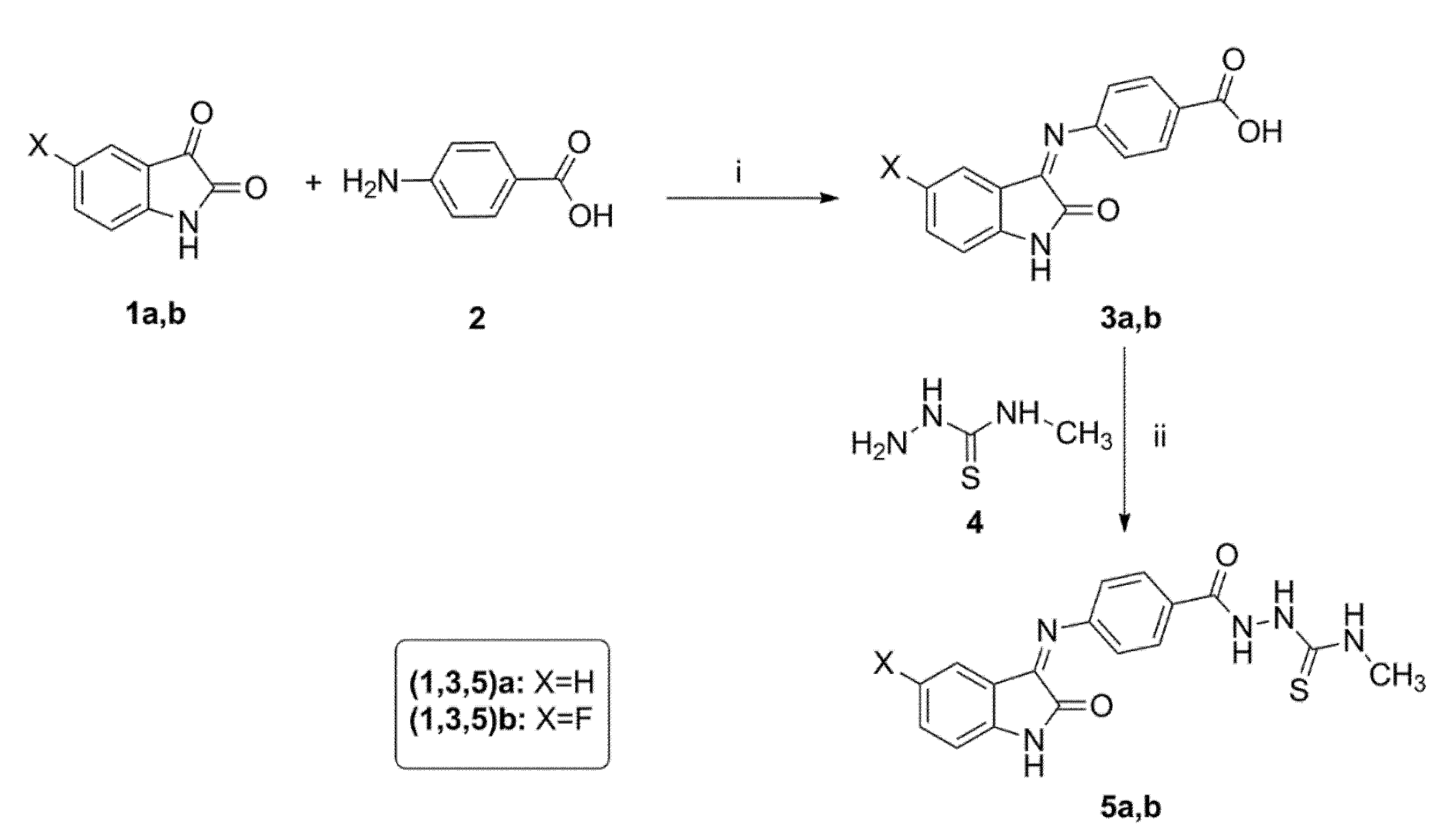
3.1.2. General Procedure for Synthesis of 2-(4-((5-Substituted-2-oxoindolin-3-ylidene)amino)benzoyl)-N-methylhydrazinecarbothioamide 5a,b
To a mixture of benzoic acid, derivatives 3a,b (3 mmol), EDC (3.3 mmol), 4-methylthiosemicarbazide 4 (3.6 mmol) and HOBt (3.9 mmol) in DMF (20 mL) were added to triethylamine (9 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for one hour. Next, the ice bath was removed, and the reaction mixture was stirred at room temperature overnight. The reaction was quenched by addition of water (40 mL). The precipitated solid was filtered and purified by flash column chromatography using (hexanes/EtOAc, 3:1) as the mobile phase system to give N-methylthiosemicarbazises 5a,b.
N-Methyl-2-(4-((2-oxoindolin-3-ylidene)amino)benzoyl)hydrazinecarbothioamide 5a
Yellow powder (yield, 78 %); mp (°C) 249-251; FT-IR (KBr, cm−1): 3235 (NH), 1691(C=O), 1669 (C=O), 1587 (C=N); 1H NMR (400 MHz, DMSO) δ 12.61 (s, 2H), 11.22 (s, 1H), 9.25 (d, J = 4.5 Hz, 1H), 7.75–7.52 (m, 2H), 7.48–7.20 (m, 2H), 7.23–7.02 (m, 2H), 6.94 (d, J = 7.8 Hz, 2H), 3.10 (d, J = 4.6 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 178.12, 163.10, 142.71, 132.05, 131.96, 131.65, 131.58, 122.80, 121.15, 121.04, 120.80, 120.56, 120.48, 111.54, 31.84; ESI-MS m/z: 354.3 [M+H]+; Anal. Calcd. for C17H15N5O2S: C, 57.78; H, 4.28; N, 19.82. Found: C, 57.62; H, 4.48; N, 19.95.
2-(4-((5-Fluoro-2-oxoindolin-3-ylidene)amino)benzoyl)-N-methylhydrazinecarbothioamide 5b
Orange powder (yield, 82 %); mp (°C) 252-254; FT-IR (KBr, cm−1): 3284 (NH), 1689 (C=O), 1654 (C=O), 1588 (CH=N); 1H NMR (400 MHz, DMSO) δ 12.48 (s, 1H), 11.21 (s, 2H), 9.31 (d, J = 4.5 Hz, 1H), 7.63–7.28 (m, 2H), 7.35–6.98 (m, 3H), 7.10–6.75 (m, 2H), 3.10 (d, J = 4.6 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 178.09, 163.17, 159.87, 157.51, 138.94, 131.38 (d, J = 3.4 Hz), 121.95, 121.86, 117.88, 117.64, 112.62, 112.54, 108.16, 107.91, 31.78; ESI-MS m/z: 372.2 [M+H]+; Anal. Calcd. for C17H14FN5O2S: C, 54.89; H, 3.80; N, 18.86. Found: C, 55.05; H, 3.94; N, 18.97.
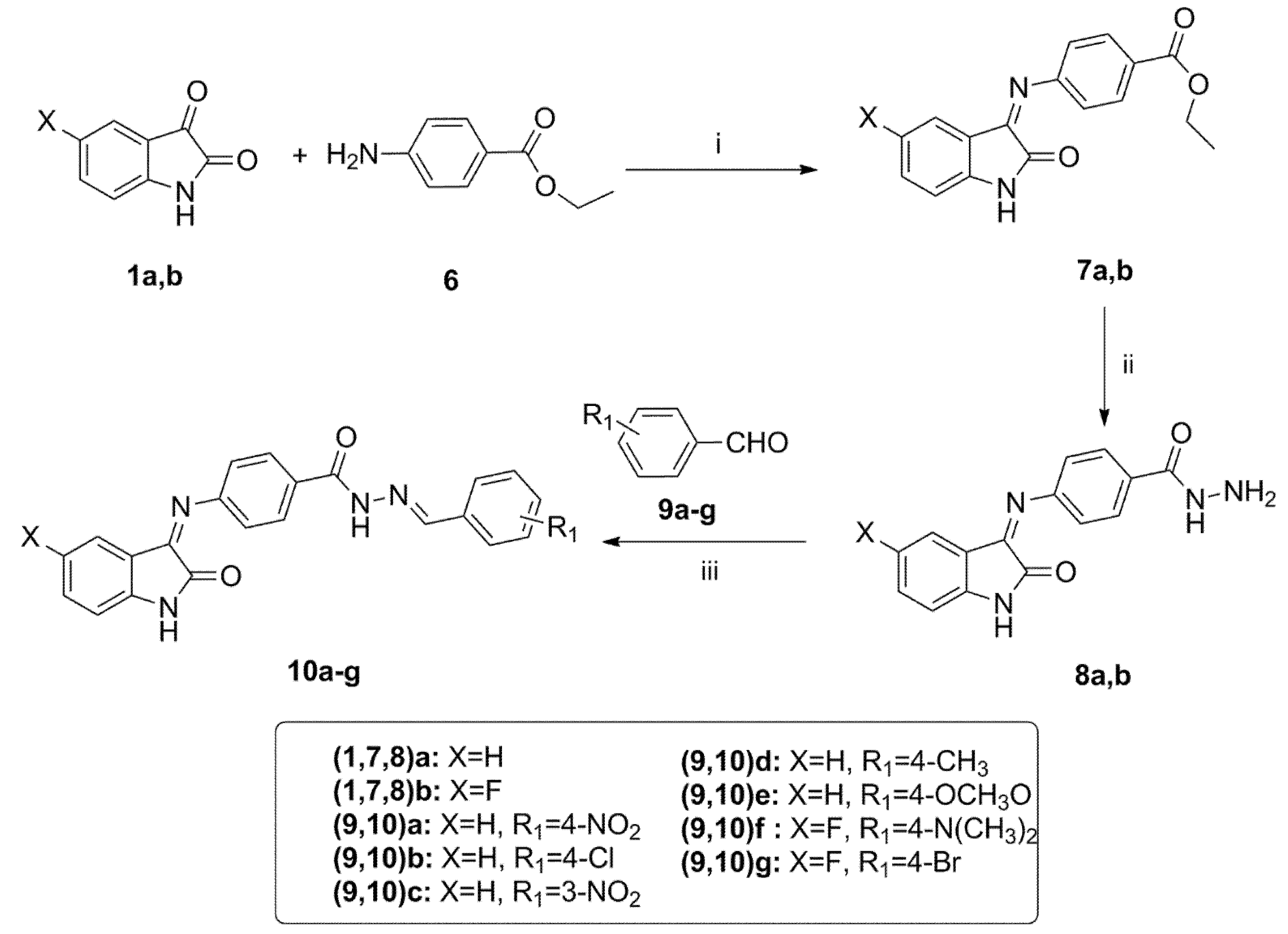
3.1.5. General Procedure for Synthesis of N′-(4-Substitutedbenzylidene)-4-((5-substituted-2-oxoindolin-3-ylidene)amino)benzohydrazide 10a–g
Benzaldehyde derivatives 9a–g (1.8 mmol) were added to a solution of hydrazides 8a,b (1.8 mmol) in absolute ethanol (10 mL). Next, 0.3 mL of glacial acetic acid was added, and the reaction mixture was heated under reflux for 6 h. After cooling, the precipitate was collected, and the crude precipitate was recrystallized from methanol to give hydrazones 10a–g.
N′-(4-Nitrobenzylidene)-4-((2-oxoindolin-3-ylidene)amino)benzohydrazide 10a
Orange powder (yield, 94%, E: Z = 5.3: 1); mp (°C) 220–222; FT-IR (KBr, cm−1): 3412 (NH), 1653 (C=O), 1636 (C=O), 1588 (C=N); 1H NMR (400 MHz, DMSO) δ 11.78 (s, 1H), 10.56–9.57 (d, J = 14.2 Hz, 1H), 8.49 (s, 1H), 8.29 (d, J = 8.8 Hz, 2H), 7.95 (d, J = 8.7 Hz, 2H), 7.71 (d, J = 8.6 Hz, 2H), 7.04–6.87 (m, 2H), 6.63 (d, J = 8.6 Hz, 2H), 5.91-5.86 (m, 2H); 13C NMR (101 MHz, DMSO) δ 164.67, 163.25, 156.87, 153.08, 147.99, 141.61, 139.11, 134.62, 130.17, 128.12, 126.69, 124.53, 122.73, 121.83, 119.46, 117.93, 113.09, 110.44; ESI-MS m/z: 414.5 [M+H]+; Anal. Calcd. for C22H15N5O4: C, 63.92; H, 3.66; N, 16.94. Found: C, 63.84; H, 3.89; N, 16.79.
N′-(4-Chlorobenzylidene)-4-((2-oxoindolin-3-ylidene)amino)benzohydrazide 10b
White powder (yield, 89%, E: Z = 8.9: 1); mp (°C) 237–239; FT-IR (KBr, cm−1): 3398 (NH), 1666 (C=O), 1646 (C=O), 1596 (C=N); 1H NMR (400 MHz, DMSO) δ 11.55 (s, 1H), 8.40 (s, 1H), 7.82–7.51 (m, 3H), 7.51 (d, J = 8.5 Hz, 2H), 6.9–7.2 (m, 4H), 6.62 (d, J = 8.6 Hz, 2H), 5.90–5.72 (m, 2H); 13C NMR (101 MHz, DMSO) δ 163.35, 152.86, 144.95, 144.76, 134.52, 134.46, 134.25, 134.16, 129.93, 129.72, 129.52, 129.36, 129.20, 129.12, 128.91, 119.87, 119.80, 119.72, 113.19, 113.08; ESI-MS m/z: 403.9 [M+H]+; Anal. Calcd. for C22H15ClN4O2: C, 65.59; H, 3.75; N, 13.91. Found: C, 65.65; H, 3.97; N, 14.11.
N′-(3-Nitrobenzylidene)-4-((2-oxoindolin-3-ylidene)amino)benzohydrazide 10c
Orange powder (yield, 93%, E: Z = 8:1); mp (°C) 200–201; FT-IR (KBr, cm−1): 3312 (NH), 1689 (C=O), 1664 (C=O), 1604 (C=N); 1H NMR (400 MHz, DMSO) δ 10.94 (s, 1H), 10.77–10.49 (m, 1H), 8.82–8.67 (m, 2H), 8.51–8.36 (m, 2H), 7.98–7.26 (m, 4H), 7.15 (t, J = 7.6 Hz, 1H), 7.14–6.73 (m, 4H); 13C NMR (101 MHz, DMSO) δ 164.72, 157.36, 150.49, 148.79, 145.73, 139.10, 135.47, 134.40, 131.29, 129.20, 126.58, 124.02, 122.93, 121.82, 117.92, 116.59, 111.48, 110.43; ESI-MS m/z: 414.3 [M+H]+; Anal. Calcd. for C22H15N5O4: C, 63.92; H, 3.66; N, 16.94. Found: C, 63.96; H, 3.71; N, 17.14.
N′-(4-Methylbenzylidene)-4-((2-oxoindolin-3-ylidene)amino)benzohydrazide 10d
Reddish orange powder (yield, 84%, E: Z = 6.7:1); mp (°C) 234–236; FT-IR (KBr, cm−1): 3358 (NH), 1661 (C=O), 1636 (C=O), 1581 (C=N); 1H NMR (400 MHz, DMSO) δ 11.03 (s, 1H), 10.71–9.56 (m, 1H), 8.67 (s, 1H), 7.99–7.87 (m, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.65–7.46 (m, 1H), 7.42–7.33 (m, 3H), 7.16 (td, J = 7.7, 1.0 Hz, 2H), 7.02–6.91 (m, 3H), 2.37 (s, 3H); 13C NMR (101 MHz, DMSO) δ 165.08, 163.26, 161.73, 145.67, 141.80, 139.11, 134.88, 131.67, 130.31, 129.98, 128.79, 127.51, 126.69, 122.73, 121.83, 117.93, 116.27, 110.44, 21.63; ESI-MS m/z: 383.7 [M+H]+; Anal. Calcd. for C23H18N4O2: C, 72.24; H, 4.74; N, 14.65. Found: C, 72.33; H, 4.81; N, 14.55.
N′-(4-Methoxybenzylidene)-4-((2-oxoindolin-3-ylidene)amino)benzohydrazide 10e
Orange powder (yield, 87%, E: Z = 5.7:1); mp (°C) 239–240; FT-IR (KBr, cm−1): 3339 (NH), 1683(C=O), 1659 (C=O), 1611 (C=N); 1H NMR (400 MHz, DMSO) δ 11.02 (s, 1H), 10.86 (s, 1H), 8.65 (d, J = 3.8 Hz, 2H), 7.98 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 8.7 Hz, 2H), 7.43–7.38 (m, 1H), 7.15 (d, J = 8.8 Hz, 2H), 7.05 (dd, J = 10.3, 6.9 Hz, 4H), 6.93 (t, J = 7.3 Hz, 1H), 3.84 (s, 3H); 13C NMR (101 MHz, DMSO) δ 165.25, 163.12, 162.15, 160.99, 151.23, 145.68, 134.90, 133.96, 131.53, 130.46, 129.37, 128.68, 127.04, 123.04, 117.13, 115.27, 114.87, 111.60, 55.86; ESI-MS m/z: 399.2 [M+H]+; Anal. Calcd. for C23H18N4O3: C, 69.34; H, 4.55; N, 14.06. Found: C, 69.39; H, 4.47; N, 14.31.
N′-(4-(Dimethylamino)benzylidene)-4-((5-fluoro-2-oxoindolin-3-ylidene)amino)benzohydrazide 10f
Reddish orange powder (yield, 91%, E: Z = 8:1); mp (°C) 202–204; FT-IR (KBr, cm−1): 3296 (NH), 1678 (C=O), 1643 (C=O), 1595 (C=N); 1H NMR (400 MHz, DMSO) δ 11.59 (s, 1H), 10.92 (m, 1H), 8.69 (s, 1H), 8.08–7.87 (m, 1H), 7.84 (d, J = 8.9 Hz, 1H), 7.44–7.30 (m, 1H), 7.30–7.18 (m, 1H), 7.18–7.04 (m, 1H), 7.07–6.93 (m, 1H), 6.95–6.77 (m, 4H), 6.81–6.68 (m, 1H), 3.07 (s, 3H), 2.92 (s, 3H); 13C NMR (101 MHz, DMSO) δ 167.11, 165.70, 163.55, 157.41, 153.86, 151.02, 141.13, 135.96 (d, J = 3.1 Hz), 132.01, 127.93, 124.92, 123.26, 120.61, 119.67, 114.67, 112.76, 112.36, 105.39, 40.45, 40.14; ESI-MS m/z: 430.6 [M+H]+; Anal. Calcd. for C24H20FN5O2: C, 67.12; H, 4.69; N, 16.31. Found: C, 67.37; H, 4.86; N, 16.52.
N′-(4-Bromobenzylidene)-4-((5-fluoro-2-oxoindolin-3-ylidene)amino)benzohydrazide 10g
Orange powder (yield, 90%, E: Z = 7.3:1); mp (°C) 227–229; FT-IR (KBr, cm−1): 3312 (NH), 1675(C=O), 1657 (C=O), 1586 (C=N); 1H NMR (400 MHz, DMSO) δ 10.94 (s, 1H), 10.75–10.57 (m, 1H), 8.69 (s, 1H), 7.95 (d, J = 8.2 Hz, 3H), 7.83 (d, J = 6.8 Hz, 3H), 7.64 (d, J = 8.1 Hz, 1H), 7.32 (t, J = 8.9 Hz, 1H), 7.22–6.71 (m, 3H); 13C NMR (101 MHz, DMSO) δ 164.91, 163.46, 161.20, 159.13, 156.77, 150.87, 142.01, 132.88, 131.26 (d, J = 1.2 Hz), 126.57, 120.93, 120.70, 117.19, 117.10, 115.95, 115.70, 113.47, 112.52; ESI-MS m/z: 467.3 [M+H]+; Anal. Calcd. for C22H14FBrN4O2: C, 56.79; H, 3.03; N, 12.04. Found: C, 56.84; H, 3.11; N, 12.27.
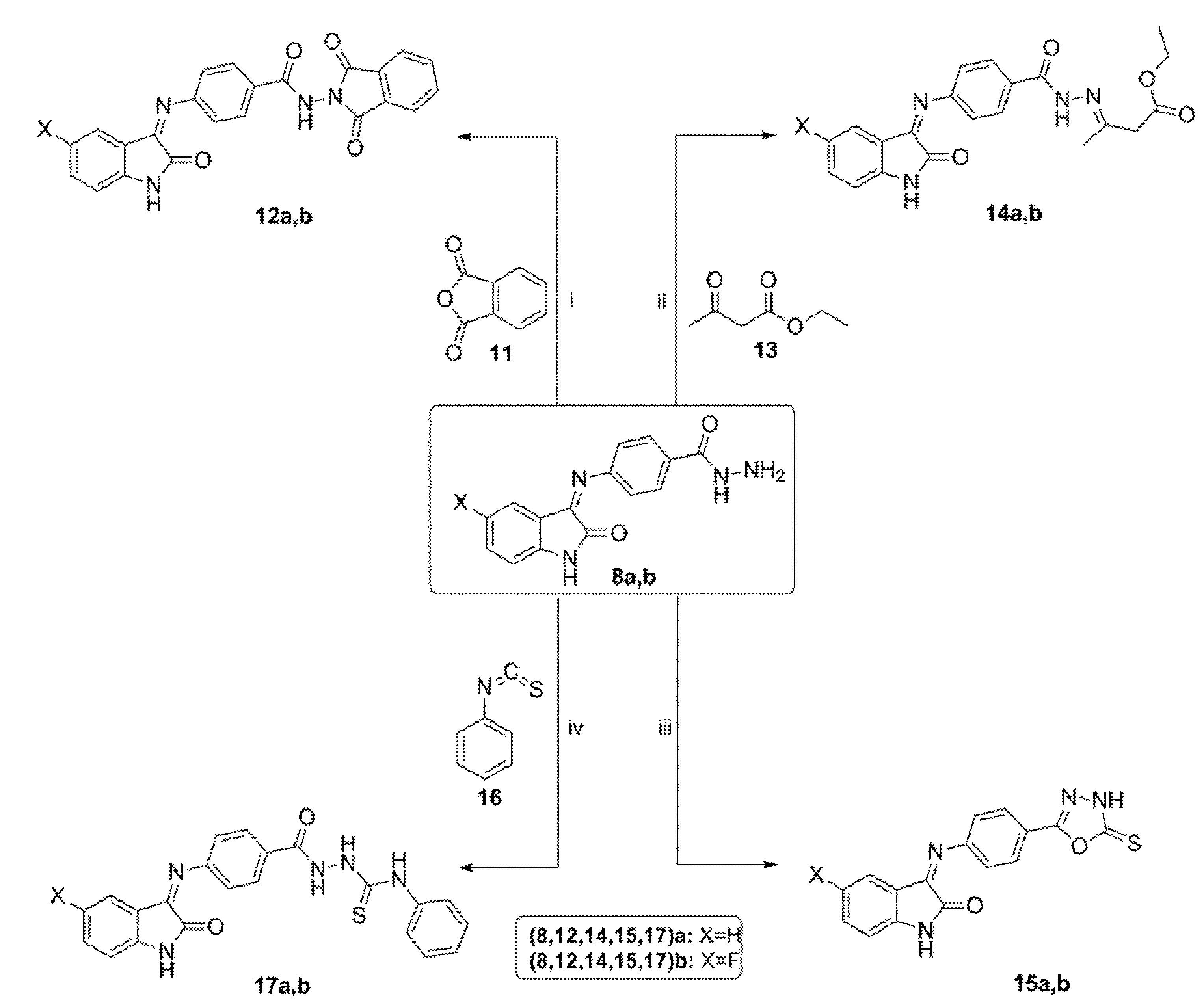
3.1.6. General Procedure for Synthesis of N-(1,3-dioxoisoindolin-2-yl)-4-((5-substituted-2-oxoindolin-3-ylidene)amino)benzamide 12a,b
A mixture of hydrazide derivatives 8a,b (2.1 mmol) and phthalic anhydride 11 (2.1 mmol) in 10 mL glacial acetic acid was stirred under sonication for 4 h at 50 °C. Next, the reaction mixture was cooled, and the reaction was quenched by addition of water (30 mL). The separated precipitate was collected and purified by flash column chromatography using (hexane/EtOAc, 4:1) as an eluent to afford cyclic imides 12a,b.
N-(1,3-Dioxoisoindolin-2-yl)-4-((2-oxoindolin-3-ylidene)amino)benzamide 12a
Orange powder (yield, 75%); mp (°C) 187–189; FT-IR (KBr, cm−1): 3365 (NH), 1696 (C=O), 1663 (C=O), 1596 (C=N); 1H NMR (400 MHz, DMSO) δ 12.84 (s, 1H), 11.25 (s, 1H), 8.43–7.85 (m, 1H), 7.86–7.61 (m, 2H), 7.70–7.46 (m, 2H), 7.47–7.25 (m, 2H), 7.25–6.77 (m, 5H); 13C NMR (101 MHz, DMSO) δ 173.14, 167.66, 162.99, 162.94, 142.74, 132.54, 131.83, 131.27, 127.49, 122.98, 121.81, 120.89, 120.31, 120.27, 117.91, 111.64, 111.54, 110.42; ESI-MS m/z: 411.2 [M+H]+; Anal. Calcd. for C23H14N4O4: C, 67.31; H, 3.44; N, 13.65. Found: C, 67.29; H, 3.70; N, 13.76.
N-(1,3-Dioxoisoindolin-2-yl)-4-((5-fluoro-2-oxoindolin-3-ylidene)amino)benzamide 12b
Orange powder (yield, 81%); mp (°C) 225–227; FT-IR (KBr, cm−1): 3342 (NH), 1689 (C=O), 1670 (C=O), 1612 (C=N); 1H NMR (400 MHz, DMSO) δ 10.73–10.65 (m, 1H), 9.81 (d, J = 14.8 Hz, 1H), 8.06–7.53 (m, 2H), 7.51–7.26 (m, 1H), 7.30–7.07 (m, 2H), 7.13–6.86 (m, 3H), 6.89–6.38 (m, 3H); 13C NMR (101 MHz, DMSO) δ 167.72, 163.47, 159.71, 157.37, 139.11, 135.23 (d, J = 1.4 Hz), 126.19, 126.16, 124.15, 124.06, 113.71, 113.47, 112.90, 111.30, 111.22, 105.08, 105.01, 104.75; ESI-MS m/z: 429.5 [M+H]+; Anal. Calcd. for C23H13FN4O4: C, 64.49; H, 3.06; N, 13.08. Found: C, 64.21; H, 2.89; N, 13.22.
3.1.7. General Procedure for Synthesis of Ethyl 3-(2-(4-((5-Substituted-2-oxoindolin-3-ylidene)amino)benzoyl)hydrazono)butanoate 14a,b
Ethyl acetoacetate 13 (2 mmol) was added to a solution of hydrazides 8a,b (2 mmol) in absolute ethanol (10 mL). After the addition of 0.2 mL of glacial acetic acid, the reaction mixture was heated under reflux for 4 h. After cooling, the formed precipitate was collected, and the crude solid was recrystallized from methanol to afford hydrazones 14a,b.
Ethyl 3-(2-(4-((2-oxoindolin-3-ylidene)amino)benzoyl)hydrazono)butanoate 14a
Yellow powder (yield, 86%); mp (°C) 206–208; FT-IR (KBr, cm−1) 3321 (NH), 1685(C=O), 1673 (C=O), 1657 (C=O), 1614 (C=N); 1H NMR (400 MHz, DMSO) δ 14.19 (s, 1H), 11.03 (s, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.43–7.24 (m, 2H), 7.16 (t, J = 7.6 Hz, 1H), 7.12–6.96 (m, 2H), 6.95–6.81 (m, 2H), 5.07 (s, 2H), 4.13 (q, J = 6.8 Hz, 2H), 2.26 (s, 3H), 1.23 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 167.61, 163.25, 162.26, 156.36, 141.77, 139.11, 132.36, 130.51, 127.50, 122.41, 121.10, 120.09, 117.92, 111.10, 110.43, 91.95, 59.49, 18.50, 14.85; ESI-MS m/z: 393.3 [M+H]+; Anal. Calcd. for C21H20N4O4: C, 64.28; H, 5.14; N, 14.28. Found: C, 63.96; H, 4.95; N, 13.95.
Ethyl 3-(2-(4-((5-Fluoro-2-oxoindolin-3-ylidene)amino)benzoyl)hydrazono)butanoate 14b
Yellow powder (yield, 88%); mp (°C) 213–215; FT-IR (KBr, cm−1): 3354 (NH), 1698(C=O), 1675 (C=O), 1648 (C=O), 1598 (C=N); 1H NMR (400 MHz, DMSO) δ 14.20 (s, 1H), 11.04 (s, 1H), 7.37–7.13 (m, 2H), 7.16–7.13 (m, 2H), 7.11–6.73 (m, 3H), 5.11 (s, 2H), 4.13 (q, J = 7.1 Hz, 2H), 2.25 (s, 3H), 1.23 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 167.56, 162.41, 159.77, 157.41, 156.20, 137.92, 131.83, 131.80 (d, J = 3.3 Hz), 122.43, 122.34, 116.80, 116.56, 112.02, 107.16, 106.91, 92.77, 59.60, 18.38, 14.82; ESI-MS m/z: 411.2 [M+H]+; Anal. Calcd. for C21H19FN4O4: C, 61.46; H, 4.67; N, 13.65. Found: C, 61.75; H, 4.88; N, 13.82.
3.1.8. General Procedure for Synthesis of 5-Substituted-3-((4-(5-thioxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)imino)indolin-2-one 15a,b
Potassium hydroxide (2 mmol) was added to a solution of hydrazides 8a,b (2 mmol) and carbon disulfide (4 mmol) in 20 mL of absolute ethanol. The reaction mixture was refluxed for 12 h. Next, the reaction mixture was cooled, and the solvent was evaporated. The residue was dissolved in water and acidified with 10% HCl. The formed precipitate was collected and recrystallized from ethanol to generate the corresponding oxadiazole derivatives 15a,b.
3-((4-(5-Thioxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)imino)indolin-2-one 15a
Yellow powder (yield, 86%); mp (°C) 219–220; FT-IR (KBr, cm−1): 3252 (NH), 1679(C=O), 1612 (C=N); 1H NMR (400 MHz, DMSO) δ 10.71 (s, 1H), 10.54 (s, 1H), 7.37 (d, J = 7.5 Hz, 2H), 7.30–7.10 (m, 2H), 7.08–6.77 (m, 4H); 13C NMR (101 MHz, DMSO) δ 186.12, 163.25, 160.80, 139.11, 127.52, 127.36, 126.67, 122.88, 122.73, 121.94, 121.84, 118.09, 117.93, 110.44; ESI-MS m/z: 323.5 [M+H]+; Anal. Calcd. for C16H10N4O2S: C, 59.62; H, 3.13; N, 17.38. Found: C, 59.68; H, 3.28; N, 17.51.
5-Fluoro-3-((4-(5-thioxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)imino)indolin-2-one 15b
Yellow powder (yield, 82%); mp (°C) 231–233; FT-IR (KBr, cm−1): 3263 (NH), 1668 (C=O), 1607 (C=N); 1H NMR (400 MHz, DMSO) δ 10.67 (d, J = 15.0 Hz, 2H), 7.17–7.14 (m, 1H), 7.08–6.91 (m, 3H), 6.91–6.73 (m, 3H); 13C NMR (101 MHz, DMSO) δ 186.84, 159.70, 157.36, 135.20 (d, J = 1.2 Hz), 135.19, 126.19, 126.16, 124.06, 113.69, 113.46, 111.29, 111.20, 105.01, 104.76; ESI-MS m/z: 341.3 [M+H]+; Anal. Calcd. for C16H9FN4O2S: C, 56.47; H, 2.67; N, 16.46. Found: C, 56.51; H, 2.70; N, 16.53.
3.1.9. General Procedures for Synthesis of 2-(4-((5-Substituted-2-oxoindolin-3-ylidene)amino)benzoyl)-N-substitutedhydrazinecarbothioamide 17a,b
Phenyl isothiocyanate 16 (1.9 mmol) was added to a solution of hydrazides 8a,b (1.9 mmol) in absolute ethanol (10 mL). The reaction mixture was refluxed for 8 h. After cooling, the precipitated solid was collected and purified by flash column chromatography using (hexanes/EtOAc, 3:2) as an eluent to yield N-phenylthiosemicarbazides 17a,b.
2-(4-((2-Oxoindolin-3-ylidene)amino)benzoyl)-N-phenylhydrazinecarbothioamide 17a
Yellow powder (yield, 93%); mp (°C) 211–213; FT-IR (KBr, cm−1): 3342 (NH), 1686 (C=O), 1654 (C=O), 1602 (C=N); 1H NMR (400 MHz, DMSO) δ 10.72 (s, 1H), 10.64–10.48 (m, 1H), 10.05 (d, J = 17.9 Hz), 9.57 (d, J = 13.4 Hz, 1H), 7.95 (d, J = 7.5 Hz, 1H), 7.89–7.60 (m, 2H), 7.56–7.30 (m, 6H), 7.17 (t, J = 7.3 Hz, 2H), 6.99 (t, J = 7.4 Hz, 1H), 6.88 (d, J = 7.6 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 179.93, 163.27, 139.68, 139.12, 129.03, 128.92, 128.45, 127.54, 126.71, 126.17, 125.18, 124.92, 124.17, 122.74, 122.50, 121.86, 117.95, 110.46; ESI-MS m/z: 415.6 [M+H]+; Anal. Calcd. for C22H17N5O2S: C, 63.60; H, 4.12; N, 16.86. Found: C, 63.52; H, 4.29; N, 16.63.
2-(4-((5-Fluoro-2-oxoindolin-3-ylidene)amino)benzoyl)-N-phenylhydrazinecarbothioamide 17b
Orange powder (yield, 92%); mp (°C) 203–205; FT-IR (KBr, cm−1): 3339 (NH), 1694 (C=O), 1663 (C=O), 1593 (C=N); 1H NMR (400 MHz, DMSO) δ 12.70 (s, 1H), 11.14 (s, 1H), 10.68 (d, J = 15.0 Hz, 1H), 9.82 (d, J = 14.9 Hz, 1H), 7.67–7.61 (m, 2H), 7.45 (t, J = 7.7 Hz, 1H), 7.38–7.29 (m, 3H), 7.25–7.13 (m, 3H), 7.02–6.92 (m, 2H), 6.87–6.81 (m, 1H); 13C NMR (101 MHz, DMSO) δ 176.78, 163.48, 159.71, 157.37, 139.23, 138.78, 135.21 (d, J = 1.2 Hz), 128.94, 126.72, 126.20, 126.16, 124.06, 113.70, 113.46, 111.29, 111.21, 105.01, 104.76; ESI-MS m/z: 435.3 [M+H]+; Anal. Calcd. for C22H16FN5O2S: C, 60.96; H, 3.72; N, 16.16. Found: C, 60.69; H, 3.56; N, 16.33.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}