
Fluorinated Protein and Peptide Materials for Biomedical Applications


Abstract
:1. Introduction
Special Properties of Fluorine
2. Incorporation of Fluorine into Protein and Peptide Materials
2.1. Solid Phase Peptide Synthesis
2.2. Non-Canonical Amino Acid Incorporation
2.3. Protein Fragment Ligation
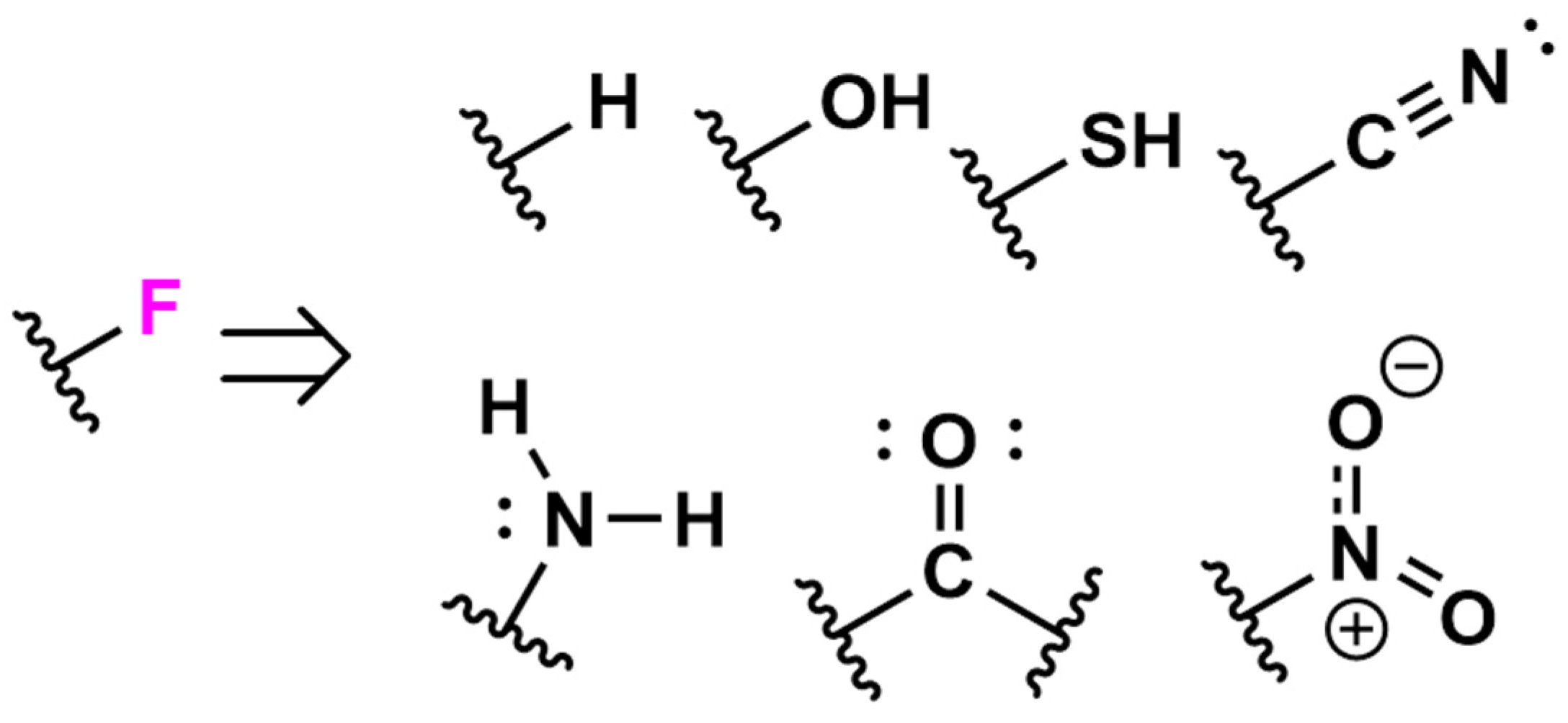
2.4. Bulk Protein Fluorination via Fluorinating Agents
2.5. Post-Translational Modifications with Fluorine
3. Impact of Fluorine on Secondary Structure
3.1. Monomeric Alpha-Helices
3.2. Beta-Sheets
3.3. Intrinsically Disordered Proteins
4. Impact of Fluorine on Supersecondary Structure
4.1. Coiled-Coils
4.2. Collagen Triple-Helices
4.3. Beta Barrels
4.4. Protein Block Copolymers
5. Protein and Peptide Mimetic Therapeutics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct Name | Fluorinated Residue(s) | Number of Fluorines | Position of Fluorines | Synthetic Method | (Super) Secondary Structure(s) | Sequence | References |
|---|---|---|---|---|---|---|---|
| BII1F2 | Leucine (L) | 6 per L (12 total) | Methyl group, δ-carbon | SPPS | Monomer α-helix | TRSSRAGLQFPVGRVHRLLRK | [70] |
| BII5F2 | Leucine (L) | 6 per L (12 total) | Methyl group, δ-carbon | SPPS | Monomer α-helix | RAGLQFPVGRVHRLLRK | [70] |
| BII6F2 | Leucine (L) | 6 per L (12 total) | Methyl group, δ-carbon | SPPS | Monomer α-helix | AGLQFPVGRVHRLLRK | [70] |
| BII10F2 | Leucine (L) | 6 per L (12 total) | Methyl group, δ-carbon | SPPS | Monomer α-helix | FPVGRVHRLLRK | [70] |
| M2F2 | Leucine (L) | 6 per L (12 total) | Methyl group, δ-carbon | SPPS | Monomer α-helix | GIGKFLHAAKKFAKAFVAELMNS | [70] |
| M2F5 | Leucine (L) | 6 per L (30 total) | Methyl group, δ-carbon | SPPS | Monomer α-helix | GIGKFLHALKKFLKAFLAELMNS | [70] |
| DOPA-Phe(4F)-Phe(4F)-OMe | Phenylalanine (F) | 4 per F (8 total) | Aryl ring, all positions | Purchased | Linear Tripeptide | 3OHYFF | [71] |
| 2ZNX (Fluorinated Antibody) | Tryptophan (W) | 1 per W (12 total) | Aryl ring, 5-carbon | Residue-specific ncAA incorporation | Antibody (Combination of α-helices and β-sheets) | Chain A: DIVLTQSPATLSVTPGNSVSLSCRASQSIGNNLHWYQQKSHESPRLLIKYASQSISGIPSRFSGSGSGTDFTLSINSVETEDFGMYFCQQSNSWPYTFGGGTKLEITGGGGSGGGGSGGGGSDIQLQESGPSLVKPSQTLSLTCSVTGDSITSDYWSWIRKFPGNRLEYMGYVSYSGSTYYNPSLKSRISITRDTSKNQYYLDLNSVTTEDTATYYCANWDGDYWGQGTLVTVSAAHHHHHH Chain B: DIVLTQSPATLSVTPGNSVSLSCRASQSIGNNLHWYQQKSHESPRLLIKYASQSISGIPSRFSGSGSGTDFTLSINSVETEDFGMYFCQQSNSWPYTFGGGTKLEITGGGGSGGGGSGGGGSDIQLQESGPSLVKPSQTLSLTCSVTGDSITSDYWSWIRKFPGNRLEYMGYVSYSGSTYYNPSLKSRISITRDTSKNQYYLDLNSVTTEDTATYYCANWDGDYWGQGTLVTVSAAHHHHHH | [72] |
| (flpHypGly)7 | Proline (P) | 1 per P (7 total) | Pyrrolidine ring, 4′-carbon | SPPS | Collagen monomer | PhPGPhPGPhPGPhPGPhPGPhPGPhPG | [73] |
5.1. Bioactive Peptides
5.1.1. Antimicrobial Bioactive Peptides
5.1.2. Antiviral Bioactive Peptides
5.2. Fluorinated Antibodies
5.3. Collagen Mimetics for Fiber Repair
6. Drug Delivery
| Construct Name | Fluorinated Residue(s) | Number of Fluorines | Position of Fluorines | Synthetic Method | Assembly Morphology | Sequence | References |
|---|---|---|---|---|---|---|---|
| F-TRAP | Leucine (L) | 3 per L (12 total) | Methyl group, δ-carbon | Residue-specific ncAA incorporation | Protein micelles | MRGSHHHHHHGSACELAARGDATATATATAACGDLAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASGLQAARGDATATATATAVDKPIAASAVPGVGVPGVGVPGFGVPGVGVPGVGVPGVGVPGVGVPGFGVPGVGVPGVGVPLEGSGTGAKLN | [81] |
| D1 | (R,R)-β-diaryl (βPhPh) | 1 per βPhPh (1 total) | Aryl ring, 2-ortho position | Ligation | Fibrous nanotubes | AβPhPh | [82] |
| D2 | (S,S)-β-diaryl (βPhPh) | 1 per βPhPh (1 total) | Aryl ring, 2-ortho position | Ligation | Fibrous nanotubes | AβPhPh | [82] |
| MfeGlyK16 | Homoalanine (hA) | 1 per hA (8 total) | Ethyl group, γ-carbon | SPPS | Hydrogelator Agent | hAKhAKhAKhAK hAKhAKhAKhAK-BzOH | [83] |
| DfeGlyK16 | Homoalanine (hA) | 2 per hA (16 total) | Ethyl group, γ-carbon | SPPS | Hydrogelator Agent | hAKhAKhAKhAK hAKhAKhAKhAK-BzOH | [83] |
| TfeGlyK16 | Homoalanine (hA) | 3 per hA (24 total) | Ethyl group, γ-carbon | SPPS | Hydrogelator Agent | hAKhAKhAKhAK hAKhAKhAKhAK-BzOH | [83] |
| Fmoc-3F-Phe-DAP | Phenylalanine (F) | 1 per F (1 total) | Aryl ring, 3-meta position | SPPS | Hydrogelator Agent | Fmoc-F-DAP | [84] |
| Fmoc-F5-Phe-DAP | Phenylalanine (F) | 5 per F (5 total) | Aryl ring, all positions | SPPS | Hydrogelator Agent | Fmoc-F-DAP | [84] |
| 4-fluorobenzyl-diphenylalanine | Phenylalanine (F) | 1 per F (1 total) | Aryl ring, 4-para position | SPPS | Hydrogelator Agent | Bz-FF | [85] |
| No name provided | Phenylalanine (F) | 5 per F (15 total) | Aryl ring, all positions | SPPS | Peptide emulsion | FFFGGGCCGGKGRGD | [86] |
6.1. Spherical, Colloidal Fluorinated Peptide and Protein Vehicles
6.1.1. Fluorinated Protein Micelles
6.1.2. Fluorinated Peptide Emulsions
6.2. Fluorinated Fibers
6.3. Fluorinated Protein and Peptide Hydrogels
7. Bioimaging
7.1. Fluorine-19 Magnetic Resonance Imaging
7.2. Fluorine-18 Radiotracers in Positron Emission Tomography Imaging
8. Perspective and Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, X.; Kokkonda, S.; El Mazouni, F.; White, J.; Burrows, J.N.; Kaminsky, W.; Charman, S.A.; Matthews, D.; Rathod, P.K.; Phillips, M.A. Fluorine Modulates Species Selectivity in the Triazolopyrimidine Class of Plasmodium falciparum Dihydroorotate Dehydrogenase Inhibitors. J. Med. Chem. 2014, 57, 5381–5394. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- El’kin, O.V.; Bushuev, A.N.; Tolstobrov, I.V.; Fomin, S.V.; Shirokova, E.S.; Sazanov, A.V.; Kozvonin, V.A.; Kozulin, D.A. Chapter 11—Organofluorine compounds in artificial blood circulation systems. In Fascinating Fluoropolymers and Their Applications; Ameduri, B., Fomin, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 401–424. [Google Scholar]
- Ebnesajjad, S. Applications of Fluorocarbon Gases and Liquids. In Concise Handbook of Fluorocarbon Gases, 1st ed.; Wiley-Scrivener: Hoboken, NJ, USA, 2021; pp. 41–86. [Google Scholar]
- Buer Benjamin, C.; Meagher Jennifer, L.; Stuckey Jeanne, A.; Marsh, E.N.G. Structural basis for the enhanced stability of highly fluorinated proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 4810–4815. [Google Scholar] [CrossRef] [PubMed]
- Tredwell, M.; Gouverneur, V. 1.5 Fluorine in Medicinal Chemistry: Importance of Chirality. In Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 70–85. [Google Scholar]
- Charity, R.J.; Webb, T.B.; Elson, J.M.; Hoff, D.E.M.; Pruitt, C.D.; Sobotka, L.G.; Brown, K.W.; Cerizza, G.; Estee, J.; Lynch, W.G.; et al. Observation of the Exotic Isotope 13F Located Four Neutrons beyond the Proton Drip Line. Phys. Rev. Lett. 2021, 126, 132501. [Google Scholar] [CrossRef]
- Rickmeier, J.; Ritter, T. Site-Specific Deoxyfluorination of Small Peptides with [18F]Fluoride. Angew. Chem. Int. Ed. 2018, 57, 14207–14211. [Google Scholar] [CrossRef]
- Leroux, F.R.; Manteau, B.; Vors, J.-P.; Pazenok, S. Trifluoromethyl ethers–synthesis and properties of an unusual substituent. Beilstein J. Org. Chem. 2008, 4, 13. [Google Scholar] [CrossRef]
- Smart, B.E. Characteristics of C-F Systems. In Organofluorine Chemistry: Principles and Commercial Applications; Banks, R.E., Smart, B.E., Tatlow, J.C., Eds.; Springer: Boston, MA, USA, 1994; pp. 57–88. [Google Scholar]
- Spartan 2018; Wavefunction, Inc.: Irvine, CA, USA, 2018.
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Hartrampf, N.; Saebi, A.; Poskus, M.; Gates, Z.P.; Callahan, A.J.; Cowfer, A.E.; Hanna, S.; Antilla, S.; Schissel, C.K.; Quartararo, A.J.; et al. Synthesis of proteins by automated flow chemistry. Science 2020, 368, 980–987. [Google Scholar] [CrossRef]
- Wang, P.; Tang, Y.; Tirrell, D.A. Incorporation of Trifluoroisoleucine into Proteins in Vivo. J. Am. Chem. Soc. 2003, 125, 6900–6906. [Google Scholar] [CrossRef]
- Son, S.; Tanrikulu, I.C.; Tirrell, D.A. Stabilization of bzip Peptides through Incorporation of Fluorinated Aliphatic Residues. ChemBioChem 2006, 7, 1251–1257. [Google Scholar] [CrossRef]
- Wang, P.; Fichera, A.; Kumar, K.; Tirrell, D.A. Alternative Translations of a Single RNA Message: An Identity Switch of (2S,3R)-4,4,4-Trifluorovaline between Valine and Isoleucine Codons. Angew. Chem. Int. Ed. 2004, 43, 3664–3666. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Herberich, B.; Schultz Peter, G. Expanding the Genetic Code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef]
- Liu, C.C.; Schultz, P.G. Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [PubMed]
- Voloshchuk, N.; Montclare, J.K. Incorporation of unnatural amino acids for synthetic biology. Mol. BioSyst. 2010, 6, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef]
- Bode, J.W. Chemical Protein Synthesis with the α-Ketoacid–Hydroxylamine Ligation. Acc. Chem. Res. 2017, 50, 2104–2115. [Google Scholar] [CrossRef]
- Schmidt, M.; Toplak, A.; Quaedflieg, P.J.L.M.; Nuijens, T. Enzyme-mediated ligation technologies for peptides and proteins. Curr. Opin. Chem. Biol. 2017, 38, 1–7. [Google Scholar] [CrossRef]
- Meyer, D.; Jangra, H.; Walther, F.; Zipse, H.; Renaud, P. A third generation of radical fluorinating agents based on N-fluoro-N-arylsulfonamides. Nat. Commun. 2018, 9, 4888. [Google Scholar] [CrossRef]
- Robalo, J.R.; Vila Verde, A. Unexpected trends in the hydrophobicity of fluorinated amino acids reflect competing changes in polarity and conformation. Phys. Chem. Chem. Phys. 2019, 21, 2029–2038. [Google Scholar] [CrossRef]
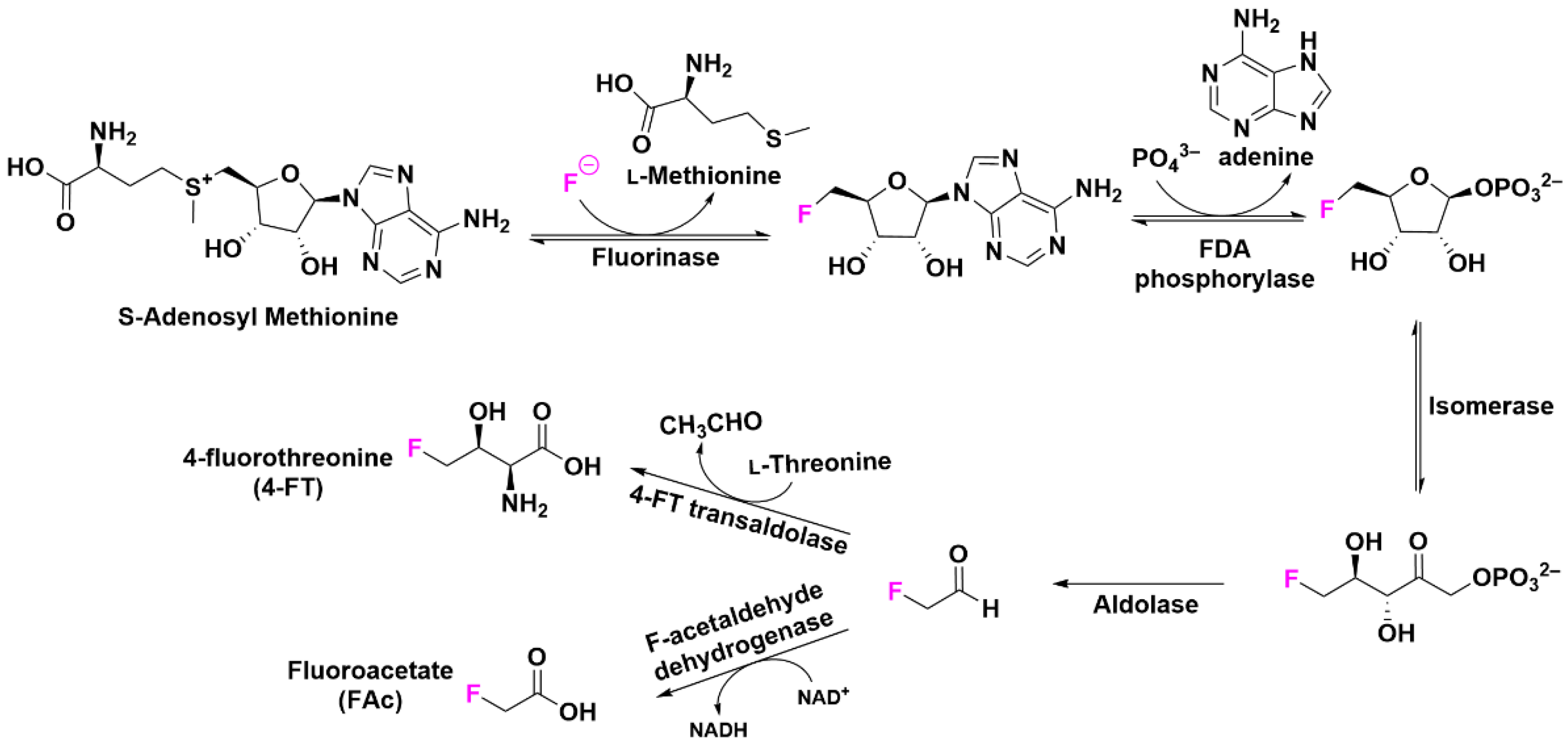
- Wang, Y.; Deng, Z.; Qu, X. Characterization of a SAM-dependent fluorinase from a latent biosynthetic pathway for fluoroacetate and 4-fluorothreonine formation in Nocardia brasiliensis [version 1; peer review: 2 approved]. F1000Research 2014, 3, 61. [Google Scholar] [CrossRef] [Green Version]
- McMurry, J.L.; Chang, M.C.Y. Fluorothreonyl-tRNA deacylase prevents mistranslation in the organofluorine producer Streptomyces cattleya. Proc. Natl. Acad. Sci. USA 2017, 114, 11920–11925. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Tong, M.H.; Raab, A.; Fang, Q.; Wang, S.; Kyeremeh, K.; Yu, Y.; Deng, H. An unusual metal-bound 4-fluorothreonine transaldolase from Streptomyces sp. MA37 catalyses promiscuous transaldol reactions. Appl. Microbiol. Biotechnol. 2020, 104, 3885–3896. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.; Zhao, Y.; Buuh, Z.Y.; Gorman, N.; Goldman, A.R.; Islam, M.S.; Tang, H.-Y.; Wang, R.E. Steric-Free Bioorthogonal Labeling of Acetylation Substrates Based on a Fluorine–Thiol Displacement Reaction. J. Am. Chem. Soc. 2021, 143, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Monde, K.; Miura, N.; Hashimoto, M.; Taniguchi, T.; Inabe, T. Conformational Analysis of Chiral Helical Perfluoroalkyl Chains by VCD. J. Am. Chem. Soc. 2006, 128, 6000–6001. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
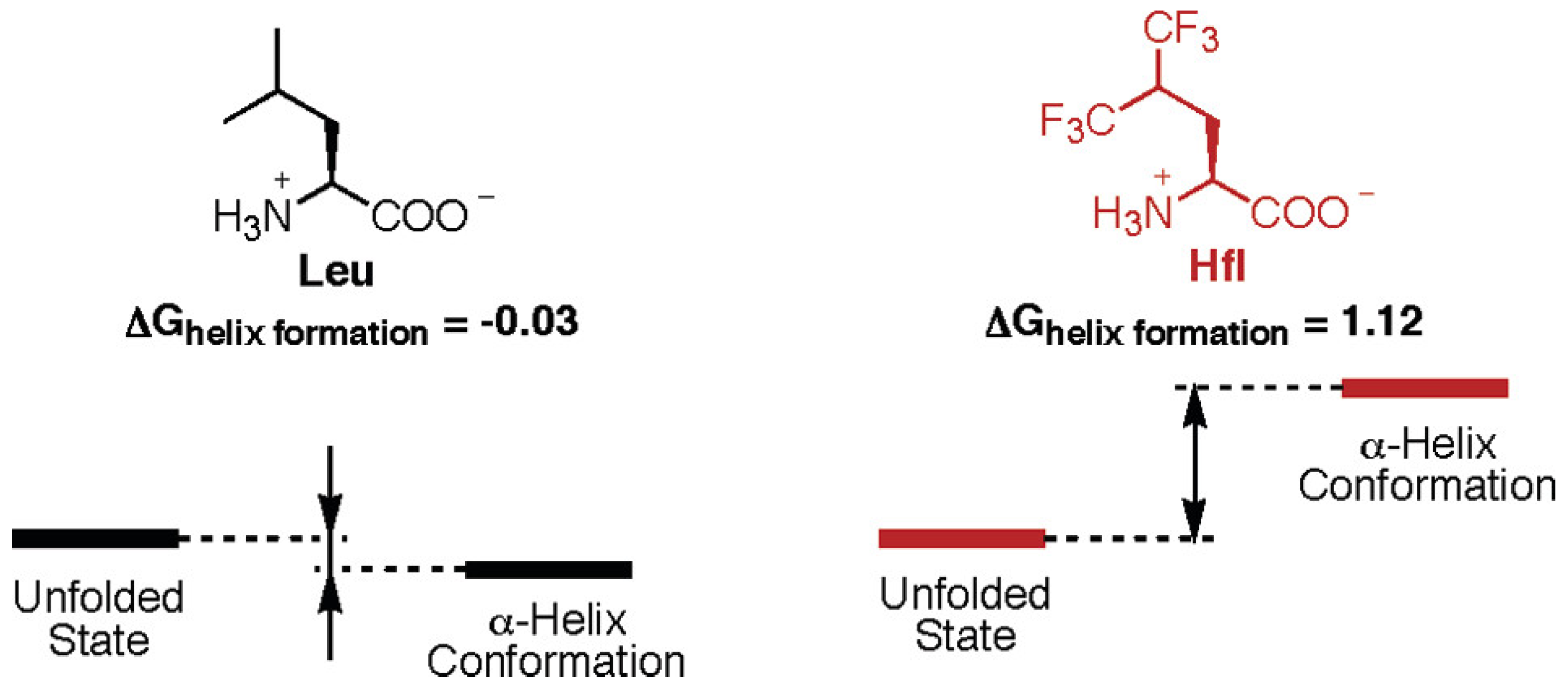
- Chiu, H.-P.; Suzuki, Y.; Gullickson, D.; Ahmad, R.; Kokona, B.; Fairman, R.; Cheng, R.P. Helix Propensity of Highly Fluorinated Amino Acids. J. Am. Chem. Soc. 2006, 128, 15556–15557. [Google Scholar] [CrossRef]
- Horng, J.-C.; Raleigh, D.P. Φ-Values beyond the Ribosomally Encoded Amino Acids: Kinetic and Thermodynamic Consequences of Incorporating Trifluoromethyl Amino Acids in a Globular Protein. J. Am. Chem. Soc. 2003, 125, 9286–9287. [Google Scholar] [CrossRef]
- Clark, G.A.; Baleja, J.D.; Kumar, K. Cross-Strand Interactions of Fluorinated Amino Acids in β-Hairpin Constructs. J. Am. Chem. Soc. 2012, 134, 17912–17921. [Google Scholar] [CrossRef]
- Chiu, H.-P.; Kokona, B.; Fairman, R.; Cheng, R.P. Effect of Highly Fluorinated Amino Acids on Protein Stability at a Solvent-Exposed Position on an Internal Strand of Protein G B1 Domain. J. Am. Chem. Soc. 2009, 131, 13192–13193. [Google Scholar] [CrossRef]
- Kirberger, S.E.; Maltseva, S.D.; Manulik, J.C.; Einstein, S.A.; Weegman, B.P.; Garwood, M.; Pomerantz, W.C.K. Synthesis of Intrinsically Disordered Fluorinated Peptides for Modular Design of High-Signal 19F MRI Agents. Angew. Chem. Int. Ed. 2017, 56, 6440–6444. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.; Brodsky, B. Supercoiled Protein Motifs: The Collagen Triple-Helix and the α-Helical Coiled Coil. J. Struct. Biol. 1998, 122, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Babu, I.R.; Hamill, E.K.; Kumar, K. A Highly Stereospecific and Efficient Synthesis of Homopentafluoro-phenylalanine. J. Org. Chem. 2004, 69, 5468–5470. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Lee, K.-H.; Al-Hashimi, H.M.; Marsh, E.N.G. Modulating Protein Structure with Fluorous Amino Acids: Increased Stability and Native-like Structure Conferred on a 4-Helix Bundle Protein by Hexafluoroleucine. J. Am. Chem. Soc. 2006, 128, 337–343. [Google Scholar] [CrossRef]
- Tang, Y.; Ghirlanda, G.; Petka, W.A.; Nakajima, T.; DeGrado, W.F.; Tirrell, D.A. Fluorinated Coiled-Coil Proteins Prepared In Vivo Display Enhanced Thermal and Chemical Stability. Angew. Chem. Int. Ed. 2001, 40, 1494–1496. [Google Scholar] [CrossRef]
- Erdbrink, H.; Nyakatura, E.K.; Huhmann, S.; Gerling, U.I.M.; Lentz, D.; Koksch, B.; Czekelius, C. Synthesis of enantiomerically pure (2S,3S)-5,5,5-trifluoroisoleucine and (2R,3S)-5,5,5-trifluoro-allo-isoleucine. Beilstein J. Org. Chem. 2013, 9, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.A.; Völler, J.-S.; Budisa, N.; Koksch, B. Deciphering the Fluorine Code—The Many Hats Fluorine Wears in a Protein Environment. Acc. Chem. Res. 2017, 50, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Reeb, J.; Rost, B. Secondary Structure Prediction. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, UK, 2019; pp. 488–496. [Google Scholar]
- Syud, F.A.; Stanger, H.E.; Gellman, S.H. Interstrand Side Chain−Side Chain Interactions in a Designed β-Hairpin: Significance of Both Lateral and Diagonal Pairings. J. Am. Chem. Soc. 2001, 123, 8667–8677. [Google Scholar] [CrossRef]
- Widyarani; Sari, Y.W.; Ratnaningsih, E.; Sanders, J.P.M.; Bruins, M.E. Production of hydrophobic amino acids from biobased resources: Wheat gluten and rubber seed proteins. Appl. Microbiol. Biotechnol. 2016, 100, 7909–7920. [Google Scholar] [CrossRef]
- Uversky, V.N. The alphabet of intrinsic disorder. Intrinsically Disord. Proteins 2013, 1, e24684. [Google Scholar] [CrossRef] [Green Version]
- Pelley, J.W. 3-Protein Structure and Function. In Elsevier’s Integrated Review Biochemistry, 2nd ed.; Pelley, J.W., Ed.; W. B. Saunders: Philadelphia, PA, USA, 2012; pp. 19–28. [Google Scholar]
- Ulrich, T.; Rapaport, D. Biogenesis of beta-barrel proteins in evolutionary context. Int. J. Med. Microbiol. 2015, 305, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Rabotyagova, O.S.; Cebe, P.; Kaplan, D.L. Protein-Based Block Copolymers. Biomacromolecules 2011, 12, 269–289. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Chen, H.; Fan, K.; Wei, P.; Guo, X.; Jin, C.; Zeng, C.; Tang, C.; Lai, L. De Novo Design of a βαβ Motif. Angew. Chem. Int. Ed. 2009, 48, 3301–3303. [Google Scholar] [CrossRef]
- Buer, B.C.; Marsh, E.N.G. Fluorine: A new element in protein design. Protein Sci. 2012, 21, 453–462. [Google Scholar] [CrossRef]
- Jäckel, C.; Salwiczek, M.; Koksch, B. Fluorine in a Native Protein Environment—How the Spatial Demand and Polarity of Fluoroalkyl Groups Affect Protein Folding. Angew. Chem. Int. Ed. 2006, 45, 4198–4203. [Google Scholar] [CrossRef]
- Tang, Y.; Ghirlanda, G.; Vaidehi, N.; Kua, J.; Mainz, D.T.; Goddard, W.A.; DeGrado, W.F.; Tirrell, D.A. Stabilization of Coiled-Coil Peptide Domains by Introduction of Trifluoroleucine. Biochemistry 2001, 40, 2790–2796. [Google Scholar] [CrossRef] [PubMed]
- Montclare, J.K.; Son, S.; Clark, G.A.; Kumar, K.; Tirrell, D.A. Biosynthesis and Stability of Coiled-Coil Peptides Containing (2S,4R)-5,5,5-Trifluoroleucine and (2S,4S)-5,5,5-Trifluoroleucine. ChemBioChem 2009, 10, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Tirrell, D.A. Biosynthesis of a Highly Stable Coiled-Coil Protein Containing Hexafluoroleucine in an Engineered Bacterial Host. J. Am. Chem. Soc. 2001, 123, 11089–11090. [Google Scholar] [CrossRef]
- More, H.T.; Zhang, K.S.; Srivastava, N.; Frezzo, J.A.; Montclare, J.K. Influence of Fluorination on Protein-Engineered Coiled-Coil Fibers. Biomacromolecules 2015, 16, 1210–1217. [Google Scholar] [CrossRef]
- Bilgiçer, B.; Fichera, A.; Kumar, K. A Coiled Coil with a Fluorous Core. J. Am. Chem. Soc. 2001, 123, 4393–4399. [Google Scholar] [CrossRef]
- Cejas, M.A.; Kinney, W.A.; Chen, C.; Vinter, J.G.; Almond, H.R., Jr.; Balss, K.M.; Maryanoff, C.A.; Schmidt, U.; Breslav, M.; Mahan, A.; et al. Thrombogenic collagen-mimetic peptides: Self-assembly of triple helix-based fibrils driven by hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 8513–8518. [Google Scholar] [CrossRef] [PubMed]
- Welte, H.; Zhou, T.; Mihajlenko, X.; Mayans, O.; Kovermann, M. What does fluorine do to a protein? Thermodynamic, and highly-resolved structural insights into fluorine-labelled variants of the cold shock protein. Sci. Rep. 2020, 10, 2640. [Google Scholar] [CrossRef] [PubMed]
- Yuvienco, C.; More, H.T.; Haghpanah, J.S.; Tu, R.S.; Montclare, J.K. Modulating Supramolecular Assemblies and Mechanical Properties of Engineered Protein Materials by Fluorinated Amino Acids. Biomacromolecules 2012, 13, 2273–2278. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Raines, R.T. Collagen Structure and Stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef]
- Chorghade, M.S.; Mohapatra, D.K.; Sahoo, G.; Gurjar, M.K.; Mandlecha, M.V.; Bhoite, N.; Moghe, S.; Raines, R.T. Practical syntheses of 4-fluoroprolines. J. Fluor. Chem. 2008, 129, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.A.; Raines, R.T. Stereoelectronic Effects on Collagen Stability: The Dichotomy of 4-Fluoroproline Diastereomers. J. Am. Chem. Soc. 2003, 125, 9262–9263. [Google Scholar] [CrossRef]
- Lalonde, J.M.; Bernlohr, D.A.; Banaszak, L.J. The up-and-down β-barrel proteins. FASEB J. 1994, 8, 1240–1247. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Therapeutic Proteins. In Therapeutic Proteins: Methods and Protocols; Voynov, V., Caravella, J.A., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 1–26. [Google Scholar]
- Murray, J.E.; Laurieri, N.; Delgoda, R. Chapter 24-Proteins. In Pharmacognosy; Badal, S., Delgoda, R., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 477–494. [Google Scholar]
- Li Petri, G.; Di Martino, S.; De Rosa, M. Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem. 2022, 65, 7438–7475. [Google Scholar] [CrossRef]
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Kumar, K. Antimicrobial Activity and Protease Stability of Peptides Containing Fluorinated Amino Acids. J. Am. Chem. Soc. 2007, 129, 15615–15622. [Google Scholar] [CrossRef]
- Hu, T.; Agazani, O.; Nir, S.; Cohen, M.; Pan, S.; Reches, M. Antiviral Activity of Peptide-Based Assemblies. ACS Appl. Mater. Interfaces 2021, 13, 48469–48477. [Google Scholar] [CrossRef] [PubMed]
- Acchione, M.; Lee, Y.-C.; DeSantis, M.E.; Lipschultz, C.A.; Wlodawer, A.; Li, M.; Shanmuganathan, A.; Walter, R.L.; Smith-Gill, S.; Barchi, J.J. Specific Fluorine Labeling of the HyHEL10 Antibody Affects Antigen Binding and Dynamics. Biochemistry 2012, 51, 6017–6027. [Google Scholar] [CrossRef] [PubMed]
- Dones, J.M.; Tanrikulu, I.C.; Chacko, J.V.; Schroeder, A.B.; Hoang, T.T.; Gibson, A.L.F.; Eliceiri, K.W.; Raines, R.T. Optimization of interstrand interactions enables burn detection with a collagen-mimetic peptide. Org. Biomol. Chem. 2019, 17, 9906–9912. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, M.; Rubini, M.; Adamo, M.F.A.; Scocchi, M.; Saviano, M.; Tossi, A.; Gennaro, R.; Caporale, A. Natural and Synthetic Halogenated Amino Acids—Structural and Bioactive Features in Antimicrobial Peptides and Peptidomimetics. Molecules 2021, 26, 7401. [Google Scholar] [CrossRef]
- Mora, L.; Aristoy, M.C.; Toldrá, F. Bioactive Peptides. In Encyclopedia of Food Chemistry; Melton, L., Shahidi, F., Varelis, P., Eds.; Academic Press: Oxford, UK, 2019; pp. 381–389. [Google Scholar]
- Lutz, H.U. Homeostatic roles of naturally occurring antibodies: An overview. J. Autoimmun. 2007, 29, 287–294. [Google Scholar] [CrossRef]
- Berger, M.; Shankar, V.; Vafai, A. Therapeutic Applications of Monoclonal Antibodies. Am. J. Med. Sci. 2002, 324, 14–30. [Google Scholar] [CrossRef]
- Ellison, A.J.; Tanrikulu, I.C.; Dones, J.M.; Raines, R.T. Cyclic Peptide Mimetic of Damaged Collagen. Biomacromolecules 2020, 21, 1539–1547. [Google Scholar] [CrossRef]
- Wen, H.; Jung, H.; Li, X. Drug Delivery Approaches in Addressing Clinical Pharmacology-Related Issues: Opportunities and Challenges. AAPS J. 2015, 17, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Wang, A.L.; Bhattacharya, A.; Montclare, J.K. Protein based biomaterials for therapeutic and diagnostic applications. Prog. Biomed. Eng. 2021, 4, 012003. [Google Scholar] [CrossRef]
- Hill, L.K.; Frezzo, J.A.; Katyal, P.; Hoang, D.M.; Gironda, Z.B.Y.; Xu, C.; Xie, X.; Delgado-Fukushima, E.; Wadghiri, Y.Z.; Montclare, J.K. Protein-Engineered Nanoscale Micelles for Dynamic 19F Magnetic Resonance and Therapeutic Drug Delivery. ACS Nano 2019, 13, 2969–2985. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, A.; Pellegrino, S.; Das, P.; Yuran, S.; Bucci, R.; Ferri, N.; Meneghetti, F.; Castellano, C.; Reches, M.; Gelmi, M.L. Dipeptide Nanotubes Containing Unnatural Fluorine-Substituted β2,3-Diarylamino Acid and l-Alanine as Candidates for Biomedical Applications. Org. Lett. 2015, 17, 4468–4471. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, S.; Schmidt, R.F.; Sahoo, A.K.; tom Dieck, T.; Hohmann, T.; Schade, B.; Brademann-Jock, K.; Thünemann, A.F.; Netz, R.R.; Gradzielski, M.; et al. Rational design of amphiphilic fluorinated peptides: Evaluation of self-assembly properties and hydrogel formation. Nanoscale 2022, 14, 10176–10189. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.M.; Abraham, B.L.; Fujita, T.; Watrous, M.J.; Toriki, E.S.; Takano, T.; Nilsson, B.L. Low-Molecular-Weight Supramolecular Hydrogels for Sustained and Localized in Vivo Drug Delivery. ACS Appl. Bio. Mater. 2019, 2, 2116–2124. [Google Scholar] [CrossRef]
- Wu, F.-Y.; Hsu, S.-M.; Cheng, H.; Hsu, L.-H.; Lin, H.-C. The effect of fluorine on supramolecular hydrogelation of 4-fluorobenzyl-capped diphenylalanine. New J. Chem. 2015, 39, 4240–4243. [Google Scholar] [CrossRef]
- Medina, S.H.; Michie, M.S.; Miller, S.E.; Schnermann, M.J.; Schneider, J.P. Fluorous Phase-Directed Peptide Assembly Affords Nano-Peptisomes Capable of Ultrasound-Triggered Cellular Delivery. Angew. Chem. Int. Ed. 2017, 56, 11404–11408. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, J.; Thomas, A.; Ou-Yang, D.; Muzykantov, V.R. The shape of things to come: Importance of design in nanotechnology for drug delivery. Ther. Deliv. 2012, 3, 181–194. [Google Scholar] [CrossRef]
- Kashapov, R.; Gaynanova, G.; Gabdrakhmanov, D.; Kuznetsov, D.; Pavlov, R.; Petrov, K.; Zakharova, L.; Sinyashin, O. Self-Assembly of Amphiphilic Compounds as a Versatile Tool for Construction of Nanoscale Drug Carriers. Int. J. Mol. Sci. 2020, 21, 6961. [Google Scholar] [CrossRef]
- Nasr, M.; Nawaz, S.; Elhissi, A. Amphotericin B lipid nanoemulsion aerosols for targeting peripheral respiratory airways via nebulization. Int. J. Pharm. 2012, 436, 611–616. [Google Scholar] [CrossRef]
- Arbain, N.H.; Basri, M.; Salim, N.; Wui, W.T.; Abdul Rahman, M.B. Development and Characterization of Aerosol Nanoemulsion System Encapsulating Low Water Soluble Quercetin for Lung Cancer Treatment. Mater. Today Proc. 2018, 5, S137–S142. [Google Scholar] [CrossRef]
- Asmawi, A.A.; Salim, N.; Abdulmalek, E.; Abdul Rahman, M.B. Modeling the Effect of Composition on Formation of Aerosolized Nanoemulsion System Encapsulating Docetaxel and Curcumin Using D-Optimal Mixture Experimental Design. Int. J. Mol. Sci. 2020, 21, 4357. [Google Scholar] [CrossRef]
- Sloand, J.N.; Nguyen, T.T.; Zinck, S.A.; Cook, E.C.; Zimudzi, T.J.; Showalter, S.A.; Glick, A.B.; Simon, J.C.; Medina, S.H. Ultrasound-Guided Cytosolic Protein Delivery via Transient Fluorous Masks. ACS Nano 2020, 14, 4061–4073. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Singh, I.; Sharma, A.K.; Kumar, P. Ultrashort Peptide Self-Assembly: Front-Runners to Transport Drug and Gene Cargos. Front. Bioeng. Biotechnol. 2020, 8, 504. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Erbas, A.; Tantakitti, F.; Palmer, L.C.; Jackman, J.A.; Olvera de la Cruz, M.; Cho, N.-J.; Stupp, S.I. Co-assembly of Peptide Amphiphiles and Lipids into Supramolecular Nanostructures Driven by Anion−π Interactions. J. Am. Chem. Soc. 2017, 139, 7823–7830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lou, S.; Yu, Z. Polar−π Interactions Promote Self-assembly of Dipeptides into Laminated Nanofibers. Langmuir 2019, 35, 4710–4717. [Google Scholar] [CrossRef]
- Hamley, I.W. Peptide Nanotubes. Angew. Chem. Int. Ed. 2014, 53, 6866–6881. [Google Scholar] [CrossRef] [PubMed]
- Handelman, A.; Lavrov, S.; Kudryavtsev, A.; Khatchatouriants, A.; Rosenberg, Y.; Mishina, E.; Rosenman, G. Nonlinear Optical Bioinspired Peptide Nanostructures. Adv. Opt. Mater. 2013, 1, 875–884. [Google Scholar] [CrossRef]
- Yuran, S.; Razvag, Y.; Reches, M. Coassembly of Aromatic Dipeptides into Biomolecular Necklaces. ACS Nano 2012, 6, 9559–9566. [Google Scholar] [CrossRef]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRX 2005, 2, 554–571. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Gong, C.; Liu, B.; Li, Y.; Wang, L.; Su, Z.; Wei, G. Recent advances in the fabrication, functionalization, and bioapplications of peptide hydrogels. Soft Matter 2020, 16, 10029–10045. [Google Scholar] [CrossRef]
- Bashir, S.; Hina, M.; Iqbal, J.; Rajpar, A.H.; Mujtaba, M.A.; Alghamdi, N.A.; Wageh, S.; Ramesh, K.; Ramesh, S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12, 2702. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, C. Advances in bioimaging—Challenges and potentials. J. Phys. D Appl. Phys. 2018, 51, 040201. [Google Scholar] [CrossRef]
- Preslar, A.T.; Tantakitti, F.; Park, K.; Zhang, S.; Stupp, S.I.; Meade, T.J. 19F Magnetic Resonance Imaging Signals from Peptide Amphiphile Nanostructures Are Strongly Affected by Their Shape. ACS Nano 2016, 10, 7376–7384. [Google Scholar] [CrossRef] [PubMed]
- Preslar, A.T.; Lilley, L.M.; Sato, K.; Zhang, S.; Chia, Z.K.; Stupp, S.I.; Meade, T.J. Calcium-Induced Morphological Transitions in Peptide Amphiphiles Detected by 19F-Magnetic Resonance Imaging. ACS Appl. Mater. Interfaces 2017, 9, 39890–39894. [Google Scholar] [CrossRef]
- Yuan, Y.; Sun, H.; Ge, S.; Wang, M.; Zhao, H.; Wang, L.; An, L.; Zhang, J.; Zhang, H.; Hu, B.; et al. Controlled Intracellular Self-Assembly and Disassembly of 19F Nanoparticles for MR Imaging of Caspase 3/7 in Zebrafish. ACS Nano 2015, 9, 761–768. [Google Scholar] [CrossRef]
- Sloand, J.N.; Miller, M.A.; Medina, S.H. Fluorinated peptide biomaterials. Pept. Sci. 2021, 113, e24184. [Google Scholar] [CrossRef]
- Fox, M.S.; Gaudet, J.M.; Foster, P.J. Fluorine-19 MRI Contrast Agents for Cell Tracking and Lung Imaging. Magn. Reson. Insights 2015, 8s1. [Google Scholar] [CrossRef]
- Shukla, A.; Kumar, U. Positron emission tomography: An overview. J. Med. Phys. 2006, 31, 13–21. [Google Scholar] [CrossRef]
- Fletcher, J.W.; Djulbegovic, B.; Soares, H.P.; Siegel, B.A.; Lowe, V.J.; Lyman, G.H.; Coleman, R.E.; Wahl, R.; Paschold, J.C.; Avril, N.; et al. Recommendations on the Use of 18F-FDG PET in Oncology. J. Nucl. Med. 2008, 49, 480–508. [Google Scholar] [CrossRef]
- Berger, A. Positron emission tomography. BMJ 2003, 326, 1449. [Google Scholar] [CrossRef]
- Awenat, S.; Piccardo, A.; Carvoeiras, P.; Signore, G.; Giovanella, L.; Prior, J.O.; Treglia, G. Diagnostic Role of 18F-PSMA-1007 PET/CT in Prostate Cancer Staging: A Systematic Review. Diagnostics 2021, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Huynh, P.T.; Soni, N.; Pal, R.; Sarkar, S.; Jung, J.-M.; Lee, W.; Yoo, J. Direct radiofluorination of a heat-sensitive antibody by Al–18F complexation. New J. Chem. 2019, 43, 15389–15395. [Google Scholar] [CrossRef]
- Krishnan, H.S.; Ma, L.; Vasdev, N.; Liang, S.H. 18F-Labeling of Sensitive Biomolecules for Positron Emission Tomography. Chem. –A Eur. J. 2017, 23, 15553–15577. [Google Scholar] [CrossRef]
- Paulus, A.; Desai, P.; Carney, B.; Carlucci, G.; Reiner, T.; Brand, C.; Weber, W.A. Development of a clickable bimodal fluorescent/PET probe for in vivo imaging. EJNMMI Res. 2015, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef] [PubMed]
- Stadmiller, S.S.; Aguilar, J.S.; Waudby, C.A.; Pielak, G.J. Rapid Quantification of Protein-Ligand Binding via 19F NMR Lineshape Analysis. Biophys. J. 2020, 118, 2537–2548. [Google Scholar] [CrossRef]


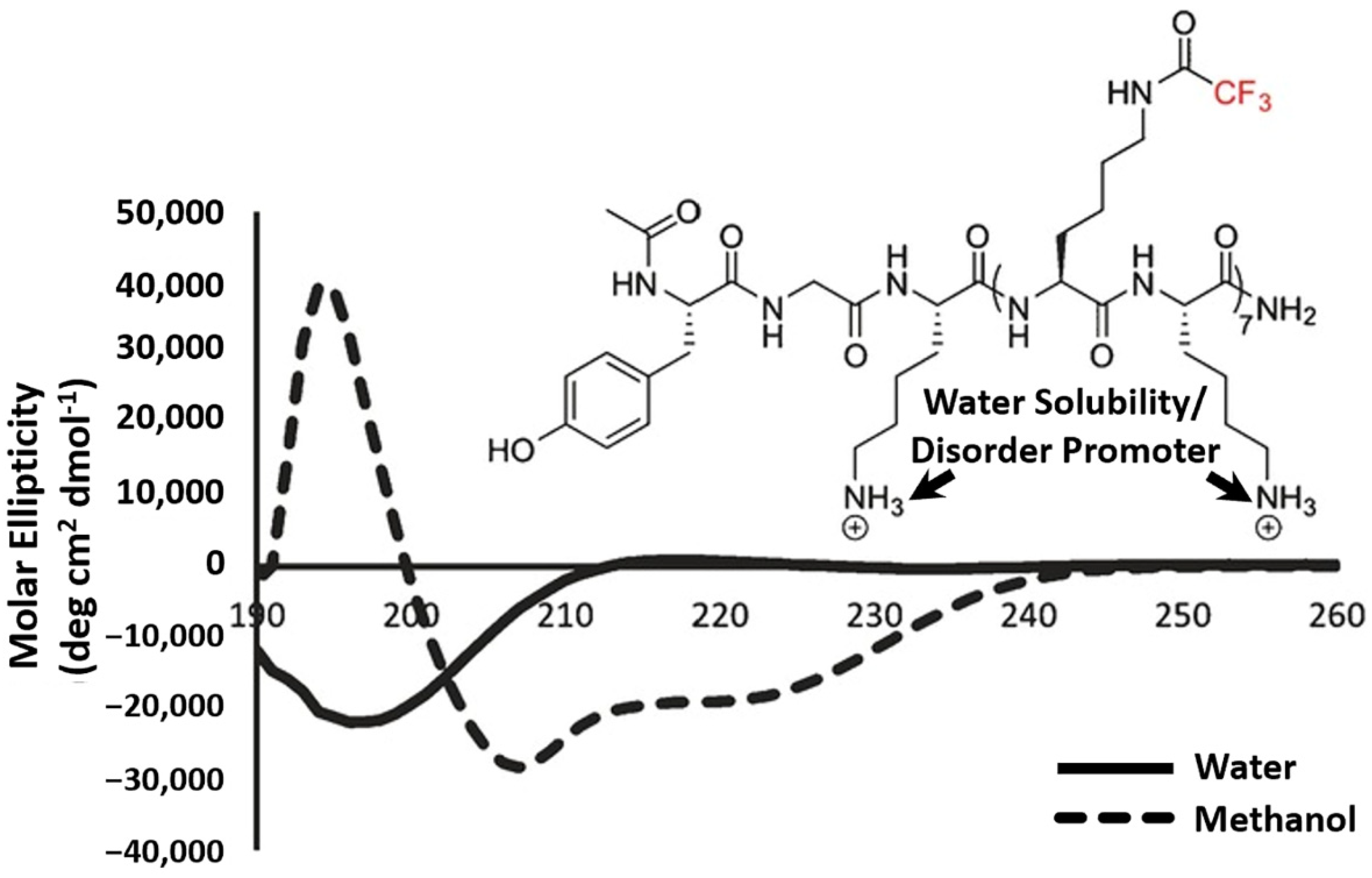
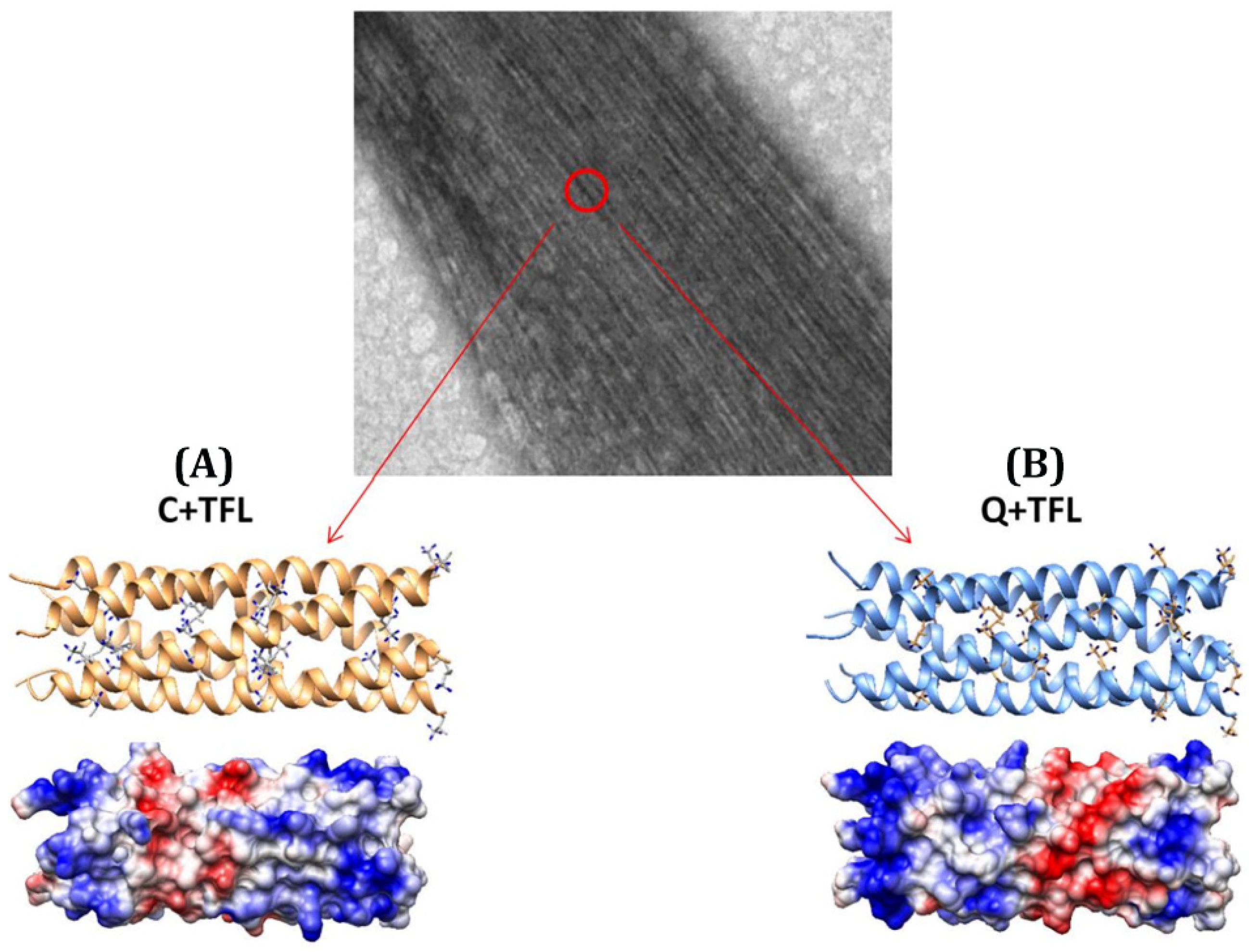

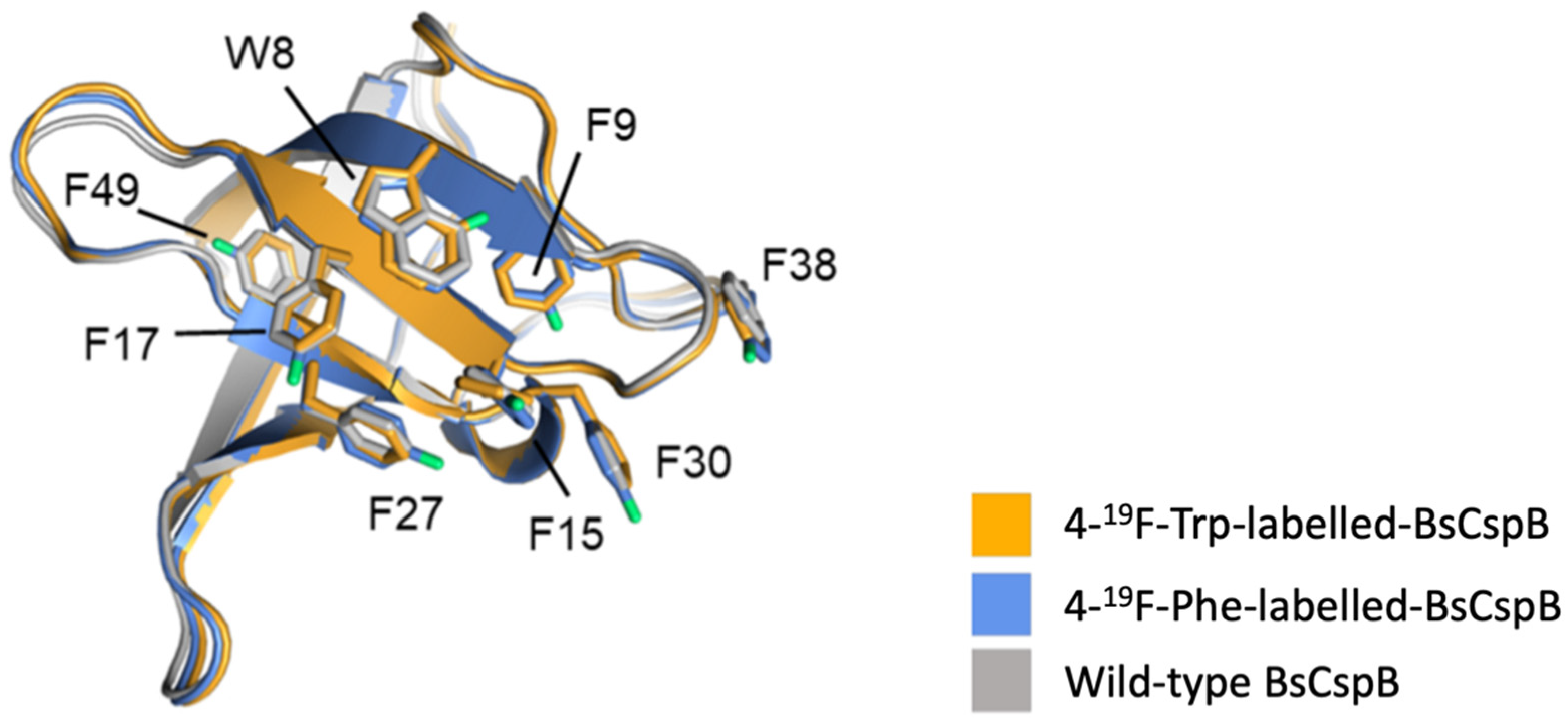
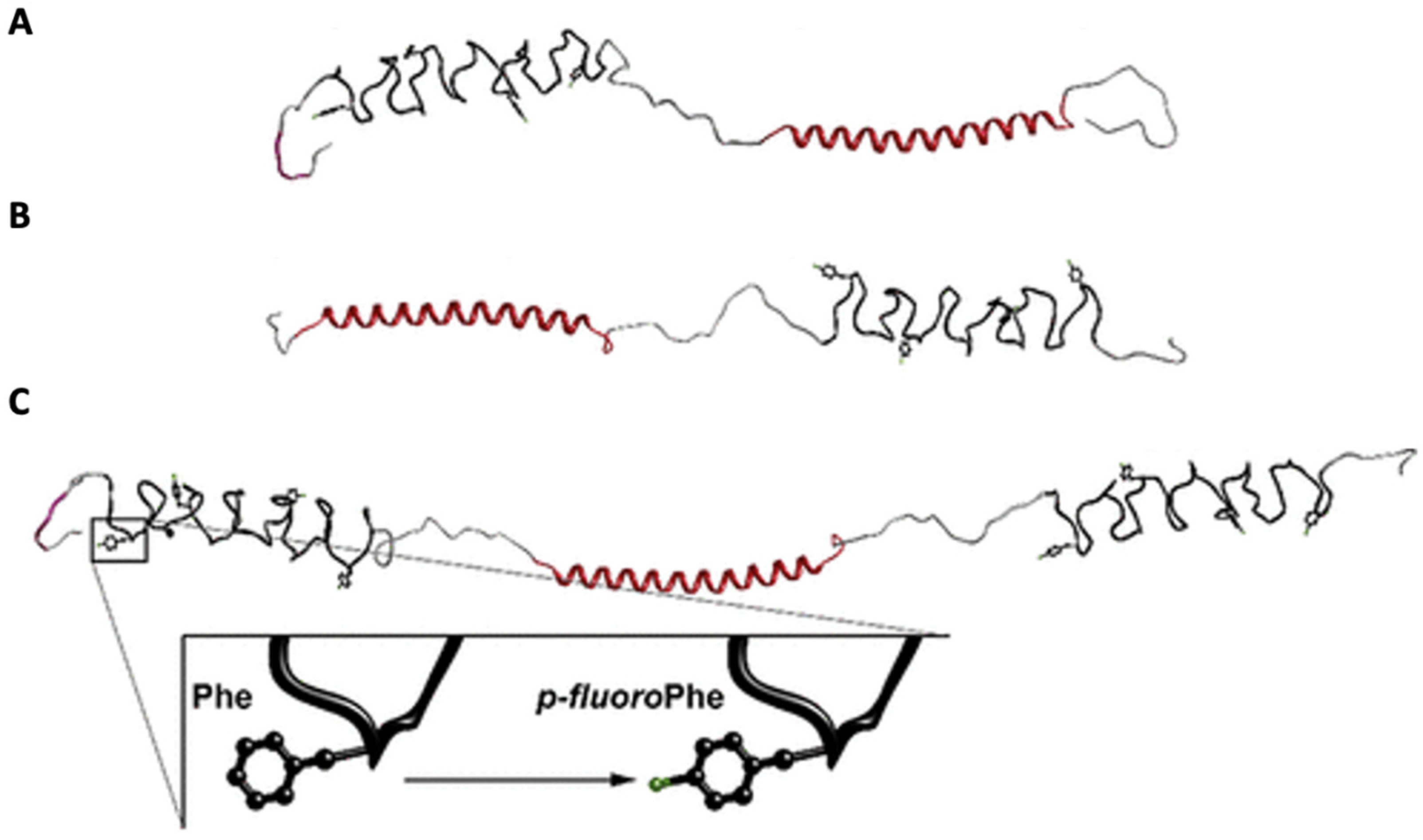



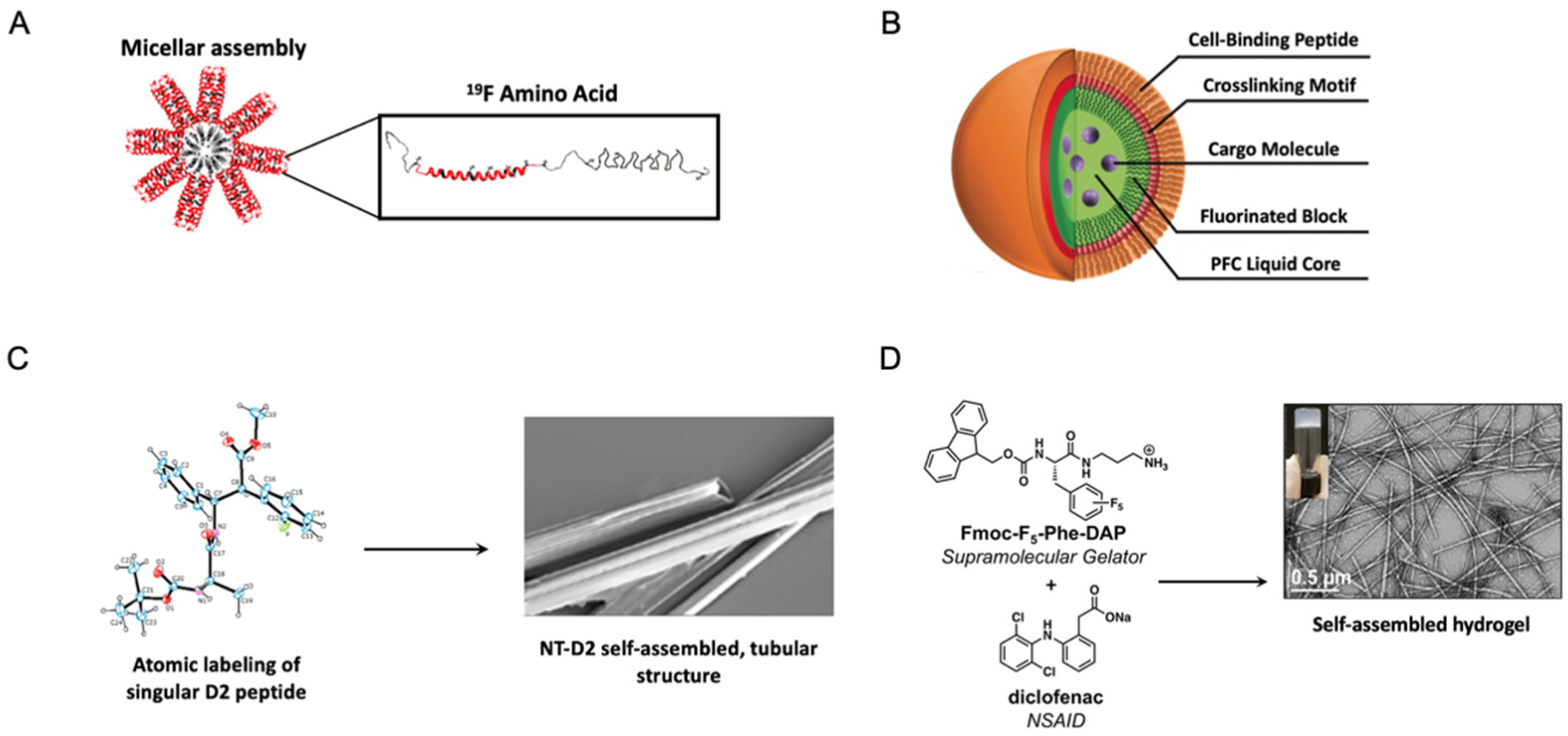
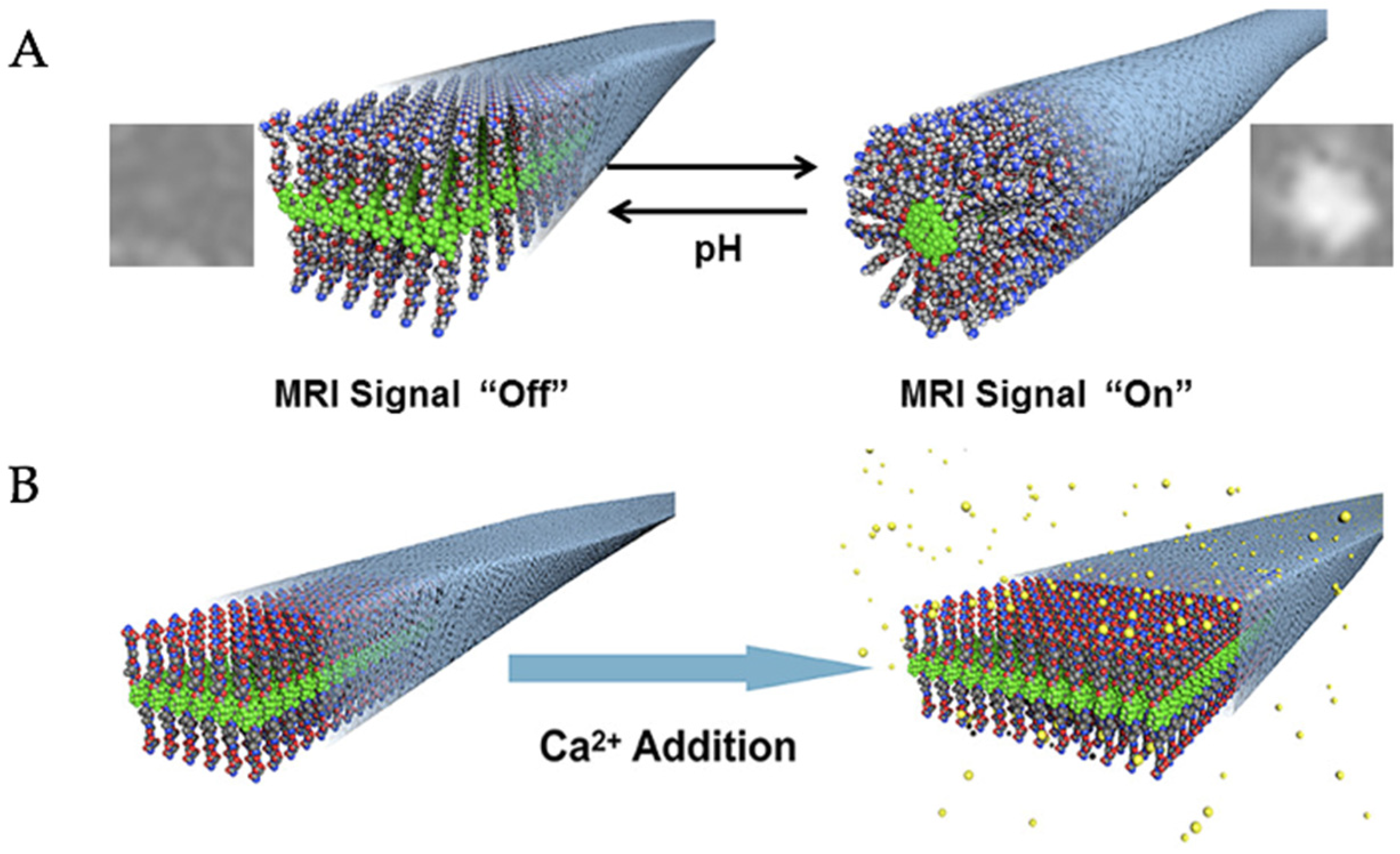
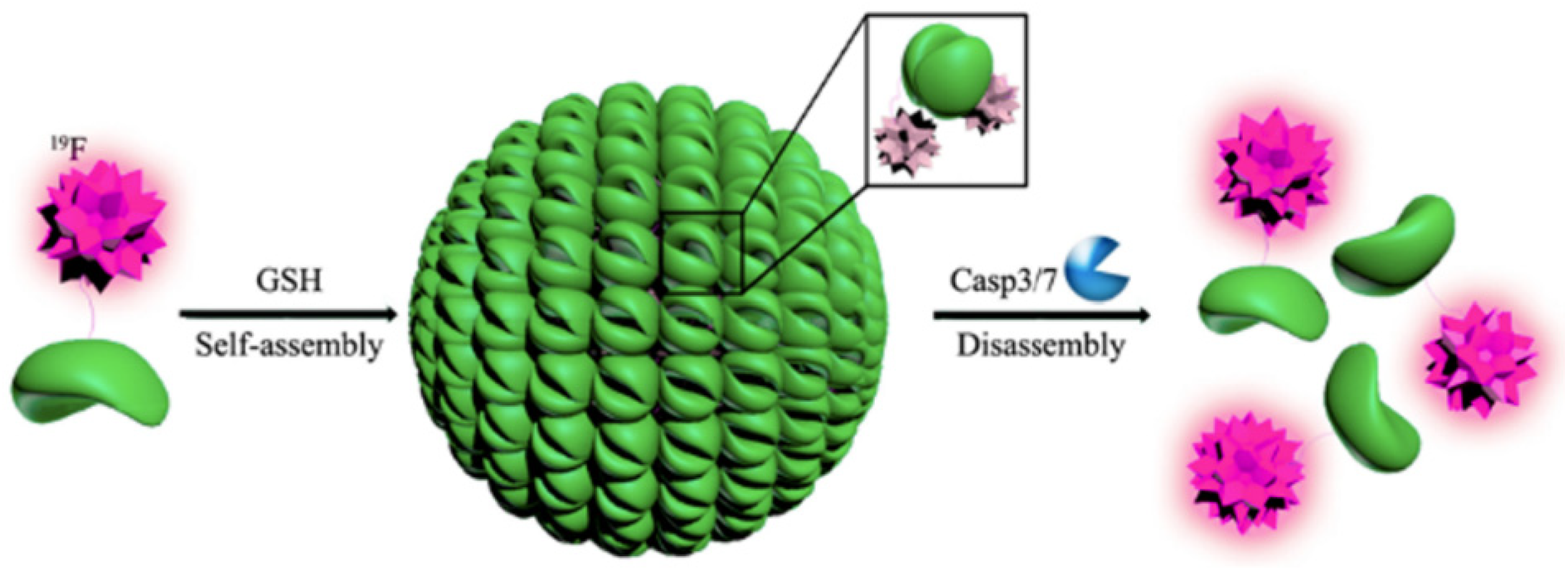
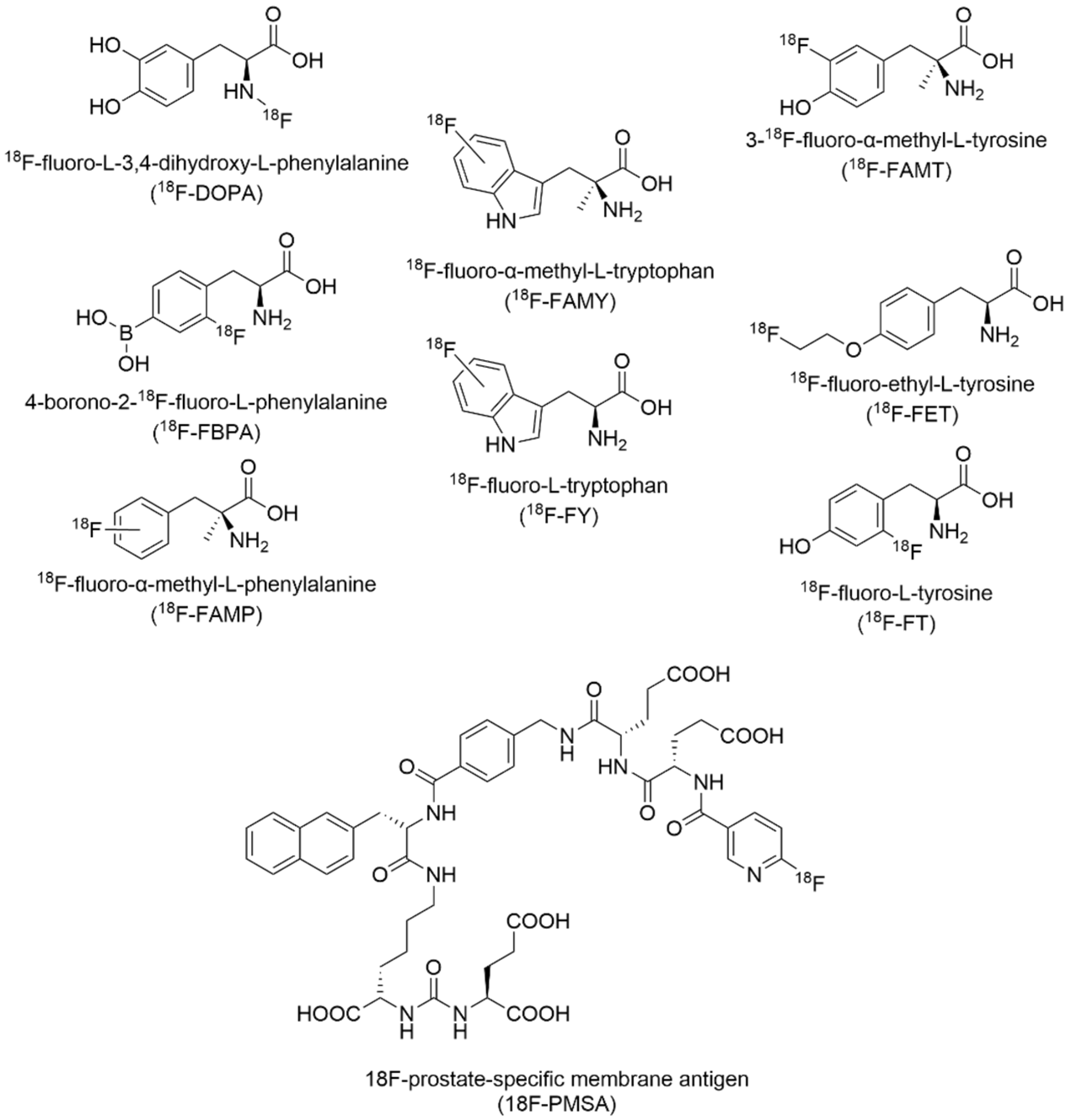
| Construct Name | Fluorinated Residue(s) | Number of Fluorines | Position of Fluorines | Synthetic Method | Secondary Structure | Sequence | References |
|---|---|---|---|---|---|---|---|
| KHfl | Leucine (L) | 6 per L (6 total) | Methyl groups, δ-carbons | SPPS | α-helix | YGGKAAAAKLAAKAAAAK | [32] |
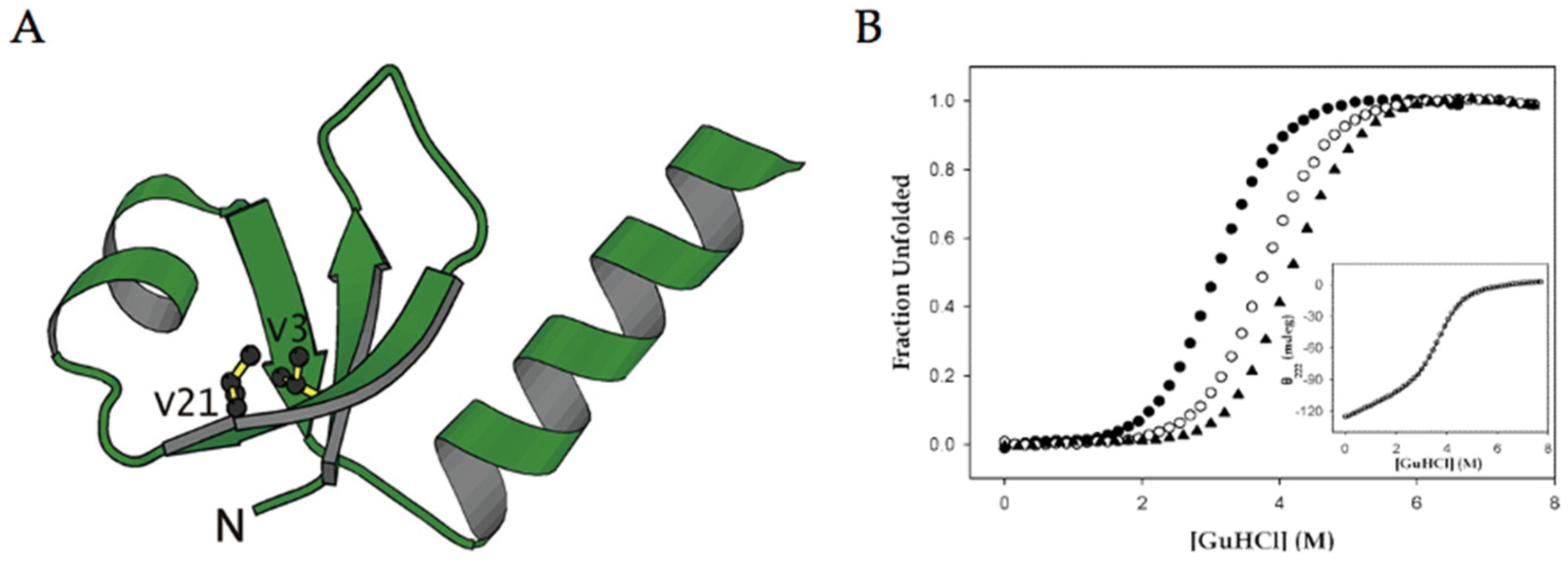
| tfV3 | Valine (V) | 3 per V (3 total) | Methyl group, γ-carbon | SPPS | β-sheet | MKVIFLKDVKGKGKKGEIK NVADGYANNFLFKQGLAIE ATPANLKALEAQK | [33] |
| tfV21 | Valine (V) | 3 per V (3 total) | Methyl group, γ-carbon | SPPS | β-sheet | MKVIFLKDVKGKGKKGEIK NVADGYANNFLFKQGLAIE ATPANLKALEAQK | [33] |
| HH1 | Leucine (L) | 6 per L (12 total) | Methyl groups, δ-carbons | SPPS | β-hairpin | QRLTNCCNTLEG (cyclic) | [34] |
| HH2 | Leucine (L) | 6 per L (12 total) | Methyl groups, δ-carbons | SPPS | β-hairpin | L@VPAVTL (cyclic) | [34] |
| GB1-QfL | Leucine (L) | 4 per L (4 total) | Methyl groups, δ-carbons | SPPS | β-sheet | MGYKLALNGKTLKGETT TEAVDAATAEKVFKQYA NDNEGEWAYDDATLKT FLVTE | [35] |
| GB1-Hfl | Leucine (L) | 6 per L (6 total) | Methyl groups, δ-carbons | SPPS | β-sheet (globular protein) | MGYKLALNGKTLKGETTTE AVDAATAEKVFKQYANDN EGEWAYDDATLKTFLVTE | [35] |
| GB1-Pff | Phenylalanine (F) | 5 per F (5 total) | Aryl ring, all positions | SPPS | β-sheet (globular protein) | MGYKLALNGKTLKGETTTE AVDAATAEKVFKQYANDN EGEWAYDDATLKTFFVTE | [35] |
| Peptide 1 (n = 7) | Lysine (K) | 3 per TFA-modified K (21 total) | Modified acetyl group | SPPS | Intrinsically disordered protein | YGKTFAKKTFAKKTFAKKTFAKK TFAKKTFAKKTFAKK | [36] |
| Construct Name | Fluorinated Residue(s) | Number of Fluorines | Position of Fluorines | Synthetic Method | (Super) Secondary Structure(s) | Sequence | References |
|---|---|---|---|---|---|---|---|
| Tfl-GCN4-p1d | Leucine (L) | 3 per Leucine (12 total) | Methyl group, δ-carbon | Residue-specific ncAA incorporation | α-helical coiled-coil | RMKQLEDKVEELLSKNYHLENEVARLKKLVGER | [53] |
| Hfl-GCN4-p1d | Leucine (L) | 6 per Leucine (24 total) | Methyl group, δ-carbon | Residue-specific ncAA incorporation | α-helical coiled-coil | RMKQLEDKVEELLSKNYHLENEVARLKKLVGER | [53] |
| TFL-Protein A1 | Leucine (L) | 3 per Leucine (24 total) | Methyl group, δ-carbon | Residue-specific ncAA incorporation | α-helical coiled-coil | MRGSHHHHHHGSASGDLENE VAQLEREVR SLEDEAAELEQKVSRLKNEIEDLKAEIGDLNNTSGIRRPAAKLN | [54] |
| HFL-Protein A1 | Leucine (L) | 6 per Leucine (48 total) | Methyl group, δ-carbon | Residue-specific ncAA incorporation | α-helical coiled-coil | MRGSHHHHHHGSASGDLENE VAQLEREVR SLEDEAAELEQKVSRLKNEIEDLKAEIGDLNNTSGIRRPAAKLN | [55] |
| C+TFL | Leucine (L) | 3 per Leucine (21 total) | Methyl group, δ-carbon | Residue-specific ncAAincorporation | α-helical coiled-coil | MRGSHHHHHHGSIEGRAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTSKL | [56] |
| Q+TFL | Leucine (L) | 3 per Leucine (21 total) | Methyl group, δ-carbon | Residue-specific ncAA incorporation | α-helical coiled-coil | MRGSHHHHHHGSIEGRVKEITFLKNTAPQMLRELQETNAALQDVRELLRQQSKL | [56] |
| Peptide 2 (GCN4 modified coiled-coil region) | Leucine (L) Valine (V) | 3 per L or V (21 total) | Methyl group, δ-carbon | SPPS | α-helical coiled-coil | HNRMKQLEDKVEELLSKNASLEYEVARLKKLVGE | [57] |
| (flpProGly)7 collagen triple helix | Proline (P) | 1 per P (7 total) | Pyrrolidine ring, 4′-carbon | SPPS | Proline Type II Collagen triple helix | PhPGPhPGPhPGPhPGPhPGPhPGPhPG | [58] |
| (GlyProHyp)10 collagen model peptide | Phenylalanine (F) | 5 per F (10 total) | Aryl ring, all positions | SPPS | Proline Type II Collagen triple helix | FGPhPGPhPGPhPGPhPGPhPGPhPGPhPGPhPGPhPGPhPF | [58] |
| 19F-Trp-labelled-BsCspB | Tryptophan (W) | 1 per W (1 total) | Aryl ring, 4-, 5-, or 6-carbon | Residue-specific ncAA incorporation | β-barrel | MLEGKVKWFNSEKGFGFIEVEGQDDVFVHFSAIQGEGFKTLEEGQAVSFEIVEGNRGPQAANVTKEA | [59] |
| 19F-Phe-labelled-BsCspB | Phenylalanine (F) | 1 per F (7 total) | Aryl ring, 2-ortho, 3-meta, or 4-para-position | Residue-specific ncAA incorporation | β-barrel | MLEGKVKWFNSEKGFGFIEVEGQDDVFVHFSAIQGEGFKTLEEGQAVSFEIVEGNRGPQAANVTKEA | [59] |
| pFF-EC | Phenylalanine (F) | 1 per F (2 total) | Aryl ring, 4-para-position | Residue-specific ncAA incorporation | Block Copolymer: Disordered elastin-like polypeptide connected to coiled coil | MRGSHHHHHHHGSKPIAASA[(VPGVG)2VPGFG(VPGVG)2]5VPLEGSELAATATATATATATAACGDLAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASGLQAATATATATATATAVDLQPS | [60] |
| pFF-CE | Phenylalanine (F) | 1 per F (2 total) | Aryl ring, 4-para-position | Residue-specific ncAA incorporation | Block Copolymer: Disordered elastin-like polypeptide connected to coiled coil | MRGSHHHHHHHGSACELAATATATATATATAACGDLAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASGLQAATATATATATATAVDKPIAASA[(VPGVG)2VPGFG(VPGVG)2]5LEGSGTGAKLN | [60] |
| pFF-ECE | Phenylalanine (F) | 1 per F (3 total) | Aryl ring, 4-para-position | Residue-specific ncAA incorporation | Block Copolymer: Disordered elastin-like polypeptide connected to coiled coil | MRGSHHHHHHHGSKPIAASA[(VPGVG)2VPGFG(VPGVG)2]5LEGSELAATATATATATATAACGDLAPQMLRELQETNAALQDVRELLRQQVKEITFLKNTVMESDASGLQAATATATATATATAVDKPIAASA[(VPGVG)2VPGFG(VPGVG)2]5LEGSGTGAKLN | [60] |
| Construct Name | Fluorinated Residue(s) | Number of Fluorines | Position of Fluorines | Synthetic Method | Assembly Morphology | Sequence | References |
|---|---|---|---|---|---|---|---|
| C7K2 | C-terminus | 13 per endcap (13 total) | C-terminal endcap | SPPS | Ribbon-like/cylindrical peptide amphiphiles | KKAAVV-CO-CF2-CF2- CF2-CF2-CF2-CF3 | [103,104] |
| C7E2 | C-terminus | 11 per endcap (11 total) | C-terminal endcap | SPPS | Ribbon-like/cylindrical peptide amphiphiles | EEAAVV-CO-CF2-CF2- CF2-CF2-CF2-CF3 | [103,104] |
| C8K2 | C-terminus | 15 per endcap (15 total) | C-terminal endcap | SPPS | Ribbon-like/cylindrical peptide amphiphiles | KKAAVV-CO-CF2-CF2- CF2-CF2-CF2-CF2-CF3 | [103] |
| C8E2 | C-terminus | 13 per endcap (13 total) | C-terminal endcap | SPPS | Ribbon-like/cylindrical peptide amphiphiles | EEAAVV-CO-CF2-CF2- CF2-CF2-CF2-CF2-CF3 | [103] |
| C7E3 | C-terminus | 11 per endcap (11 total) | C-terminal endcap | SPPS | Ribbon-like/cylindrical peptide amphiphiles | EEEAAVV-CO-CF2-CF2- CF2-CF2-CF2-CF3 | [103] |
| Compound 1 | Lysine (K) | 6 per FMBA-modified K (6 total) | Modified side chain aryl ring (3′ and 5′ positions) | SPPS | Linear Peptide | CDQVDFMBAKC | [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monkovic, J.M.; Gibson, H.; Sun, J.W.; Montclare, J.K. Fluorinated Protein and Peptide Materials for Biomedical Applications. Pharmaceuticals 2022, 15, 1201. https://doi.org/10.3390/ph15101201
Monkovic JM, Gibson H, Sun JW, Montclare JK. Fluorinated Protein and Peptide Materials for Biomedical Applications. Pharmaceuticals. 2022; 15(10):1201. https://doi.org/10.3390/ph15101201
Chicago/Turabian StyleMonkovic, Julia M., Halle Gibson, Jonathan W. Sun, and Jin Kim Montclare. 2022. "Fluorinated Protein and Peptide Materials for Biomedical Applications" Pharmaceuticals 15, no. 10: 1201. https://doi.org/10.3390/ph15101201
APA StyleMonkovic, J. M., Gibson, H., Sun, J. W., & Montclare, J. K. (2022). Fluorinated Protein and Peptide Materials for Biomedical Applications. Pharmaceuticals, 15(10), 1201. https://doi.org/10.3390/ph15101201