1. Introduction
The anticancer drug cyclophosphamide is widely used for the treatment of numerous childhood cancer malignancies, including neonates and infants, and is likely to remain an important chemotherapeutic for the years to come [
1,
2]. Cyclophosphamide, like other oxazaphosphorines, is a prodrug and is required to undergo a relatively complex pathway of metabolism to generate its active alkylating form phosphoramide mustard via 4-hydroxycyclophosphamide (
Figure 1), which can then target DNA replication in tumour cells, alongside the formation of numerous inactive or toxic metabolites [
2].
While many studies have been published investigating the pharmacokinetics and pharmacogenetics of cyclophosphamide in both adult and childhood cancer settings, there remains a dearth of information relating to the clinical pharmacology of cyclophosphamide in neonates and infants [
3,
4]. This leads to uncertainties as to the most appropriate dosing regimens to use in these patient populations, which currently vary between tumour type and clinical treatment protocols, and are commonly supported by relatively limited scientific rationale. For example, cyclophosphamide dose may be based on mg/kg up to 1 year of age or up to a weight of 10 kg in one clinical study protocol, as compared to body surface area-based dosing in another. In addition, commonly implemented dose reductions of 25–50% are applied inconsistently between studies and with different body weight or age cut-off points for a wide range of drugs, as previously discussed [
5,
6]. These factors can lead to substantial differences in the dose of drug administered to neonates and infants as compared to older children, particularly for infants marginally above or below defined dosing cut-offs.
Figure 1.
Cyclophosphamide metabolic pathway (adapted from Jonge et al., 2005 [
7]). Active metabolites are bold, metabolites measured in this study are blue. CYP; Cytochrome P450, ADH; Alcohol dehydrogenase, ALDH; aldehyde dehydrogenase, GST; Glutathione-S-transferase.
Figure 1.
Cyclophosphamide metabolic pathway (adapted from Jonge et al., 2005 [
7]). Active metabolites are bold, metabolites measured in this study are blue. CYP; Cytochrome P450, ADH; Alcohol dehydrogenase, ALDH; aldehyde dehydrogenase, GST; Glutathione-S-transferase.
Although such differences and inconsistencies in dosing regimens for neonates and infants are not uncommon for chemotherapeutics used in paediatric oncology, it may be particularly important for cyclophosphamide due to its complex metabolism and potential maturational differences in the ontogeny of key enzymes involved in its activation. Indeed, a recently published study from the US has highlighted a relationship between exposure to the active metabolite 4-hydroxycyclophosphamide and toxicity, and proposed a reduced cyclophosphamide dosage for young infants who experienced higher exposures, as compared to older children in a brain tumour setting [
8].
The current study was designed to investigate the pharmacokinetics (plasma concentrations and observed level of inter-patient variation in drug exposure) and pharmacogenetics of cyclophosphamide in children <2 years of age at diagnosis. Patients recruited to the study received cyclophosphamide according to the defined dosing regimen for the clinical trial on which they were being treated. Patients were recruited into each of three defined age groups (0–6 months, 7–12 months, and 13–24 months), in order to investigate potential differences in neonate and infant populations where very limited clinical pharmacology data currently exist. The data generated were compared to previously published data generated by our group, using the same population pharmacokinetic model in older children receiving cyclophosphamide.
3. Discussion
As limited data currently exist relating to the pharmacokinetics of cyclophosphamide in neonates and infants, we have assessed the pharmacokinetics of cyclophosphamide and its metabolites in 25 patients under the age of 24 months. The collection of pharmacokinetic data for a specific drug in neonate and infant patient populations is a challenging initiative, as highlighted by the length of time taken to complete the current study. However, the availability of clinical pharmacology data in this patient population has the potential to provide valuable insights which may help to guide future dosing regimens [
11,
12]. This is particularly pertinent in the case of cyclophosphamide, a commonly use anticancer prodrug which requires metabolic activation into its active metabolites.
With patients categorized by age (0–6, 7–12, and 13–24 months) or weight (above and below 10 kg), there were no significant differences in cyclophosphamide clearance observed between patient groups. This was due to large inter-patient variability (CV 41%) in clearance for these infants and neonates, consistent with other drugs used in this age group such as carboplatin and etoposide [
13]. This would suggest that, based on a purely pharmacological rationale, there is no clear evidence for cyclophosphamide dose reductions, when comparing younger patients above and below the commonly used age or body weight cut-off values for reduced dosing (commonly one year of age or 10 kg body weight). Across the patient cohort studied a wide range of cyclophosphamide doses (100–1500 mg/m
2) were administered, as a result of differences in the dosing protocols being followed. Although, there were differences in dosing across protocols, there were no significant differences in clearance or exposure observed between tumour types (
Figure S4). Although there was a trend towards lower exposures in the neuroblastoma group, for those dosed at 5–10 mg/kg/day, these patients are dosed over five consecutive days and their cumulative exposure would therefore be comparable to the other dosing regimens studied.
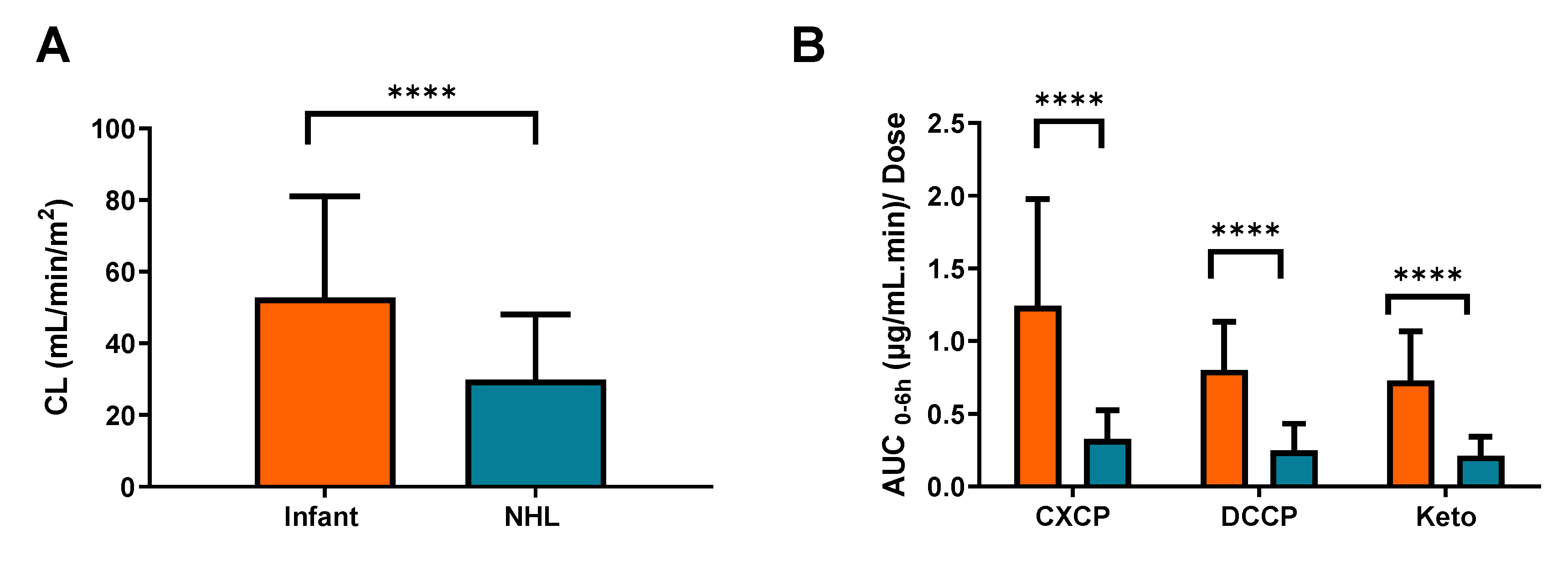
While in the current study no significant differences in cyclophosphamide clearance were reported across tumour types, marked differences in drug clearance and metabolism are observed between the current data and recently published results from a comparable study conducted by our group in a paediatric Non-Hodgkin’s Lymphoma (NHL) patient population. Data from the previous study showed significantly lower cyclophosphamide clearances and metabolite exposures than the data generated from infants recruited to the current study. These findings suggest that age may well be an important factor to take into account when considering cyclophosphamide pharmacokinetics and drug disposition in a childhood cancer setting. Additional factors which may be important to consider include the potential for induction or saturation of cyclophosphamide metabolism, which may impact on the differences between the studies. However, clearance values were determined following the first cyclophosphamide dose in both studies, therefore there is not sufficient time for auto-induction of metabolic enzymes. Furthermore, although patients were generally administered higher relative doses in the infant study (100–1500 mg/m2) than the NHL study (250 mg/m2), no differences in clearance were observed across the dose range investigated in the current study.
Although there are very limited published data available relating to cyclophosphamide pharmacokinetics in infants, a study recently carried out at St Jude Children’s Hospital in the US, indicated that younger infants exhibited higher exposures to 4-hydroxy-cyclophosphamide, an intermediate metabolite in the activation pathway, than older children [
8]. While our study involved measurement of the inactive metabolites of cyclophosphamide as opposed to this intermediate activation metabolite, our findings that cyclophosphamide clearance is higher in infants, with a relative increase in metabolite production observed, are in line with these recent findings from the St. Jude group.
Pharmacogenetic variation has been shown to play a key role in determining cyclophosphamide clearance, with CYP2B6*6 arguably being the most important minor allele investigated [
10,
14,
15,
16]. In the current study a 30% reduction in cyclophosphamide clearance in the *1/*6 group (
n = 7) was observed relative to the *1/*1 group (
n = 18). Although not significant, this finding is consistent with the previously discussed paediatric NHL study, which showed a comparable 34% reduction in cyclophosphamide clearance in the *1/*6 genotype group [
10]. In addition, these data are supported by the subsequent trend of increased cyclophosphamide AUC and decreased metabolite AUC
0–6 for *1/*6 patients. This suggests that this decrease in cyclophosphamide clearance in the *1/*6 genotype is consistent across age groups. Indeed, when the clearance data for the two studies is combined, a significant reduction in clearance associated with the *1/*6 genotype is observed (
Figure S3). In contrast, the other SNPs investigated (CYP2B6*5 and CYP2C19*2) in the current study were associated with less pronounced changes in clearance.
When investigating ontogeny of drug metabolizing enzymes, CYP2B6 is not of primary focus, likely due to its low contribution to metabolism (8%) of clinically relevant drugs. However, from a paediatric oncology perspective, cyclophosphamide is used as part of first line treatment in a variety of childhood cancers such as ALL, Ewings sarcoma, NHL, retinoblastoma and Wilms tumour. Therefore, the potential for childhood cancer patients to receive cyclophosphamide drug is high. Greater numbers of patients are required to determine significant differences in clearance due to ontogeny and genetic variation, to better understand when dose modifications should be applied.
In conclusion, whilst this study demonstrates no significant differences in cyclophosphamide clearance in patients during the first two years of life, it does highlight the large differences in dosing protocols across tumour types. Future studies should focus on establishing the best practice for cyclophosphamide administration, whether it be a low dose over several days or a higher dose on a single day. Furthermore, the study highlights clear differences in cyclophosphamide clearance in children less than two years of age as compared to older children. A more focused and expansive study looking at the full spectrum of ages of children, with a single tumour type and a standardized dosing regimen, would be desirable to determine the best dosing practices for cyclophosphamide in a paediatric setting.
4. Materials and Methods
4.1. Patients and Treatment
The study was approved by the UK Trent Multicentre Ethics Committee and registered through the appropriate clinical trial registries (REC: 06/MRE04/46; CTA: 17136/0245/001; EUDRACT: 2006-002845-36) ahead of patient recruitment. Participating centres were required to obtain local ethical approval and written informed consent from the parents of patients recruited to the study. Patients aged 0–2 years of age at diagnosis, receiving cyclophosphamide as standard treatment according to the relevant clinical trial protocol, were eligible. The various cyclophosphamide dosing regimens in place across active clinical trial treatment protocols during patient recruitment to this pharmacology study are described in
Table S1. All patients were required to have a central venous catheter in place to participate in the study, which involved multiple blood draws for pharmacokinetic analysis. At the time of patient registration baseline toxicity values prior to cyclophosphamide administration were recorded. This included key hematological toxicities such as neutrophil, white blood cell (WBC), hemoglobin and platelet count.
Cyclophosphamide was administered at doses ranging from 100–1500 mg/m2 (5–75 mg/kg), with infusion times ranging from a slow bolus infusion to a 90 min infusion as part of the standard treatment regimen that each patient was receiving. Details of co-medications that patients were receiving on their chemotherapy regimens were recorded. Toxicity was assessed by the National Cancer Institute Common Toxicity Criteria (CTC), version 2.0, following cyclophosphamide treatment. Only toxicities of grade 3 or above were recorded. Patient status was assessed during follow up at six and 12 months after initial cyclophosphamide sampling.
4.2. Blood Sampling and Analysis
Blood samples for quantification of cyclophosphamide and the inactive metabolites, 4-ketocyclophosphamide (Keto), dechloroethylcyclophosphamide (DCCP) and carboxyphosphamide (CXCP) were obtained from a central line prior to drug administration and at multiple time points following the beginning of infusion, commonly 1, 2, 4, 6 and 24 h. Only inactive metabolites were measured due to the challenges associated with measuring 4-hydroxycyclophosphamide metabolite [
8], where additional immediate stabilisation and analysis procedures are required to measure this metabolite. Given that these samples were taken from multiples sites across the UK, measurement of this metabolite was not feasible. All samples were taken from a different lumen to that used for drug administration following a standardized procedure, with the actual drug administration and sampling times recorded for all patients for pharmacokinetic analysis. Plasma was obtained from whole blood samples (2 mL) by centrifugation at 1200 g for 5 min at 4 °C and was frozen at −20 °C prior to analysis. Samples were sent by overnight courier, on dry ice and in an insulated container, to the Newcastle University Centre for Cancer, for analysis.
Sample analysis was carried out using a fully validated LC/MS assay as previously described, with a limit of quantification of 0.5 µg/mL for cyclophosphamide and 0.05 µg/mL for the metabolites [
9,
10]. Appropriate parent drug and metabolite quality control samples were included in each run and the assay exhibited within- and between-run coefficients of variation and bias <15%. Standard curves were linear between 0.5–10 µg/mL and 0.05–1.0 µg/mL for cyclophosphamide and its metabolites, respectively, with samples diluted as required for those containing higher drug concentrations.
4.3. Pharmacokinetic Analysis
Data generated for cyclophosphamide were analysed using a pharmacokinetic model previously published by our group using nonlinear mixed effects modelling (NONMEM version 7.2) in a childhood cancer patient population aged 3–19 years of age [
10]. The first order conditional estimation method with η/ε interaction was used, together with the ADVAN3 and TRANS4 routines. A composite error model was most appropriate to describe within-subject error. An additive error model, on the logarithmic scale, was used for interindividual variability in pharmacokinetic parameters. One thousand data sets were simulated to provide visual predictive checks (VPC) using the population pharmacokinetic model. The median and 90% prediction intervals were plotted with the original data. Empirical Bayes estimates of pharmacokinetic parameters including clearance (CL), volume (V1) and area under the plasma concentration-time curve (AUC) were generated, with the covariates creatinine and CYP2B6*6 genotype due to their relationship with cyclophosphamide CL. To allow for differences in body size, population parameters were scaled to body surface area using the same allometric scaling approach as previously published [
17].
For the cyclophosphamide metabolites, CXCP, DCCP, and Keto, concentrations were not included in the model for cyclophosphamide. Instead AUC values from time 0–6 h were calculated using the trapezoidal rule (WinNonLin v 8.0, Certara L.P. Pharsight, St. Louis, MO, USA), to allow comparisons with previously published data.
Pharmacokinetic data generated on both the parent drug and metabolites were compared with previous data generated using the same analytical techniques and analysed using the same population pharmacokinetic model in a population of 49 childhood cancer population aged 3–19 years [
10].
4.4. Pharmacogenetic Analysis
Genomic DNA was obtained from whole blood samples using Qiagen QIAamp® DNA Blood Maxi kits according to the manufacturer’s instructions. DNA purity and concentration were measured using a NanoDrop ND-1000 (Thermo Scientific, Rockford, USA) and stored at −20 °C prior to pharmacogenetic analysis. Genotyping was carried out for SNPs CYP2B6*4 785A > G (rs2279343), CYP2B6*5 1459C > T (rs3211371), CYP2B6*6 785A > G (rs2279343) & 516G > A (rs3745274), CYP2C19*2 681G > A (rs4244285), CYP2C19*17 806C > T (rs12248560), GSTP1 313A > G (rs1695), CAR 540C > T (rs2307424), and PXR-25385C > T (rs3814055) using TaqMan® probes and an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Allelic discrimination was performed using sequence detection software (Applied Biosystems).
4.5. Statistical Analysis
Differences in cyclophosphamide and metabolite exposure or clearance were assessed in terms of age, weight and genotype. Age was separated into three groups: 0–6, 7–12, and 13–24 months. To investigate the effect of weight on clearance and exposure patients were separated into two groups, above or below 10 kg. These cut off values were selected as dose reductions are typically applied to patients below 10 kg or one year of age. Differences in clearance and exposure were also compared between CYP2B6 genotype groups (SNPs *1/*1 and *1/*6). Overall differences between groups were assessed with the Mann–Whitney and Kruskal–Wallis tests (GraphPad Software, Inc., San Diego, CA, USA, version 8). Statistical significance was associated with p values < 0.05. Inter-patient variability was described using the coefficient of variation (CV), calculated using the geometric mean and standard deviation.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}