6. Immune Cell Engagers in Solid Tumors
7. Immune Checkpoint Blockade/Inhibition
8. Future Perspectives on Bispecific Targeting of Solid Tumors
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Couzin-Frankel, J. Breakthrough of the Year 2013. Cancer Immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [Green Version]
- Dougan, M.; Dranoff, G.; Dougan, S.K. Cancer Immunotherapy: Beyond Checkpoint Blockade. Annu. Rev. Cancer Biol. 2019, 3, 55–75. [Google Scholar] [CrossRef]
- Greenbaum, U.; Mahadeo, K.M.; Kebriaei, P.; Shpall, E.J.; Saini, N.Y. Chimeric Antigen Receptor T-Cells in B-Acute Lymphoblastic Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1594. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA Approves Fourth CAR-T Cell Therapy. Nat. Rev. Drug Discov. 2021, 20, 166. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody Therapy of Cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Scott, A.M.; Allison, J.P.; Wolchok, J.D. Monoclonal Antibodies in Cancer Therapy. Cancer Immun. 2012, 12, 14. [Google Scholar] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Riechmann, L.; Clark, M.; Waldmann, H.; Winter, G. Reshaping Human Antibodies for Therapy. Nature 1988, 332, 323–327. [Google Scholar] [CrossRef]
- Klein, C.; Sustmann, C.; Thomas, M.; Stubenrauch, K.; Croasdale, R.; Schanzer, J.; Brinkmann, U.; Kettenberger, H.; Regula, J.T.; Schaefer, W. Progress in Overcoming the Chain Association Issue in Bispecific Heterodimeric IgG Antibodies. MAbs 2012, 4, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.W.; Zhang, W. Current Trends and Challenges in the Downstream Purification of Bispecific Antibodies. Antib 2021, 4, 73–88. [Google Scholar] [CrossRef]
- Milstein, C.; Cuello, A.C. Hybrid Hybridomas and Their Use in Immunohistochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An Efficient Route to Human Bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Pyo, K.-H.; Soo, R.A.; Cho, B.C. The Promise of Bispecific Antibodies: Clinical Applications and Challenges. Cancer Treat. Rev. 2021, 99, 102240. [Google Scholar] [CrossRef] [PubMed]
- Poussin, M.; Sereno, A.; Wu, X.; Huang, F.; Manro, J.; Cao, S.; Carpenito, C.; Glasebrook, A.; Power, D.J., Jr.; Demarest, S. Dichotomous Impact of Affinity on the Function of T Cell Engaging Bispecific Antibodies. J. Immunother. Cancer 2021, 9, e002444. [Google Scholar] [CrossRef]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K.; et al. Relative Target Affinities of T-Cell–Dependent Bispecific Antibodies Determine Biodistribution in a Solid Tumor Mouse Model. Mol. Cancer 2018, 17, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Kotanides, H.; Jimenez, X.; Zhou, Q.; Persaud, K.; Bohlen, P.; Witte, L.; Zhu, Z. Acquired Antagonistic Activity of a Bispecific Diabody Directed against Two Different Epitopes on Vascular Endothelial Growth Factor Receptor 2. J Immunol. Methods 1999, 230, 159–171. [Google Scholar] [CrossRef]
- Li, B.; Meng, Y.; Zheng, L.; Zhang, X.; Tong, Q.; Tan, W.; Hu, S.; Li, H.; Chen, Y.; Song, J.; et al. Bispecific Antibody to ErbB2 Overcomes Trastuzumab Resistance through Comprehensive Blockade of ErbB2 Heterodimerization. Cancer Res. 2013, 73, 6471–6483. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Krippendorff, B.-F.; Sharma, S.; Walz, A.C.; Lavé, T.; Shah, D.K. Influence of Molecular Size on Tissue Distribution of Antibody Fragments. MAbs 2016, 8, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Elshiaty, M.; Schindler, H.; Christopoulos, P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int. J. Mol. Sci. 2021, 22, 5632. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for Extended Serum Half-Life of Protein Therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Jen, E.Y.; Xu, Q.; Schetter, A.; Przepiorka, D.; Shen, Y.L.; Roscoe, D.; Sridhara, R.; Deisseroth, A.; Philip, R.; Farrell, A.T.; et al. FDA Approval: Blinatumomab for Patients with B-Cell Precursor Acute Lymphoblastic Leukemia in Morphologic Remission with Minimal Residual Disease. Clin. Cancer Res. 2019, 25, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Fucà, G.; Spagnoletti, A.; Ambrosini, M.; de Braud, F.; Di Nicola, M. Immune Cell Engagers in Solid Tumors: Promises and Challenges of the next Generation Immunotherapy. ESMO Open 2021, 6, 100046. [Google Scholar] [CrossRef] [PubMed]
- Kellner, C.; Derer, S.; Valerius, T.; Peipp, M. Boosting ADCC and CDC Activity by Fc Engineering and Evaluation of Antibody Effector Functions. Methods 2014, 65, 105–113. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umaña, P. Engineering Therapeutic Bispecific Antibodies Using CrossMab Technology. Methods 2019, 154, 21–31. [Google Scholar] [CrossRef]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential Species-Restricted Heavy/Light Chain Pairing in Rat/Mouse Quadromas. Implications for a Single-Step Purification of Bispecific Antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar]
- Labrijn, A.F.; Meesters, J.I.; de Goeij, B.E.C.G.; van den Bremer, E.T.J.; Neijssen, J.; van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient Generation of Stable Bispecific IgG1 by Controlled Fab-Arm Exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, S.; Wu, C. Generation of Fabs-in-Tandem Immunoglobulin Molecules for Dual-Specific Targeting. Methods 2019, 154, 87–92. [Google Scholar] [CrossRef] [PubMed]
- DiGiammarino, E.; Ghayur, T.; Liu, J. Design and Generation of DVD-IgTM Molecules for Dual-Specific Targeting. Methods Mol. Biol. 2012, 899, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Beeram, M.; Mayordomo, J.I.; Hanna, D.L.; Ajani, J.A.; Blum Murphy, M.A.; Murthy, R.K.; Piha-Paul, S.A.; Bauer, T.M.; Bendell, J.C.; et al. Single Agent Activity of ZW25, a HER2-Targeted Bispecific Antibody, in Heavily Pretreated HER2-Expressing Cancers. J. Clin. Oncol. 2018, 36 (Suppl. 15), 2500. [Google Scholar] [CrossRef]
- Neijssen, J.; Cardoso, R.M.F.; Chevalier, K.M.; Wiegman, L.; Valerius, T.; Anderson, G.M.; Moores, S.L.; Schuurman, J.; Parren, P.W.H.I.; Strohl, W.R.; et al. Discovery of Amivantamab (JNJ-61186372), a Bispecific Antibody Targeting EGFR and MET. J. Biol. Chem. 2021, 296, 100641. [Google Scholar] [CrossRef]
- Shim, H. Bispecific Antibodies and Antibody–Drug Conjugates for Cancer Therapy: Technological Considerations. Biomolecules 2020, 10, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblett, K.J.; Barnscher, S.D.; Davies, R.H.; Hammond, P.W.; Hernandez, A.; Wickman, G.R.; Fung, V.K.; Ding, T.; Garnett, G.; Galey, A.S.; et al. Abstract P6-17-13: ZW49, a HER2 Targeted Biparatopic Antibody Drug Conjugate for the Treatment of HER2 Expressing Cancers. Cancer Res. 2019, 79 (Suppl. 4), P6-17-13. [Google Scholar] [CrossRef]
- de Goeij, B.E.C.G.; Vink, T.; Ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody-Drug Conjugate Targeting HER2 and CD63. Mol. Cancer 2016, 15, 2688–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, E.A.; Rossi, D.L.; Stein, R.; Goldenberg, D.M.; Chang, C.-H. A Bispecific Antibody-IFNalpha2b Immunocytokine Targeting CD20 and HLA-DR Is Highly Toxic to Human Lymphoma and Multiple Myeloma Cells. Cancer Res. 2010, 70, 7600–7609. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Cai, M.; Bigner, D.D.; Mehta, A.I.; Kuan, C.-T.; Sampson, J.H. Bispecific Antibodies Engage T Cells for Antitumor Immunotherapy. Expert Opin. Biol. 2011, 11, 843–853. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T Cell Engagers for Cancer Immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Dovedi, S.J.; Elder, M.J.; Yang, C.; Sitnikova, S.I.; Irving, L.; Hansen, A.; Hair, J.; Jones, D.C.; Hasani, S.; Wang, B.; et al. Design and Efficacy of a Monovalent Bispecific PD-1/CTLA4 Antibody That Enhances CTLA4 Blockade on PD-1+ Activated T Cells. Cancer Discov. 2021, 11, 1100–1117. [Google Scholar] [CrossRef]
- Yun, J.; Lee, S.-H.; Kim, S.-Y.; Jeong, S.-Y.; Kim, J.-H.; Pyo, K.-H.; Park, C.-W.; Heo, S.G.; Yun, M.R.; Lim, S.; et al. Antitumor Activity of Amivantamab (JNJ-61186372), an EGFR-MET Bispecific Antibody, in Diverse Models of EGFR Exon 20 Insertion-Driven NSCLC. Cancer Discov. 2020, 10, 1194–1209. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Lipfert, L.; Chevalier, K.; Bushey, B.S.; Henley, B.; Lenhart, R.; Sendecki, J.; Beqiri, M.; Millar, H.J.; Packman, K.; et al. Amivantamab (JNJ-61186372), an Fc Enhanced EGFR/CMet Bispecific Antibody, Induces Receptor Downmodulation and Antitumor Activity by Monocyte/Macrophage Trogocytosis. Mol. Cancer 2020, 19, 2044–2056. [Google Scholar] [CrossRef]
- Grugan, K.D.; Dorn, K.; Jarantow, S.W.; Bushey, B.S.; Pardinas, J.R.; Laquerre, S.; Moores, S.L.; Chiu, M.L. Fc-Mediated Activity of EGFR x c-Met Bispecific Antibody JNJ-61186372 Enhanced Killing of Lung Cancer Cells. mAbs 2017, 9, 114–126. [Google Scholar] [CrossRef] [Green Version]
- FDA Grants Accelerated Approval to Amivantamab-Vmjw for Metastatic Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-amivantamab-vmjw-metastatic-non-small-cell-lung-cancer (accessed on 23 July 2021).
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming Therapy Resistance in EGFR -Mutant Lung Cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Park, K.; John, T.; Kim, S.-W.; Lee, J.S.; Shu, C.A.; Kim, D.-W.; Viteri Ramirez, S.; Spira, A.I.; Sabari, J.K.; Han, J.-Y.; et al. Amivantamab (JNJ-61186372), an Anti-EGFR-MET Bispecific Antibody, in Patients with EGFR Exon 20 Insertion (Exon20ins)-Mutated Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2020, 38 (Suppl. 15), 9512. [Google Scholar] [CrossRef]
- Bauml, J.; Cho, B.C.; Park, K.; Lee, K.H.; Cho, E.K.; Kim, D.-W.; Kim, S.-W.; Haura, E.B.; Sabari, J.K.; Sanborn, R.E.; et al. Amivantamab in Combination with Lazertinib for the Treatment of Osimertinib-Relapsed, Chemotherapy-Naïve EGFR Mutant (EGFRm) Non-Small Cell Lung Cancer (NSCLC) and Potential Biomarkers for Response. J. Clin. Oncol. 2021, 39 (Suppl. 15), 9006. [Google Scholar] [CrossRef]
- Shreeve, S.M.; Martinez, M.; Verheijen, R.B.; Xie, J.; Sun, T.; Haddish-Berhane, N.; Curtin, J.C.; Karkera, J.; Pang, D.; Roshak, A.; et al. P76.73 MARIPOSA: Randomized Phase 3 Study of First-Line Amivantamab + Lazertinib vs Osimertinib vs. Lazertinib in EGFR-Mutant NSCLC. J. Thorac. Oncol. 2021, 16, S620–S621. [Google Scholar] [CrossRef]
- Shu, C.A.; Goto, K.; Cho, B.C.; Griesinger, F.; Yang, J.C.-H.; Felip, E.; Xie, J.; Chen, J.; Mahoney, J.; Thayu, M.; et al. CHRYSALIS-2: A Phase 1/1b Study of Lazertinib as Monotherapy and in Combination with Amivantamab in Patients with EGFR-Mutant NSCLC. J. Clin. Oncol. 2021, 39 (Suppl. 15), TPS9132. [Google Scholar] [CrossRef]
- Weisser, N.E.; Wickman, G.; Abraham, L.; O’Toole, J.; Harbourne, B.; Guedia, J.; Cheng, C.W.; Chan, P.; Browman, D.; Gold, M.R.; et al. Abstract 1005: The Bispecific Antibody Zanidatamab’s (ZW25’s) Unique Mechanisms of Action and Durable Anti-Tumor Activity in HER2-Expressing Cancers. Cancer Res. 2021, 81 (Suppl. 13), 1005. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hanna, D.L.; El-Khoueiry, A.B.; Kang, Y.-K.; Oh, D.-Y.; Chaves, J.M.; Rha, S.Y.; Hamilton, E.P.; Pant, S.; Javle, M.M.; et al. Zanidatamab (ZW25) in HER2-Positive Biliary Tract Cancers (BTCs): Results from a Phase I Study. J. Clin. Oncol. 2021, 39 (Suppl. 3), 299. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hamilton, E.P.; Beeram, M.; Hanna, D.L.; El-Khoueiry, A.B.; Kang, Y.-K.; Lee, K.W.; Lee, J.; Rha, S.Y.; Chaves, J.M.; et al. Zanidatamab (ZW25) in HER2-Expressing Gastroesophageal Adenocarcinoma (GEA): Results from a Phase I Study. J. Clin. Oncol. 2021, 39 (Suppl. 3), 164. [Google Scholar] [CrossRef]
- Lee, K.W.; Im, Y.-H.; Lee, K.S.; Cho, J.Y.; Oh, D.-Y.; Chung, H.C.C.; Chao, Y.; Bai, L.-Y.; Yen, C.J.; Kim, I.-H.; et al. Zanidatamab, an Anti-HER2 Bispecific Antibody, plus Chemotherapy with/without Tislelizumab as First-Line Treatment for Patients with Advanced HER2-Positive Breast Cancer or Gastric/Gastroesophageal Junction Adenocarcinoma: A Phase 1B/2 Trial-in-Progress. J. Clin. Oncol. 2021, 39 (Suppl. 15), TPS2656. [Google Scholar] [CrossRef]
- Ji, D.; Zhang, J.; Shen, W.; Du, Y.; Xu, J.; Yang, J.; Luo, X.; Kong, P.; Yang, F.; Hu, X.-C. Preliminary Safety, Efficacy and Pharmacokinetics (PK) Results of KN026, a HER2 Bispecific Antibody in Patients (Pts) with HER2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2020, 38 (Suppl. 15), 1041. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Wu, J.; Xu, N.; Ying, J.; Xiang, X.; Zhang, Y.; Wang, J.; Zhao, R.; Ye, F.; et al. The Preliminary Efficacy of KN026 (Anti-HER2 BsAb) in Advanced Gastric and Gastroesophageal Junction Cancer Patients with HER2 Expression. J. Clin. Oncol. 2021, 39 (Suppl. 15), e16005. [Google Scholar] [CrossRef]
- Gong, J.; Dong, Z.; Liu, D.; Xu, J.; Yang, J.; Yang, Y.; Qi, Y.; Men, J.; Kong, P.; Xu, T.; et al. 339 Preliminary Safety, Tolerability and Efficacy Results of KN026 (a HER2-Targeted Bispecific Antibody) in Combination with KN046 (an Anti-PD-L1/CTLA-4 Bispecific Antibody) in Patients (Pts) with HER2 Aberrated Solid Tumors. J. Immunother. Cancer 2020, 8 (Suppl. 3). [Google Scholar] [CrossRef]
- Watanabe, S.; Yonesaka, K.; Tanizaki, J.; Nonagase, Y.; Takegawa, N.; Haratani, K.; Kawakami, H.; Hayashi, H.; Takeda, M.; Tsurutani, J.; et al. Targeting of the HER2/HER3 Signaling Axis Overcomes Ligand-Mediated Resistance to Trastuzumab in HER2-Positive Breast Cancer. Cancer Med. 2019, 8, 1258–1268. [Google Scholar] [CrossRef] [Green Version]
- Alsina, M.; Varga, A.; Amatu, A.; Schellens, J.H.M.; Witteveen, P.O.; Boni, V.; Moreno, V.; Bol, K.; Lourbakos, A.; Ferrer, M.M.; et al. Phase I/II Study of Single Agent MCLA-128, a Full Length IgG1 Bispecific Antibody Targeting the HER3 Pathway: Overall Safety at the Recommended Phase II Dose (R2PD) and Preliminary Activity in HER2+ Metastatic Gastric/Gastroesophageal Junction Cancer (GC/GEJ). Ann. Oncol. 2018, 29, viii223–viii224. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Petit, T.; Pistilli, B.; Goncalves, A.; Ferreira, A.A.; Dalenc, F.; Cardoso, F.; Mita, M.M.; Dezentjé, V.O.; Manso, L.; et al. Clinical Activity of MCLA-128 (Zenocutuzumab), Trastuzumab, and Vinorelbine in HER2 Amplified Metastatic Breast Cancer (MBC) Patients (Pts) Who Had Progressed on Anti-HER2 ADCs. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3093. [Google Scholar] [CrossRef]
- Pistilli, B.; Wildiers, H.; Hamilton, E.P.; Ferreira, A.A.; Dalenc, F.; Vidal, M.; Gavilá, J.; Goncalves, A.; Murias, C.; Mouret-Reynier, M.-A.; et al. Clinical Activity of MCLA-128 (Zenocutuzumab) in Combination with Endocrine Therapy (ET) in ER+/HER2-Low, Non-Amplified Metastatic Breast Cancer (MBC) Patients (Pts) with ET-Resistant Disease Who Had Progressed on a CDK4/6 Inhibitor (CDK4/6i). J. Clin. Oncol. 2020, 38 (Suppl. 15), 1037. [Google Scholar] [CrossRef]
- Liu, S.V. NRG1 Fusions: Biology to Therapy. Lung Cancer 2021, 158, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; O’Reilly, E.M.; O’Kane, G.M.; Goto, K.; Kim, D.-W.; Neuzillet, C.; Martin-Romano, P.; Duruisseaux, M.; Nagasaka, M.; Rodon, J.; et al. Efficacy and Safety of Zenocutuzumab in Advanced Pancreas Cancer and Other Solid Tumors Harboring NRG1 Fusions. J. Clin. Oncol. 2021, 39 (Suppl. 15), 3003. [Google Scholar] [CrossRef]
- McBride, W.J.; Zanzonico, P.; Sharkey, R.M.; Norén, C.; Karacay, H.; Rossi, E.A.; Losman, M.J.; Brard, P.-Y.; Chang, C.-H.; Larson, S.M.; et al. Bispecific Antibody Pretargeting PET (ImmunoPET) with an 124I-Labeled Hapten-Peptide. J. Nucl. Med. 2006, 47, 1678–1688. [Google Scholar]
- Frampas, E.; Rousseau, C.; Bodet-Milin, C.; Barbet, J.; Chatal, J.-F.; Kraeber-Bodéré, F. Improvement of Radioimmunotherapy Using Pretargeting. Front. Oncol. 2013, 3, 159. [Google Scholar] [CrossRef] [Green Version]
- Ku, G.; Bendell, J.C.; Tolcher, A.W.; Hurvitz, S.A.; Krishnamurthy, A.; El-Khoueiry, A.B.; Patnaik, A.; Shroff, R.T.; Noonan, A.; Hahn, N.M.; et al. 525O A Phase I Dose Escalation Study of PRS-343, a HER2/4-1BB Bispecific Molecule, in Patients with HER2-Positive Malignancies. Ann. Oncol. 2020, 31, S462–S463. [Google Scholar] [CrossRef]
- Anonymous. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/removab (accessed on 17 July 2021).
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab. mAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The Trifunctional Antibody Catumaxomab for the Treatment of Malignant Ascites Due to Epithelial Cancer: Results of a Prospective Randomized Phase II/III Trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Burges, A.; Wimberger, P.; Kümper, C.; Gorbounova, V.; Sommer, H.; Schmalfeldt, B.; Pfisterer, J.; Lichinitser, M.; Makhson, A.; Moiseyenko, V.; et al. Effective Relief of Malignant Ascites in Patients with Advanced Ovarian Cancer by a Trifunctional Anti-EpCAM × Anti-CD3 Antibody: A Phase I/II Study. Clin. Cancer Res. 2007, 13, 3899–3905. [Google Scholar] [CrossRef] [Green Version]
- Knödler, M.; Körfer, J.; Kunzmann, V.; Trojan, J.; Daum, S.; Schenk, M.; Kullmann, F.; Schroll, S.; Behringer, D.; Stahl, M.; et al. Randomised Phase II Trial to Investigate Catumaxomab (Anti-EpCAM × Anti-CD3) for Treatment of Peritoneal Carcinomatosis in Patients with Gastric Cancer. Br. J. Cancer 2018, 119, 296–302. [Google Scholar] [CrossRef]
- Sebastian, M.; Passlick, B.; Friccius-Quecke, H.; Jäger, M.; Lindhofer, H.; Kanniess, F.; Wiewrodt, R.; Thiel, E.; Buhl, R.; Schmittel, A. Treatment of Non-Small Cell Lung Cancer Patients with the Trifunctional Monoclonal Antibody Catumaxomab (Anti-EpCAM × Anti-CD3): A Phase I Study. Cancer Immunol. Immunother. 2007, 56, 1637–1644. [Google Scholar] [CrossRef]
- Zhao, H.; Ma, Y.; Zhang, Y.; Hong, S.; Yang, Y.; Fang, W.; Xu, J.; Van, H.; Kong, P.; Yang, F.; et al. The Preliminary Efficacy and Safety Data of KN046 in Patients Failed on Prior Immune Checkpoint Inhibitors Therapy. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3020. [Google Scholar] [CrossRef]
- Xu, B.; Li, Q.; Zhang, Q.; Zhang, Y.; Ouyang, Q.; Zhang, Y.; Liu, Q.; Sun, T.; Xu, J.; Yang, J.; et al. Abstract 1660: Preliminary Safety Tolerability & Efficacy Results of KN046 (an Anti-PD-L1/CTLA-4 Bispecific Antibody) in Combination with Nab-Paclitaxel in Patients with Metastatic Triple-Negative Breast Cancer (MTNBC). Cancer Res. 2021, 81 (Suppl. 13), 1660. [Google Scholar] [CrossRef]
- Richardson, G.; Kichenadasse, G.; Ganju, V.; Xu, J.; Van, H.; Kong, P.; Yang, F.; Wei, Y.; Lu, Y.; Guo, K.; et al. MA06.09 Preliminary Safety, Efficacy Results of KN046 (Bispecific Anti-PD-L1/CTLA4) in Subjects With Rare Thoracic Tumors. J. Thorac. Oncol. 2021, 16, S154–S155. [Google Scholar] [CrossRef]
- Ji, J.; Shen, L.; Li, Z.; Xu, N.; Liu, T.; Chen, Y.; Li, C.; Gao, X.; Ji, K.; Mao, C.; et al. AK104 (PD-1/CTLA-4 Bispecific) Combined with Chemotherapy as First-Line Therapy for Advanced Gastric (G) or Gastroesophageal Junction (GEJ) Cancer: Updated Results from a Phase Ib Study. J. Clin. Oncol. 2021, 39 (Suppl. 3), 232. [Google Scholar] [CrossRef]
- Bai, L.; Sun, M.; Xu, A.; Bai, Y.; Wu, J.; Shao, G.; Song, L.; Jin, X.; Song, W.; Li, B.; et al. Phase 2 Study of AK104 (PD-1/CTLA-4 Bispecific Antibody) plus Lenvatinib as First-Line Treatment of Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2021, 39 (Suppl. 15), 4101. [Google Scholar] [CrossRef]
- Correnti, C.E.; Laszlo, G.S.; de van der Schueren, W.J.; Godwin, C.D.; Bandaranayake, A.; Busch, M.A.; Gudgeon, C.J.; Bates, O.M.; Olson, J.M.; Mehlin, C.; et al. Simultaneous Multiple Interaction T-Cell Engaging (SMITE) Bispecific Antibodies Overcome Bispecific T-Cell Engager (BiTE) Resistance via CD28 Co-Stimulation. Leukemia 2018, 32, 1239–1243. [Google Scholar] [CrossRef]
- Damato, B.E.; Dukes, J.; Goodall, H.; Carvajal, R.D. Tebentafusp: T cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers 2019, 11, 971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperno-Neumann, S.; Hassel, J.C.; Rutkowski, P.; Baurain, J.-F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Abstract CT002: Phase 3 Randomized Trial Comparing Tebentafusp with Investigator’s Choice in First Line Metastatic Uveal Melanoma. Cancer Res. 2021, 81 (Suppl. 13), CT002. [Google Scholar] [CrossRef]
- Middleton, M.R.; McAlpine, C.; Woodcock, V.K.; Corrie, P.; Infante, J.R.; Steven, N.M.; Evans, T.R.J.; Anthoney, A.; Shoushtari, A.N.; Hamid, O.; et al. Tebentafusp, A TCR/Anti-CD3 Bispecific Fusion Protein Targeting Gp100, Potently Activated Antitumor Immune Responses in Patients with Metastatic Melanoma. Clin. Cancer Res. 2020, 26, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef]
- Skora, A.D.; Douglass, J.; Hwang, M.S.; Tam, A.J.; Blosser, R.L.; Gabelli, S.B.; Cao, J.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; et al. Generation of MANAbodies Specific to HLA-Restricted Epitopes Encoded by Somatically Mutated Genes. Proc. Natl. Acad. Sci. USA 2015, 112, 9967–9972. [Google Scholar] [CrossRef] [Green Version]
- Lowe, D.B.; Bivens, C.K.; Mobley, A.S.; Herrera, C.E.; McCormick, A.L.; Wichner, T.; Sabnani, M.K.; Wood, L.M.; Weidanz, J.A. TCR-like Antibody Drug Conjugates Mediate Killing of Tumor Cells with Low Peptide/HLA Targets. MAbs 2017, 9, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Douglass, J.; Hsiue, E.H.-C.; Mog, B.J.; Hwang, M.S.; DiNapoli, S.R.; Pearlman, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific Antibodies Targeting Mutant RAS Neoantigens. Sci. Immunol. 2021, 6, eabd5515. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a Neoantigen Derived from a Common TP53 Mutation. Science 2021, 371. [Google Scholar] [CrossRef]
- Fajardo, C.A.; Guedan, S.; Rojas, L.A.; Moreno, R.; Arias-Badia, M.; de Sostoa, J.; June, C.H.; Alemany, R. Oncolytic Adenoviral Delivery of an EGFR-Targeting T-Cell Engager Improves Antitumor Efficacy. Cancer Res. 2017, 77, 2052–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus–Driven Production of a Bispecific T-Cell Engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T Cells Secreting BiTEs Circumvent Antigen Escape without Detectable Toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Blanco, B.; Ramírez-Fernández, Á.; Alvarez-Vallina, L. Engineering Immune Cells for in Vivo Secretion of Tumor-Specific T Cell-Redirecting Bispecific Antibodies. Front. Immunol. 2020, 11, 1792. [Google Scholar] [CrossRef]
- Blanco, B.; Compte, M.; Lykkemark, S.; Sanz, L.; Alvarez-Vallina, L. T Cell-Redirecting Strategies to “STAb” Tumors: Beyond CARs and Bispecific Antibodies. Trends Immunol. 2019, 40, 243–257. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Zhang, L.; Lu, Y.; Zhang, Q.; Fan, D.; Zhang, Y.; Zhang, Y.; Ye, Z.; Xiong, D. Mesenchymal Stromal Cells as Vehicles of Tetravalent Bispecific Tandab (CD3/CD19) for the Treatment of B Cell Lymphoma Combined with IDO Pathway Inhibitor D-1-Methyl-Tryptophan. J. Hematol. Oncol. 2017, 10, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.-P.; et al. Long-Term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Campbell, C.; Savas, P.; Nuciforo, P.; Aura, C.; de Azambuja, E.; Eidtmann, H.; Ellis, C.E.; Baselga, J.; et al. Tumor-Infiltrating Lymphocytes and Associations With Pathological Complete Response and Event-Free Survival in HER2-Positive Early-Stage Breast Cancer Treated With Lapatinib and Trastuzumab. JAMA Oncol. 2015, 1, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-Analysis of Tumor- and T Cell-Intrinsic Mechanisms of Sensitization to Checkpoint Inhibition. Cell 2021, 184, 596–614.e14. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonarelli, G.; Giugliano, F.; Corti, C.; Repetto, M.; Tarantino, P.; Curigliano, G. Research and Clinical Landscape of Bispecific Antibodies for the Treatment of Solid Malignancies. Pharmaceuticals 2021, 14, 884. https://doi.org/10.3390/ph14090884
Antonarelli G, Giugliano F, Corti C, Repetto M, Tarantino P, Curigliano G. Research and Clinical Landscape of Bispecific Antibodies for the Treatment of Solid Malignancies. Pharmaceuticals. 2021; 14(9):884. https://doi.org/10.3390/ph14090884
Chicago/Turabian StyleAntonarelli, Gabriele, Federica Giugliano, Chiara Corti, Matteo Repetto, Paolo Tarantino, and Giuseppe Curigliano. 2021. "Research and Clinical Landscape of Bispecific Antibodies for the Treatment of Solid Malignancies" Pharmaceuticals 14, no. 9: 884. https://doi.org/10.3390/ph14090884
APA StyleAntonarelli, G., Giugliano, F., Corti, C., Repetto, M., Tarantino, P., & Curigliano, G. (2021). Research and Clinical Landscape of Bispecific Antibodies for the Treatment of Solid Malignancies. Pharmaceuticals, 14(9), 884. https://doi.org/10.3390/ph14090884