
Effects of Magnesium Orotate, Benfotiamine and a Combination of Vitamins on Mitochondrial and Cholinergic Function in the TgF344-AD Rat Model of Alzheimer’s Disease

 , ,
, ,
Abstract
:1. Introduction
2. Results
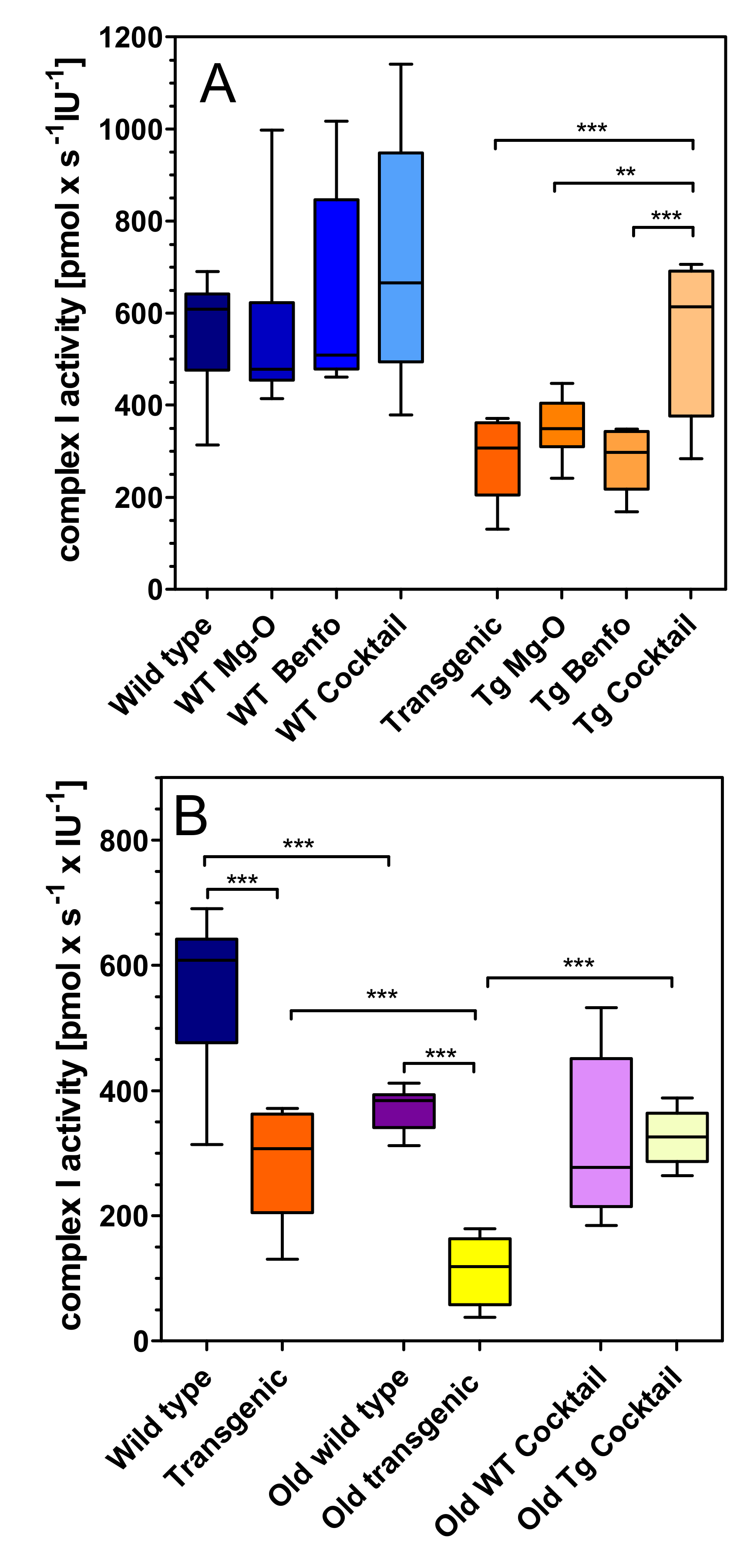
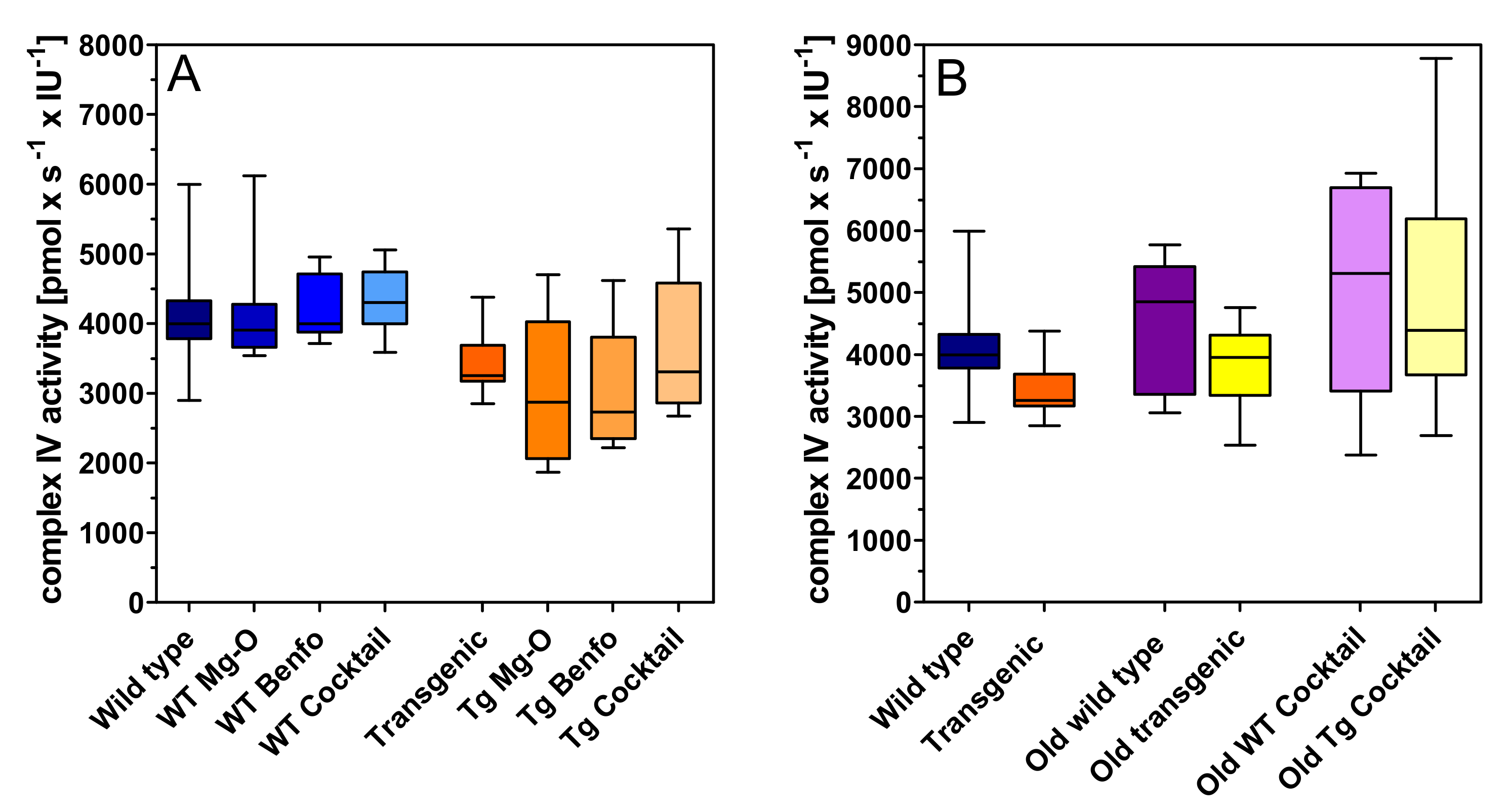
2.1. Mitochondrial Respiration
2.2. Energy Metabolites
2.3. Extracellular Magnesium Levels
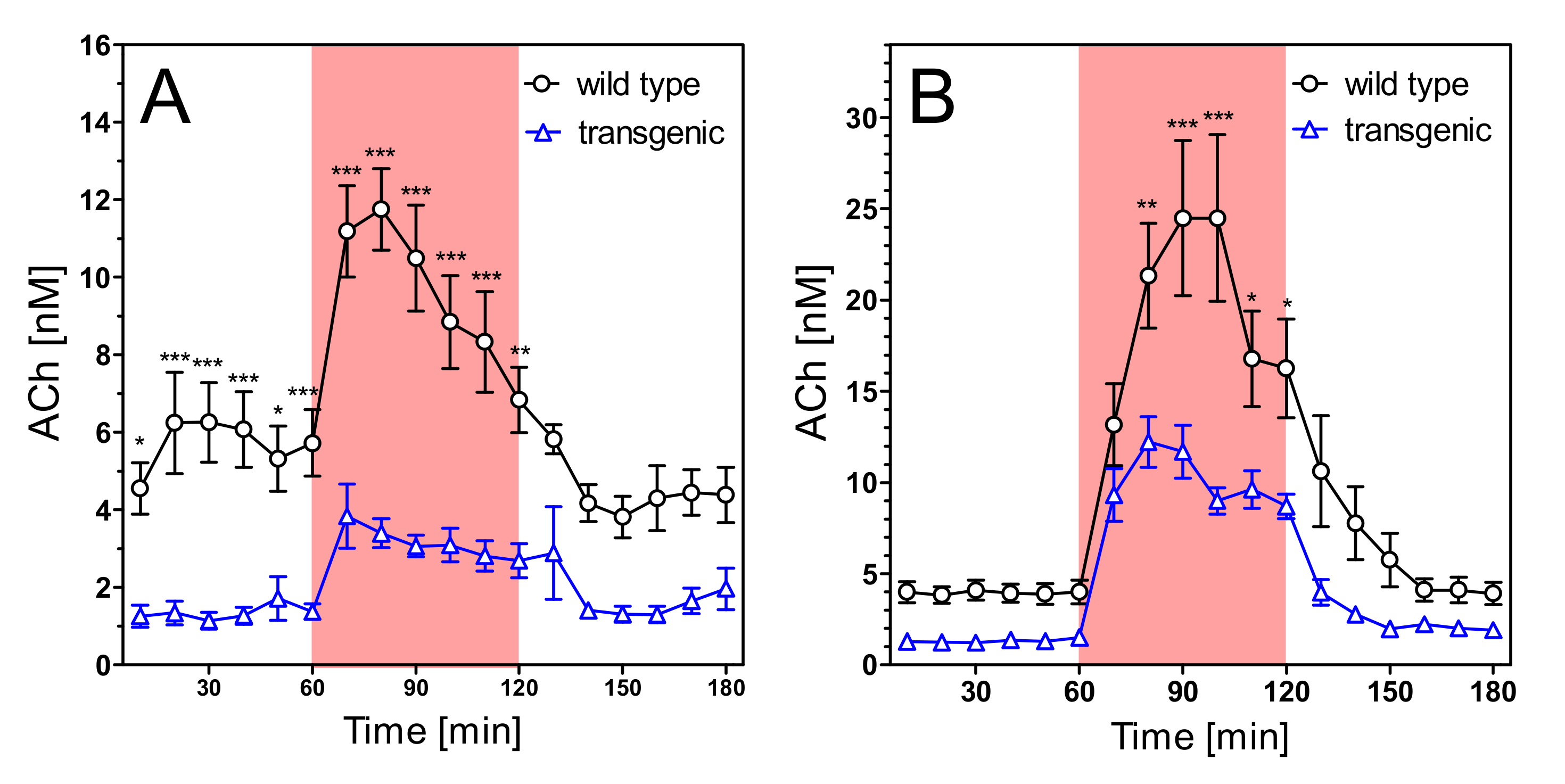
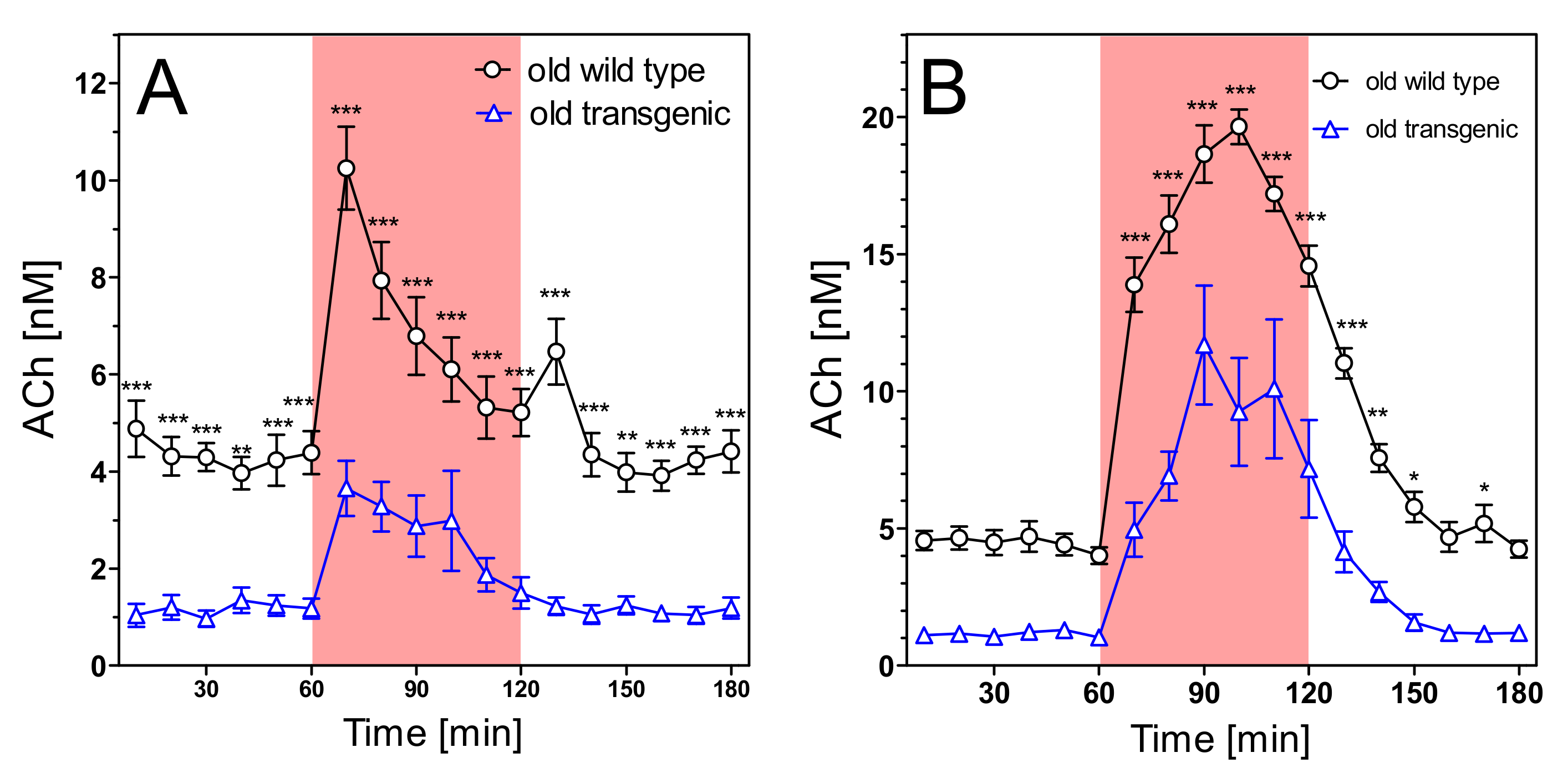
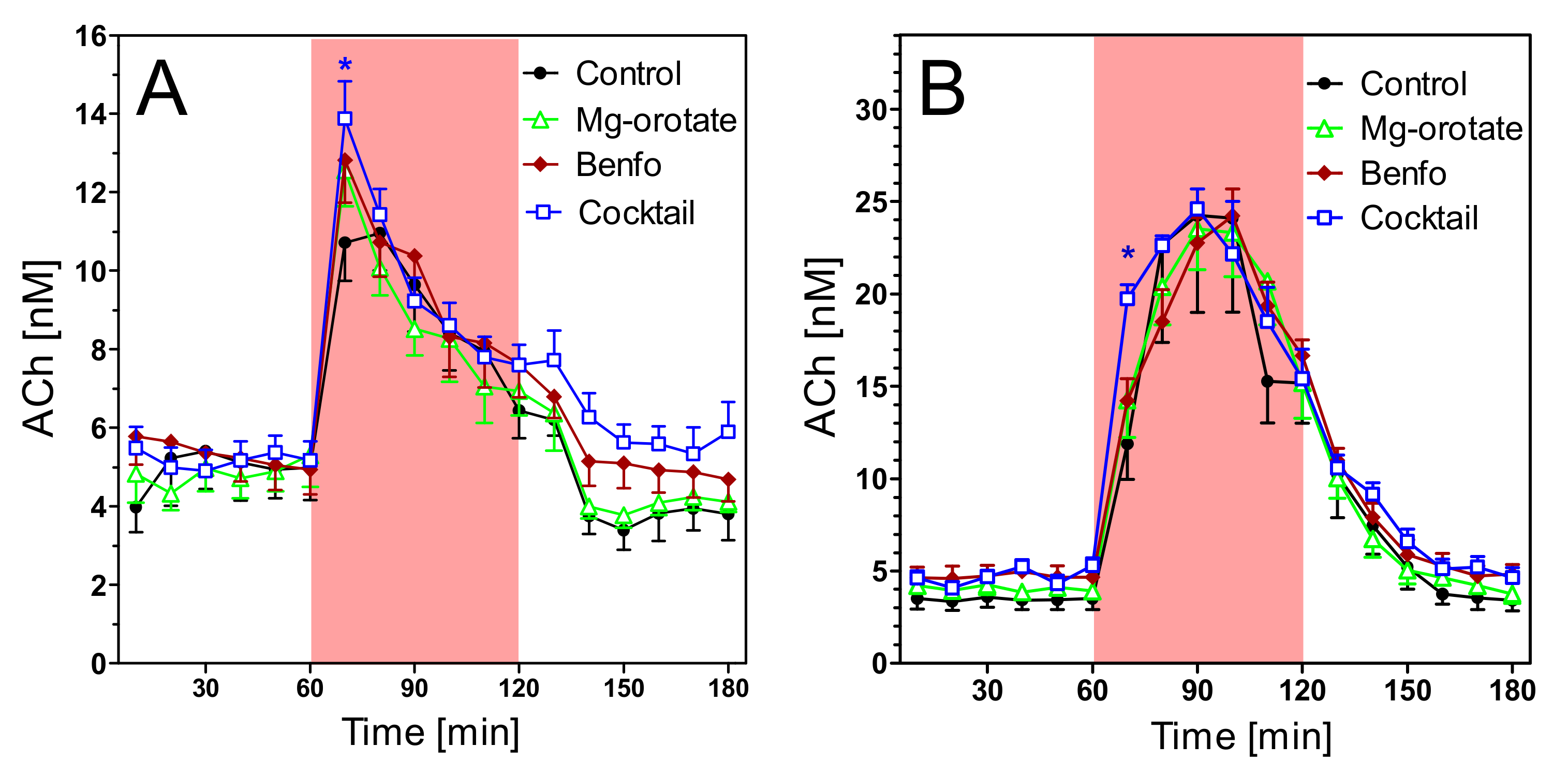
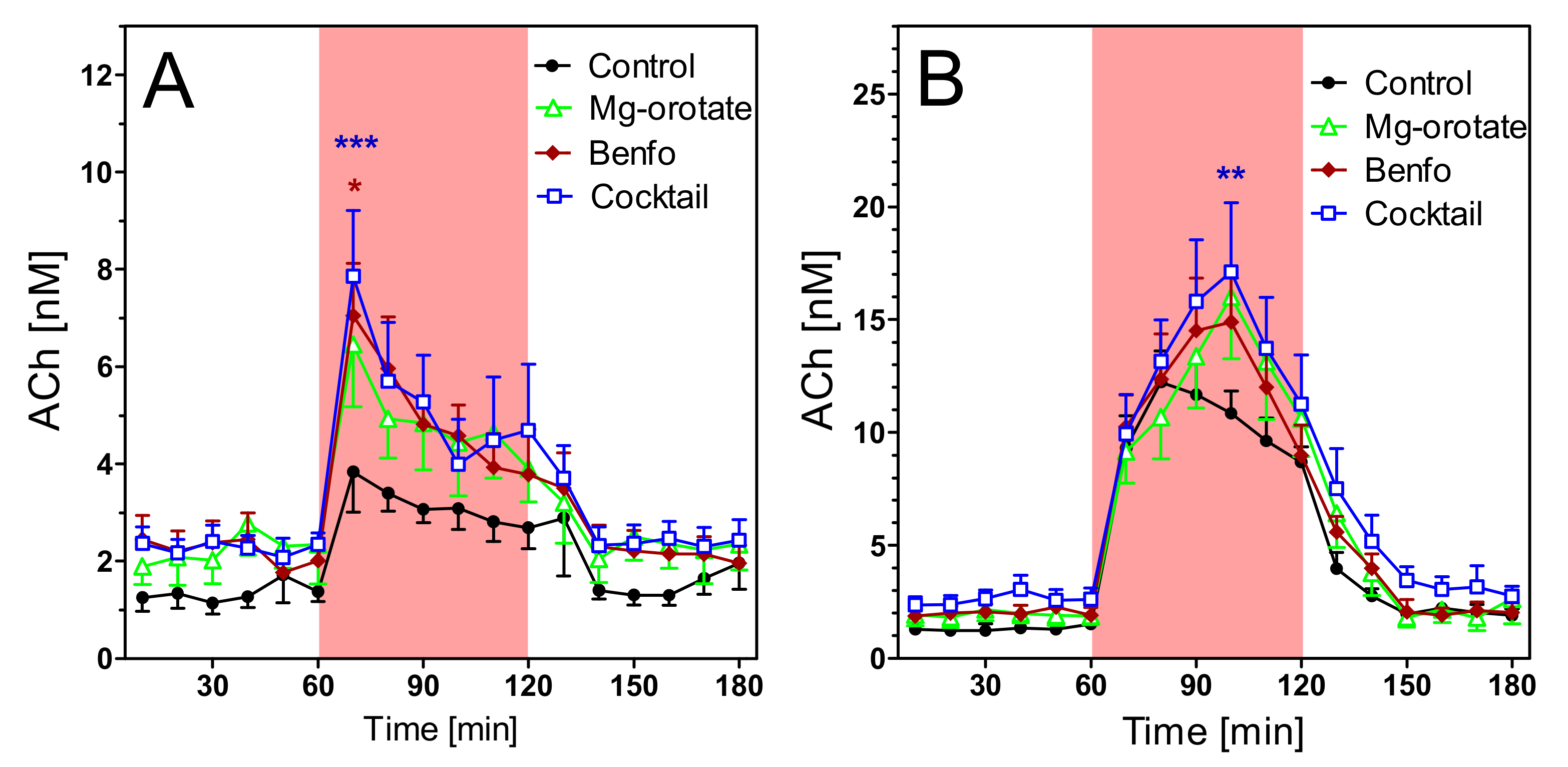
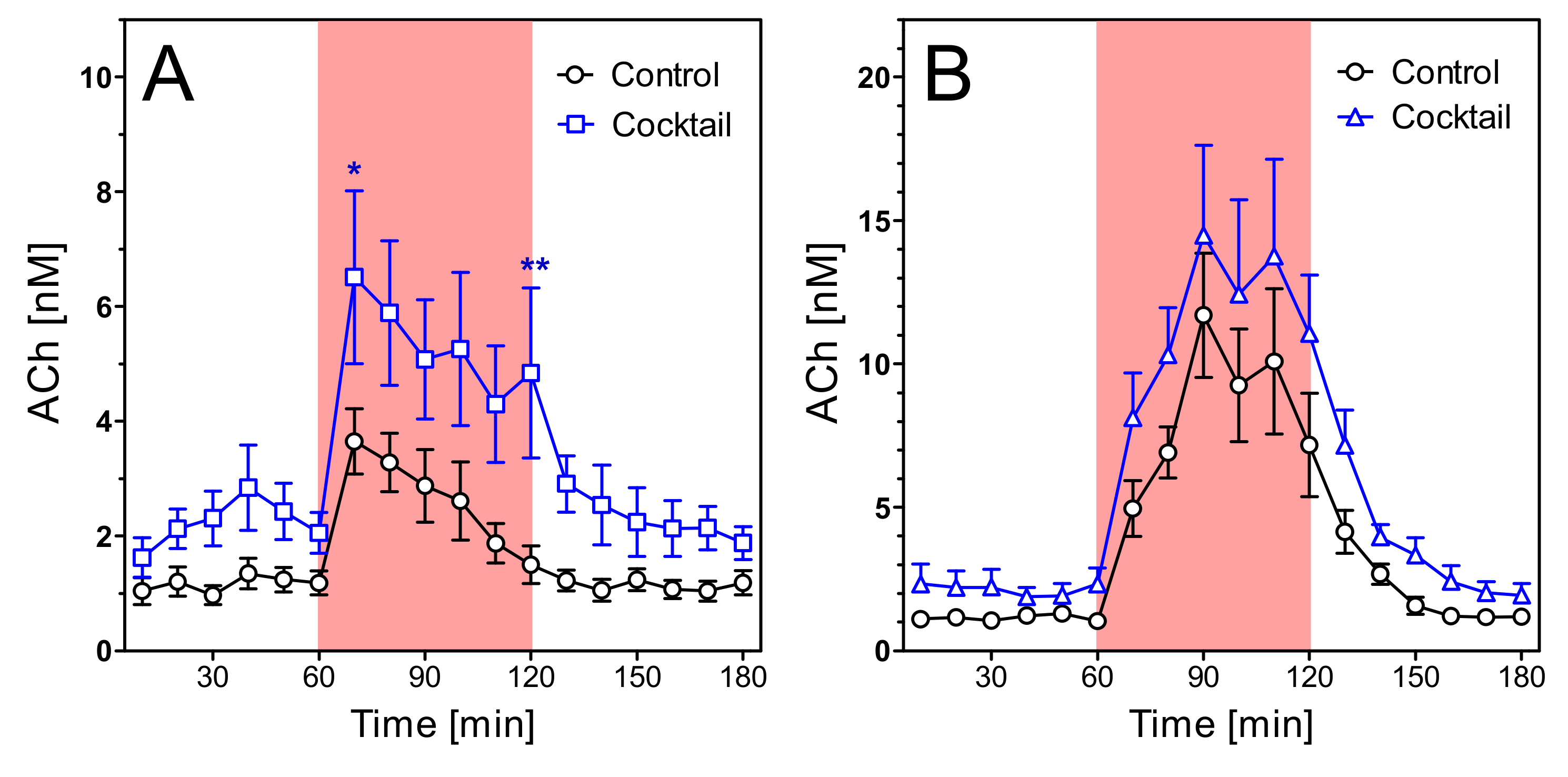
2.4. Acetylcholine Levels
2.5. Cholinergic Enzymes
3. Discussion
4. Materials and Methods
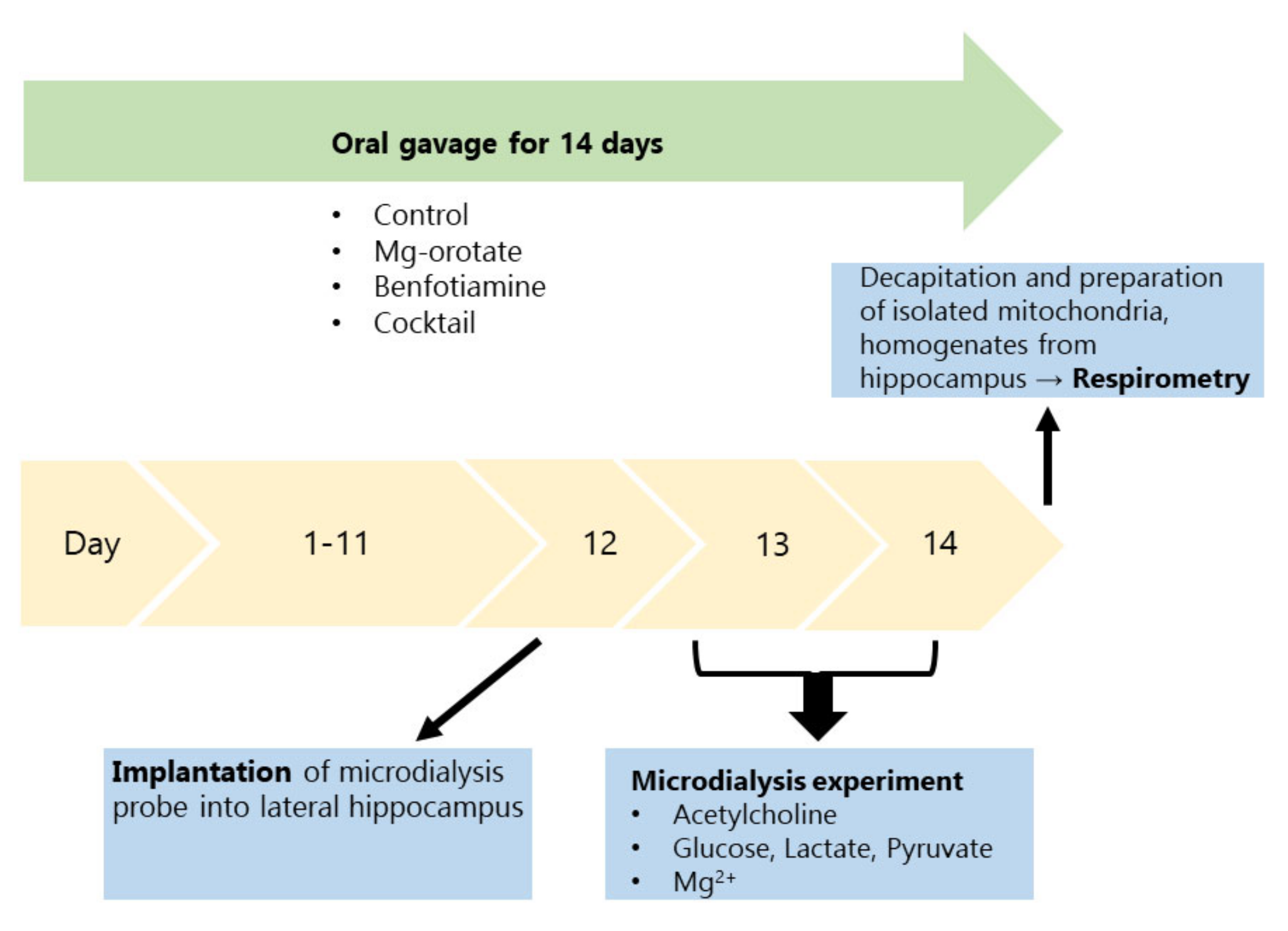
4.1. Animals and Treatments
4.2. Mitochondrial Respirometry
4.3. Microdialysis
4.4. Cholinergic Function
4.5. Analytical Measurements
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s Dement. 2018, 4, 195–214. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Bowen, D.M.; Neary, D.; Davison, A.N. Metabolic processes in Alzheimer’s disease: Adenine nucleotide content and production of 14CO2 from [U-14C]glucose in vitro in human neocortex. J. Neurochem. 1983, 41, 1329–1334. [Google Scholar] [CrossRef]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer. Brain Ann. Neurol. 1983, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.; Filley, C.M.; Kleinschmidt-DeMasters, B.K. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Stoiljkovic, M.; Kelley, C.; Stutz, B.; Horvath, T.L.; Hajós, M. Altered cortical and hippocampal excitability in TgF344-AD rats modeling Alzheimer’s disease pathology. Cereb. Cortex 2019, 29, 2716–2727. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Ladd, A.C.; Bennett, J.P. Postmortem Alzheimer’s disease hippocampi show oxidative phosphorylation gene expression opposite that of isolated pyramidal neurons. J. Alzheimer’s Dis. 2015, 45, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimer’s Dement. 2017, 13, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Pickett, E.K.; Rose, J.; McCrory, C.; McKenzie, C.-A.; King, D.; Smith, C.; Gillingwater, T.; Henstridge, C.; Spires-Jones, T.L. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 2018, 136, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Abolhassan, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech. Ageing Dev. 2017, 161 Pt A, 95–104. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Han, S.; Jeong, Y.Y.; Sheshadri, P.; Su, X.; Cai, Q. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 2020, 21, e49801. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Song, C.Y.; Li, M.Y.; Xu, L.L.; Shen, Y.; Yang, H.; Ding, M.; Liu, X.T.; Xie, Z.H. Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport 2018, 29, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondrial dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Gibson, G.E.; Sheu, K.-F.R.; Blass, J.P.; Baker, A.; Carlson, K.C.; Harding, B.; Perrino, P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol. 1988, 45, 836–840. [Google Scholar] [CrossRef]
- Sang, S.; Pan, X.; Chen, Z.; Zeng, F.; Pan, S.; Liu, H.; Jin, L.; Fei, G.; Wang, C.; Ren, S.; et al. Thiamine diphosphate reduction strongly correlates with brain glucose hypometabolism in Alzheimer’s disease, whereas amyloid deposition does not. Alzheimer’s Res. Ther. 2018, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Fei, G.; Lu, J.; Jin, L.; Pan, S.; Chen, Z.; Wang, C.; Sang, S.; Liu, H.; Hu, W.; et al. Measurement of blood thiamine metabolites for Alzheimer’s disease diagnosis. EBioMedicine 2016, 3, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Baaij, J.H.F.; Hoenderop, J.G.J.; Bindels, R.J.M. Magnesium in man: Implications for health and disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef]
- Li, W.; Yu, J.; Liu, Y.; Huang, X.; Abumaria, N.; Zhu, Y.; Huang, X.; Yiong, W.; Ren, C. Elevation of brain magnesium prevents synaptic loss and reverses cognitive deficits in Alzheimer’s disease mouse model. Mol. Brain 2014, 7, 1–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löffler, M.; Carrey, E.A.; Zameitat, E. Orotic acid, more than just an intermediate of pyrimidine de novo synthesis. J. Genet. Genom. 2015, 42, 207–219. [Google Scholar] [CrossRef]
- Pesini, A.; Iglesias, E.; Bayona-Bafaluy, M.; Garrido-Pérez, N.; Meade, P.; Gaudó, P.; Jiménez-Salvador, I.; Benito, P.A.; Montoya, J.; Ferrer, I.; et al. Brain pyrimidine nucleotide synthesis and Alzheimer disease. Aging 2019, 11, 19. [Google Scholar] [CrossRef]
- Looi, J.C.; Sachdev, P.S. Geriatric psychiatry: Is the jury still out on the cognitive effects of homocysteine and one-carbon metabolism? Curr. Opin. Psychol. 2003, 16, 649–658. [Google Scholar] [CrossRef]
- Collerton, D. Cholinergic function and intellectual decline in Alzheimer’s disease. Neuroscience 1986, 19, 1–28. [Google Scholar] [CrossRef]
- Pepeu, G.; Giovannini, M.G. The fate of the brain cholinergic neurons in neurodegenerative diseases. Brain Res. 2017, 1670, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. 2006, 113, 1625–1644. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Cansev, M.; van Wijk, N.; Turkyilmaz, M.; Orhan, F.; Sijben, J.W.; Broersen, L.M. Specific multi-nutrient enriched diet enhances hippocampal cholinergic transmission in aged rats. Neurobiol. Aging 2015, 36, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Di Meco, A.; Li, J.-G.; Barrero, C.; Merali, S.; Praticò, D. Elevated levels of brain homocysteine directly modulate the pathological phenotype of a mouse model of tauopathy. Mol. Psychiatry 2019, 24, 1696–1706. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; von Bernhardi, R.; Godoy, J.A.; Inestrosa, N.C.; Johnson, G.V. Phosphorylated tau potentiates Aß-induced mitochondrial damage in mature neurons. Neurobiol. Dis. 2014, 71, 260–269. [Google Scholar] [CrossRef]
- Da Silva, S.L.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimer’s Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Bredesen, D.E. Reversal of cognitive decline: A novel therapeutic program. Aging 2014, 6, 707–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Waal, H.; Stam, C.J.; Lansbergen, M.M.; Wieggers, R.L.; Kamphuis, P.J.G.H.; Scheltens, P.; Maestu, F.; Van Straaten, E.C.W. The effect of Souvenaid on functional brain network organisation in patients with mild Alzheimer’s disease: A randomised controlled study. PLoS ONE 2014, 9, e86558. [Google Scholar] [CrossRef] [Green Version]
- Scheltens, P.; Kamphuis, P.J.; Verhey, F.R.; Olde Rikkert, M.G.; Wurtman, R.J.; Wilkinson, D.; Twisk, J.W.; Kurz, A. Efficacy of a medical food in mild Alzheimer’s disease: A randomized, controlled trial. Alzheimer’s Dement. 2010, 6, 1–10. [Google Scholar] [CrossRef]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T. 36-month LipiDiDiet multinutrient clinical trial in prodromal Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 29–40. [Google Scholar] [CrossRef]
- Smith, A.D.; Smith, S.; De Jager, C.A.; Whitbread, P.; Johnston, C.; Agacinski, G.; Oulhaj, A.; Bradley, K.M.; Jacoby, R.; Refsum, H. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS ONE 2010, 5, e12244. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control: An Introduction to OXPHOS Analysis; Oroboros Instruments Corp.: Innsbruck, Austria, 2014. [Google Scholar]
- Hagl, S.; Kocher, A.; Schiborr, C.; Eckert, S.H.; Ciobanu, I.; Birringer, M.; El-Askary, H.; Helal, A.; Khayyal, M.T.; Frank, J.; et al. Rice bran extract protects from mitochondrial dysfunction in guinea pig brains. Pharmacol. Res. 2013, 76, 17–27. [Google Scholar] [CrossRef]
- Schwarzkopf, T.M.; Koch, K.; Klein, J. Reduced severity of ischemic stroke and improvement of mitochondrial function after dietary treatment with the anaplerotic substance triheptanoin. Neuroscience 2015, 300, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O.; Oscai, L.B.; Don, I.J.; Molé, P.A. Mitochondrial citric acid cycle and related enzymes: Adaptive response to exercise. Biochem. Biophys. Res. Commun. 1970, 40, 1368–1373. [Google Scholar] [CrossRef]
- Eigentler, A.; Draxl, A.; Gnaiger, E. Citrate synthase, a mitochondrial marker ernzyme. Mitochond. Physiol. Netw. 2020, 17, 1–12. [Google Scholar]
- Lietsche, J.; Gorka, J.; Hardt, S.; Karas, M.; Klein, J. Self-built microdialysis probes with improved recoveries of ATP and neuropeptides. J. Neurosci. Methods 2014, 237, 1–8. [Google Scholar] [CrossRef]
- Kopf, S.R.; Buchholzer, M.L.; Hilgert, M.; Löffelholz, K.; Klein, J. Glucose plus choline improve passive avoidance behaviour and increase hippocampal acetylcholine release in mice. Neuroscience 2001, 103, 365–371. [Google Scholar] [CrossRef]
- Hartmann, J.; Kiewert, C.; Klein, J. Neurotransmitters and energy metabolites in amyloid-bearing APPSWExPSEN1dE9 mouse brain. J. Pharmacol. Exp. Ther. 2010, 332, 364–370. [Google Scholar] [CrossRef] [Green Version]
- Yuliani, T.; Lobentanzer, S.; Klein, J. Central cholinergic function and metabolic changes in streptozotocin-induced brain injury. J. Neurochem. 2021, 158, 1307–1319. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Glucose [µM] | Lactate [µM] | Lactate/Pyruvate Ratio |
|---|---|---|---|
| Wild type control | 157 ± 16 | 183 ± 19 | 15 ± 2.5 |
| Transgenic control | 180 ± 21 | 194 ± 8 | 13.1 ± 0.8 |
| Wt Mg-orotate | 161 ± 6 | 205 ± 19 | 17.3 ± 2.2 |
| Tg Mg-orotate | 152 ± 23 | 207 ± 23 | 14 ± 1.6 |
| Wt Benfotiamine | 172 ± 11 | 186 ± 8 | 15 ± 1.4 |
| Tg Benfotiamine | 160 ± 26 | 163 ± 15 | 12.7 ± 1.5 |
| Wt Cocktail | 167 ± 18 | 183 ± 18 | 14.6 ± 0.9 |
| Tg Cocktail | 155 ± 18 | 175 ± 20 | 14.3 ± 0.7 |
| Old wt control | 194 ± 11 | 210 ± 12 | 15 ± 0.9 |
| Old tg control | 177 ± 10 | 198 ± 10 | 14.9 ± 0.7 |
| Old wt Cocktail | 174 ± 7 | 195 ± 10 | 15.9 ± 1.4 |
| Old tg Cocktail | 172 ± 10 | 197 ± 12 | 14.1 ± 1.2 |
| Substance | Weighed Portion | % Percentage | Function |
|---|---|---|---|
| Water, deionised | 18.75 | 75 | Vehicle |
| Soy oil | 5 | 20 | Vehicle |
| Glycerol | 0.625 | 2.5 | Cosolvent |
| Lecithin | 0.5 | 2 | Emulsifier |
| Methylcellulose | 0.125 | 0.5 | Pseudo-emulsifier |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viel, C.; Brandtner, A.T.; Weißhaar, A.; Lehto, A.; Fuchs, M.; Klein, J. Effects of Magnesium Orotate, Benfotiamine and a Combination of Vitamins on Mitochondrial and Cholinergic Function in the TgF344-AD Rat Model of Alzheimer’s Disease. Pharmaceuticals 2021, 14, 1218. https://doi.org/10.3390/ph14121218
Viel C, Brandtner AT, Weißhaar A, Lehto A, Fuchs M, Klein J. Effects of Magnesium Orotate, Benfotiamine and a Combination of Vitamins on Mitochondrial and Cholinergic Function in the TgF344-AD Rat Model of Alzheimer’s Disease. Pharmaceuticals. 2021; 14(12):1218. https://doi.org/10.3390/ph14121218
Chicago/Turabian StyleViel, Christian, Adrian T. Brandtner, Alexander Weißhaar, Alina Lehto, Marius Fuchs, and Jochen Klein. 2021. "Effects of Magnesium Orotate, Benfotiamine and a Combination of Vitamins on Mitochondrial and Cholinergic Function in the TgF344-AD Rat Model of Alzheimer’s Disease" Pharmaceuticals 14, no. 12: 1218. https://doi.org/10.3390/ph14121218
APA StyleViel, C., Brandtner, A. T., Weißhaar, A., Lehto, A., Fuchs, M., & Klein, J. (2021). Effects of Magnesium Orotate, Benfotiamine and a Combination of Vitamins on Mitochondrial and Cholinergic Function in the TgF344-AD Rat Model of Alzheimer’s Disease. Pharmaceuticals, 14(12), 1218. https://doi.org/10.3390/ph14121218