Abstract
The antiproliferative effects played by benzothiazoles in different cancers have aroused the interest for these molecules as promising antitumor agents. In this work, a library of phenylacetamide derivatives containing the benzothiazole nucleus was synthesized and compounds were tested for their antiproliferative activity in paraganglioma and pancreatic cancer cell lines. The novel synthesized compounds induced a marked viability reduction at low micromolar concentrations both in paraganglioma and pancreatic cancer cells. Derivative 4l showed a greater antiproliferative effect and higher selectivity index against cancer cells, as compared to other compounds. Notably, combinations of derivative 4l with gemcitabine at low concentrations induced enhanced and synergistic effects on pancreatic cancer cell viability, thus supporting the relevance of compound 4l in the perspective of clinical translation. A target prediction analysis was also carried out on 4l by using multiple computational tools, identifying cannabinoid receptors and sentrin-specific proteases as putative targets contributing to the observed antiproliferative activity.
1. Introduction
Heterocycles represent precious scaffolds in natural and synthetic molecules, endowed with a great variety of biological activities. Benzothiazoles are members of the bicyclic heteroaromatic family, and they are widely used in medicinal chemistry as the scaffolds of several drugs, including antimicrobial, anti-inflammatory, anticonvulsant, neuroprotective and many others [1,2]. The easy functionalization of the aromatic ring of benzothiazole and the 2-amino or 2-mercapto substituents, which are frequently used as building blocks, makes them attractive and reactive components that are useful in organic and medicinal chemistry programs [3,4]. The development of benzothiazole-based drugs has led to a number of derivatives currently marketed to treat different pathologies, such as the neuroprotective agent riluzole, the diuretic ethoxzolamide, the antidiabetic zopolrestat, the immunosuppressant frentizole, or the diagnostic tool thioflavin T (Figure 1A).
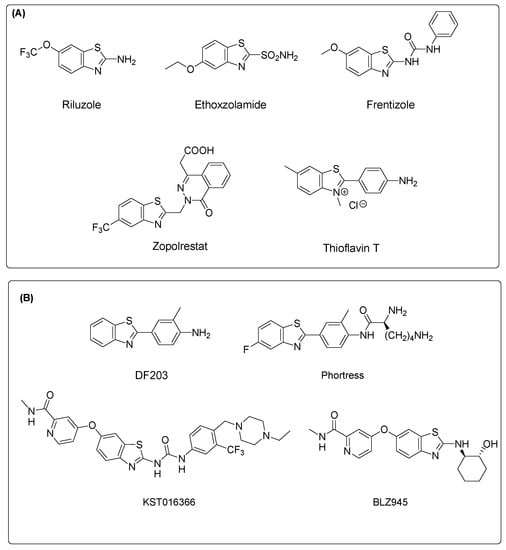
Figure 1.
(A) Selected marketed drugs containing the benzothiazole nucleus; (B) Chemical structures of some benzothiazole derivatives endowed with strong antiproliferative effects in cancer cell models.
The potent antiproliferative effects exerted by benzothiazoles in different cancer models has elicited interest in these molecules as promising antitumor agents (Figure 1B) [5,6]. Considering that 2-phenylbenzothiazoles have been recognized as highly potent cytotoxic compounds, several derivatives were identified and tested in different cancer cell lines. In particular, the chemical manipulation of the lead compound DF203 [7], a 2-phenylbenzothiazole derivative, led to the development of the clinic candidate prodrug Phortress [8]. More recently, the derivative BLZ945, acting as a CSF-1R kinase inhibitor [9,10], has been identified, and it is currently under evaluation as single agent and in combination with spartalizumab for the treatment of advanced solid tumors in adults (ClinicalTrials.gov, NCT02829723) [11]. The 2-ureidobenzothiazole derivative KST016366 has been reported as a potent multikinase inhibitor, displaying a broad-spectrum antiproliferative activity against a wide panel of cancer cell lines [12].
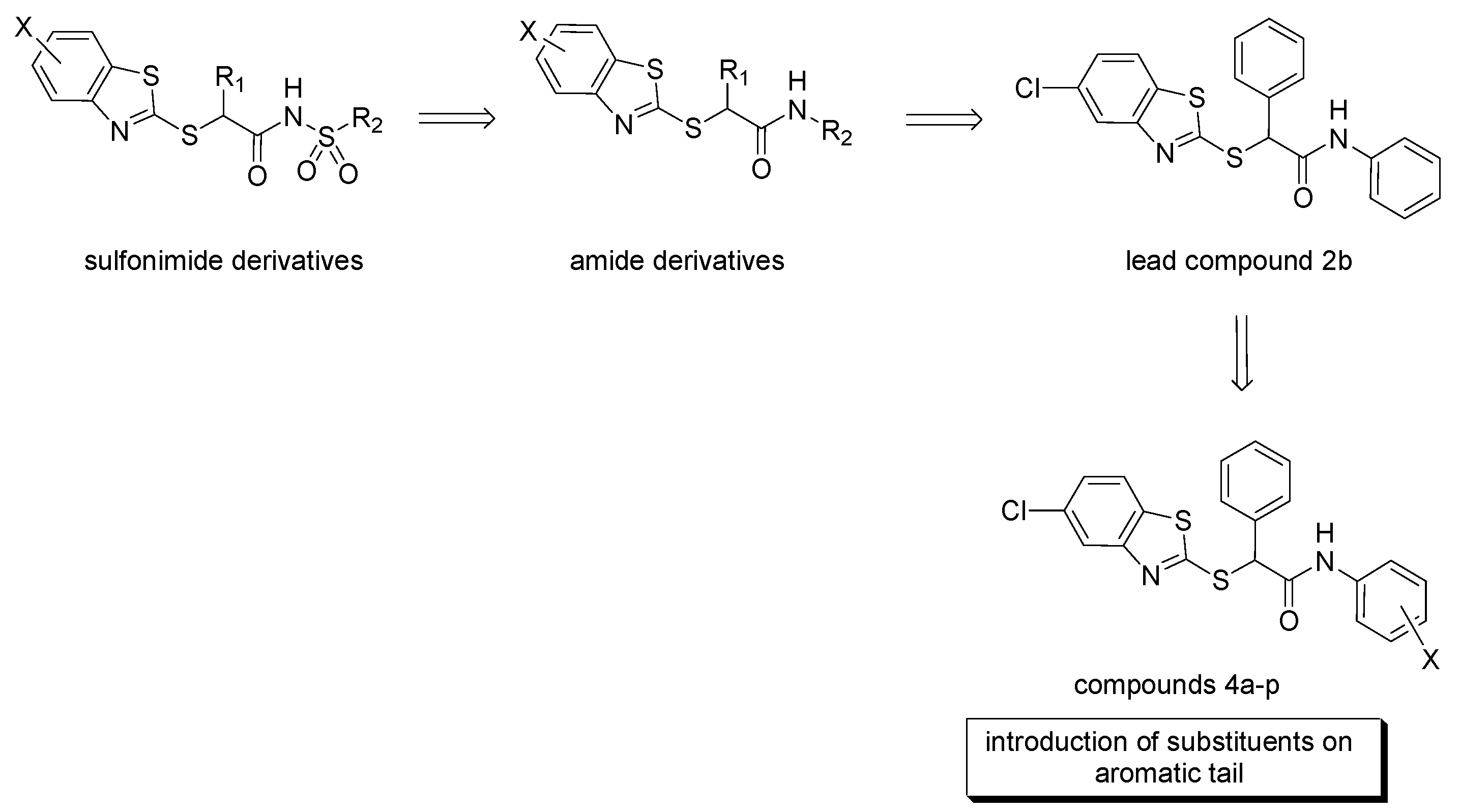
In previous studies, our research group synthesized 2-mercaptobenzothiazole derivatives as Peroxisome Proliferator-Activated Receptor Alpha (PPARα) antagonists [13] (Figure 2). These compounds were able to antagonize PPARα at low micromolar concentrations, and displayed also interesting antiproliferative effects when tested in paraganglioma, glioblastoma, colorectal and pancreatic cancer cell models [14,15]. Interestingly, the benzothiazole derivative 2b showed a marked cytotoxic effect, mainly in paraganglioma cells, with a dose-dependent inhibition profile and a potency comparable to that of the commercially available PPARα antagonist GW6471 [16,17]. However, it cannot be excluded that other mechanisms of action, or molecular targets, in addition to PPARα inhibition, may emerge for explaining the antiproliferative activity of this compound.
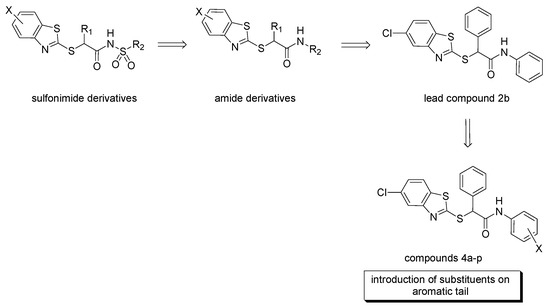
Figure 2.
Cytotoxic 2-mercaptobenzothiazole derivatives containing sulfonimide or amide groups, and novel derivatives 4a–p synthesized in this study.
To further explore the potential of this class of molecules as antiproliferative agents, we synthetized novel derivatives of the lead compound 2b by keeping unaltered the benzothiazole scaffold and the amide functional group, and by introducing substituents on the distal aromatic ring. In particular, substituents with different electronic properties and steric hindrance were selected to obtain a series of para-substituted (4a–f), meta-substituted (4g–l) and disubstituted analogs that bear electron-withdrawing substituents (4m–p). These modifications were performed to test how this molecular portion could modulate the cytotoxic activity in different cancer cell lines.
In this regard, the synthesized compounds 4a–p were tested for their antiproliferative activity in three distinct pancreatic cancer cell lines (AsPC-1, Capan-2, BxPC-3) and two paraganglioma cell lines (PTJ64i, PTJ86i) that have been established by Prof. Cama’s research group at the University of Chieti [17]. Compounds of the series that consistently affected cell viability across the tested cancer cell lines more potently than the lead compound 2b were further analyzed against normal HFF-1 fibroblast cells, to evaluate their toxicity. In the perspective of clinical translation, we also tested whether the most potent and less toxic derivatives could be usefully combined with already approved drugs by analyzing the effects of combined treatments on cancer and normal cell viability.
A target prediction study was also performed to shed light on the putative mechanisms of action contributing to the antiproliferative effects of novel compounds.
2. Results and Discussion
2.1. Chemistry
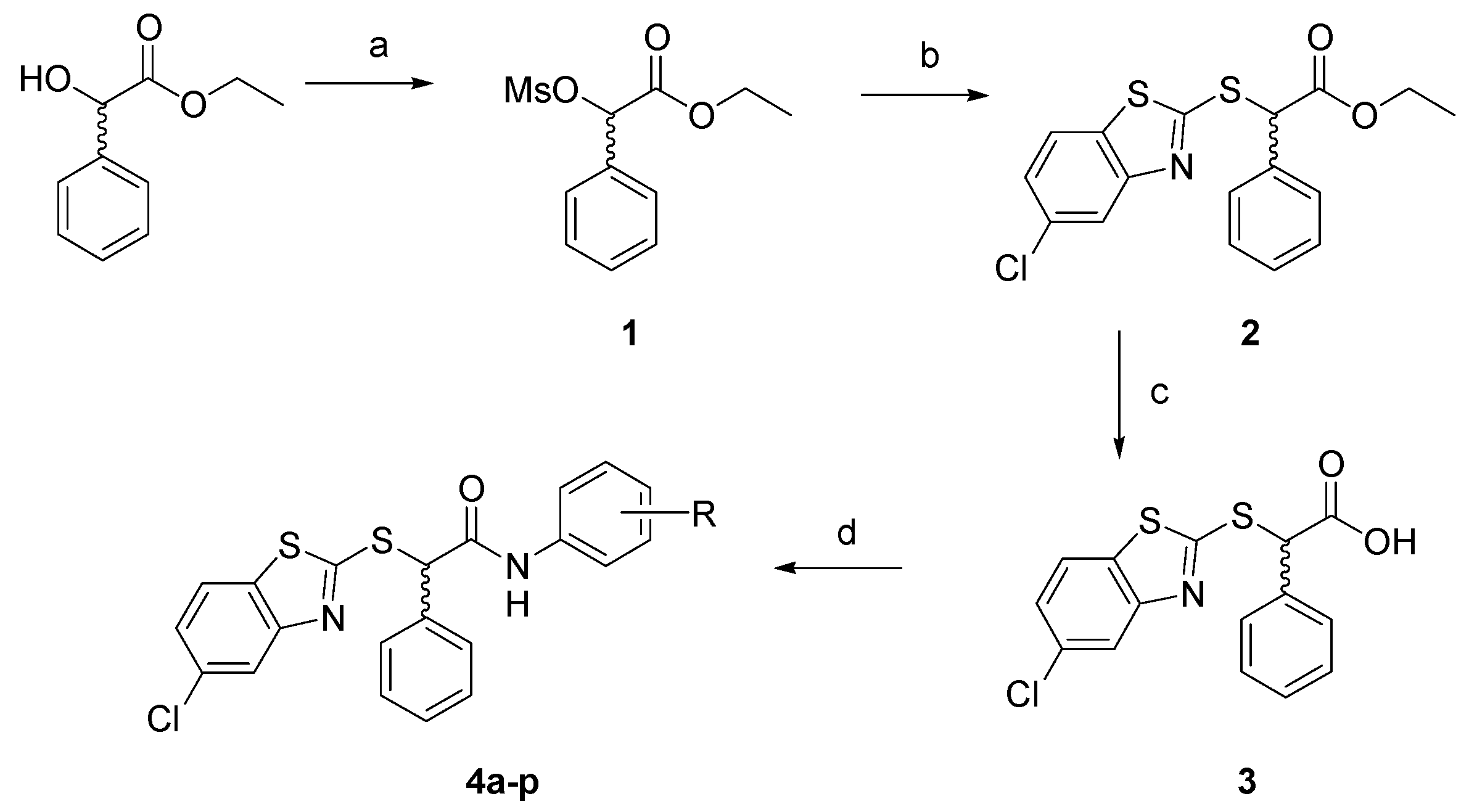
The synthesis of the planned compounds 4a–p was carried out as depicted in Scheme 1. Briefly, the commercially available ethyl mandelate was treated with mesyl chloride and triethylamine in THF at 0 °C. The resulting mesylate 1 was reacted with 5-chloro-2-mercaptobenzothiazole and triethylamine in THF at 50 °C for 24 h; the ester 2 was hydrolyzed in a basic medium to afford acid 3. The direct coupling of 3 with the proper amines HOBt, DCC, and N-methylmorpholine in DMF led to the amides 4a–p. Crude products were purified by column chromatography or crystallization, obtaining desired amides in good purity and discrete yields.
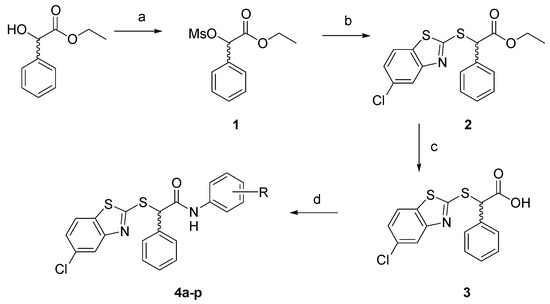
Scheme 1.
Synthetic route to final compounds 4a–p. Reagents and conditions: (a) mesyl chloride, TEA, THF, 0 °C-r.t.; (b) 5-chloro-2-mercaptobenzothiazole, TEA, THF, 0°-r.t.−50 °C; (c) NaOH 2N, THF, r.t.; (d) substituted aniline, DCC, HOBt, NMM, DMF, 0 °C-r.t.
Final compounds, including para-substituted 4a–f, meta-substituted 4g–l and a group of m–p disubstituted analogues 4m–p bearing electron-withdrawing substituents, are reported in Table 1. For each compound, the method of purification, the yield, and the melting point are also specified.

Table 1.
Final compounds 4a–p synthesized in this study.
2.2. Antiproliferative Activity
We analyzed by MTT assays the effects of synthesized compounds 4a–p on the viability of three pancreatic cancer cell lines (AsPC-1, Capan-2, BxPC-3) and two paraganglioma cell models (PTJ64i, PTJ86i), based on relevant antiproliferative effects previously shown by the lead compound 2b in the same cells [16].
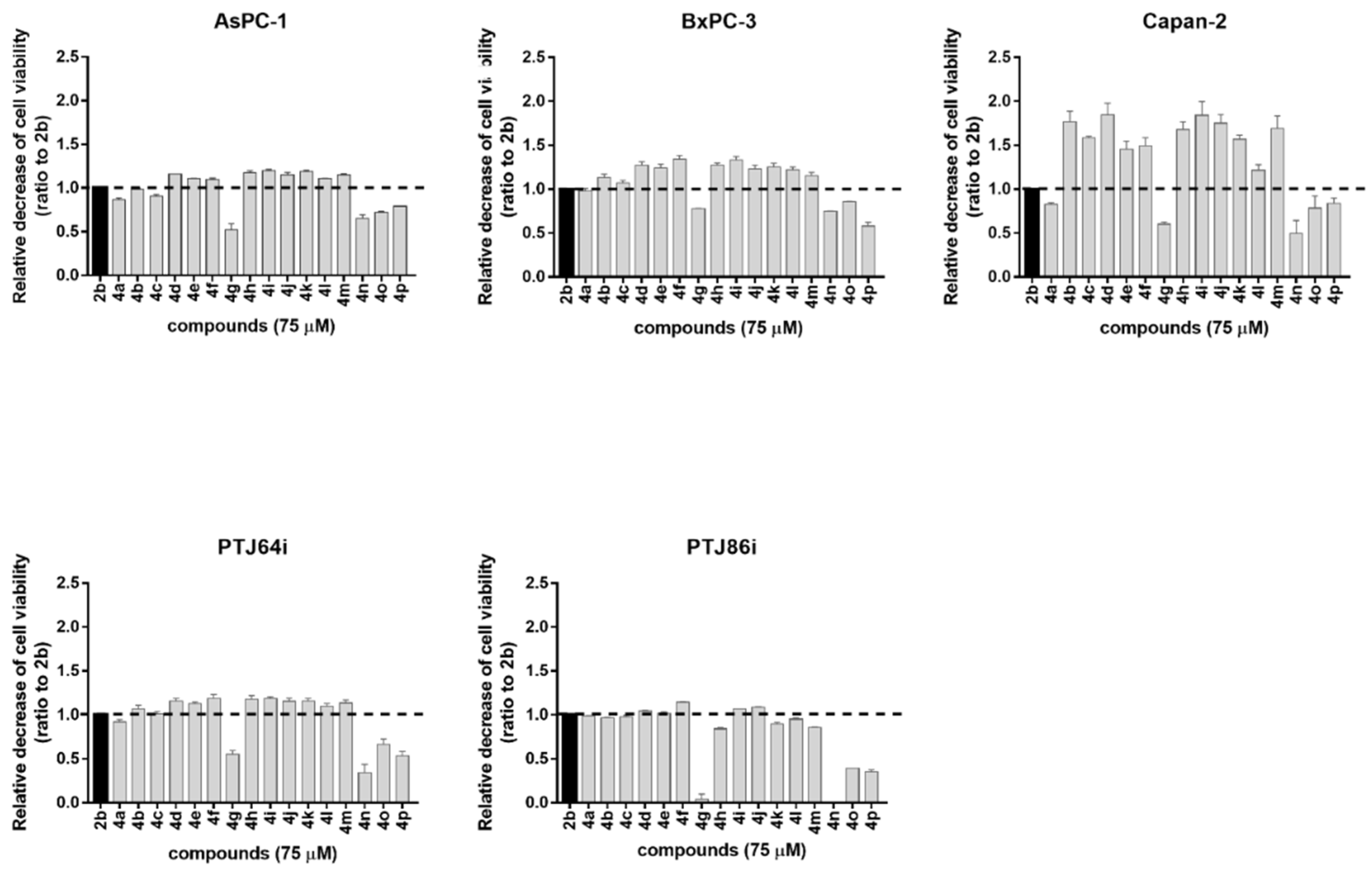
Compounds 4a–p were submitted to a preliminary MTT assay at a one-point screening concentration of 75 μM for 72 h, including 2b as a reference compound (Figure 3). Overall, the majority of the tested compounds affected cancer cell viability, with a potency comparable, or superior to, 2b with the exception of 4g (m-methoxy) and the 2-bromo-substituted derivatives 4n (2-Br, 5-NO2), 4o (2-Br, 4-CF3), and 4p (2-Br, 5-CF3). The presence of the electron-donating methoxy group decreased the activity of 2b, in both the para and meta positions. Conversely, the introduction of electron-withdrawing substituents, such as halogens, nitro, trifluoromethyl and acetylamino groups, improved the antiproliferative activity of the lead compound 2b, and this effect was observed for meta and para positions, including the m,p-dichloro derivative 4m. This general trend was observed for 4a–p in all the selected cancer cell lines. Starting from these data, we selected nine compounds displaying a greater antiproliferative activity than 2b to perform concentration–response curves. Specifically, these compounds, namely 4d, 4e, 4f, 4h, 4i, 4j, 4k, 4l, and 4m, were tested at concentrations of 3, 6, 12 and 24 µM for 72 h. In addition, the nine compounds, together with 2b, were tested against human fibroblasts HFF-1, in order to evaluate their selectivity against tumor cells, as compared to normal cells. Effects on cancer or normal cell viability were extrapolated from concentration–response curves and are expressed as IC50 values in Table 2.
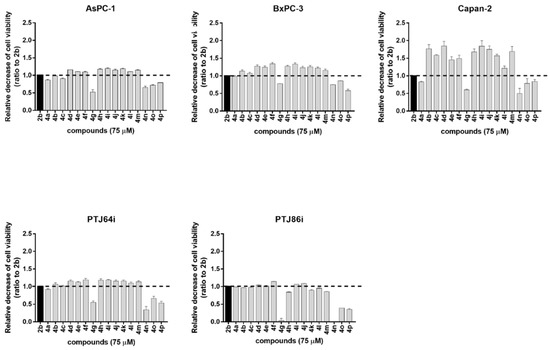
Figure 3.
Screening of the effects of novel derivatives (4a–p) on the viability of pancreatic (AsPC-1, BxPC-3, and Capan-2) and paraganglioma (PTJ64i and PTJ86i) cancer cell lines. The lead compound 2b was included as a reference and the histograms show the relative decrease of cancer cell viability observed after treatments, as compared to 2b. Cell viability was assessed by an MTT assay using compounds at 75 µM for 72 h. Data shown are the means ± SD of duplicate experiments with quintuplicate determinations and are calculated as ratios relative to the reference compound 2b (dashed line).
Table 2.
IC50 values of 2b and its nine most active derivatives on cancer and normal cell lines.
Notably, the compounds 4k and 4l displayed the greatest and most consistent selectivity index (SI) values across the tested cancer cell lines (Table 3 and Table S1), which supported their potential as effective and safe anticancer agents in pancreatic cancer and paraganglioma treatment.
Table 3.
Selectivity index values for compounds 4k and 4l.
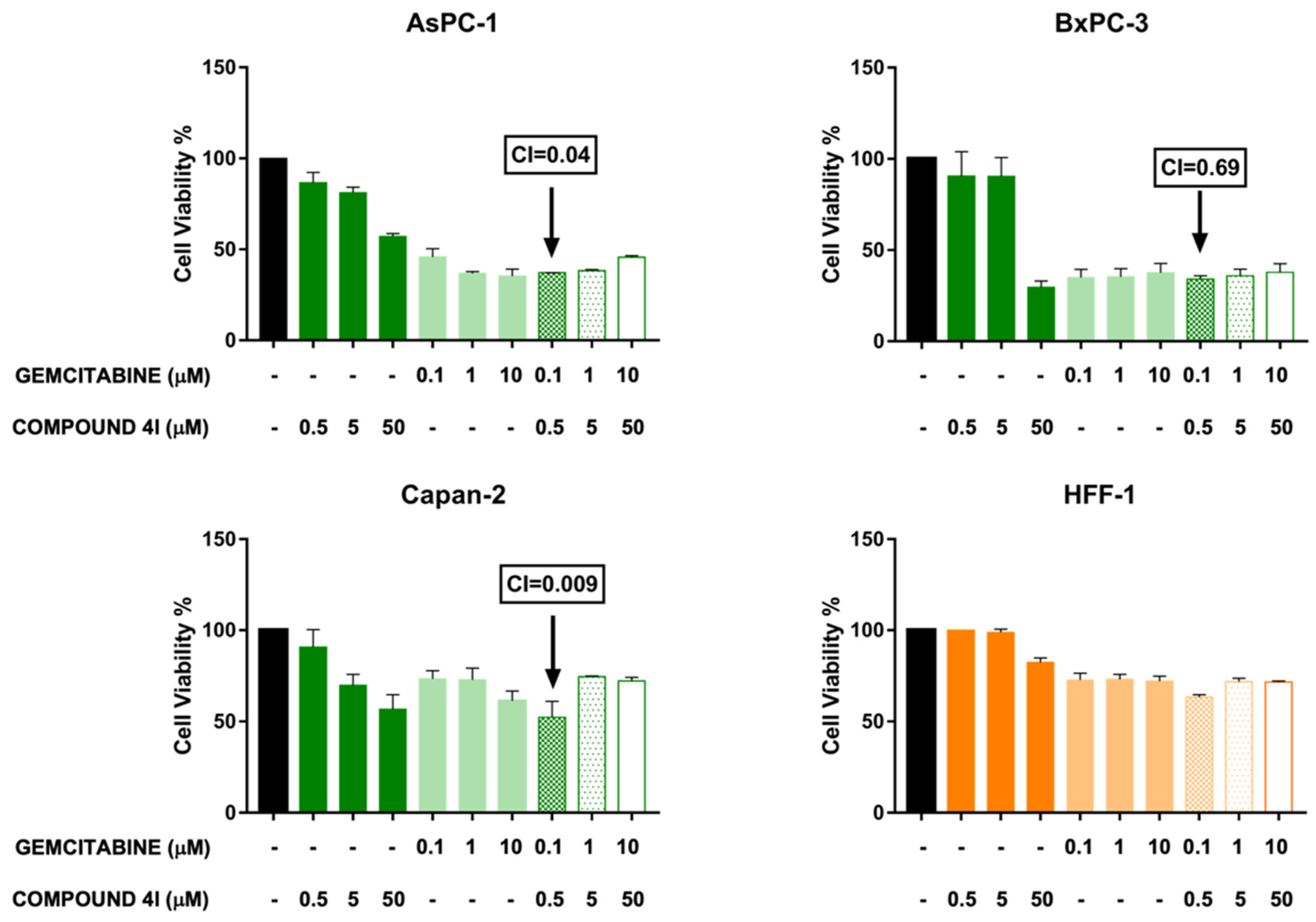
Considering that gemcitabine is one of the first-line therapies in pancreatic cancer, to deepen the potential clinical relevance of compounds 4k and 4l we further analyzed the effects of the combinations between each of them and gemcitabine on pancreatic cancer cell line viability (Figure 4 and Figure S1, and Table 4).
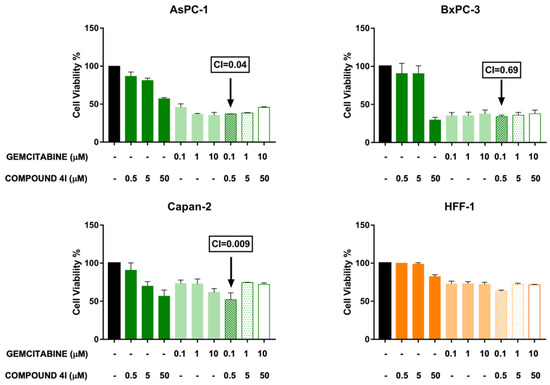
Figure 4.
Effect of combined treatments with compound 4l and gemcitabine on the viability of pancreatic cancer and normal fibroblast cells. Cell viability was assessed by MTT assays after a 72 h incubation of cells with 4l and gemcitabine at the indicated concentrations, as single agents or in combination. Histograms represent the means ± SD of two independent experiments with quintuplicate determinations. Combination indexes (CIs) were calculated by CompuSyn software. Combinations assessed as synergistic by CIs < 1 are indicated.
Table 4.
Percentage of cell viability inhibition after compound 4l and gemcitabine treatments in PC and normal fibroblast cell lines.
Remarkably, the combination between the lowest concentrations of gemcitabine and 4l (0.1 and 0.5 µM, respectively) decreased pancreatic cancer cell viability in a more marked and synergistic manner (CIs < 1) across the three pancreatic cancer cell lines, as compared to combinations with higher concentrations of the two agents (Figure 4 and Table 4). Notably, the combination with the lowest concentrations of the two compounds appeared safer in normal fibroblast HFF-1 cells, as compared with the pancreatic cancer cell lines (inhibition rate 37% in HFF-1, as compared with 63% in AsPC-1, 66% in BxPC-3 and 48% in Capan-2), supporting the relevance of compound 4l in the perspective of clinical translation (Table 4).
2.3. Target Prediction Studies
In principle, there could be multiple putative molecular targets for explaining the cytotoxic activity of derivatives 4a–p, and their identification may contribute to a more comprehensive understanding of their bioactivity. Since the high-throughput in vivo target profiling of compounds to identify a potential binding protein for a specific molecule could be expensive and time-consuming, an in silico target fishing study was applied to identify novel proteins possibly involved in the network of molecular events underlying the cytotoxic activity against the cancer cell lines. In silico target fishing (also known as target prediction or target identification) is emerging as an efficient alternative to predict the macromolecular target speedily [18]. A primary distinction divides these methods into ligand-based, i.e., comparing the characteristics (fingerprints) of the query molecule with those present in the databases, and structure-based, i.e., comparing the putative ligand with the features of the target protein’s active site [19]. Various tools, freely accessible or not, have been designed to this aim.
In the present work, all query molecules were analyzed using different web-based tools including ligand-based methods such as SEA SEARCH, PLATO, PPB2, and SuperPred, and structure-based tools such as PharmMapper (Table 5). The resulting targets were filtered, selecting only those expressed in the human organism. Furthermore, as most tools retrieve many possible targets, only the highest-ranked (more reliable) were selected and analyzed. Results for the selected derivative 4l are shown in Table 6, and the prediction results for other analogue compounds are very similar to those presented.
Table 5.
Description, database, and target ranking criterion of the used web tools. The URL of each tool is indicated. All sites were accessed in 1 March 2022.
Table 6.
Predicted targets for compound 4l.
By analyzing the obtained results, to reduce the risk of false-positive prediction [30] we focused on the target protein classes more frequently retrieved by multiple tools, namely cannabinoid receptors (CBR1, CBR2, and G-protein coupled receptor 55—GPCR55—an orphan GPCR binding cannabinoid), which were predicted by four over six programs, as well as sentrin-specific proteases (SENP6, SENP7, SENP8), which were suggested by three over six programs (Table 6). To further assess the significance of our prediction, the potential role of these two targets in pancreatic cancer was explored by a literature research.
Cannabinoid receptors belong to the Class A GPCR family and regulate several functions, such as neurotransmission, immune and inflammatory responses [31]. It is worth noting that the cannabinoid system is a well-known player in cancer biology [32]. The overexpression of both CB1 and CB2 receptors on pancreatic cancer cells, as compared to a very limited expression in healthy pancreatic cells, was revealed by a study in patients [33]. In addition, antagonists to these receptors seem to be extremely promising as antitumor agents, since they are selective in interacting with pancreatic tumor cells, as compared to healthy cells [34].
Sentrin-specific proteases, also known as SUMO-specific proteases, are cysteine proteases responsible for the DeSUMOylation of target proteins, an essential post-translational modification process [35]. To date, seven SENP isoforms have been identified, showing different localization and substrate preference [36]. Considering that SENPs regulate proteins involved in DNA repair, cell cycle, and neovascularization, the deregulation (overexpression or downregulation) of SENP activity results in cellular dysfunction associated with the development of different diseases, including prostate, thyroid, colon, lung, and pancreatic cancer [37]. For instance, the upregulation of SENP3 is a prognostic marker of pancreatic cancer according to the Cancer Genome Atlas (TCGA) [38]. For these reasons, in recent years SENP proteases have emerged as potential targets for cancer therapy [39]; the high sequence homology of members of human SENPs, along with the differences in substrate specificity and subcellular localization might be an interesting way to discover selective SENP inhibitors [40].
2.4. Docking Studies
To further validate our predictions, docking calculations were carried out on two representative target proteins. Ligands were flexibly docked in the binding site of the Cannabinoid CB1 receptor (CBR1) using the crystallographic structure of CBR1 retrieved by the Protein Data Bank (PDB: 6KPG) [41].
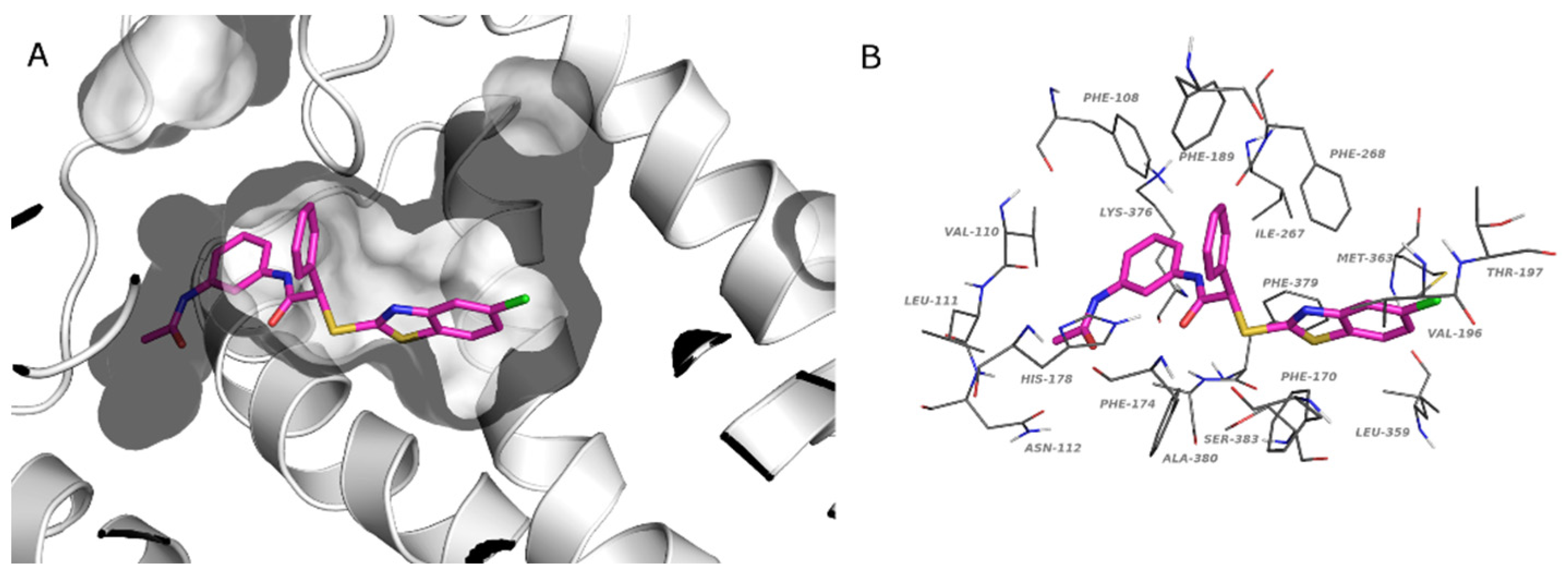
Docked poses in the CBR1 binding site are pretty well conserved among studied compounds: Figure 5a reports the binding mode of compound 4l highlighting the optimal fitting of the ligand in the CB1 binding pocket. The reported ligand establishes interactions with Phe108, Val110, Leu111, Asn112, Phe170, Phe174, His178, Phe189, Val196, Thr197, Ile267, Phe268, Leu359, Met363, Lys376, Phe379, Ala380, and Ser383 (Figure 5b). These contacts are common to the other ligands as demonstrated by the interaction diagram reported in the Supplementary Materials (Supplementary Figure S2).
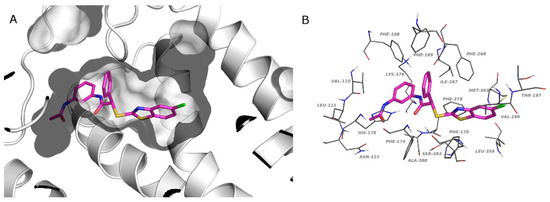
Figure 5.
Predicted binding pose of 4l in the catalytic site of CBR1 (PDB:6KPG). Panel (A) CBR1: gray cartoon, binding cavity: gray surface. Panel (B) Residues that interact with 4l: gray lines. (A,B) Compound 4l, magenta stick. (O, red; N, blue; S, yellow; Cl, green).
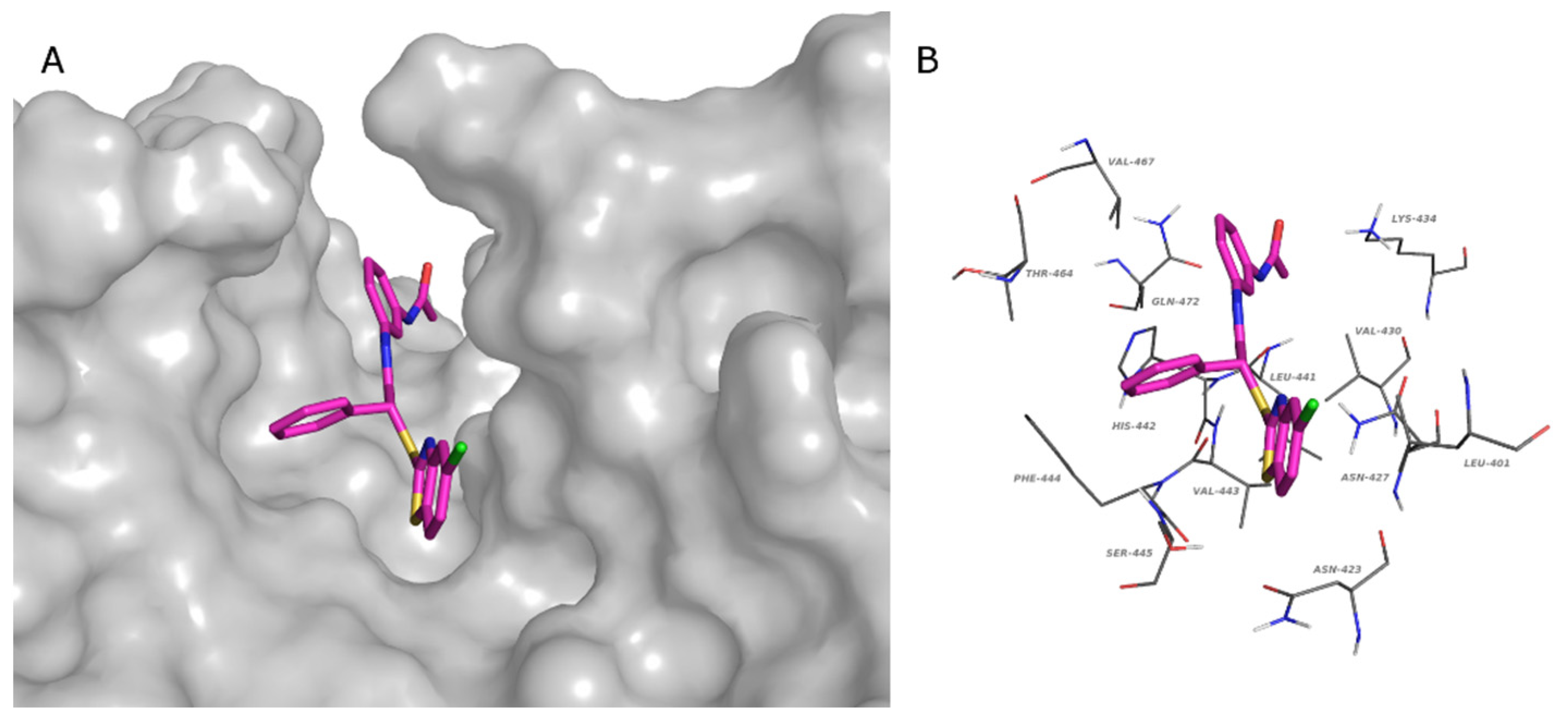
Since the crystallographic structure of SENP6 and SENP7 is not available, the chimeric form of SENP2 containing the SENP6 characteristic loop 1 (similar to SENP7) was used for the structure-based studies (PDB: 3ZO5) [42]. To explore the possible binding sites of the protein, the Sitemap tool was used. Among the identified sites, the one that was superimposable to the most studied SENPs was chosen for the grid generation. This binding site possesses a characteristic form that is able to host all studied compounds. The tridentate structure of the compounds fits well with the structure of the binding site. In Figure 6a, the binding mode of 4l is shown by the surface representation of the protein, while in Figure 6b, the residues that make contacts with the ligand are reported. In detail, these residues are Leu401, Asn 423, Asn427, Val430, Lys434, Leu441, His442, Val443, Phe444, Ser445, Thr464, Val467, and Gln472. The interaction diagram of all compounds and residues is reported in Supplementary Figure S3.
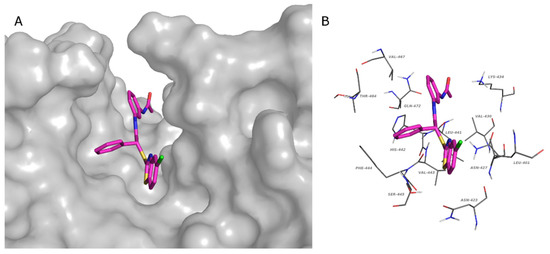
Figure 6.
Predicted binding pose of 4l in the catalytic site of the chimera SENP2-loop1 of SENP6 (PDB:3ZO5). (A) Chimera SENP2: gray surface. (B) Residues that interact with 4l: gray lines. (A,B) Compound 4l, magenta stick. (O, red; N, blue; S, yellow; Cl, green).
Although the identified protein classes seem to be promising targets of our benzothiazole derivatives, further in vitro and in vivo studies will be necessary to validate our prediction.
2.5. Physicochemical and Pharmacokinetic Properties Calculation
QikProp [43] calculations were carried out to predict the parameters affecting the drug-likeness and bioavailability of the studied benzothiazoles (Table 7). The calculated physicochemical and pharmacokinetic properties denote that the studied compounds have a drug-like profile, presenting at most one violation of the rule of five due to their lipophilicity (logPoct/water > 5). The predicted low solubility and possible HERG inhibition represent limiting aspects that need further optimization. On the other hand, our compounds present excellent oral absorption and remarkable cell permeability. Moreover, some of the analyzed compounds show good CNS activity and a promising brain/blood partition coefficient.
Table 7.
Physicochemical and pharmacokinetic properties of the studied ligands.
3. Materials and Methods
3.1. Chemistry
Melting points were determined with a Buchi Melting Point B-450 and were uncorrected. NMR spectra were recorded on a Varian Mercury 300 spectrometer with 1H at 300.060 MHz and 13C at 75.475 MHz. Proton chemical shifts were referenced to the TMS internal standard. Chemical shifts are reported in parts per million (ppm, δ units). Coupling constants are reported in units of Hertz (Hz). Splitting patterns are designed as s, singlet; d, doublet; t, triplet; q, quartet; dd, double doublet; m, multiplet; b, broad. Elemental analyses were carried out on a PerkinElmer 240B micro-analyzer, obtaining results within ± 0.4 % of the theoretical values. The purity of all compounds was over 98%. All commercial chemicals and solvents were reagent grade and were obtained from Merck; they were used without further purification, unless otherwise specified. Chemical reactions were monitored by thin layer chromatography on silica gel plates (60F-254, Sigma Aldrich, Italy) and the analysis of the plates was carried out using a UV lamp 254/365 nm. Flash chromatography was performed on silica gel 60 (Merck).
3.1.1. Synthesis of Ethyl 2-[(Methylsulfonyl)oxy]-2-phenylacetate 1
Triethylamine (834 μL, 6 mmol) was added to a solution of ethyl mandelate (162 mg, 1 mmol) in THF (8 mL) at 0 °C under stirring. Then methanesulfonyl chloride (232 μL, 3 mmol) was added dropwise, and the reaction was allowed to stir for two hours at room temperature. A saturated solution of ammonium chloride was added to the mixture, that was extracted with dichloromethane. Combined organic phases were washed with a saturated sodium chloride solution, dried on sodium sulfate, and the solvent was evaporated under reduced pressure. The crude material was purified by column chromatography on silica gel, with petroleum ether/ethyl acetate 8:2 as eluent. White solid, 93% yield, m.p. 60–61 °C; 1H NMR (CDCl3) δ 1.23 (t, 3H, J 7.2 Hz), 3.08 (s, 3H), 4.16–4.30 (m, 2H), 5.91 (s, 1H), 7.39–7.46 (m, 5H); 13C NMR (CDCl3) δ 13.9, 39.4, 62.3, 79.0, 127.7, 129.0, 129.9, 132.7, 167.7.
3.1.2. Synthesis of Ethyl 2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-2-phenylacetate 2
Triethylamine (278 μL, 2 mmol) was added to a solution of 5-cloro-2-mercaptobenzothiazole (202 mg, 1 mmol) in THF (10 mL) at 0 °C under stirring. After 30 min mesylate 1 (258 mg, 1 mmol) was added dropwise, and the temperature was allowed to reach room temperature. The mixture was then heated at 50 °C and stirred for 48 h. After the removal of THF, the resulting oil was dissolved in distilled water and extracted with dichloromethane. The organic phase was washed with a saturated solution of sodium chloride, dried on sodium sulfate, and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel, with petroleum ether/ethyl acetate 8:1 as eluent. Pale yellow solid, 87% yield; 1H NMR (CDCl3) δ 1.26 (t, 3H, J 6.9 Hz), 4.19 and 4.28 (dq, 2H, J 6.9 Hz), 5.76 (s, 1H), 7.25–7.38 (m, 5H), 7.52 (dd, 1H, J 8.4, 1.8 Hz), 7.64 (d, 1H, J 8.4 Hz), 7.83 (d, 1H, J 1.8 Hz); 13C NMR (CDCl3) δ 14.3, 54.8, 62.5, 121.7, 121.9, 125.0, 128.7, 129.1, 129.2, 132.3, 133.9, 153.9, 167.0, 169.6.
3.1.3. Synthesis of 2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-2-phenylacetic Acid 3
NaOH 2N (4 mL, 8 mmol) was added to ester 2 (363 mg, 1 mmol) in THF (5 mL) and the solution was stirred at room temperature overnight. THF was removed under reduced pressure, and the aqueous phase was acidified by HCl 2N, obtaining a precipitate that was collected by filtration under vacuum and recrystallized from cyclohexane. White crystals, 99% yield, m.p. 186–188 °C; 1H NMR (CD3OD) δ 5.79 (s, 1H), 7.31–7.41 (m, 5H), 7.54 (dd, 1H, J 8.4, 1.8 Hz), 7.81 (d, 1H, J 8.4 Hz), 7.84 (d, 1H, J 1.8 Hz); 13C NMR (CD3OD) δ 55.0, 120.9, 122.2, 124.8, 128.4, 128.7, 128.9, 132.2, 133.8, 135.0, 153.8, 167.9, 171.2.
3.1.4. General Procedure for the Synthesis of Amides 4a–p
To a stirred solution of acid 3 (168 mg, 0.5 mmol) in DMF (5 mL) at 0 °C, N,N’-dicyclohexylcarbodiimide (DCC, 103 mg, 0.5 mmol) and hydroxybenzotriazole (HOBt, 77 mg, 0.5 mmol) were added. After 15 min, N-methylmorpholine (NMM, 55 μL, 0.5 mmol) and the selected amine (0.5 mmol) were added in sequence to the reaction mixture. The resulting solution was then stirred at room temperature for 24 h, concentrated and the residue was dissolved in dichloromethane, and washed with a saturated solution of sodium bicarbonate and brine. The combined organic layers were dried on sodium sulfate and concentrated under vacuum to provide the crude products, which were purified by column chromatography or crystallization.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(4-methoxyphenyl)-2-phenylacetamide 4a
Pale yellow solid (silica gel, chloroform), 58% yield; m.p. 190 °C (dec); 1H NMR (CDCl3) δ 3.76 (s, 3H), 5.81 (s, 1H), 6.82 (d, 2H, J 8.7 Hz), 7.30–7.42 (m, 6H), 7.54–7.57 (m, 2H), 7.67 (d, 1H, J 8.4 Hz), 7.91 (d, 1H, J 1.8 Hz), 9.01 (bs, 1H); 13C NMR (CDCl3) δ 55.0, 55.4, 114.1, 121.1, 121.4, 121.9, 125.3, 128.8, 128.9, 129.0, 130.8, 132.6, 133.5, 134.5, 152.9, 159.5, 166.4, 168.3. Calcd for C22H17ClN2O2S2: C, 59.92; H, 3.89; N, 6.35. Found: C, 59.81; H, 3.90; N, 6.33.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(4-chlorophenyl)-2-phenylacetamide 4b
Pale yellow solid (silica gel, chloroform), 63% yield; m.p. 178–180 °C; 1H NMR (CDCl3) δ 5.83 (s, 1H), 7.26 (d, 1H, J 8.7 Hz), 7.34–7.56 (m, 9H), 7.70 (d, 1H, J 8.7 Hz), 7.95 (d, 1H, J 1.8 Hz), 9.39 (bs, 1H); 13C NMR (CDCl3) δ 54.8, 120.9, 121.0, 125.5, 128.8, 129.0, 129.1, 129.5, 132.7, 133.5, 134.1, 136.3, 152.8, 166.9, 168.5. Calcd for C21H14Cl2N2OS2: C, 56.63; H, 3.17; N, 6.29. Found: C, 56.77; H, 3.18; N, 6.27.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(4-fluorophenyl)-2-phenylacetamide 4c
Pale yellow solid (silica gel, dichloromethane), 52% yield; m.p. 168–170 °C; 1H NMR (CDCl3) δ 5.77 (s, 1H), 6.98 (t, 2H, J 8.4 Hz), 7.32–7.55 (m, 8H), 7.67 (d, 1H, J 8.7 Hz), 7.91 (d, 1H, J 2.4 Hz), 9.23 (bs, 1H); 13C NMR (CDCl3) δ 54.8, 115.5, 115.8, 121.1, 121.3, 121.5, 122.0, 125.4, 128.8, 128.9, 129.0, 132.6, 133.7, 134.2, 153.1, 157.9, 161.4, 166.8. Calcd for C21H14ClFN2OS2: C, 58.80; H, 3.29; N, 6.53. Found: C, 58.69; H, 3.30; N, 6.54.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-2-phenyl-N-[4-(trifluoromethyl)phenyl] Acetamide 4d
Pale yellow solid (silica gel, dichloromethane), 47% yield; m.p. 183–185 °C; 1H NMR (CDCl3) δ 5.79 (s, 1H), 7.34–7.41 (m, 4H), 7.51–7.56 (m, 4H), 7.64 (d, 2H, J 8.1 Hz), 7.70 (d, 1H, J 8.1 Hz), 7.93 (d, 1H, J 1.5 Hz), 9.71 (bs, 1H); 13C NMR (CDCl3) δ 54.6, 119.2, 121.0, 122.1, 125.6, 126.3 (q), 128.9, 129.0, 132.8, 133.5, 133.8, 140.8, 152.7, 167.3, 168.7. Calcd for C22H14ClF3N2OS2: C, 55.17; H, 2.95; N, 5.85. Found: C, 55.22; H, 2.94; N, 5.84.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(4-nitrophenyl)-2-phenylacetamide 4e
Pale yellow solid (silica gel, dichloromethane), 43% yield, m.p. 191–193 °C; 1H NMR (CDCl3) δ 5.79 (s, 1H), 6.61 (d, 2H, J 9.3 Hz), 7.33–7.41 (m, 4H), 7.50–7.54 (m, 2H), 7.70 (d, 2H, J 8.7 Hz), 7.91 (d, 1H, J 1.8 Hz), 8.05 (d, 2H, J 9 Hz), 8.18 (d, 2H, J 9.3 Hz), 10.05 (bs, 1H); 13C NMR (CDCl3) δ 54.5, 113.3, 119.1, 120.9, 122.2, 125.1, 125.7, 126.3, 128.9, 129.1, 129.2, 133.0, 133.4, 143.6, 143.7, 152.5, 167.6, 168.9. Calcd for C21H14ClN3O3S2: C, 55.32; H, 3.09; N, 9.22. Found: C, 55.57; H, 3.10; N, 9.24.
N-(4-Acetamidophenyl)-2-[(5-chlorobenzo[d]thiazol-2-yl)thio]-2-phenylacetamide 4f
White cristals (from ethyl acetate/methanol), 59% yield; m.p. 243 °C (dec); 1H NMR (DMSO-d6) δ 1.98 (s, 3H), 5.96 (s, 1H), 7.29–7.51 (m, 8H), 7.64–7.69 (m, 2H,), 7.86 (d, 1H, J 2.4 Hz), 8.03 (d, 1H, J 8.4 Hz), 9.89 (bs, 1H) 10.64 (bs, 1H); 13C NMR (DMSO-d6) δ 24.3, 42.5, 110.0, 119.7, 120.2, 121.0, 123.9, 125.2, 128.7, 129.2, 131.7, 134.0, 135.9, 136.5, 153.7, 166.4, 167.0, 168.0, 168.4. Calcd for C23H18ClN3O2S2: C, 59.03; H, 3.88; N, 8.98. Found: C, 59.08; H, 3.87; N, 8.96.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(3-methoxyphenyl)-2-phenylacetamide 4g
White solid (silica gel, cyclohexane/ethyl acetate 7:1), 44% yield; m.p. 160–162 °C; 1H NMR (CDCl3) δ 3.75 (s, 3H), 5.78 (s, 1H), 6.64 (dd, 1H, J 8.1, 2.4 Hz), 6.92 (d, 1H, J 8.1 Hz), 7.17 (t, 1H, J 8.1 Hz), 7.30–7.43 (m, 5H), 7.53 (dd, 2H, J 8.1, 2.4 Hz), 7.67 (d, 1H, J 8.1 Hz), 7.93 (d, 1H, J 1.8 Hz), 9.30 (bs, 1H); 13C NMR (CDCl3) δ 54.8, 55.2, 104.9, 110.8, 111.6, 121.1, 122.0, 125.3, 128.8, 128.9, 129.0, 129.7, 132.6, 133.6, 134.2, 139.0, 153.0, 160.1, 166.8, 168.4. Calcd for C22H17ClN2O2S2: C, 59.92; H, 3.89; N, 6.35. Found: C, 59.80; H, 3.89; N, 6.37.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(3-chlorophenyl)-2-phenylacetamide 4h
White solid (silica gel, dichloromethane), 76% yield; m.p. 175–177 °C; 1H NMR (CDCl3) δ 5.77 (s, 1H), 7.08 (d, 1H, J 8.1 Hz), 7.20–7.41 (m, 6H), 7.52 (dd, 2H, J 8.1, 2.4 Hz), 7.69 (d, 1H, J 8.1 Hz), 7.72 (s, 1H), 7.93 (d, 1H, J 1.8 Hz), 9.42 (bs, 1H); 13C NMR (CDCl3) δ 54.7, 171.5, 119.8, 121.1, 122.0, 124.5, 125.5, 128.8, 129.0, 129.1, 130.0, 132.8, 133.5, 133.9, 134.7, 138.9, 152.8, 167.0. Calcd for C21H14Cl2N2OS2: C, 56.63; H, 3.17; N, 6.29. Found: C, 56.67; H, 3.16; N, 6.27.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(3-fluorophenyl)-2-phenylacetamide 4i
White solid (silica gel, dichloromethane), 51% yield; m.p. 151–153 °C; 1H NMR (CDCl3) δ 5.77 (s, 1H), 6.79 (dt, 1H), 7.08–7.55 (m, 9H), 7.68 (d, 1H, J 8.1 Hz), 7.92 (d, 1H, J 2.4 Hz), 9.44 (bs, 1H); 13C NMR (CDCl3) δ 54.8, 107.1, 107.4, 111.1, 111.4, 114.8, 114.9, 121.1, 122.0, 125.4, 128.8, 128.9, 129.0, 130.0, 130.1, 132.7, 133.6, 134.1, 139.2, 152.9, 161.3, 164.5, 167.1. Calcd for C21H14ClFN2OS2: C, 58.80; H, 3.29; N, 6.53. Found: C, 58.63; H, 3.30; N, 6.55.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-2-phenyl-N-[3-(trifluoromethyl) phenyl]acetamide 4j
White solid (silica gel, cyclohexane/diethyl ether 4:1), 45% yield; m.p. 155–157 °C; 1H NMR (CDCl3) δ 5.77 (s, 1H), 7.33–7.41 (m, 7H), 7.53 (d, 2H, J 6.3 Hz), 7.61 (d, 1H, J 8.1 Hz), 7.70 (d, 1H, J 8.7 Hz), 7.93 (s, 2H); 13C NMR (CDCl3) δ 54.6, 116.4 (q), 112.0, 121.0, 122.0, 122.4, 125.5, 128.8, 129.0, 129.5, 132.8, 133.6, 133.8, 138.3, 152.9, 167.2. Calcd for C22H14ClF3N2OS2: C, 55.17; H, 2.95; N, 5.85. Found: C, 55.21; H, 2.96; N, 5.83.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(3-nitrophenyl)-2-phenylacetamide 4k
Pale yellow solid (silica gel, dichloromethane), 48% yield; m.p. 193–195 °C; 1H NMR (CDCl3) δ 5.77 (s, 1H), 7.34–7.54 (m, 7H), 7.70 (d, 1H, J 8.7 Hz), 7.83 (dd, 1H, J 8.1, 1.2 Hz), 7.92–7.95 (m, 2H), 8.47 (t, 1H, J 2.1 Hz), 9.88 (bs, 1H); 13C NMR (CDCl3) δ 54.6, 110.0, 114.5, 119.0, 121.0, 122.1, 125.1, 125.7, 128.8, 129.1, 129.8, 133.0, 133.6, 139.0, 148.5, 167.4. Calcd for C21H14ClN3O3S2: C, 55.32; H, 3.09; N, 9.22. Found: C, 55.45; H, 3.08; N, 9.22.
N-(3-Acetamidophenyl)-2-[(5-chlorobenzo[d]thiazol-2-yl)thio]-2-phenylacetamide 4l
White crystals (from chloroform), 41% yield; m.p. 197–199 °C; 1H NMR (CDCl3) δ 2.11 (s, 3H), 5.76 (s, 1H), 7.21 (d, 2H, J 5.4 Hz), 7.25–7.39 (m, 6H), 7.52 (dd, 2H, J 8.1, 2.4 Hz), 7.65 (d, 1H, J 8.7 Hz), 7.80 (s, 1H), 7.94 (d, 1H, J 1.8 Hz), 9.18 (bs, 1H); 13C NMR (CDCl3) δ 24.6, 55.2, 110.0, 110.9, 115.2, 115.7, 121.4, 121.9, 125.3, 128.8, 128.9, 129.0, 129.6, 132.6, 133.6, 134.2, 138.2, 138.5, 153.1, 166.9, 168.4. Calcd for C23H18ClN3O2S2: C, 59.03; H, 3.88; N, 8.98. Found: C, 59.08; H, 3.89; N, 9.00.
2-[(5-Chlorobenzo[d]thiazol-2-yl)thio]-N-(3,4-dichlorophenyl)-2-phenyl Acetamide 4m
White solid (silica gel, dichloromethane), 44% yield; m.p. 203–204 °C; 1H NMR (DMSO-d6) δ 5.96 (s, 1H), 7.33–7.47 (m, 5H), 7.54–7.64 (m, 3H), 7.85 (d, 1H, J 1.5 Hz), 7.94 (d, 1H, J 2.4 Hz), 8.03 (d, 1H, J 8.1 Hz), 11.03 (bs, 1H); 13C NMR (DMSO-d6) δ 56.2, 119.8, 120.9, 123.9, 125.1, 125.9, 128.7, 129.2, 129.4, 131.3, 131.6, 131.7, 134.0, 135.7, 138.9, 153.6, 167.3, 167.7. Calcd for C21H13Cl3N2OS2: C, 52.57; H, 2.73; N, 5.84. Found: C, 52,44; H, 2.72; N, 5.85.
N-(2-Bromo-5-nitrophenyl)-2-[(5-chlorobenzo[d]thiazol-2-yl)thio]-2-phenyl Acetamide 4n
White crystals (from cyclohexane/methanol), 48% yield; m.p. 176–178 °C; 1H NMR (CDCl3) δ 5.98 (s, 1H), 7.31–7.57 (m, 7H), 7.69 (d, 1H, J 8.7 Hz), 7.87–7.92 (m, 2H), 9.32 (d, 1H, J 3 Hz), 9.51 (bs, 1H); 13C NMR (CDCl3) δ 54.5, 116.7, 119.4, 121.5, 122.0, 125.4, 128.8, 129.2, 129.3, 129.6, 132.6, 133.5, 133.6, 135.5, 152.9, 167.7 Calcd for C21H13BrClN3O3S2: C, 47.16; H, 2.45; N, 7.86. Found: C, 47.07; H, 2.46; N, 7.85.
N-[2-Bromo-4-(trifluoromethyl)phenyl]-2-[(5-chlorobenzo[d]thiazol-2-yl)thio]-2-phenylacetamide 4o
White crystals (from petroleum ether/methanol), 51% yield; m.p. 179–180 °C; 1H NMR (CDCl3) δ 5.95 (s, 1H), 7.24–7.28 (m, 1H), 7.34–7.44 (m, 3H), 7.53–7.60 (m, 3H), 7.67 (d, 1H, J 8.7 Hz), 7.75 (s, 1H), 7.91 (d, 1H, J 1.8 Hz), 8.49 (d, 1H, J 8.7 Hz), 9.14 (bs, 1H); 13C NMR (CDCl3) δ 55.5, 113.3, 121.6, 121.7, 121.8, 125.3, 125.5, 125.6, 128.7, 129.2, 129.3, 129.4, 132.4, 133.6, 133.8, 153.2, 166.6, 167.3. Calcd for C22H13BrClF3N2OS2: C, 47.37; H, 2.35; N, 5.02. Found: C, 47.39; H, 2.35; N, 5.01.
N-[2-Bromo-5-(trifluoromethyl)phenyl]-2-[(5-chlorobenzo[d]thiazol-2-yl)thio]-2-phenylacetamide 4p
White crystals (from petroleum ether), 46% yield; m.p. 157–159 °C; 1H NMR (CDCl3) δ 5.96 (s, 1H), 7.20–7.32 (m, 2H), 7.39–7.44 (m, 3H), 7.56 (dd, 2H, J 7.5, 1.2 Hz), 7.61 (d, 1H, J 8.1 Hz), 7.67 (d, 1H, J 8.7 Hz), 7.92 (d, 1H, J 1.8 Hz), 8.67 (s, 1H), 9.16 (bs, 1H); 13C NMR (CDCl3) δ 55.2, 119.0, 121.7, 121.8, 121.9, 122.0, 125.2, 128.7, 129.2, 129.3, 132.4, 132.8, 133.9, 136.1, 153.2, 167.4. Calcd for C22H13BrClF3N2OS2: C, 47.37; H, 2.35; N, 5.02. Found: C, 47.27; H, 2.35; N, 5.04.
3.2. Cell Lines, Treatments, and Cell Viability Assay
Pancreatic cancer (AsPC-1, BxPC-3, and Capan-2), paraganglioma (PTJ86i and PTJ64i) and normal fibroblast (HFF-1) cell lines were cultured as previously described [17]. All compounds were dissolved in DMSO (stock solutions) and then diluted in culture media to the final working concentrations. In this way, the solutions were completely clear and devoid of any undissolved material by microscopic inspection. The final concentration of DMSO in the experiments was at most 0.18% and showed no cell toxicity. The effects of compounds 4a-p and 2b on cancer cell viability were tested by an MTT assay (Sigma-Aldrich, St. Louis, MO, USA), as described by Florio et al. [17]. In the initial screening all compounds were tested at a one-point screening concentration of 75 μM for 72 h (five replica wells per each condition). Then, concentration–response curves were generated by incubating cancer or normal fibroblast cell lines for 72 h with compounds showing a greater antiproliferative activity than 2b, at concentrations ranging from 0 μ M to 24 μM (five replica wells per each condition). For combined treatments, the experimental design was made according to the Chou–Talalay method for drug combination studies, as previously described [44].
3.3. Calculation of Half Maximal Inhibitory Concentration (IC50), Selectivity Index (SI), Combination Index (CI) Values and Statistical Analysis
IC50 values were extrapolated from concentration–response curves and calculated using the CompuSyn software [45]. The interactions between compound 4l and gemcitabine were assessed by calculating the CI values using the CompuSyn software. Based on this analysis, a CI < 1 indicates synergism, a CI = 1 indicates additive effects and a CI > 1 indicates antagonism. SI values were calculated as previously described [46]. Comparisons of mean values were performed using an unpaired Student’s t-test. For multiple comparisons, a one-way ANOVA followed by Dunnett’s test were employed. A p-value < 0.05 was estimated as statistically significant.
3.4. In Silico Studies
The SMILE string of the selected compound was obtained from Marvin Sketch (ChemAxon). Swiss Target Prediction database (http://www.swisstargetprediction.ch/), PLATO (http://platomussel.uniba.it/), SEASearch (https://sea.bkslab.org/), PPB2 (http://ppb2.gdb.tools/), SuperPred (https://prediction.charite.de/subpages/target_prediction.php), ChemMapper (http://www.lilab-ecust.cn/chemmapper), and PharmMapper (http://www.lilab-ecust.cn/pharmmapper/) were used to predict potential targets. “Homo sapiens” as the organism and “cancer target” were chosen as an option when possible (SuperPred). In all other cases, the predicted targets were screened by UniProt database (http://www.uniprot.org/uniprot/) by entering the target protein names and selecting only those belonging the “Homo sapiens (Human)” organism and “cancer” as protein name or tissue. Finally, the targets that did not meet the setting parameters were removed. Since these programs use different methods to score the targets, only a max of 20 targets was taken into consideration. All used programs were accessed on 1 March 2022.
The structure-based analysis was carried out using the Schrödinger Life-Sciences Suite 2021–4 [43]. The 2D sketcher in Maestro was used to construct ligand structures that were submitted to LigPrep to obtain ligand 3D geometry, identify all potential tautomers, and protonation states at pH 7.0 ± 0.4. The resulting structures were minimized utilizing MacroModel, the OPLS4 force field, and 5000 steps of the PRCG algorithm with a convergence criterion of 0.05 KJ/mol. Docking calculations were carried out on the 3D coordinates of CBR1 with PDB ID 6KPG and the chimeric form of SENP2 containing the SENP7 loop, PDB ID 3ZO5. Before docking calculations, protein structures were prepared using the Protein Preparation routine in Maestro [47] that fixes the protein structure and relaxes it through a constrained minimization. For the CBR1 the docking protocol was validated by reobtaining the X-ray geometry of the crystallographic ligand (RMSD 0.5135Å). To define the more proper binding site in SENP6, a SiteMap [48,49] calculation was carried out and the subsequent Glide Grid was generated on the predicted site. Glide [50,51] SP flexible docking calculations were carried out on both proteins
4. Conclusions
In conclusion, a library of phenylacetamide derivatives containing the benzothiazole nucleus was synthesized and tested for its antiproliferative activity in three pancreatic and two paraganglioma cancer cell lines. Most of the derivatives showed an improved antiproliferative activity, as compared to the lead compound 2b, allowing us to trace some preliminary structure–activity relationships. Considering both the antiproliferative activities and selectivity profiles, 4l was further analyzed in combination with gemcitabine on the three pancreatic cancer cell lines. Notably, the combination of the two compounds when used at the lowest concentrations (0.1 μM gemcitabine and 0.5 μM 4l, respectively) affected pancreatic cancer cell viability in a potent and synergistic manner. This synergistic effect supports the interest in this class of benzothiazoles in view of a possible translation into cancer treatment. A computational study was also conducted to predict the putative targets involved in the observed antiproliferative activity in pancreatic cancer cells: the different tools used to this aim identified the cannabinoid receptors and the sentrin-specific proteases as potential targets contributing to the bioactivity of this family of compounds. Docking studies on representative targets (CBR1 and SENP6) show that all compounds fit well in their binding site. Future in vitro and in vivo studies will be necessary to validate the predicted molecular targets of our compounds and gain novel insights into the underexplored potential of these molecules as antitumor agents in paraganglioma and pancreatic cancer treatment.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph15080937/s1.
Author Contributions
Conceptualization, A.A. and R.A.; methodology, A.A., R.F., L.D.L., M.F. and N.M.; validation, M.A., B.D.F., C.M. and L.G.; data curation, R.A., L.D.L., I.F., R.R. and A.C.; writing—original draft preparation, A.A., L.D.L. and M.F.; writing—review and editing, A.A., M.F., L.D.L., M.A. and I.F.; supervision, A.A.; funding acquisition, A.A. and A.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by FAR funds (Ministero dell’Istruzione, dell’Università e della Ricerca) assigned to Alessandra Ammazzalorso and by PRIN funds (Ministero dell’Istruzione, dell’Università e della Ricerca—grant number PRIN 2017EKMFTN_005) assigned to Alessandro Cama.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Acknowledgments
A.A. dedicates this work to her friend Ercole, who bravely fought against pancreatic cancer.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sharma, P.C.; Sinhmar, A.; Sharma, A.; Rajak, H.; Pathak, D.P. Medicinal significance of benzothiazole scaffold: An insight view. J. Enzyme Inhib. Med. Chem. 2013, 28, 240–266. [Google Scholar] [CrossRef]
- Weekes, A.A.; Westwell, A.D. 2-Arylbenzothiazole as a privileged scaffold in drug discovery. Curr. Med. Chem. 2009, 16, 2430–2440. [Google Scholar] [CrossRef]
- Zhilitskaya, L.V.; Shainyan, B.A.; Yarosh, N.O. Modern approaches to the synthesis and transformations of practically valuable benzothiazole derivatives. Molecules 2021, 26, 2190. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagumpi, S. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. [Google Scholar] [CrossRef]
- Irfan, A.; Batool, F.; Zahra Naqvi, S.A.; Islam, A.; Osman, S.M.; Nocentini, A.; Alissa, S.A.; Supuran, C.T. Benzothiazole derivatives as anticancer agents. J. Enzyme Inhib. Med. Chem. 2020, 35, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Ammazzalorso, A.; Carradori, S.; Amoroso, R.; Fernández, I.F. 2-substituted benzothiazoles as antiproliferative agents: Novel insights on structure-activity relationships. Eur. J. Med. Chem. 2020, 207, 112762. [Google Scholar] [CrossRef]
- Bradshaw, T.D.; Stevens, M.F.; Westwell, A.D. The discovery of the potent and selective antitumour agent 2-(4-amino-3-methylphenyl)benzothiazole (DF 203) and related compounds. Curr. Med. Chem. 2001, 8, 203–210. [Google Scholar] [CrossRef]
- Bradshaw, T.D.; Westwell, A.D. The development of the antitumour benzothiazole prodrug, Phortress, as a clinical candidate. Curr. Med. Chem. 2004, 11, 1009–1021. [Google Scholar] [CrossRef]
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology 2013, 2, e26968. [Google Scholar] [CrossRef] [Green Version]
- Krauser, J.A.; Jin, Y.; Walles, M.; Pfaar, U.; Sutton, J.; Wiesmann, M.; Graf, D.; Pflimlin-Fritschy, V.; Wolf, T.; Camenisch, G.; et al. Phenotypic and metabolic investigation of a CSF-1R kinase receptor inhibitor (BLZ945) and its pharmacologically active metabolite. Xenobiotica 2015, 45, 107–123. [Google Scholar] [CrossRef]
- Lin, C.-C.; Gil-Martin, M.; Bauer, T.M.; Naing, A.; Wan-Teck Lim, D.; Sarantopoulos, J.; Geva, R.; Ando, Y.; Fan, L.; Choudhury, S.; et al. Abstract nr CT171: Phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors [abstract]. Cancer Res. 2020, 80 (Suppl. 16), CT171. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Cho, N.C.; Nam, G.; Pae, A.N.; Keum, G. Discovery of a nanomolar multikinase inhibitor (KST016366): A new benzothiazole derivative with remarkable broad-spectrum antiproliferative activity. ChemMedChem 2016, 11, 1587–1595. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; D’Angelo, A.; Giancristofaro, A.; De Filippis, B.; Di Matteo, M.; Fantacuzzi, M.; Giampietro, L.; Linciano, P.; Maccallini, C.; Amoroso, R. Fibrate-derived N-(methylsulfonyl)amides with antagonistic properties on PPARα. Eur. J. Med. Chem. 2012, 58, 317–322. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; De Lellis, L.; Florio, R.; Bruno, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Perconti, S.; Verginelli, F.; et al. Cytotoxic effect of a family of peroxisome proliferator-activated receptor antagonists in colorectal and pancreatic cancer cell lines. Chem. Biol. Drug Des. 2017, 90, 1029–1035. [Google Scholar] [CrossRef]
- Benedetti, E.; d’Angelo, M.; Ammazzalorso, A.; Gravina, G.L.; Laezza, C.; Antonosante, A.; Panella, G.; Cinque, B.; Cristiano, L.; Dhez, A.C.; et al. PPARα antagonist AA452 triggers metabolic reprogramming and increases sensitivity to radiation therapy in human glioblastoma primary cells. J. Cell Physiol. 2017, 232, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; De Lellis, L.; Florio, R.; Laghezza, A.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Tortorella, P.; Veschi, S.; et al. Synthesis of novel benzothiazole amides: Evaluation of PPAR activity and anti-proliferative effects in paraganglioma, pancreatic and colorectal cancer cell lines. Bioorg. Med. Chem. Lett. 2019, 29, 2302–2306. [Google Scholar] [CrossRef]
- Florio, R.; De Lellis, L.; di Giacomo, V.; Di Marcantonio, M.C.; Cristiano, L.; Basile, M.; Verginelli, F.; Verzilli, D.; Ammazzalorso, A.; Prasad, S.C.; et al. Effects of PPARα inhibition in head and neck paraganglioma cells. PLoS ONE 2017, 12, e0178995. [Google Scholar] [CrossRef] [Green Version]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallve, S. Tools for in silico target fishing. Methods 2015, 71, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Sydow, D.; Burggraaff, L.; Szengel, A.; Van Vlijmen, H.W.T.; Ijzerman, A.P.; Van Westen, G.J.P.; Volkamer, A. Advances and challenges in computational target prediction. J. Chem. Inf. Model. 2019, 59, 1728–1742. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. Swiss Target Prediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriaco, F.; Gambacorta, N.; Alberga, D.; Nicolotti, O. Quantitative polypharmacology profiling based on a multifingerprint similarity predictive approach. J. Chem. Inf. Model. 2021, 61, 4868–4876. [Google Scholar] [CrossRef]
- Alberga, D.; Trisciuzzi, D.; Montaruli, M.; Leonetti, F.; Mangiatordi, G.F.; Nicolotti, O. A new approach for drug target and bioactivity prediction: The Multifingerprint Similarity Search Algorithm (MuSSeL). J. Chem. Inf. Model. 2019, 59, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Montaruli, M.; Alberga, D.; Ciriaco, F.; Trisciuzzi, D.; Tondo, A.R.; Mangiatordi, G.F.; Nicolotti, O. Accelerating drug discovery by early protein drug target prediction based on a multi-fingerprint similarity search. Molecules 2019, 24, 2233. [Google Scholar] [CrossRef] [Green Version]
- Ciriaco, F.; Gambacorta, N.; Trisciuzzi, D.; Nicolotti, O. PLATO: A predictive drug discovery web platform for efficient target fishing and bioactivity profiling of small molecules. Int. J. Mol. Sci. 2022, 23, 5245. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awale, M.; Reymond, J.L. Polypharmacology browser PPB2: Target prediction combining nearest neighbors with machine learning. J. Chem. Inf. Model. 2019, 59, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunkel, M.; Günther, S.; Ahmed, J.; Wittig, B.; Preissner, R. SuperPred: Drug classification and target prediction. Nucleic Acids Res. 2008, 36, W55–W59. [Google Scholar] [CrossRef]
- Gong, J.; Cai, C.; Liu, X.; Ku, X.; Jiang, H.; Gao, D.; Li, H. ChemMapper: A versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method. Bioinformatics 2013, 29, 1827–1829. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Di Guan, Y.; Zhang, L.X.; Liu, S.; Lu, A.P.; Cheng, Y.; Cao, D.S. A combinatorial target screening strategy for deorphaning macromolecular targets of natural product. Eur. J. Med. Chem. 2020, 204, 112644. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K. Cannabinoid receptors: Where they are and what they do. J. Neuroendocrinol. 2008, 20, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Cavic, M.; Krivokuca, A.; Casadó, V.; Canela, E. The endocannabinoid system as a target in cancer diseases: Are we there yet? Front. Pharmacol. 2019, 10, 339. [Google Scholar] [CrossRef] [Green Version]
- Michalski, C.W.; Oti, F.E.; Erkan, M.; Sauliunaite, D.; Bergmann, F.; Pacher, P.; Batkai, S.; Müller, M.W.; Giese, N.A.; Friess, H. Cannabinoids in pancreatic cancer: Correlation with survival and pain. Int. J. Cancer 2008, 122, 742–750. [Google Scholar] [CrossRef] [Green Version]
- Garmpis, N.; Damaskos, C.; Dimitroulis, D.; Garmpi, A.; Diamantis, E.; Sarantis, P.; Georgakopoulou, V.E.; Patsouras, A.; Prevezanos, D.; Syllaios, A.; et al. Targeting the endocannabinoid system: From the need for new therapies to the development of a promising strategy. What about pancreatic cancer? In Vivo 2022, 36, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Jia, Y.; Claessens, L.A.; Vertegaal, A.C.O.; Ovaa, H. Chemical tools and biochemical assays for SUMO specific proteases (SENPs). ACS Chem. Biol. 2019, 14, 2389–2395. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, P.; Woźniak, K. SENP proteases as potential targets for cancer therapy. Cancers 2021, 13, 2059. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://cancergenome.nih.gov/ (accessed on 1 March 2022).
- Schneeweis, C.; Hassan, Z.; Schick, M.; Keller, U.; Schneider, G. The SUMO pathway in pancreatic cancer: Insights and inhibition. Br. J. Cancer 2021, 124, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Zhang, J.; Ullah, S.; Kang, N.; Zhao, Y.; Zhou, H. Benzothiophene-2-carboxamide derivatives as SENPs inhibitors with selectivity within SENPs family. Eur. J. Med. Chem. 2020, 204, 112553. [Google Scholar] [CrossRef]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and signaling mechanism revealed by cannabinoid receptor-Gi complex structures. Cell 2020, 180, 655–665. [Google Scholar] [CrossRef]
- Alegre, K.O.; Reverter, D. Structural insights into the SENP6 Loop1 structure in complex with SUMO2. Protein Sci. 2014, 23, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2021-4: Maestro, Glide, Protein Preparation Wizard, Epik, SiteMap, QikProp, MacroModel; Schrödinger, LLC.: New York, NY, USA, 2021.
- Florio, R.; Veschi, S.; di Giacomo, V.; Pagotto, S.; Carradori, S.; Verginelli, F.; Cirilli, R.; Casulli, A.; Grassadonia, A.; Tinari, N.; et al. The benzimidazole-based anthelmintic parbendazole: A repurposed drug candidate that synergizes with gemcitabine in pancreatic cancer. Cancers 2019, 11, 2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammazzalorso, A.; Bruno, I.; Florio, R.; De Lellis, L.; Laghezza, A.; Cerchia, C.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; et al. Sulfonimide and amide derivatives as novel PPARα antagonists: Synthesis, antiproliferative activity, and docking studies. ACS Med. Chem. Lett. 2020, 11, 624–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
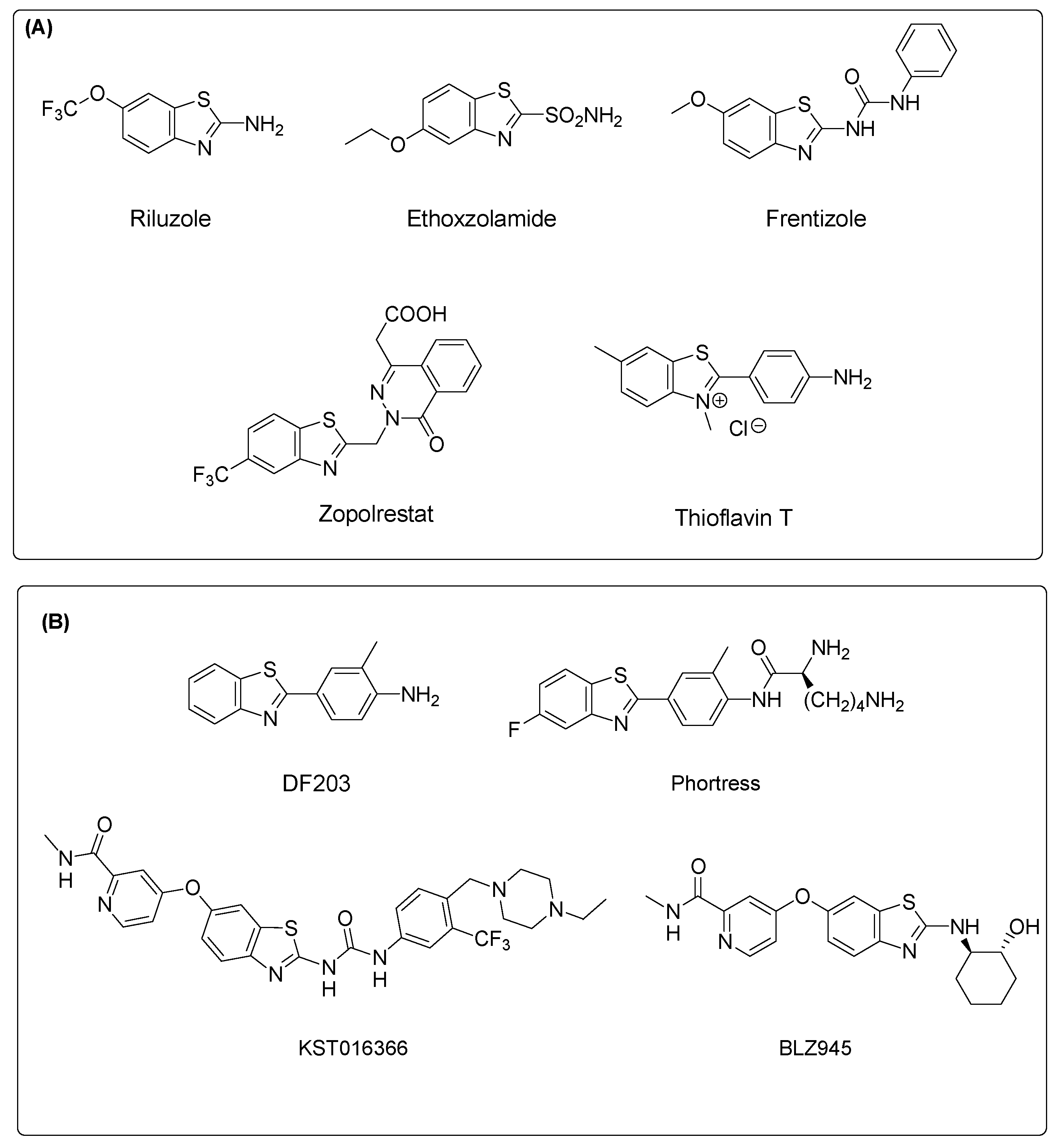
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).