
Small Molecule Inhibitors of Influenza Virus Entry



Abstract
1. Introduction
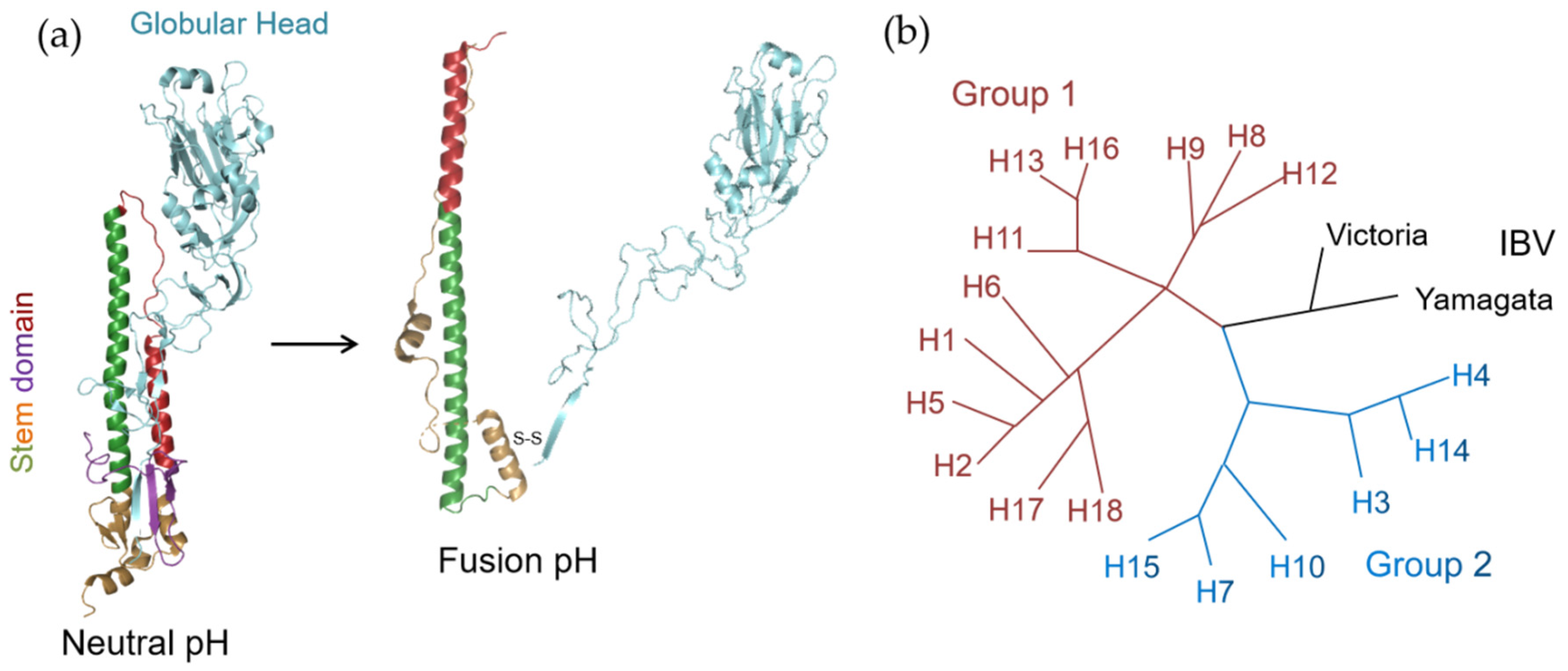
2. The Structure and Function of HA
3. Small Molecule Inhibitors Targeting HA Mediated Receptor Binding
4. Small Molecule Inhibitors Targeting HA Mediated Fusion
4.1. Group 1 Specific Influenza Fusion Inhibitors
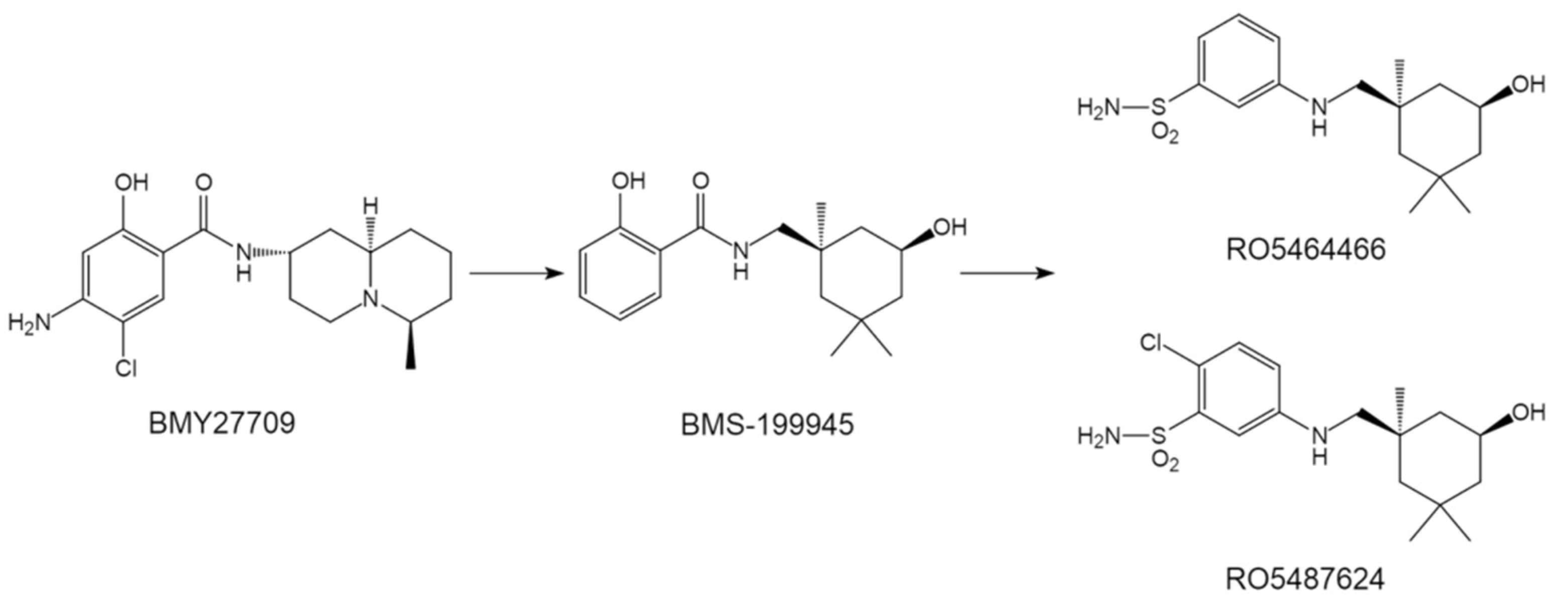
4.1.1. Benzenesulfonamides—The First Generation Orally Active HA Inhibitors
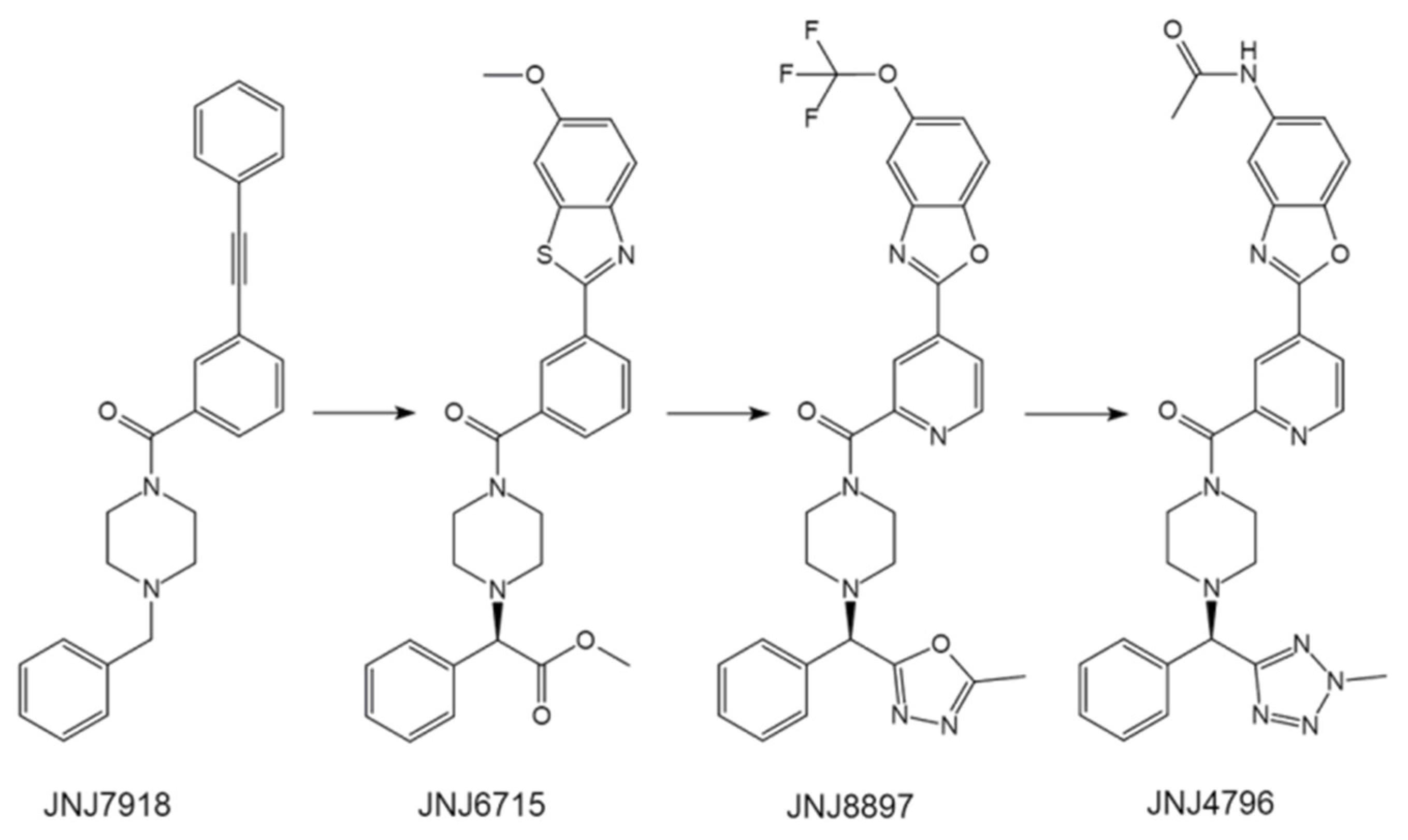
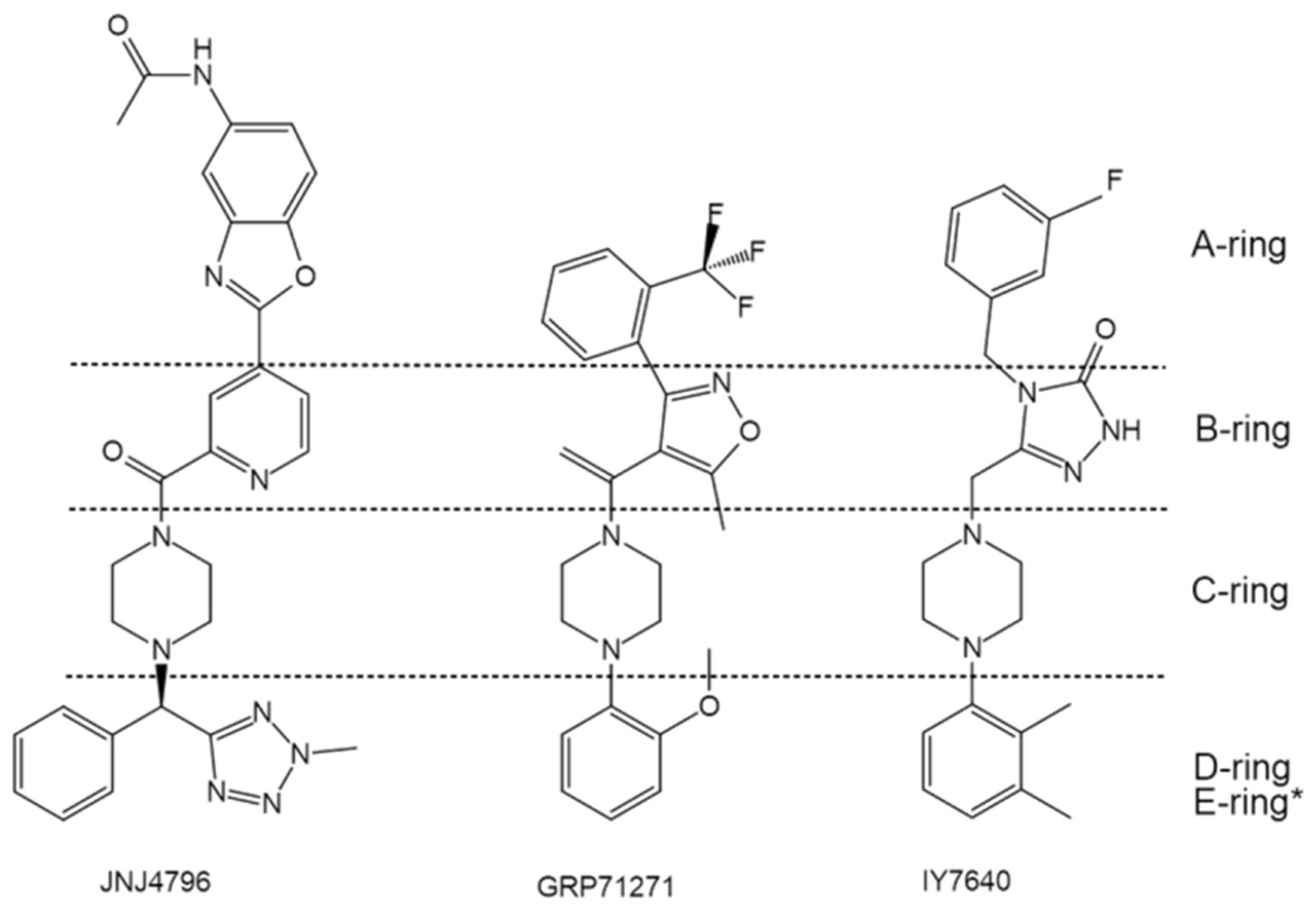
4.1.2. JNJ4796—One of the Most Potent Drug Candidates
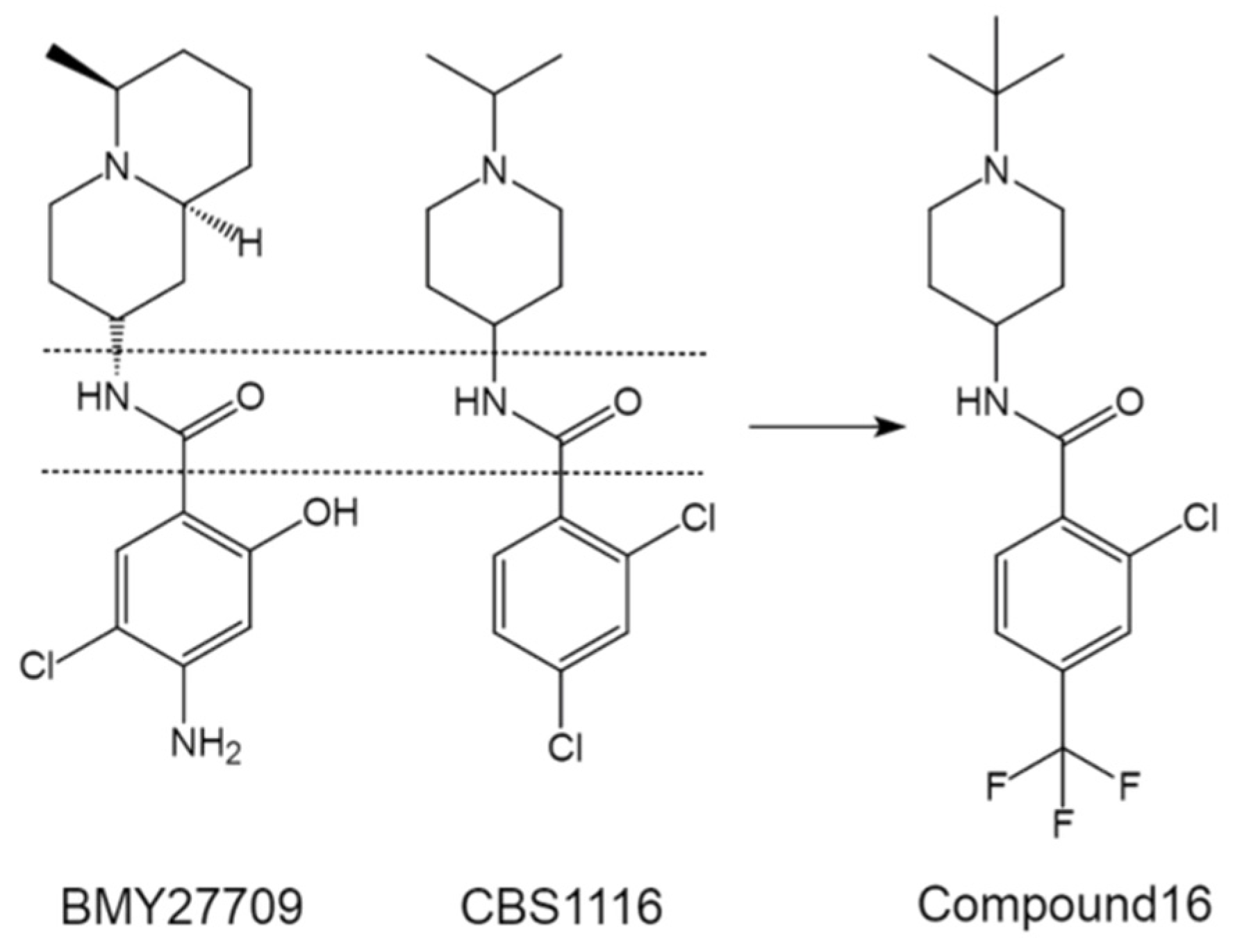
4.1.3. CBS1116—Variable Directions to Chemical Optimization
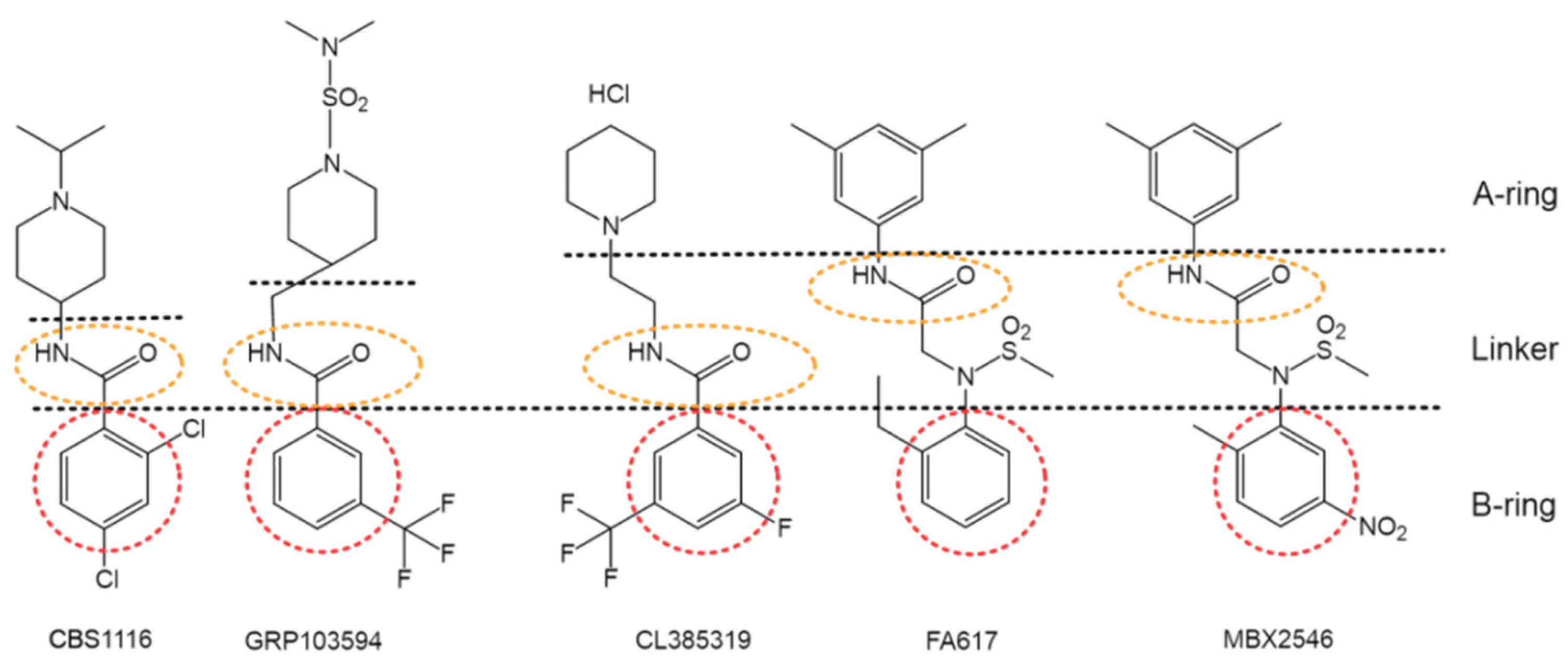
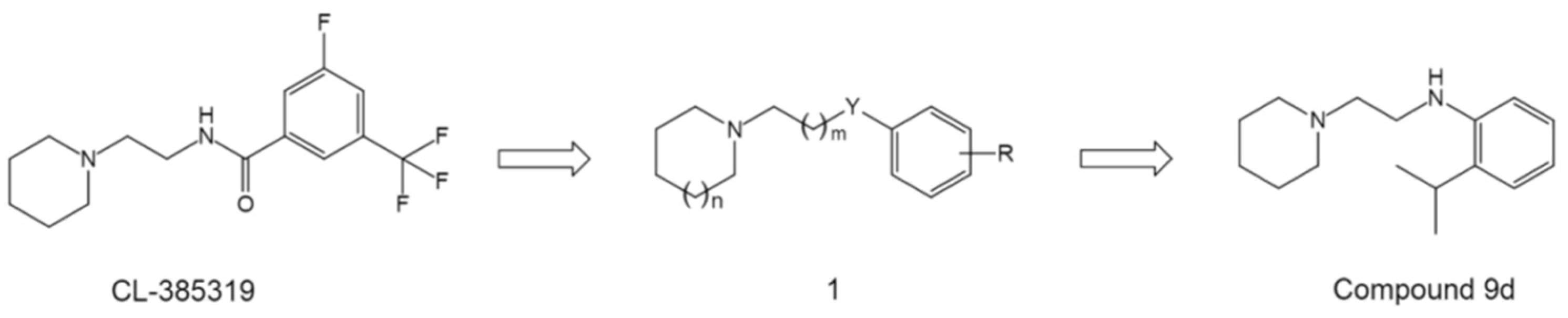
4.1.4. Others—Diverse Scaffolds toward Potent Inhibition against Group 1 HA Fusion
4.2. Group 2 Specific Influenza Fusion Inhibitors
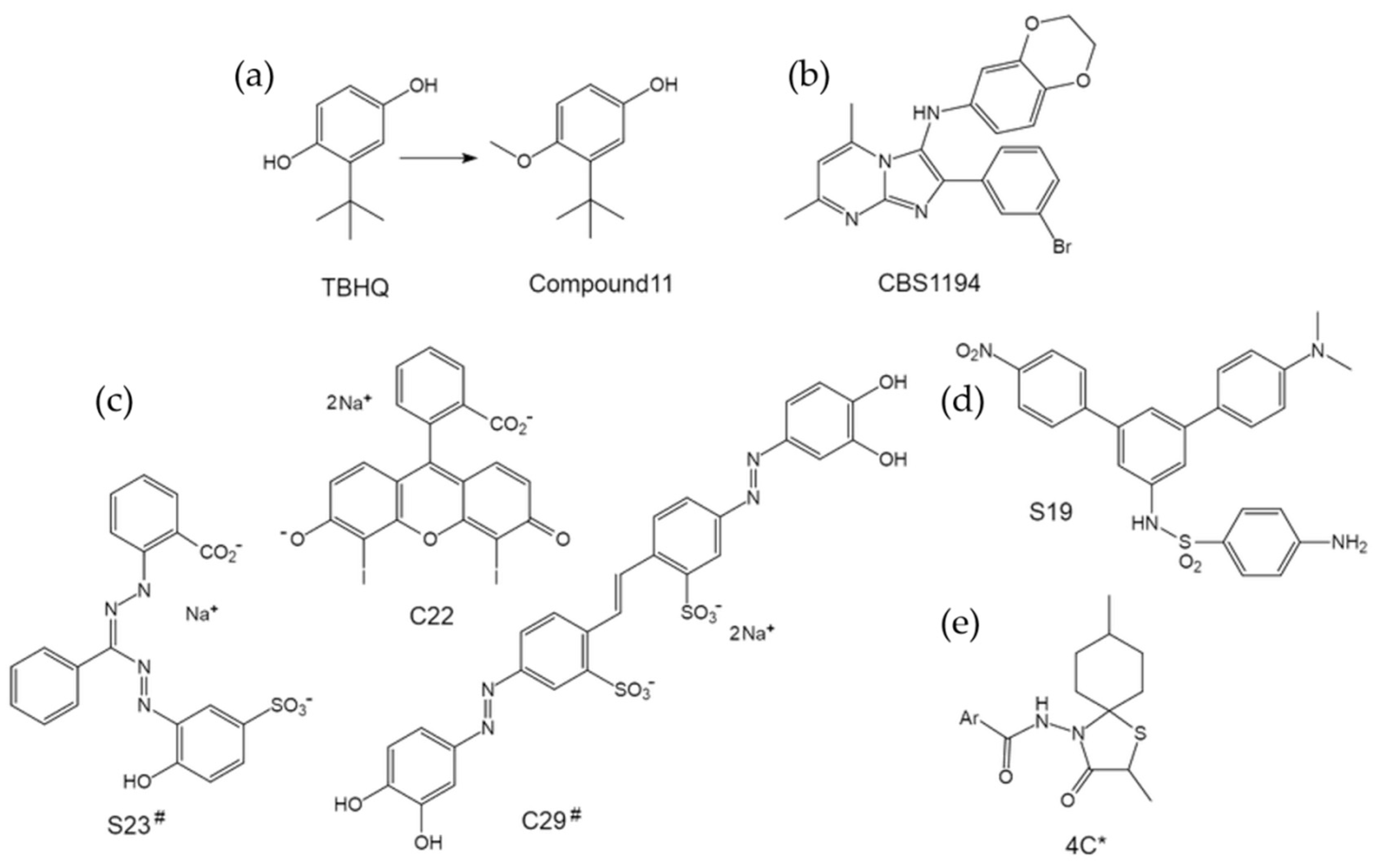
4.2.1. TBHQ—One of the Well-known Lead Molecules
4.2.2. CBS1194—A Novel Scaffold That Deserves Further Optimization
4.2.3. C22—A Facilitator of HA Conformational Change
4.2.4. Others—A Long Way to Go
4.3. Broad-Spectrum Influenza Fusion Inhibitors
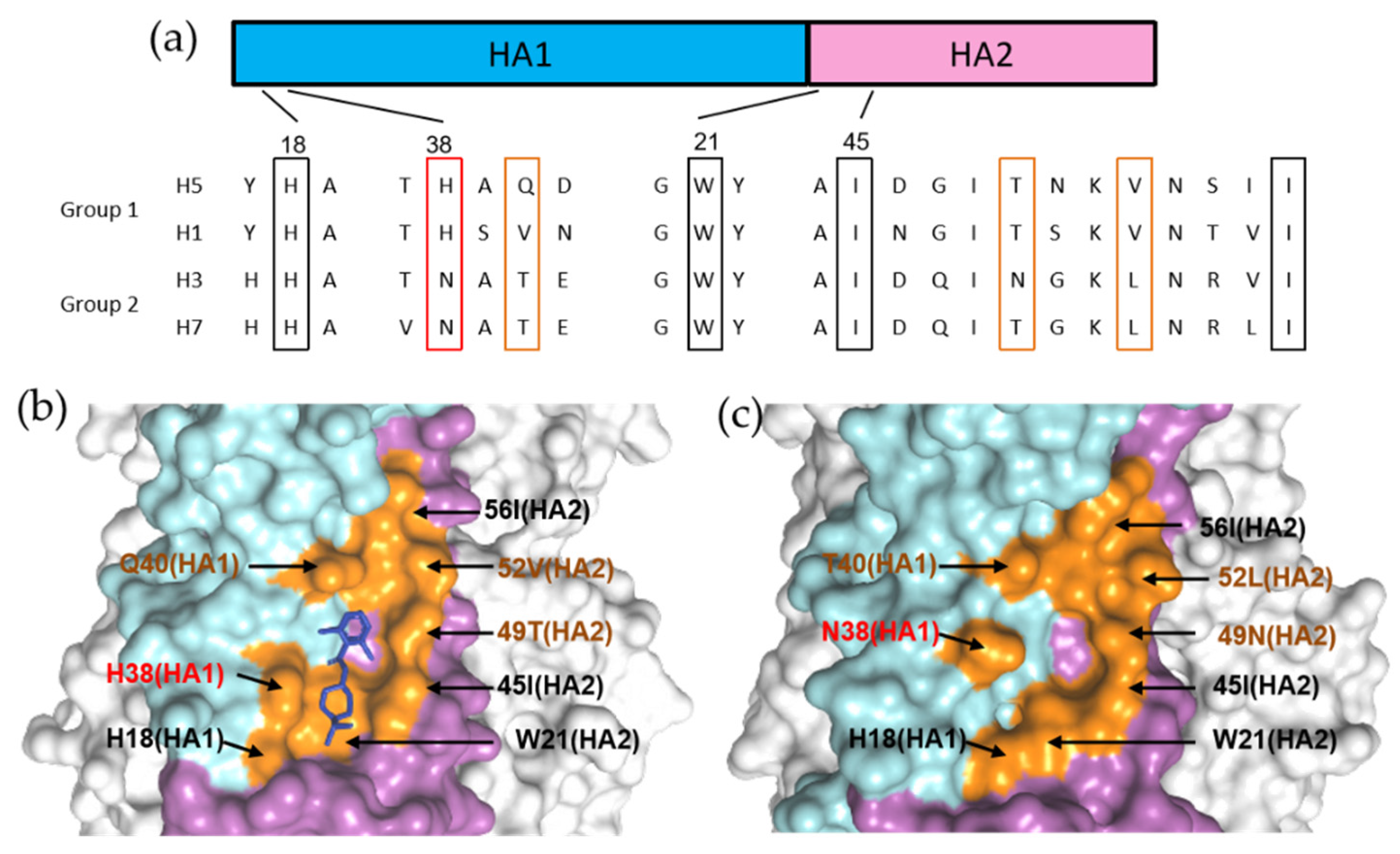
5. Structure-Based Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Group Specificity | In Vitro Activity EC50 (μM) a/CC50 (μM) b | In Vivo Effective | Ref. |
|---|---|---|---|---|
| RO5487624 | 1 | 0.09–0.45/71 | Yes | [46,47] |
| JNJ4796 | 1 | 0.012–3.24/- c | Yes | [48] |
| CBS1116 | 1 | 0.4/160 | - | [21] |
| compound 16 | 1 | 0.072/>100 | Yes d | [60] |
| GRP-103594 | 1 | 0.45/>100 | - | [65] |
| CL-385319 | 1 | 0.38–2.66/47.5 | - | [61,62] |
| 9d | 1 | 1.7–4.6/>100 | - | [75] |
| FA-617 | 1 | 0.007–0.087/- | - | [64] |
| MBX2546 | 1 | 0.3–5.8/>100 | - | [63] |
| GRP-71271 | 1 | 0.28/>100 | - | [65] |
| IY-7640 | 1 (H1N1) | 0.07–7.1/- | Yes | [66] |
| MBX2329 | 1 | 0.29–0.53/>100 | - | [63] |
| LY180299 | 1 | 0.03–0.1/>66 | - | [67] |
| Triperiden | 1 | -/>100 | - | [70,71] |
| GRP-115249 | 1 | 0.33/>100 | - | [65] |
| FA-583 | 1 | 0.044–0.080/- | - | [64] |
| S20 | 1 | 0.08–0.15/40 | - | [72] |
| Nylidirin | 1 | 1.7–3.5/549.2 | Yes | [73] |
| F0045(S) | 1 | 0.5–16.2/>157 | - | [74] |
| TBHQ | 2 | 6/>50 | - | [76,77,78] |
| TBHQ derived compound 11 | 2 | 0.6/>50 | - | [80] |
| CBS1194 | 2 | 0.36–3.7/>100 | - | [24] |
| C22 | 2 | 8.0/1000 | - | [81] |
| S19 | 2 | 0.8/>500 | - | [81] |
| 4c | 2 (H3N2) | 3.3–23/>100 | - | [82] |
| 5f | 2 (H3N2) | 0.001/1.5 | - | [83] |
| Arbidol | Broad spectrum | 8.4–62.8/- | Approved in Russia and China | [84,88] |
| M090 | Broad spectrum | 0.10–6.85/25 | - | [91] |
| camphecene | Broad spectrum | 3.6–83.8/701 | Yes | [92] |
References
- WHO. 2018 Influenza (Seasonal) Fact Sheet; WHO: Geneva, Switzerland, 2018; Available online: http://www.who.int/mediacentre/factsheets/fs211/en/ (accessed on 10 May 2021).
- Sutton, T.C. The Pandemic Threat of Emerging H5 and H7 Avian Influenza Viruses. Viruses 2018, 10, 461. [Google Scholar] [CrossRef]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- Gao, H.N.; Lu, H.Z.; Cao, B.; Du, B.; Shang, H.; Gan, J.H.; Lu, S.H.; Yang, Y.D.; Fang, Q.; Shen, Y.Z.; et al. Clinical findings in 111 cases of influenza A (H7N9) virus infection. N. Engl. J. Med. 2013, 368, 2277–2285. [Google Scholar] [CrossRef]
- Du, R.; Cui, Q.; Rong, L. Flu Universal Vaccines: New Tricks on an Old Virus. Virol. Sin. 2021, 36, 13–24. [Google Scholar] [CrossRef]
- Li, T.C.M.; Chan, M.C.W.; Lee, N. Clinical Implications of Antiviral Resistance in Influenza. Viruses 2015, 7, 4929–4944. [Google Scholar] [CrossRef]
- Lackenby, A.; Besselaar, T.G.; Daniels, R.S.; Fry, A.; Gregory, V.; Gubareva, L.V.; Huang, W.; Hurt, A.C.; Leang, S.-K.; Lee, R.T.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors and status of novel antivirals, 2016–2017. Antivir. Res. 2018, 157, 38–46. [Google Scholar] [CrossRef]
- Noshi, T.; Kitano, M.; Taniguchi, K.; Yamamoto, A.; Omoto, S.; Baba, K.; Hashimoto, T.; Ishida, K.; Kushima, Y.; Hattori, K.; et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antivir. Res. 2018, 160, 109–117. [Google Scholar] [CrossRef]
- Han, J.; Perez, J.; Schafer, A.; Cheng, H.; Peet, N.; Rong, L.; Manicassamy, B. Influenza Virus: Small Molecule Therapeutics and Mechanisms of Antiviral Resistance. Curr. Med. Chem. 2019, 25, 5115–5127. [Google Scholar] [CrossRef] [PubMed]
- Takashita, E.; Kawakami, C.; Ogawa, R.; Morita, H.; Fujisaki, S.; Shirakura, M.; Miura, H.; Nakamura, K.; Kishida, N.; Kuwahara, T.; et al. Influenza A(H3N2) virus exhibiting reduced susceptibility to baloxavir due to a polymerase acidic subunit I38T substitution detected from a hospitalised child without prior baloxavir treatment, Japan, January 2019. Eurosurveillance 2019, 24, 1900170. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; De Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. BaloxavirMarboxil for Uncomplicated Influenza in Adults and Adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Goldhill, D.H.; Velthuis, A.J.W.T.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef] [PubMed]
- Takashita, E. Influenza Polymerase Inhibitors: Mechanisms of Action and Resistance. Cold Spring Harb. Perspect. Med. 2021, 11, a038687. [Google Scholar] [CrossRef]
- Davidson, S. Treating Influenza Infection, From Now and Into the Future. Front. Immunol. 2018, 9, 1946. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.-Y.; Yang, J.; Liu, S. Investigational hemagglutinin-targeted influenza virus inhibitors. Expert Opin. Investig. Drugs 2016, 26, 63–73. [Google Scholar] [CrossRef]
- Du, R.; Cui, Q.; Rong, L. Competitive Cooperation of Hemagglutinin and Neuraminidase during Influenza A Virus Entry. Viruses 2019, 11, 458. [Google Scholar] [CrossRef]
- Rosenthal, P.B.; Zhang, X.; Formanowski, F.; Fitz, W.; Wong, C.-H.; Meier-Ewert, H.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 1998, 396, 92–96. [Google Scholar] [CrossRef]
- Su, S.; Fu, X.; XinLiang, F.; Kerlin, F.; Veit, M. Novel Influenza D virus: Epidemiology, pathology, evolution and biological characteristics. Virulence 2017, 8, 1580–1591. [Google Scholar] [CrossRef]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Luo, M. Influenza Virus Entry. Adv. Exp. Med. Biol. 2012, 726, 201–221. [Google Scholar] [CrossRef]
- Hussein, A.F.; Cheng, H.; Tundup, S.; Antanasijevic, A.; Varhegyi, E.; Perez, J.; Abdulrahman, E.M.; Elenany, M.G.; Helal, S.; Caffrey, M.; et al. Identification of entry inhibitors with 4-aminopiperidine scaffold targeting group 1 influenza A virus. Antivir. Res. 2020, 177, 104782. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Ng, K.H.; Tsang, K.-Y.; Tsang, A.K.; Chen, H.; Yuen, K.-Y. Avian influenza A H5N1 virus: A continuous threat to humans. Emerg. Microbes Infect. 2012, 1, e25. [Google Scholar] [CrossRef] [PubMed]
- Lazniewski, M.; Dawson, W.K.; Szczepinska, T.; Plewczynski, D. The structural variability of the influenza A hemagglutinin receptor-binding site. Brief Funct. Genom. 2018, 17, 415–427. [Google Scholar]
- Du, R.; Cheng, H.; Cui, Q.; Peet, N.P.; Gaisina, I.N.; Rong, L. Identification of a novel inhibitor targeting influenza A virus group 2 hemagglutinins. Antivir. Res. 2021, 186, 105013. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, D.K.; Skehel, J.J.; Wiley, D.C. A surface plasmon resonance assay for the binding of influenza virus hemagglutinin to its sialic acid receptor. Virology 1996, 217, 452–458. [Google Scholar] [CrossRef]
- Stencel-Baerenwald, J.E.; Reiss, K.; Reiter, D.M.; Stehle, T.; Dermody, T.S. The sweet spot: Defining virus-sialic acid interactions. Nat. Rev. Microbiol. 2014, 12, 739–749. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.; Orwenyo, J.; Banerjee, A.; Vasta, G.R.; Wang, L.-X. Design and synthesis of glycoprotein-based multivalent glyco-ligands for influenza hemagglutinin and human galectin-3. Bioorg. Med. Chem. 2013, 21, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Hilsch, M.; Cuellar-Camacho, J.L.; Ludwig, K.; Nie, C.; Parshad, B.; Wallert, M.; Block, S.; Lauster, D.; Böttcher, C.; et al. Adaptive Flexible Sialylated Nanogels as Highly Potent Influenza A Virus Inhibitors. Angew. Chem. Int. Ed. Engl. 2020, 59, 12417–12422. [Google Scholar] [CrossRef]
- Lu, W.; Du, W.; Somovilla, V.J.; Yu, G.; Haksar, D.; De Vries, E.; Boons, G.-J.; de Vries, R.; De Haan, C.A.M.; Pieters, R.J. Enhanced Inhibition of Influenza A Virus Adhesion by Di- and Trivalent Hemagglutinin Inhibitors. J. Med. Chem. 2019, 62, 6398–6404. [Google Scholar] [CrossRef]
- Nie, C.; Stadtmüller, M.; Parshad, B.; Wallert, M.; Ahmadi, V.; Kerkhoff, Y.; Bhatia, S.; Block, S.; Cheng, C.; Wolff, T.; et al. Heteromultivalent topology-matched nanostructures as potent and broad-spectrum influenza A virus inhibitors. Sci. Adv. 2021, 7, eabd3803. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Si, L.; Wang, Y.; Wu, Y.; Yu, F.; Jiao, P.; Shi, Y.; Wang, H.; Xiao, S.; Fu, G.; et al. Discovery of Pentacyclic Triterpenoids as Potential Entry Inhibitors of Influenza Viruses. J. Med. Chem. 2014, 57, 10058–10071. [Google Scholar] [CrossRef]
- Li, W.; Yang, F.; Meng, L.; Sun, J.; Su, Y.; Shao, L.; Zhou, D.; Yu, F. Synthesis, Structure Activity Relationship and An-ti-influenza A Virus Evaluation of Oleanolic Acid-Linear Amino Derivatives. Chem. Pharm. Bull. 2019, 67, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Meng, L.; Sun, J.; Li, W.; Shao, L.; Chen, K.; Zhou, D.; Yang, F.; Yu, F. Design, synthesis of oleanolic acid-saccharide conjugates using click chemistry methodology and study of their anti-influenza activity. Eur. J. Med. Chem. 2019, 182, 111622. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Su, Y.; Yang, F.; Xiao, S.; Yin, Z.; Liu, J.; Zhong, J.; Zhou, D.; Yu, F. Design, synthesis and biological evaluation of amino acids-oleanolic acid conjugates as influenza virus inhibitors. Bioorg. Med. Chem. 2019, 27, 115147. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, R.; Shi, Y.; Si, L.; Jiao, P.; Fan, Z.; Han, X.; Wu, X.; Zhou, X.; Yu, F.; et al. Design, synthesis and biological evaluation of novel l-ascorbic acid-conjugated pentacyclic triterpene derivatives as potential influenza virus entry inhibitors. Eur. J. Med. Chem. 2016, 110, 376–388. [Google Scholar] [CrossRef]
- Song, G.; Shen, X.; Li, S.; Li, Y.; Si, H.; Fan, J.; Li, J.; Gao, E.; Liu, S. Structure-activity relationships of 3-O-β-chacotriosyl oleanane-type triterpenoids as potential H5N1 entry inhibitors. Eur. J. Med. Chem. 2016, 119, 109–121. [Google Scholar] [CrossRef]
- Sacramento, C.Q.; Marttorelli, A.; Fintelman-Rodrigues, N.; de Freitas, C.S.; de Melo, G.R.; Rocha, M.E.; Kaiser, C.R.; Rodrigues, K.F.; da Costa, G.L.; Alves, C.M.; et al. Aureonitol, a Fungi-Derived Tetrahydrofuran, Inhibits Influenza Replication by Targeting Its Surface Glycoprotein Hemagglutinin. PLoS ONE 2015, 10, e0139236. [Google Scholar]
- Chen, X.; Si, L.; Liu, D.; Proksch, P.; Zhang, L.; Zhou, D.; Lin, W. Neoechinulin B and its analogues as potential entry inhibitors of influenza viruses, targeting viral hemagglutinin. Eur. J. Med. Chem. 2015, 93, 182–195. [Google Scholar] [CrossRef]
- Kadam, R.U.; Wilson, I.A. A small-molecule fragment that emulates binding of receptor and broadly neutralizing anti-bodies to influenza A hemagglutinin. Proc. Natl. Acad. Sci. USA 2018, 115, 4240–4245. [Google Scholar] [CrossRef]
- Lee, Y.; Youn, H.-S.; Lee, J.-G.; An, J.Y.; Park, K.R.; Kang, J.Y.; Ryu, Y.B.; Jin, M.S.; Park, K.H.; Eom, S.H. Crystal structure of the catalytic domain of Clostridium perfringens neuraminidase in complex with a non-carbohydrate-based inhibitor, 2-(cyclohexylamino)ethanesulfonic acid. Biochem. Biophys. Res. Commun. 2017, 486, 470–475. [Google Scholar] [CrossRef]
- Luo, G.; Colonno, R.; Krystal, M. Characterization of a Hemagglutinin-Specific Inhibitor of Influenza A Virus. Virology 1996, 226, 66–76. [Google Scholar] [CrossRef]
- Luo, G.; Torri, A.; Harte, W.E.; Danetz, S.; Cianci, C.; Tiley, L.; Day, S.; Mullaney, D.; Yu, K.L.; Ouellet, C.; et al. Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J. Virol. 1997, 71, 4062–4070. [Google Scholar] [CrossRef]
- Deshpande, M.S.; Wei, J.; Luo, G.; Cianci, C.; Danetz, S.; Torri, A.; Tiley, L.; Krystal, M.; Yu, K.-L.; Huang, S.; et al. An approach to the identification of potent inhibitors of influenza virus fusion using parallel synthesis methodology. Bioorg. Med. Chem. Lett. 2001, 11, 2393–2396. [Google Scholar] [CrossRef]
- Cianci, C.; Yu, K.L.; Dischino, D.D.; Harte, W.; Deshpande, M.; Luo, G.; Colonno, R.J.; Meanwell, N.A.; Krystal, M. pH-dependent changes in photoaffinity labeling patterns of the H1 influenza virus hemagglutinin by using an inhibitor of viral fusion. J. Virol. 1999, 73, 1785–1794. [Google Scholar] [CrossRef]
- Tang, G.; Lin, X.; Qiu, Z.; Li, W.; Zhu, L.; Wang, L.; Li, S.; Li, H.; Lin, W.; Yang, M.; et al. Design and Synthesis of Benzenesulfonamide Derivatives as Potent Anti-Influenza Hemagglutinin Inhibitors. ACS Med. Chem. Lett. 2011, 2, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, Y.; Li, S.; Li, H.; Qiu, Z.; Lee, C.; Lu, H.; Lin, X.; Zhao, R.; Chen, L.; et al. Inhibition of Influenza A Virus (H1N1) Fusion by Benzenesulfonamide Derivatives Targeting Viral Hemagglutinin. PLoS ONE 2011, 6, e29120. [Google Scholar] [CrossRef]
- Van Dongen, M.J.P.; Kadam, R.U.; Juraszek, J.; Lawson, E.; Brandenburg, B.; Schmitz, F.; Schepens, W.B.G.; Stoops, B.; Van Diepen, H.A.; Jongeneelen, M.; et al. A small-molecule fusion inhibitor of influenza virus is orally active in mice. Science 2019, 363, eaar6221. [Google Scholar] [CrossRef] [PubMed]
- Throsby, M.; Brink, E.V.D.; Jongeneelen, M.; Poon, L.L.M.; Alard, P.; Cornelissen, L.; Bakker, A.; Cox, F.; Van Deventer, E.; Guan, Y.; et al. Heterosubtypic Neutralizing Monoclonal Antibodies Cross-Protective against H5N1 and H1N1 Recovered from Human IgM+ Memory B Cells. PLoS ONE 2008, 3, e3942. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.; Bhabha, G.; Elsliger, M.-A.; Friesen, R.H.E.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody Recognition of a Highly Conserved Influenza Virus Epitope. Science 2009, 324, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, T.A.; Chevalier, A.; Song, Y.; Dreyfus, C.; Fleishman, S.J.; De Mattos, C.; Myers, C.A.; Kamisetty, H.; Blair, P.; Wilson, I.A.; et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 2012, 30, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Koday, M.T.; Nelson, J.; Chevalier, A.; Koday, M.; Kalinoski, H.; Stewart, L.; Carter, L.; Nieusma, T.; Lee, P.S.; Ward, A.B.; et al. A Computationally Designed Hemagglutinin Stem-Binding Protein Provides In Vivo Protection from Influenza Independent of a Host Immune Response. PLOS Pathog. 2016, 12, e1005409. [Google Scholar] [CrossRef][Green Version]
- Kadam, R.U.; Juraszek, J.; Brandenburg, B.; Buyck, C.; Schepens, W.B.G.; Kesteleyn, B.; Stoops, B.; Vreeken, R.J.; Vermond, J.; Goutier, W.; et al. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. [Google Scholar] [CrossRef]
- Bajic, G.; Maron, M.; Adachi, Y.; Onodera, T.; McCarthy, K.R.; McGee, C.E.; Sempowski, G.D.; Takahashi, Y.; Kelsoe, G.; Kuraoka, M.; et al. Influenza Antigen Engineering Focuses Immune Responses to a Subdominant but Broadly Protective Viral Epitope. Cell Host Microbe 2019, 25, 827–835.e6. [Google Scholar] [CrossRef] [PubMed]
- Bangaru, S.; Lang, S.; Schotsaert, M.; VanderVen, H.A.; Zhu, X.; Kose, N.; Bombardi, R.; Finn, J.A.; Kent, S.J.; Gilchuk, P.; et al. A Site of Vulnerability on the Influenza Virus Hemagglutinin Head Domain Trimer Interface. Cell 2019, 177, 1136–1152.e18. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; McCarthy, K.R.; Kuraoka, M.; Schmidt, A.G.; Adachi, Y.; Onodera, T.; Tonouchi, K.; Caradonna, T.; Bajic, G.; Song, S.; et al. Antibodies to a Conserved Influenza Head Interface Epitope Protect by an IgG Subtype-Dependent Mechanism. Cell 2019, 177, 1124–1135.e16. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A Neutralizing Antibody Selected from Plasma Cells That Binds to Group 1 and Group 2 Influenza A Hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef]
- Wu, N.C.; Wilson, I.A. Influenza Hemagglutinin Structures and Antibody Recognition. Cold Spring Harb. Perspect. Med. 2019, 10, a038778. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, H.; Ratia, K.; Varhegyi, E.; Hendrickson, W.G.; Li, J.; Rong, L. A Comparative High-Throughput Screening Protocol to Identify Entry Inhibitors of Enveloped Viruses. J. Biomol. Screen. 2014, 19, 100–107. [Google Scholar] [CrossRef]
- Gaisina, I.N.; Peet, N.P.; Cheng, H.; Li, P.; Du, R.; Cui, Q.; Furlong, K.; Manicassamy, B.; Caffrey, M.; Thatcher, G.R.J.; et al. Optimization of 4-Aminopiperidines as Inhibitors of Influenza a Viral Entry That Are Synergistic with Oseltamivir. J. Med. Chem. 2020, 63, 3120–3130. [Google Scholar] [CrossRef]
- Liu, S.; Li, R.; Zhang, R.; Chan, C.C.; Xi, B.; Zhu, Z.; Yang, J.; Poon, V.K.; Zhou, J.; Chen, M.; et al. CL-385319 inhibits H5N1 avian influenza A virus infection by blocking viral entry. Eur. J. Pharmacol. 2011, 660, 460–467. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, R.; Xiao, G.; Chen, Z.; Yang, J.; Zhu, Q.; Liu, S. Design, synthesis and structure–activity relationship of novel inhibitors against H5N1 hemagglutinin-mediated membrane fusion. Eur. J. Med. Chem. 2012, 57, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Antanasijevic, A.; Wang, M.; Li, B.; Mills, D.M.; Ames, J.A.; Nash, P.J.; Williams, J.D.; Peet, N.P.; Moir, D.T.; et al. New Small Molecule Entry Inhibitors Targeting Hemagglutinin-Mediated Influenza A Virus Fusion. J. Virol. 2014, 88, 1447–1460. [Google Scholar] [CrossRef]
- Lai, K.K.; Cheung, N.N.; Yang, F.; Dai, J.; Liu, L.; Chen, Z.; Sze, K.H.; Chen, H.L.; Yuen, K.-Y.; Kao, R.Y.T. Identification of Novel Fusion Inhibitors of Influenza A Virus by Chemical Genetics. J. Virol. 2016, 90, 2690–2701. [Google Scholar] [CrossRef]
- Weisshaar, M.; Cox, R.; Morehouse, Z.; Kyasa, S.K.; Yan, D.; Oberacker, P.; Mao, S.; Golden, J.E.; Lowen, A.C.; Natchus, M.G.; et al. Identification and Characterization of Influenza Virus Entry Inhibitors through Dual Myxovirus High-Throughput Screening. J. Virol. 2016, 90, 7368–7387. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Lee, S.; Lee, G.Y.; Park, S.; Bae, J.Y.; Heo, J.; Kim, H.Y.; Woo, S.H.; Lee, H.U.; Ahn, C.A.; et al. Novel Small Molecule Targeting the Hemagglutinin Stalk of Influenza Viruses. J. Virol. 2019, 93, e00878-19. [Google Scholar] [CrossRef] [PubMed]
- Staschke, K.; Hatch, S.; Tang, J.; Hornback, W.; Munroe, J.; Colacino, J.; Muesing, M. Inhibition of Influenza Virus Hemagglutinin-Mediated Membrane Fusion by a Compound Related to Podocarpic Acid. Virology 1998, 248, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Ishiwata, Y.; Iwata, N.; Honda, N.; Kakigami, T. Synthesis and anti-influenza virus activity of tricyclic compounds with a unique amine moiety. Chem. Pharm. Bull. 2001, 49, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Dang, Z.; Jung, K.; Zhu, L.; Xie, H.; Lee, K.-H.; Chen, C.-H.; Huang, L. Phenolic Diterpenoid Derivatives as Anti-Influenza A Virus Agents. ACS Med. Chem. Lett. 2015, 6, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Qiu, Z.; Lin, X.; Li, W.; Zhu, L.; Li, S.; Li, H.; Wang, L.; Chen, L.; Wu, J.Z.; et al. Discovery of novel 1-phenyl-cycloalkane carbamides as potent and selective influenza fusion inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3507–3510. [Google Scholar] [CrossRef]
- White, K.M.; De Jesus, P.; Chen, Z.; Abreu, P.; Barile, E.; Mak, P.A.; Anderson, P.; Nguyen, Q.T.; Inoue, A.; Stertz, S.; et al. A Potent Anti-influenza Compound Blocks Fusion through Stabilization of the Prefusion Conformation of the Hemagglutinin Protein. ACS Infect. Dis. 2015, 1, 98–109. [Google Scholar] [CrossRef]
- Jang, Y.; Shin, J.S.; Lee, J.-Y.; Shin, H.; Kim, S.J.; Kim, M. In Vitro and In Vivo Antiviral Activity of Nylidrin by Targeting the Hemagglutinin 2-Mediated Membrane Fusion of Influenza A Virus. Viruses 2020, 12, 581. [Google Scholar] [CrossRef]
- Yao, Y.; Kadam, R.U.; Lee, C.-C.D.; Woehl, J.L.; Wu, N.C.; Zhu, X.; Kitamura, S.; Wilson, I.A.; Wolan, D.W. An influenza A hemagglutinin small-molecule fusion inhibitor identified by a new high-throughput fluorescence polarization screen. Proc. Natl. Acad. Sci. USA 2020, 117, 18431–18438. [Google Scholar] [CrossRef]
- Leiva, R.; Barniol-Xicota, M.; Codony, S.; Ginex, T.; Vanderlinden, E.; Montes, M.; Caffrey, M.; Luque, F.J.; Naesens, L.; Vázquez, S. Aniline-Based Inhibitors of Influenza H1N1 Virus Acting on Hemagglutinin-Mediated Fusion. J. Med. Chem. 2018, 61, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Bodian, D.L.; Yamasaki, R.B.; Buswell, R.L.; Stearns, J.F.; White, J.M.; Kuntz, I.D. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 1993, 32, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gamblin, S.J.; Skehel, J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar] [CrossRef] [PubMed]
- Antanasijevic, A.; Cheng, H.; Wardrop, D.J.; Rong, L.; Caffrey, M. Inhibition of Influenza H7 Hemagglutinin-Mediated Entry. PLoS ONE 2013, 8, e76363. [Google Scholar] [CrossRef]
- Van Esch, G. Toxicology of tert-butylhydroquinone (TBHQ). Food Chem. Toxicol. 1986, 24, 1063–1065. [Google Scholar] [CrossRef]
- Antanasijevic, A.; Hafeman, N.J.; Tundup, S.; Kingsley, C.; Mishra, R.K.; Rong, L.; Manicassamy, B.; Wardrop, D.; Caffrey, M. Stabilization and Improvement of a Promising Influenza Antiviral: Making a PAIN PAINless. ACS Infect. Dis. 2016, 2, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.R.; Kuntz, I.D.; White, J.M. Structure-based identification of an inducer of the low-pH conformational change in the influenza virus hemagglutinin: Irreversible inhibition of infectivity. J. Virol. 1997, 71, 8808–8820. [Google Scholar] [CrossRef]
- Vanderlinden, E.; Göktaş, F.; Cesur, Z.; Froeyen, M.; Reed, M.L.; Russell, C.J.; Cesur, N.; Naesens, L. Novel Inhibitors of Influenza Virus Fusion: Structure-Activity Relationship and Interaction with the Viral Hemagglutinin. J. Virol. 2010, 84, 4277–4288. [Google Scholar] [CrossRef]
- Cihan-Üstündağ, G.; Zopun, M.; Vanderlinden, E.; Ozkirimli, E.; Persoons, L.; Çapan, G.; Naesens, L. Superior inhibition of influenza virus hemagglutinin-mediated fusion by indole-substituted spirothiazolidinones. Bioorg. Med. Chem. 2020, 28, 115130. [Google Scholar] [CrossRef]
- Boriskin, Y.S.; Leneva, I.A.; Pecheur, E.-I.; Polyak, S.J. Arbidol: A Broad-Spectrum Antiviral Compound that Blocks Viral Fusion. Curr. Med. Chem. 2008, 15, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Blaising, J.; Polyak, S.J.; Pécheur, E.-I. Arbidol as a broad-spectrum antiviral: An update. Antivir. Res. 2014, 107, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Pécheur, E.-I.; Borisevich, V.; Halfmann, P.; Morrey, J.D.; Smee, D.F.; Prichard, M.; Mire, C.E.; Kawaoka, Y.; Geisbert, T.W.; Polyak, S.J. The Synthetic Antiviral Drug Arbidol Inhibits Globally Prevalent Pathogenic Viruses. J. Virol. 2016, 90, 3086–3092. [Google Scholar] [CrossRef] [PubMed]
- Kadam, R.U.; Wilson, I.A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. USA 2017, 114, 206–214. [Google Scholar] [CrossRef]
- Brancato, V.; Peduto, A.; Wharton, S.; Martin, S.; More, V.; Di Mola, A.; Massa, A.; Perfetto, B.; Donnarumma, G.; Schiraldi, C.; et al. Design of inhibitors of influenza virus membrane fusion: Synthesis, structure–activity relationship and in vitro antiviral activity of a novel indole series. Antivir. Res. 2013, 99, 125–135. [Google Scholar] [CrossRef]
- Wright, Z.V.; Wu, N.C.; Kadam, R.U.; Wilson, I.A.; Wolan, D.W. Structure-based optimization and synthesis of antiviral drug Arbidol analogues with significantly improved affinity to influenza hemagglutinin. Bioorg. Med. Chem. Lett. 2017, 27, 3744–3748. [Google Scholar] [CrossRef]
- Zhao, X.; Li, R.; Zhou, Y.; Xiao, M.; Ma, C.; Yang, Z.; Zeng, S.; Du, Q.; Yang, C.; Jiang, H.; et al. Discovery of Highly Potent Pinanamine-Based Inhibitors against Amantadine- and Oseltamivir-Resistant Influenza A Viruses. J. Med. Chem. 2018, 61, 5187–5198. [Google Scholar] [CrossRef]
- Zarubaev, V.; Garshinina, A.; Tretiak, T.; Fedorova, V.; Shtro, A.; Sokolova, A.; Yarovaya, O.; Salakhutdinov, N. Broad range of inhibiting action of novel camphor-based compound with anti-hemagglutinin activity against influenza viruses in vitro and in vivo. Antivir. Res. 2015, 120, 126–133. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Korchagina, D.V.; Zarubaev, V.V.; Tretiak, T.S.; Anfimov, P.M.; Kiselev, O.I.; Salakhutdinov, N.F. Camphor-based symmetric diimines as inhibitors of influenza virus reproduction. Bioorg. Med. Chem. 2014, 22, 2141–2148. [Google Scholar] [CrossRef]
- Wu, N.C.; Wilson, I.A. Structural Biology of Influenza Hemagglutinin: An Amaranthine Adventure. Viruses 2020, 12, 1053. [Google Scholar] [CrossRef]
- Russell, R.; Gamblin, S.; Haire, L.; Stevens, D.; Xiao, B.; Ha, Y.; Skehel, J. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology 2004, 325, 287–296. [Google Scholar] [CrossRef]
- Thoennes, S.; Li, Z.-N.; Lee, B.-J.; Langley, W.A.; Skehel, J.J.; Russell, R.J.; Steinhauer, D.A. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 2008, 370, 403–414. [Google Scholar] [CrossRef]
- Antanasijevic, A.; Durst, M.A.; Cheng, H.; Gaisina, I.N.; Perez, J.T.; Manicassamy, B.; Rong, L.; Lavie, A.; Caffrey, M. Structure of avian influenza hemagglutinin in complex with a small molecule entry inhibitor. Life Sci. Alliance 2020, 3, e202000724. [Google Scholar] [CrossRef] [PubMed]
- Leneva, I.A.; Russell, R.J.; Boriskin, Y.S.; Hay, A.J. Characteristics of arbidol-resistant mutants of influenza virus: Implications for the mechanism of anti-influenza action of arbidol. Antivir. Res. 2009, 81, 132–140. [Google Scholar] [CrossRef]
- Basu, A.; Komazin-Meredith, G.; McCarthy, C.; Antanasijevic, A.; Cardinale, S.C.; Mishra, R.K.; Barnard, D.L.; Caffrey, M.; Rong, L.; Bowlin, T.L. Molecular Mechanism Underlying the Action of Influenza A Virus Fusion Inhibitor MBX2546. ACS Infect. Dis. 2017, 3, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Lampejo, T. Influenza and antiviral resistance: An overview. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1–8. [Google Scholar] [CrossRef] [PubMed]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Cui, Q.; Caffrey, M.; Rong, L.; Du, R. Small Molecule Inhibitors of Influenza Virus Entry. Pharmaceuticals 2021, 14, 587. https://doi.org/10.3390/ph14060587
Chen Z, Cui Q, Caffrey M, Rong L, Du R. Small Molecule Inhibitors of Influenza Virus Entry. Pharmaceuticals. 2021; 14(6):587. https://doi.org/10.3390/ph14060587
Chicago/Turabian StyleChen, Zhaoyu, Qinghua Cui, Michael Caffrey, Lijun Rong, and Ruikun Du. 2021. "Small Molecule Inhibitors of Influenza Virus Entry" Pharmaceuticals 14, no. 6: 587. https://doi.org/10.3390/ph14060587
APA StyleChen, Z., Cui, Q., Caffrey, M., Rong, L., & Du, R. (2021). Small Molecule Inhibitors of Influenza Virus Entry. Pharmaceuticals, 14(6), 587. https://doi.org/10.3390/ph14060587