
Effect of Dihydroartemisinin-Piperaquine on the Pharmacokinetics of Praziquantel for Treatment of Schistosoma mansoni Infection

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Sociodemographic and Baseline Characteristics
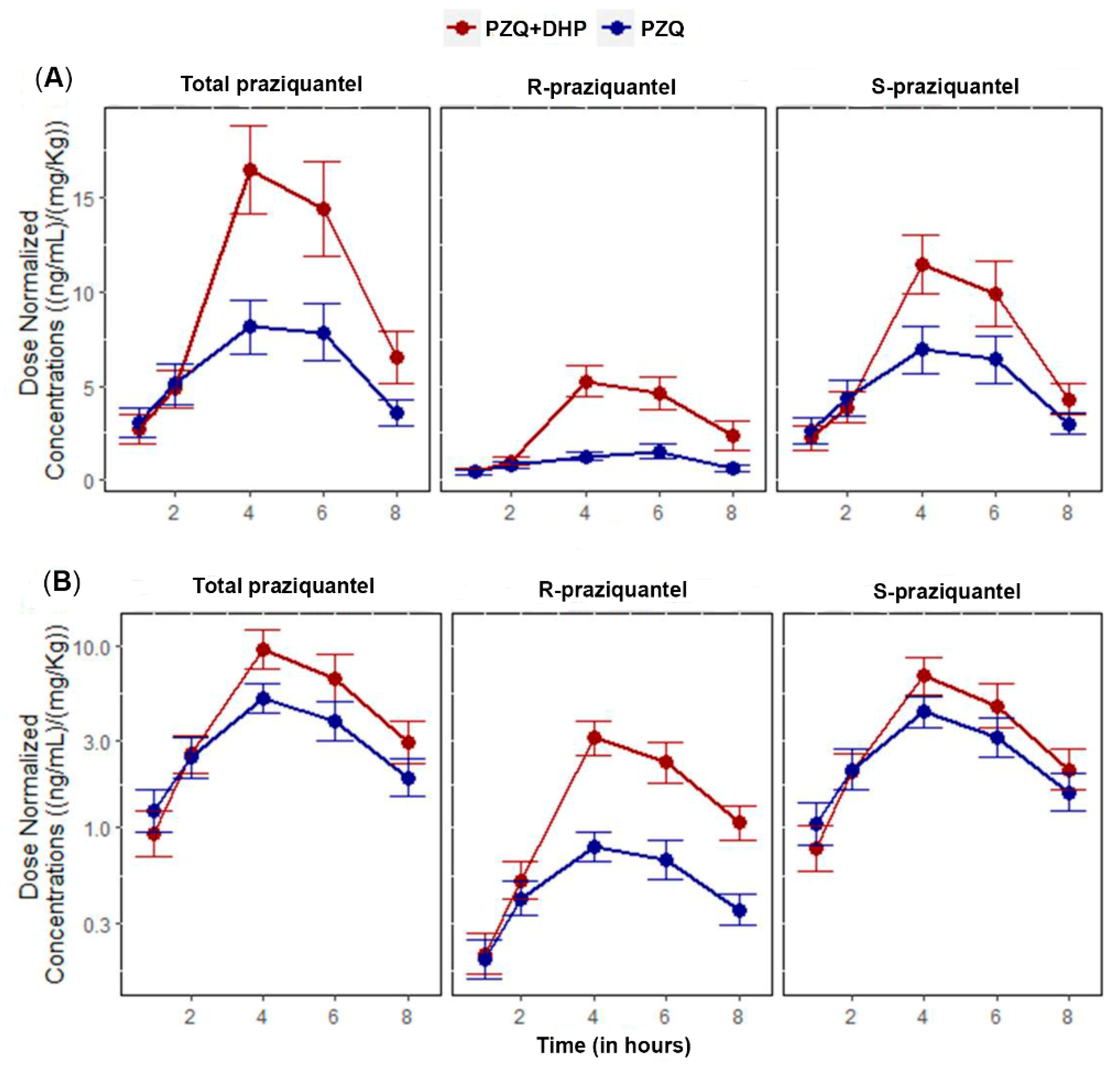
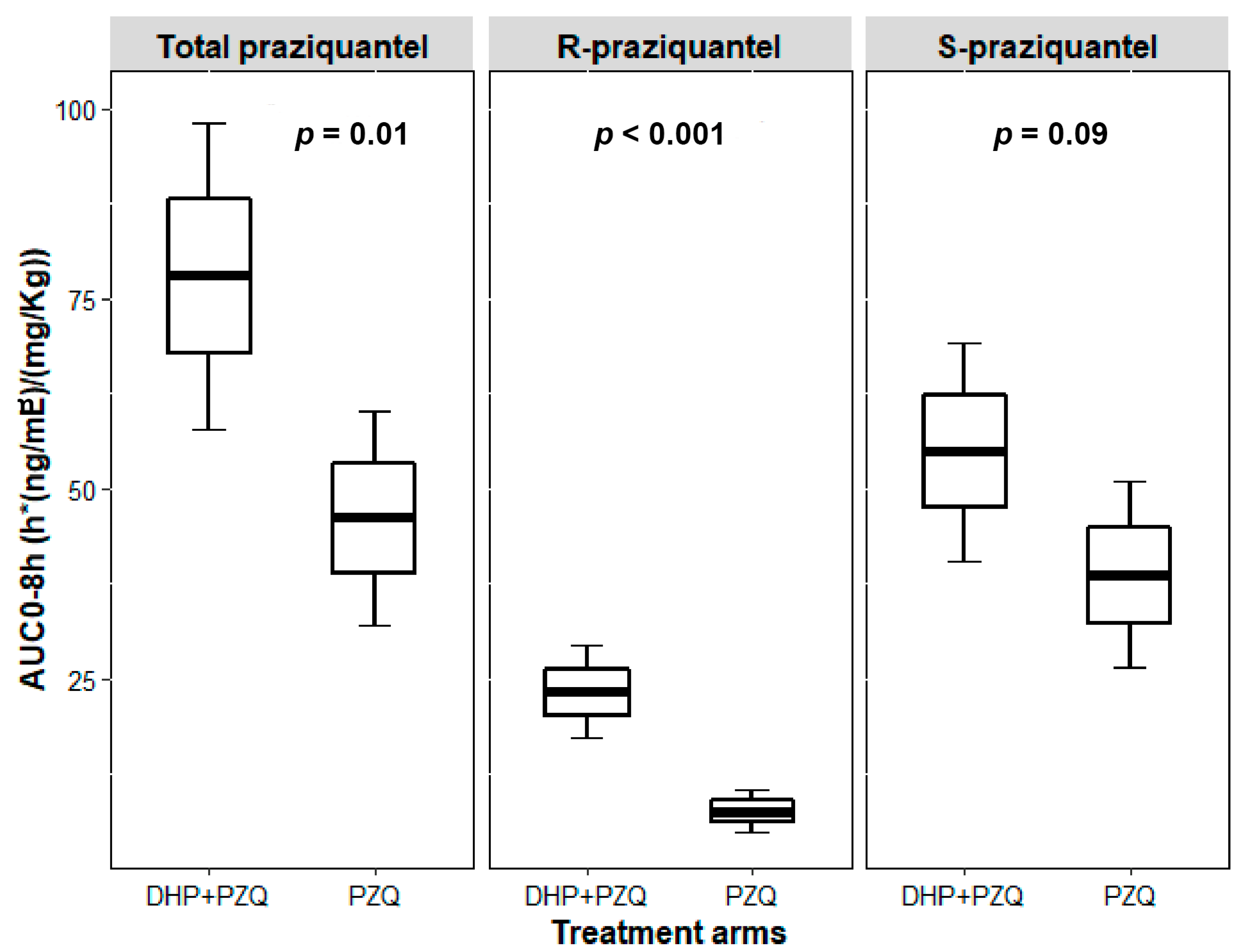
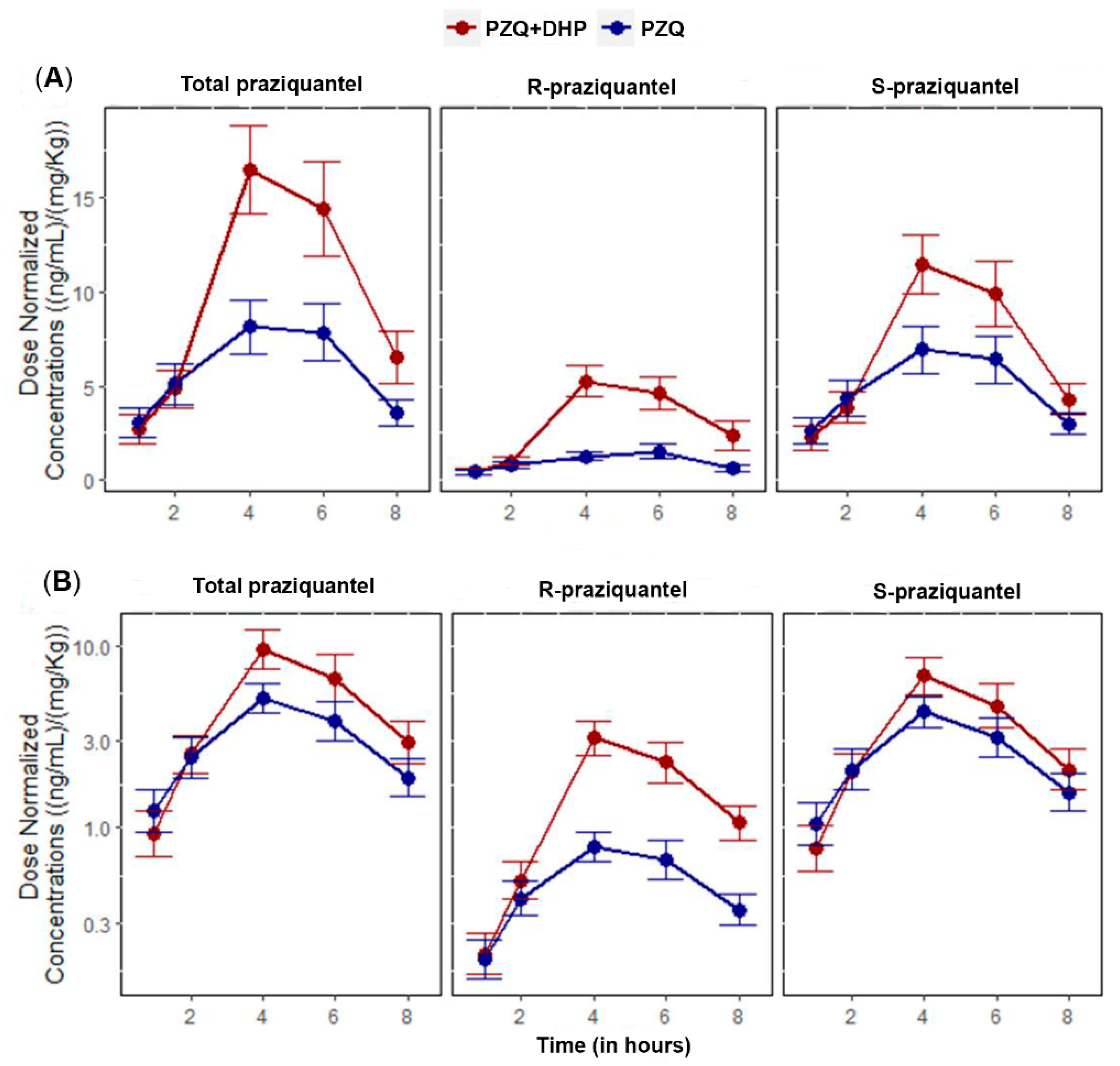
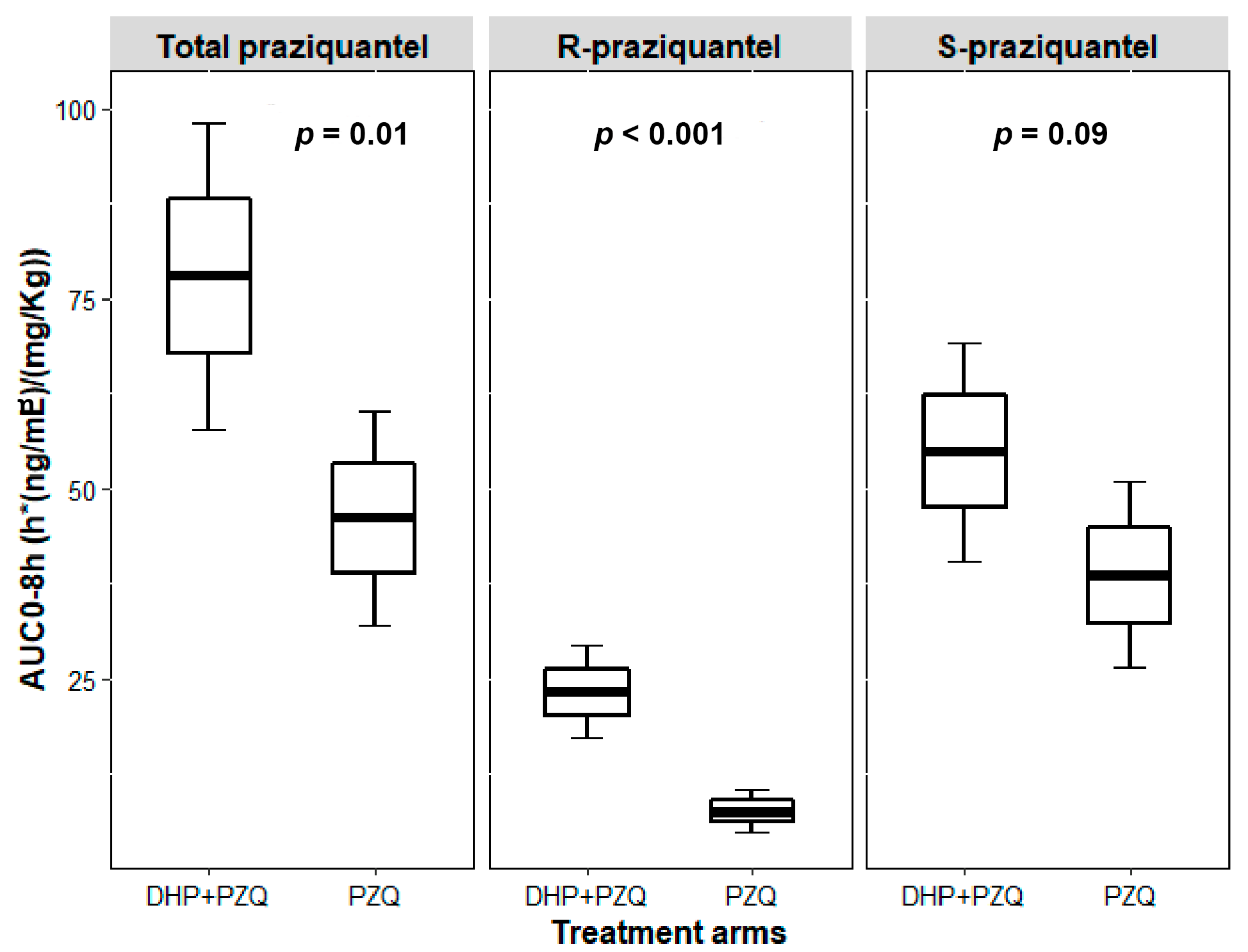
2.2. Effect of DHP on the Pharmacokinetic Parameters of Total PZQ
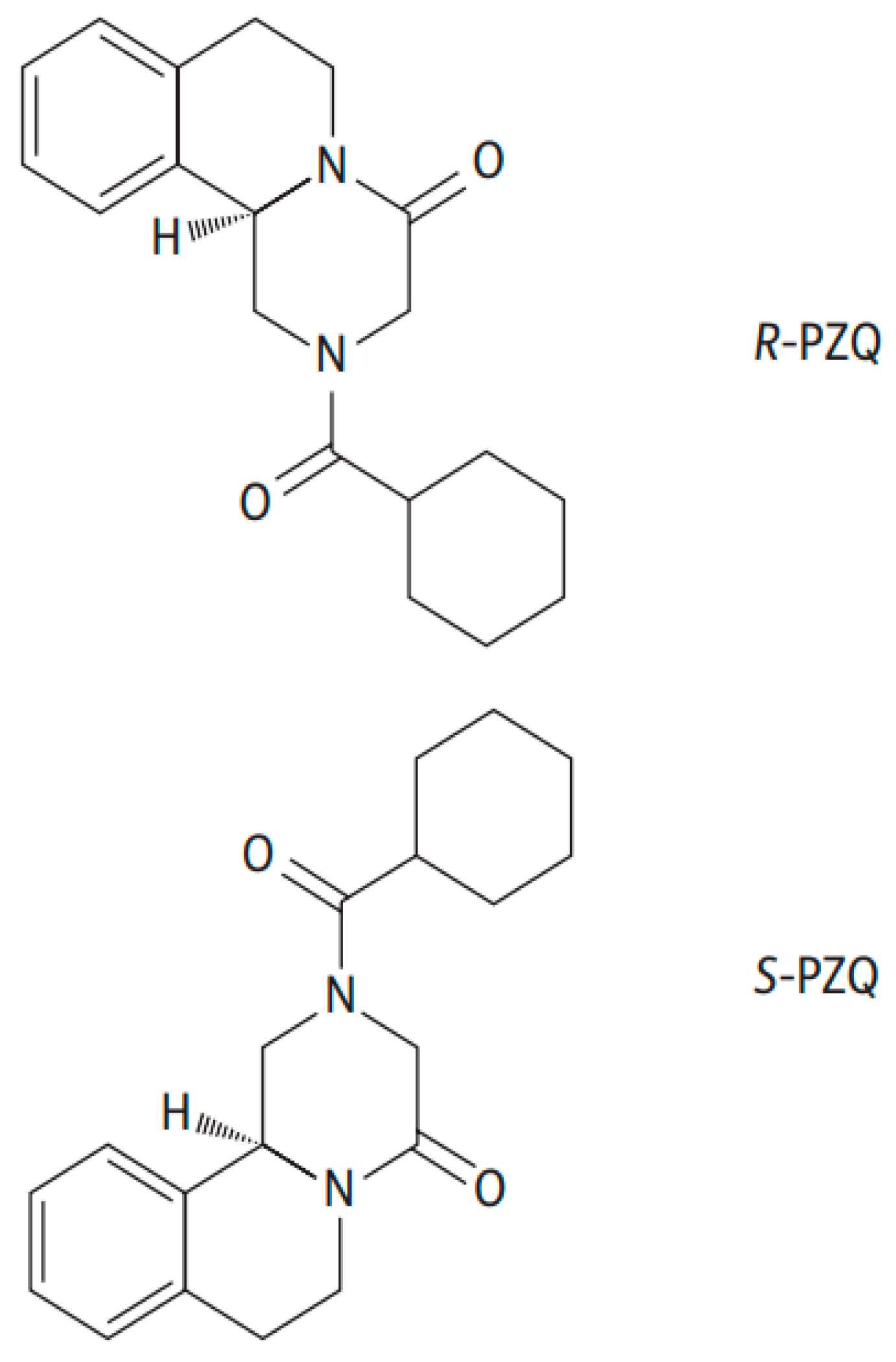
2.3. Effect of DHP on the Pharmacokinetic Parameters of R-PZQ and S-PZQ
3. Discussion
4. Materials and Methods
4.1. Study Design, Area, and Population
4.2. Treatment and Samples Collection for Pharmacokinetic Study
4.3. Chemicals and Reagents
4.4. Analytical Method and Validation
4.5. Pharmacokinetic Data Analysis
4.6. Statistical Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chitsulo, L.; Engels, D.; Montresor, A.; Savioli, L. The global status of schistosomiasis and its control. Acta Trop. 2000, 77, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Hotez, P.J.; Alvarado, M.; Basanez, M.G.; Bolliger, I.; Bourne, R.; Boussinesq, M.; Brooker, S.J.; Brown, A.S.; Buckle, G.; Budke, C.M.; et al. The global burden of disease study 2010: Interpretation and implications for the neglected tropical diseases. PLoS Negl. Trop. Dis. 2014, 8, e2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Schistosomiasis Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs115/en/index.html (accessed on 13 February 2020).
- Mazigo, H.D. Participatory integrated control strategies and elimination of schistosomiasis in sub-Saharan Africa. Lancet Glob. Health 2019, 7, e998–e999. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Neglected Tropical Diseases, Hidden Successes, Emerging Opportunities; World Health Organization: Geneva, Switzerland, 2009; Available online: https://apps.who.int/iris/handle/10665/44214 (accessed on 15 February 2020).
- World Health Organization. Schistosomiasis and Soiltransmitted Helminthiases: Numbers of People Treated in 2017; World Health Organization: Geneva, Switzerland, 2018; Available online: https://www.who.int/neglected_diseases/resources/who_wer9350/en/ (accessed on 30 January 2020).
- Frigerio, S.; Macario, M.; Iacovone, G.; Dussey-Comlavi, K.J.; Narcisi, P.; Ndiaye, A.T.; Moramarco, S.; Alvaro, R.; Palombi, L.; Buonomo, E. Schistosoma haematobium infection, health and nutritional status in school-age children in a rural setting in Northern Senegal. Minerva Pediatr. 2016, 68, 282–287. [Google Scholar]
- Kinung’hi, S.M.; Mazigo, H.D.; Dunne, D.W.; Kepha, S.; Kaatano, G.; Kishamawe, C.; Ndokeji, S.; Angelo, T.; Nuwaha, F. Coinfection of intestinal schistosomiasis and malaria and association with haemoglobin levels and nutritional status in school children in Mara region, Northwestern Tanzania: A cross-sectional exploratory study. BMC Res. Notes 2017, 10, 583. [Google Scholar] [CrossRef]
- Koukounari, A.; Fenwick, A.; Whawell, S.; Kabatereine, N.B.; Kazibwe, F.; Tukahebwa, E.M.; Stothard, J.R.; Donnelly, C.A.; Webster, J.P. Morbidity indicators of Schistosoma mansoni: Relationship between infection and anemia in Ugandan schoolchildren before and after praziquantel and albendazole chemotherapy. Am. J. Trop. Med. Hyg. 2006, 75, 278–286. [Google Scholar] [CrossRef]
- Stoltzfus, R.J.; Albonico, M.; Tielsch, J.M.; Chwaya, H.M.; Savioli, L. Linear growth retardation in Zanzibari school children. J. Nutr. 1997, 127, 1099–1105. [Google Scholar] [CrossRef]
- Hotez, P.J.; Fenwick, A.; Savioli, L.; Molyneux, D.H. Rescuing the bottom billion through control of neglected tropical diseases. Lancet 2009, 373, 1570–1575. [Google Scholar] [CrossRef]
- Hotez, P.J.; Herricks, J.R. Helminth elimination in the pursuit of sustainable development goals: A “worm index” for human development. PLoS Negl. Trop. Dis. 2015, 9, e0003618. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Investing to Overcome the Global Impact of Neglected Tropical Diseases, Third WHO Report on Neglected Tropical Diseases; World Health Organization: Geneva, Switzerland, 2015; Available online: https://apps.who.int/iris/bitstream/handle/10665/152781/9789241564861_eng.pdf?sequence=1 (accessed on 6 November 2019).
- Mnkugwe, R.H.; Minzi, O.S.; Kinung’hi, S.M.; Kamuhabwa, A.A.; Aklillu, E. Prevalence and correlates of intestinal schistosomiasis infection among school-aged children in North-Western Tanzania. PLoS ONE 2020, 15, e0228770. [Google Scholar] [CrossRef] [Green Version]
- Munisi, D.Z.; Buza, J.; Mpolya, E.A.; Kinung’hi, S.M. Schistosoma mansoni infections, undernutrition and anaemia among primary schoolchildren in two onshore villages in Rorya district, North-Western Tanzania. PLoS ONE 2016, 11, e0167122. [Google Scholar] [CrossRef] [Green Version]
- Inobaya, M.T.; Olveda, R.M.; Chau, T.N.; Olveda, D.U.; Ross, A.G. Prevention and control of schistosomiasis: A current perspective. Res. Rep. Trop. Med. 2014, 2014, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Gryseels, B.; Mbaye, A.; De Vlas, S.J.; Stelma, F.F.; Guisse, F.; Van Lieshout, L.; Faye, D.; Diop, M.; Ly, A.; Tchuem-Tchuente, L.A.; et al. Are poor responses to praziquantel for the treatment of Schistosoma mansoni infections in Senegal due to resistance? An overview of the evidence. Trop. Med. Int. Health 2001, 6, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Bergquist, R.; Elmorshedy, H. Artemether and Praziquantel: Origin, Mode of Action, Impact, and Suggested Application for Effective Control of Human Schistosomiasis. Trop. Med. Infect. Dis. 2018, 3, 125. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Prevention and Control of Schistosomiasis and Soil-Transmitted Helminthiasis: Report of a WHO Expert Committee; Technical Report Series 912; World Health Organization: Geneva, Switzerland, 2002; Available online: https://apps.who.int/iris/handle/10665/42588 (accessed on 6 November 2019).
- Liu, R.; Dong, H.F.; Guo, Y.; Zhao, Q.P.; Jiang, M.S. Efficacy of praziquantel and artemisinin derivatives for the treatment and prevention of human schistosomiasis: A systematic review and meta-analysis. Parasites Vectors 2011, 4, 201. [Google Scholar] [CrossRef] [Green Version]
- Perez del Villar, L.; Burguillo, F.J.; Lopez-Aban, J.; Muro, A. Systematic review and meta-analysis of artemisinin based therapies for the treatment and prevention of schistosomiasis. PLoS ONE 2012, 7, e45867. [Google Scholar] [CrossRef]
- Mnkugwe, R.H.; Minzi, O.; Kinung’hi, S.; Kamuhabwa, A.; Aklillu, E. Efficacy and safety of praziquantel and dihydroartemisinin piperaquine combination for treatment and control of intestinal schistosomiasis: A randomized, non-inferiority clinical trial. PLoS Negl. Trop. Dis. 2020, 14, e0008619. [Google Scholar] [CrossRef]
- Ilett, K.F.; Ethell, B.T.; Maggs, J.L.; Davis, T.M.; Batty, K.T.; Burchell, B.; Binh, T.Q.; Thu Le, T.A.; Hung, N.C.; Pirmohamed, M.; et al. Glucuronidation of dihydroartemisinin in vivo and by human liver microsomes and expressed UDP-glucuronosyltransferases. Drug Metab. Dispos. 2002, 30, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.M.; Huang, L.; Johnson, M.K.; Lizak, P.; Kroetz, D.; Aweeka, F.; Parikh, S. In vitro metabolism of piperaquine is primarily mediated by CYP3A4. Xenobiotica 2012, 42, 1088–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Fang, Z.Z.; Zheng, Y.; Zhou, K.; Hu, C.; Krausz, K.W.; Sun, D.; Idle, J.R.; Gonzalez, F.J. Metabolic profiling of praziquantel enantiomers. Biochem. Pharmacol. 2014, 90, 166–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunajeewa, H.A.; Ilett, K.F.; Mueller, I.; Siba, P.; Law, I.; Page-Sharp, M.; Lin, E.; Lammey, J.; Batty, K.T.; Davis, T.M. Pharmacokinetics and efficacy of piperaquine and chloroquine in Melanesian children with uncomplicated malaria. Antimicrob. Agents Chemother. 2008, 52, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masimirembwa, C.M.; Naik, Y.S.; Hasler, J.A. The effect of chloroquine on the pharmacokinetics and metabolism of praziquantel in rats and in humans. Biopharm. Drug Dispos. 1994, 15, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Kovac, J.; Meister, I.; Neodo, A.; Panic, G.; Coulibaly, J.T.; Falcoz, C.; Keiser, J. Pharmacokinetics of praziquantel in Schistosoma mansoni- and Schistosoma haematobium-infected school- and preschool-aged children. Antimicrob. Agents Chemother. 2018, 62, e02253-17. [Google Scholar] [CrossRef] [Green Version]
- Meister, I.; Ingram-Sieber, K.; Cowan, N.; Todd, M.; Robertson, M.N.; Meli, C.; Patra, M.; Gasser, G.; Keiser, J. Activity of praziquantel enantiomers and main metabolites against Schistosoma mansoni. Antimicrob. Agents Chemother. 2014, 58, 5466–5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.H. Interpretation of drug interaction using systemic and local tissue exposure changes. Pharmaceutics 2020, 12, 417. [Google Scholar] [CrossRef]
- Coulibaly, J.T.; Panic, G.; Silue, K.D.; Kovac, J.; Hattendorf, J.; Keiser, J. Efficacy and safety of praziquantel in preschool-aged and school-aged children infected with Schistosoma mansoni: A randomised controlled, parallel-group, dose-ranging, phase 2 trial. Lancet Glob. Health 2017, 5, e688–e698. [Google Scholar] [CrossRef] [Green Version]
- Bustinduy, A.L.; Waterhouse, D.; de Sousa-Figueiredo, J.C.; Roberts, S.A.; Atuhaire, A.; Van Dam, G.J.; Corstjens, P.L.; Scott, J.T.; Stanton, M.C.; Kabatereine, N.B.; et al. Population pharmacokinetics and pharmacodynamics of praziquantel in Ugandan children with intestinal schistosomiasis: Higher dosages are required for maximal efficacy. mBio 2016, 7, e00227-16. [Google Scholar] [CrossRef] [Green Version]
- White, N.J. Intermittent presumptive treatment for malaria. PLoS Med. 2005, 2, e3. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.; Sekljic, H.; Fuchs, S.; Bothe, H.; Schollmeyer, D.; Miculka, C. Taste, a new incentive to switch to (R)-praziquantel in schistosomiasis treatment. PLoS Negl. Trop. Dis. 2009, 3, e357. [Google Scholar] [CrossRef] [Green Version]
- el-Lakkany, N.M.; el-Din, S.H.; Sabra, A.N.; Hammam, O.A. Pharmacodynamics of mefloquine and praziquantel combination therapy in mice harbouring juvenile and adult Schistosoma mansoni. Memorias do Instituto Oswaldo Cruz 2011, 106, 814–822. [Google Scholar] [CrossRef]
- Mnkugwe, R.H.; Minzi, O.S.; Kinung’hi, S.M.; Kamuhabwa, A.A.; Aklillu, E. Efficacy and safety of praziquantel for treatment of Schistosoma mansoni infection among school children in Tanzania. Pathogens 2019, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Castro, N.; Medina, R.; Sotelo, J.; Jung, H. Bioavailability of praziquantel increases with concomitant administration of food. Antimicrob. Agents Chemother. 2000, 44, 2903–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olliaro, P.; Delgado-Romero, P.; Keiser, J. The little we know about the pharmacokinetics and pharmacodynamics of praziquantel (racemate and R-enantiomer). J. Antimicrob. Chemother. 2014, 69, 863–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015; Available online: https://www.who.int/malaria/publications/atoz/9789241549127/en/ (accessed on 6 November 2019).
- European Medicine Agency. Guideline on Bioanalytical Method Validation EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 10 March 2021).
- R Core Team. R: A language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 10 February 2021).
- Denney, W.; Duvvuri, S.; Buckeridge, C. Simple, Automatic Noncompartmental Analysis: The PKNCA R Package. J. Pharmacokinet. Pharmacodyn. 2015, 42, S65. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Variable | Treatment Arm | p-Value | ||
|---|---|---|---|---|
| PZQ | PZQ + DHP | |||
| Age (years) | Mean ± SD Range | 12.8 ± 1.6 10–17 | 12.7 ± 2.0 9–16 | 0.79 a |
| Sex | Male N (%) | 17 (53.1) | 18 (56.3) | 0.85 |
| Female N (%) | 15 (46.9) | 14 (43.8) | 0.87 | |
| Weight (kg) | Mean ± SD | 33.1 ± 6.2 | 33.7 ± 9.3 | 0.74 a |
| Height (cm) | Mean ± SD | 141.0 ± 9.7 | 143.9 ± 12.9 | 0.32 a |
| Haemoglobin (g/dL) | Mean ± SD | 12.4 ± 1.7 | 12.2 ± 1.9 | 0.71 a |
| Infection intensity | Eggs per gran Median (range) | 246 (12–1452) | 165 (6–1722) | 0.32 b |
| Light N (%) | 9 (28.1) | 14 (43.8) | 0.46 | |
| Moderate N (%) | 11 (34.4) | 8 (25.0) | 0.67 | |
| Heavy N (%) | 12 (37.5) | 10 (31.3) | 0.77 | |
| Parameters | Treatment Arm | (PZQ + DHP)|PZQ | |
|---|---|---|---|
| PZQ + DHP | PZQ | ||
| Geometric mean (CV %) | Geometric mean (CV %) | Geometric Mean Ratio (90% CI) | |
| AUC (0-Inf) | 66.8 (112.1) | 33.5 (106.4) | 2.18 (1.27–3.76) |
| AUC (0–8h) | 50.4 (181.8) | 32.4 (111.9) | 1.73 (1.12–2.69) |
| Cmax (ng/mL) | 14.3 (169) | 8.9 (95.1) | 1.75 (1.15–2.65) |
| Half-life (h) | 1.5 (29.7) | 2.2 (47.5) | 0.7 (0.57–0.86) |
| Tmax (h) | 3.6 (55.7) | 3.5 (61.8) | |
| Parameters | R-PZQ | (PZQ + DHP)|PZQ GMR (90% CI) | S-PZQ | (PZQ + DHP)|PZQ GMR (90% CI) | ||
|---|---|---|---|---|---|---|
| Treatment Arm | Treatment Arm | |||||
| PZQ + DHP GM (CV%) | PZQ GM (CV%) | PZQ + DHP GM (CV%) | PZQ GM (CV%) | |||
| AUC (0-Inf) |
19.5 (118.1) |
4.9 (84.2) | 3.98 (2.27–7.0) |
44.8 (114.6) |
27.1 (119.6) | 1.86 (1.06–3.28) |
| AUC (0–8h) |
14.2 (235.3) |
4.8 (144.1) | 2.94 (1.75–4.92) |
35.6 (174.9) |
26.7 (114.9) | 1.5 (0.97–2.31) |
| Cmax (ng/mL) |
4.5 (206.5) |
1.5 (119.5) | 3.08 (1.91–4.96) |
9.9 (160.1) |
7.3 (96.3) | 1.5 (1.0–2.25) |
| Half-life (h) |
1.6 (34.3) |
2 (35.9) | 0.79 (0.63–0.98) |
1.7 (26.7) |
2.1 (43.5) | 0.77 (0.63–0.94) |
| Tmax (h) |
3.8 (51.4) |
3.5 (62.8) |
3.6 (55.2) |
3.5 (61.8) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minzi, O.M.; Mnkugwe, R.H.; Ngaimisi, E.; Kinung’hi, S.; Hansson, A.; Pohanka, A.; Kamuhabwa, A.; Aklillu, E. Effect of Dihydroartemisinin-Piperaquine on the Pharmacokinetics of Praziquantel for Treatment of Schistosoma mansoni Infection. Pharmaceuticals 2021, 14, 400. https://doi.org/10.3390/ph14050400
Minzi OM, Mnkugwe RH, Ngaimisi E, Kinung’hi S, Hansson A, Pohanka A, Kamuhabwa A, Aklillu E. Effect of Dihydroartemisinin-Piperaquine on the Pharmacokinetics of Praziquantel for Treatment of Schistosoma mansoni Infection. Pharmaceuticals. 2021; 14(5):400. https://doi.org/10.3390/ph14050400
Chicago/Turabian StyleMinzi, Omary Mashiku, Rajabu Hussein Mnkugwe, Eliford Ngaimisi, Safari Kinung’hi, Anna Hansson, Anton Pohanka, Appolinary Kamuhabwa, and Eleni Aklillu. 2021. "Effect of Dihydroartemisinin-Piperaquine on the Pharmacokinetics of Praziquantel for Treatment of Schistosoma mansoni Infection" Pharmaceuticals 14, no. 5: 400. https://doi.org/10.3390/ph14050400
APA StyleMinzi, O. M., Mnkugwe, R. H., Ngaimisi, E., Kinung’hi, S., Hansson, A., Pohanka, A., Kamuhabwa, A., & Aklillu, E. (2021). Effect of Dihydroartemisinin-Piperaquine on the Pharmacokinetics of Praziquantel for Treatment of Schistosoma mansoni Infection. Pharmaceuticals, 14(5), 400. https://doi.org/10.3390/ph14050400