
Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery

 , and
, and
Abstract
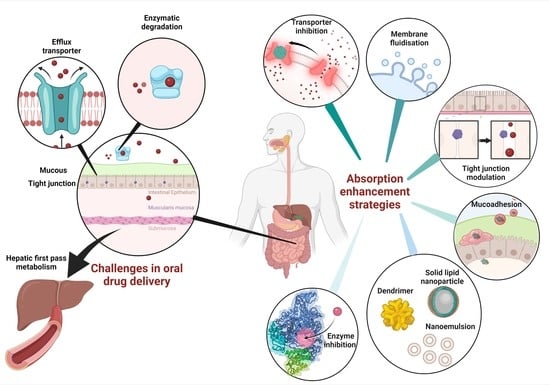
:
1. Introduction
2. The Challenges in Oral Drug Delivery
2.1. Mucous
2.2. Tight Junction
2.3. Efflux Transporters
2.4. Enzymes
2.5. First-Pass Metabolism
2.6. Intestinal Lymphatic Transport
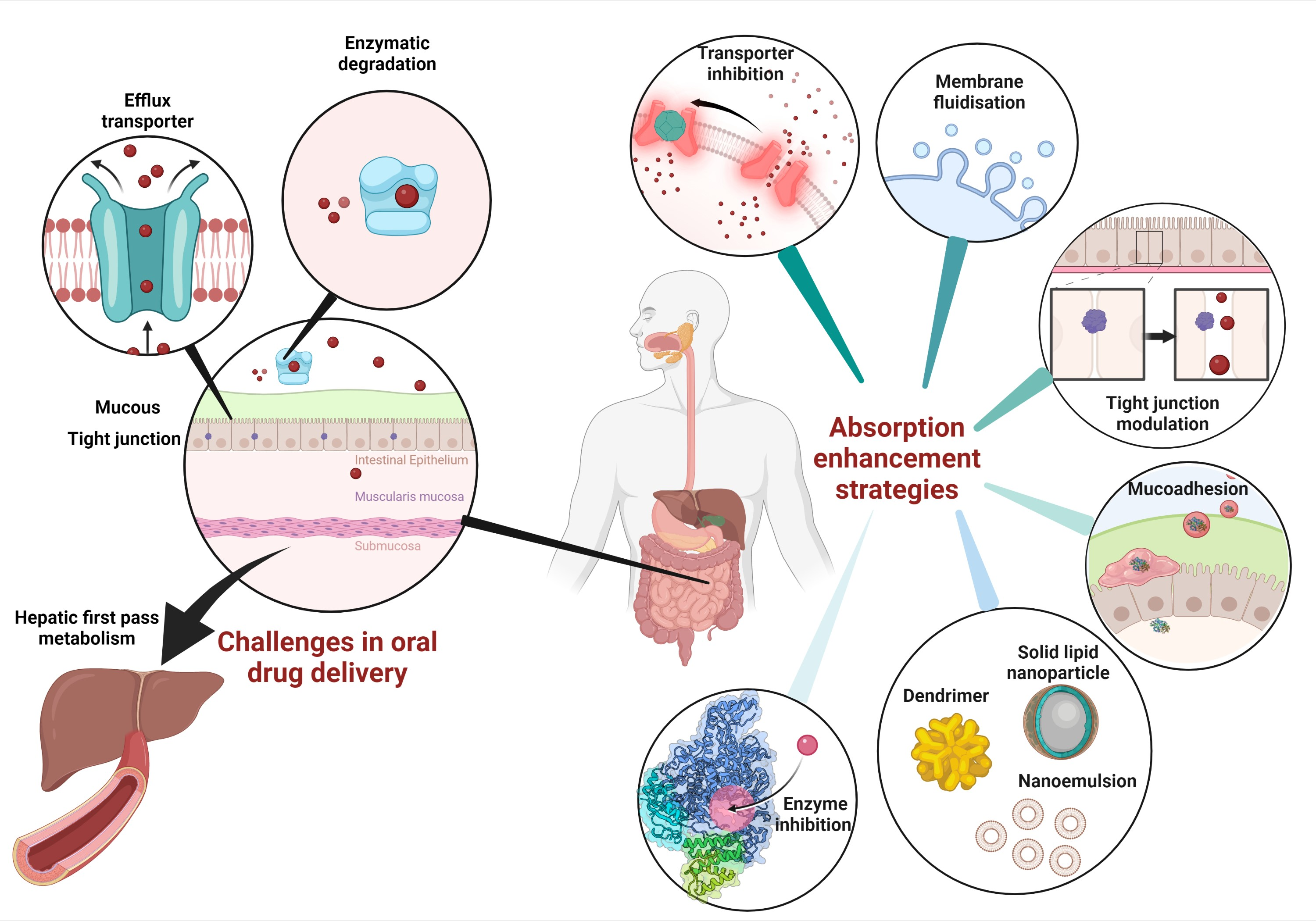
3. Current Absorption Enhancers and Their Absorption-Enhancing Mechanisms to Improve the Pharmacokinetic Profile
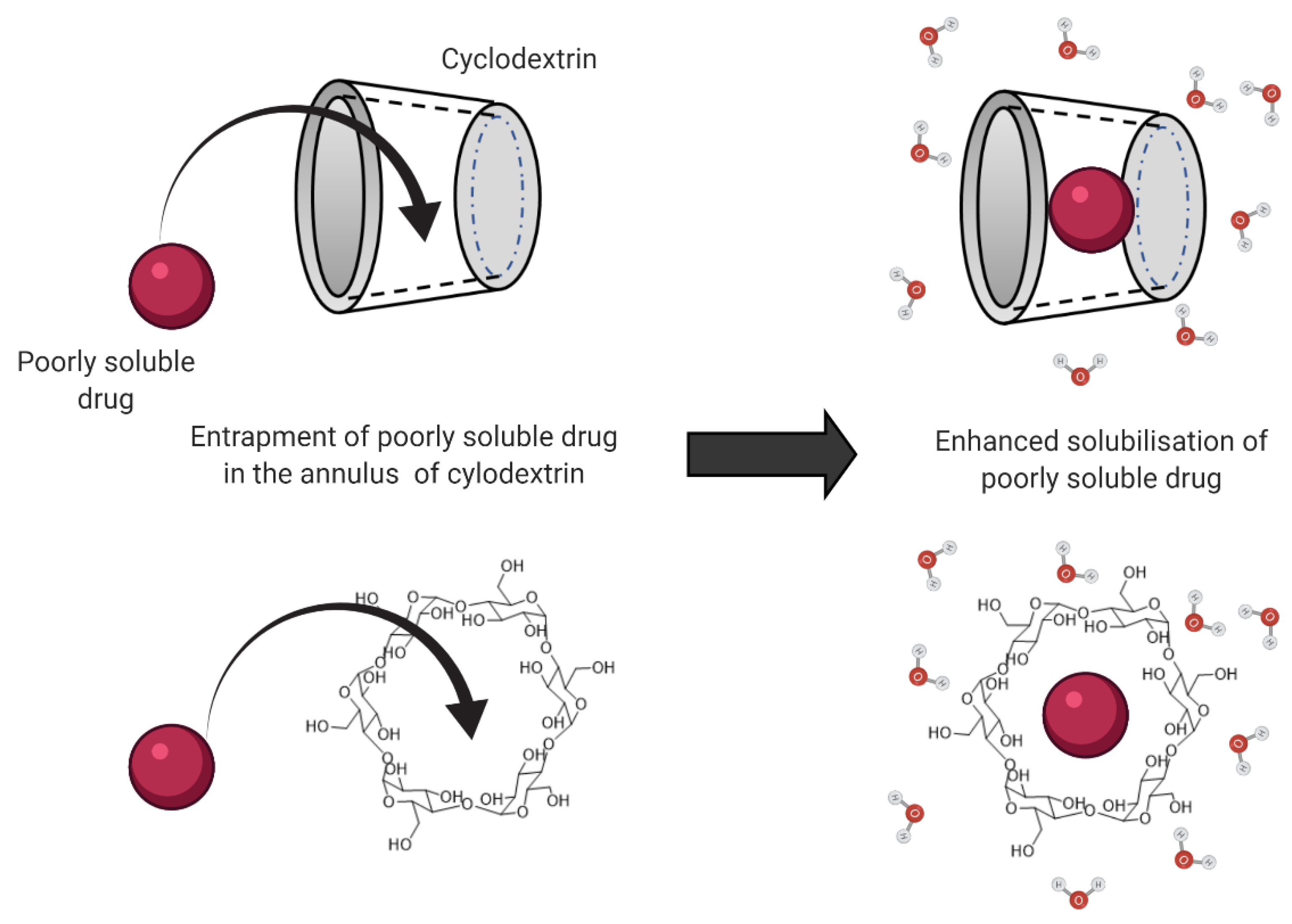
3.1. Solubilizing Agents
3.2. Bile Salts

{kind=link}
{kind=link}
| Drug (s) | Absorption Enhancer | Model | Results | Ref. |
|---|---|---|---|---|
| 5(6)-carboxyfluorescein | Sodium glycocholate (SGC) and sodium taurodeoxycholate (STDC) | In vitro: Caco-2 cell | SGC was a slightly better absorption enhancer for the 5(6)-carboxyfluorescein than STDC but not significant (p > 0.05). | [73] |
| Cefquinome | Sodium taurocholate | In vitro: Caco-2 cell | At 2 mmol/L sodium taurocholate, the transportation of cefquinome substantially increased. | [72] |
| In vivo: rat intestine | At 10 and 20 mmol/L sodium taurocholate, the absorption of the drug increased in a concentration-dependent manner. | |||
| Berberine chloride | Sodium deoxycholate | In vivo: rat intestine | AUC0–36h: 35.3-fold increase | [70] |
| Gliclazide | Taurocholic acid | In vivo: rat intestine | The microcapsules containing taurocholic acid increased the gliclazide absorption (p < 0.01). | [71] |
| EGFR2R-lytic hybrid peptide | Sodium taurodeoxycholate | In vitro: Caco-2 cell | Papp: 5.0-fold increase | [74] |
3.3. Chitosan
| Drug (s) | Absorption Enhancer | Model | Results | Ref. |
|---|---|---|---|---|
| Acyclovir | Chitosan | In vitro: Caco-2 cell | Papp: 124- and 143-fold increase | [83] |
| In vivo: rat intestine | AUC0–12 and AUC0–∞: 0.70- and 0.74-fold decrease Cmax: 0.56- and 0.63-fold decrease Tmax: 1.25- and 1.50-fold increase | |||
| In vitro: Ussing chamber | Papp: 1.08- and 2.33-fold increase | |||
| Glucosamine hydrochloride | Chitosan | In vitro: Caco-2 cell | Papp: 1.9, 2.5 and 4.0-fold increase | [88] |
| In vivo: rat intestine | Cmax: 2.8-fold increase Tmax: no change AUC0−∞: 2.5-fold increase | |||
| Salvianolic acid B | Chitosan | In vitro: Caco-2 cell | Papp: 4.43-fold increase | [79] |
| In vivo: rat intestine | AUC0–∞: 4.25-fold increase | |||
| Berberine | Chitosan hydrochloride | In vivo: rat intestine | AUC0–36: no improvement Cmax: no improvement | [86] |
| Chitosan | In vivo: rat intestine | AUC0–36: maximum 2.5-fold increase | ||
| Amphotericin B | Trimethyl chitosan | In vitro: Caco-2 cell | Papp: 1.11-fold increase | [87] |
4. Formulation Strategies to Improve Pharmacokinetics Profile
4.1. Solid Lipid Nanoparticles (SLN)
4.2. Dimers
| Drug (s) | Model | Results | Ref. |
|---|---|---|---|
| 5(6)-carboxyfluorescein (CF), fluorescein isothiocyanate-labeled dextrans (FD4, FD10) and alendronate | In vitro: diffusion chamber | Papp: increased except for FD10. | [98] |
| In vivo: rat intestine | The greatest AUC achieved in the presence of Ac50-G2 (0.5%, w/v). | ||
| Camptothecin | In vivo: rat intestine | AUC: 2- to 3-fold increase Cmax: increased Tmax: no change | [100] |
| Simvastatin | In vivo: rat intestine | AUC: increased Cmax: increased Tmax: 1.5-fold increase | [99] |
| In vitro: Caco-2 cell | Papp: increased | ||
| Propranolol | In vitro Release Study (dialysis sac) | Papp: increased | [102] |
| In vitro: Caco-2 cell | AUC: increased | [101] |
4.3. Nanoemulsions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quante, M.; Thate-Waschke, I.; Schofer, M. What are the reasons for patient preference? A comparison between oral and subcutaneous administration. Z. Orthop. Unfall. 2012, 150, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, P.A.; Kratz, J.M.; Hernández, A.; Costa, G.; Ospina, L.F.; Baena, Y.; Simões, C.M.O.; Jimenez-Kairuz, Á.; Aragon, M. In vitro intestinal permeability studies, pharmacokinetics and tissue distribution of 6-methylcoumarin after oral and intraperitoneal administration in Wistar rats. Braz. J. Pharm. Sci. 2017, 53. [Google Scholar] [CrossRef] [Green Version]
- Nunes, R.; Silva, C.; Chaves, L. Tissue-based in vitro and ex vivo models for intestinal permeability studies 4.2 4.2.1 Introduction 4.2.1.1 Anatomy, histology, and physiology of the intestine. In Concepts and Models for Drug Permeability Studies; Elsevier: Amsterdam, The Netherlands, 2016; pp. 203–236. [Google Scholar]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jambhekar, S.S.; Breen, P.J. Drug dissolution: Significance of physicochemical properties and physiological conditions. Drug Discov. Today 2013, 18, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Benet, L.Z. Predicting Drug Disposition via Application of BCS: Transport/Absorption/ Elimination Interplay and Development of a Biopharmaceutics Drug Disposition Classification System. Pharm. Res. 2005, 22, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Garjani, A. Biopharmaceutics: Absorption Aspects. Pharm. Sci. 1961, 50, 359–387. [Google Scholar] [CrossRef]
- Wagner, J.G. Method of Estimating Relative Absorption of a Drug in a Series of Clinical Studies in Which Blood Levels are Measured After Single and/or Multiple Doses. J. Pharm. Sci. 1967, 56, 652–653. [Google Scholar] [CrossRef]
- Disanto, A.R.; Wagner, J.G. Pharmacokinetics of Highly Ionized Drugs II: Methylene Blue—Absorption, Metabolism, and Excretion in Man and Dog after Oral Administration. J. Pharm. Sci. 1972, 61, 1086–1090. [Google Scholar] [CrossRef]
- Bornhorst, G.M.; Singh, R.P. Bolus Formation and Disintegration during Digestion of Food Carbohydrates. Compr. Rev. Food Sci. Food Saf. 2012, 11, 101–118. [Google Scholar] [CrossRef]
- Murakami, T.; Takano, M. Intestinal efflux transporters and drug absorption. Expert Opin. Drug Metab. Toxicol. 2008, 4, 923–939. [Google Scholar] [CrossRef]
- Kiela, P.R.; Ghishan, F.K. Physiology of Intestinal Absorption and Secretion. Best Pr. Res. Clin. Gastroenterol. 2016, 30, 145–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babadi, D.; Dadashzadeh, S.; Osouli, M.; Daryabari, M.S.; Haeri, A. Nanoformulation strategies for improving intestinal permeability of drugs: A more precise look at permeability assessment methods and pharmacokinetic properties changes. J. Control. Release 2020, 321, 669–709. [Google Scholar] [CrossRef] [PubMed]
- Shaikh Permeability Enhancement Techniques for Poorly Permeable Drugs: A Review. J. Appl. Pharm. Sci. 2012, 2, 34–39. [CrossRef] [Green Version]
- Bansil, R.; Turner, B.S. Mucin structure, aggregation, physiological functions and biomedical applications. Curr. Opin. Colloid Interface Sci. 2006, 11, 164–170. [Google Scholar] [CrossRef]
- Behrens, I.; Stenberg, P.; Artursson, P.; Kissel, T. Transport of Lipophilic Drug Molecules in a New Mucus-Secreting Cell Culture Model Based on HT29-MTX Cells. Pharm. Res. 2001, 18, 1138–1145. [Google Scholar] [CrossRef]
- Boegh, M.; Nielsen, H.M. Mucus as a Barrier to Drug Delivery—Understanding and Mimicking the Barrier Properties. Basic Clin. Pharmacol. Toxicol. 2014, 116, 179–186. [Google Scholar] [CrossRef]
- George, C.F. Drug Metabolism by the Gastrointestinal Mucosa. Clin. Pharmacokinet. 1981, 6, 259–274. [Google Scholar] [CrossRef]
- Lechanteur, A.; das Neves, J.; Sarmento, B. The role of mucus in cell-based models used to screen mucosal drug delivery. Adv. Drug Deliv. Rev. 2018, 124, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Jia, L. The Conduct of Drug Metabolism Studies Considered Good Practice (II): In Vitro Experiments. Curr. Drug Metab. 2007, 8, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.D.; Thummel, K.E.; Watkins, P.B.; Lown, K.S.; Benet, L.Z.; Paine, M.F.; Mayo, R.R.; Turgeon, D.K.; Bailey, D.G.; Fontana, R.J.; et al. Molecular and physical mechanisms of first-pass extraction. Drug Metab. Dispos. 1999, 27, 161–166. [Google Scholar]
- Lock, J.Y.; Carlson, T.L.; Carrier, R.L. Mucus models to evaluate the diffusion of drugs and particles. Adv. Drug Deliv. Rev. 2017, 124, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Falavigna, M.; Stein, P.C.; Flaten, G.E.; di Cagno, M.P. Impact of Mucin on Drug Diffusion: Development of a Straightforward In Vitro Method for the Determination of Drug Diffusivity in the Presence of Mucin. Pharmaceutics 2020, 12, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Zhang, E.; Shi, Y.; Song, B.; Du, H.; Cao, Z. Biomaterial–tight junction interaction and potential impacts. J. Mater. Chem. B 2019, 7, 6310–6320. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel Integral Membrane Proteins Localizing at Tight Junctions with No Sequence Similarity to Occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Lemmer, H.J.; Hamman, J.H. Paracellular drug absorption enhancement through tight junction modulation. Expert Opin. Drug Deliv. 2012, 10, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, Y.; Kishimoto, H.; Nakagawa, M.; Sakamoto, N.; Tobe, Y.; Furuya, T.; Tomita, M.; Hayashi, M. Effects of pharmaceutical excipients on membrane permeability in rat small intestine. Int. J. Pharm. 2013, 453, 363–370. [Google Scholar] [CrossRef]
- Anderson, J.M.; Van Itallie, C.M. Physiology and Function of the Tight Junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Shashikanth, N.; Rizzo, H.E.; Pongkorpsakol, P.; Heneghan, J.F.; Turner, J.R. Electrophysiologic Analysis of Tight Junction Size and Charge Selectivity. Curr. Protoc. 2021, 1, e143. [Google Scholar] [CrossRef]
- Maiti, S. Nanometric Biopolymer Devices for Oral Delivery of Macromolecules with Clinical Significance. In Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics; Elsevier: Amsterdam, The Netherlands, 2017; pp. 109–138. [Google Scholar] [CrossRef]
- Tam, K.Y.; Avdeef, A.; Tsinman, O.; Sun, N. The Permeation of Amphoteric Drugs through Artificial Membranes − An in Combo Absorption Model Based on Paracellular and Transmembrane Permeability. J. Med. Chem. 2009, 53, 392–401. [Google Scholar] [CrossRef]
- Thakkar, H.; Desai, J. Influence of excipients on drug absorption via modulation of intestinal transporters activity. Asian J. Pharm. 2015, 9, 69. [Google Scholar] [CrossRef]
- Daood, M.; Tsai, C.; Ahdab-Barmada, M.; Watchko, J.F. ABC Transporter (P-gp/ABCB1, MRP1/ABCC1, BCRP/ABCG2) Expression in the Developing Human CNS. Neuropediatrics 2008, 39, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Zakeri-Milani, P.; Valizadeh, H. Intestinal transporters: Enhanced absorption through P-glycoprotein-related drug interactions. Expert Opin. Drug Metab. Toxicol. 2014, 10, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Mendes, C.; Meirelles, G.C.; Silva, M.A.; Ponchel, G. Intestinal permeability determinants of norfloxacin in Ussing chamber model. Eur. J. Pharm. Sci. 2018, 121, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.R. Cytochrome P4503A (CYP3A) metabolism: Prediction ofIn Vivo activity in humans. J. Pharmacokinet. Biopharm. 1996, 24, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Yan, Y.; Sun, Y.; Zhao, B.; Hu, W.; Li, G. Contribution of TRPV1 and multidrug resistance proteins in the permeation of capsaicin across different intestinal regions. Int. J. Pharm. 2013, 445, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Mouly, S.; Paine, M.F. P-Glycoprotein Increases from Proximal to Distal Regions of Human Small Intestine. Pharm. Res. 2003, 20, 1595–1599. [Google Scholar] [CrossRef]
- Bruyère, A.; Declèves, X.; Bouzom, F.; Ball, K.; Marques, C.; Treton, X.; Pocard, M.; Valleur, P.; Bouhnik, Y.; Panis, Y.; et al. Effect of Variations in the Amounts of P-Glycoprotein (ABCB1), BCRP (ABCG2) and CYP3A4 along the Human Small Intestine on PBPK Models for Predicting Intestinal First Pass. Mol. Pharm. 2010, 7, 1596–1607. [Google Scholar] [CrossRef]
- Lee, H.J. Protein drug oral delivery: The recent progress. Arch. Pharmacal. Res. 2002, 25, 572–584. [Google Scholar] [CrossRef]
- Hetal, T.; Bindesh, P.; Sneha, T. A review on techniques for oral bioavailability enhancement of drugs. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 33. [Google Scholar]
- Roberts, M.S.; Magnusson, B.M.; Burczynski, F.J.; Weiss, M. Enterohepatic Circulation. Clin. Pharmacokinet. 2002, 41, 751–790. [Google Scholar] [CrossRef] [PubMed]
- Choonara, B.F.; Choonara, Y.; Kumar, P.; Bijukumar, D.; du Toit, L.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.; Charman, W.N.A.; Naringrekar, V.H.; Stella, V.J. Prodrugs. Do they have advantages in clinical practice? Drugs 1985, 29, 455–473. [Google Scholar] [CrossRef]
- Oz, H.S.; Ebersole, J.L. Application of Prodrugs to Inflammatory Diseases of the Gut. Molecules 2008, 13, 452–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.Q.; Aasmundstad, T.A.; Christophersen, A.S.; Mørland, J.; Bjørneboe, A. Evidence for CYP2D1-mediated primary and secondary O-dealkylation of ethylmorphine and codeine in rat liver microsomes. Biochem. Pharmacol. 1997, 53, 603–609. [Google Scholar] [CrossRef]
- Zhang, Y.; Benet, L.Z. The Gut as a Barrier to Drug Absorption: Combined role of cytochrome P450 3A and P-glycoprotein. Clin. Pharmacokinet. 2001, 40, 159–168. [Google Scholar] [CrossRef]
- Gavhane, Y.N.; Yadav, A.V. Loss of orally administered drugs in GI tract. Saudi Pharm. J. 2012, 20, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.; Park, J.-H. Liposomal delivery systems for intestinal lymphatic drug transport. Biomater. Res. 2016, 20, 36. [Google Scholar] [CrossRef] [Green Version]
- Yáñez, J.A.; Wang, S.W.J.; Knemeyer, I.W.; Wirth, M.A.; Alton, K.B. Intestinal lymphatic transport for drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 923–942. [Google Scholar] [CrossRef]
- Ryšánek, P.; Grus, T.; Šíma, M.; Slanař, O. Lymphatic Transport of Drugs after Intestinal Absorption: Impact of Drug Formulation and Physicochemical Properties. Pharm. Res. 2020, 37, 166. [Google Scholar] [CrossRef]
- Savla, R.; Browne, J.; Plassat, V.; Wasan, K.M.; Wasan, E.K. Review and analysis of FDA approved drugs using lipid-based formulations. Drug Dev. Ind. Pharm. 2017, 43, 1743–1758. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Navarro, M.; Garcia, J.; Giralt, E.; Teixidó, M. Using peptides to increase transport across the intestinal barrier. Adv. Drug Deliv. Rev. 2016, 106, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharm. 2013, 2013, 848043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, S.; Heade, J.; McCartney, F.; Waters, S.; Bleiel, S.B.; Brayden, D. Effects of surfactant-based permeation enhancers on mannitol permeability, histology, and electrogenic ion transport responses in excised rat colonic mucosae. Int. J. Pharm. 2018, 539, 11–22. [Google Scholar] [CrossRef]
- McCartney, F.; Rosa, M.; Brayden, D.J. Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies. Pharmaceutics 2019, 11, 565. [Google Scholar] [CrossRef] [Green Version]
- Maroni, A.; Zema, L.; Del Curto, M.D.; Foppoli, A.; Gazzaniga, A. Oral colon delivery of insulin with the aid of functional adjuvants. Adv. Drug Deliv. Rev. 2012, 64, 540–556. [Google Scholar] [CrossRef]
- Devasari, N.; Dora, C.P.; Singh, C.; Paidi, S.R.; Kumar, V.; Sobhia, M.E.; Suresh, S. Inclusion complex of erlotinib with sulfobutyl ether-β-cyclodextrin: Preparation, characterization, in silico, in vitro and in vivo evaluation. Carbohydr. Polym. 2015, 134, 547–556. [Google Scholar] [CrossRef]
- Rubim, A.M.; Rubenick, J.B.; Maurer, M.; LaPorta, L.V.; Rolim, C.M.B. Inclusion complex of amiodarone hydrochloride with cyclodextrins: Preparation, characterization and dissolution rate evaluation. Braz. J. Pharm. Sci. 2017, 53. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, A. Cyclodextrins as Drug Carrier Molecule: A Review. Sci. Pharm. 2008, 76, 567–598. [Google Scholar] [CrossRef]
- Wiedmann, T.S.; Kamel, L. Examination of the Solubilization of Drugs by Bile Salt Micelles. J. Pharm. Sci. 2002, 91, 1743–1764. [Google Scholar] [CrossRef]
- Loftsson, T.; Moya-Ortega, M.D.; Alvarez-Lorenzo, C.; Concheiro, A. Pharmacokinetics of cyclodextrins and drugs after oral and parenteral administration of drug/cyclodextrin complexes. J. Pharm. Pharmacol. 2015, 68, 544–555. [Google Scholar] [CrossRef]
- Pavlović, N.; Goločorbin-Kon, S.; Đanić, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile Acids and Their Derivatives as Potential Modifiers of Drug Release and Pharmacokinetic Profiles. Front. Pharmacol. 2018, 9, 1283. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Hellinger, É.; Pilbat, A.M.; Kittel, Á.; Török, Z.; Füredi, A.; Szakács, G.; Veszelka, S.; Sipos, P.; Ózsvári, B.; et al. Sucrose Esters Increase Drug Penetration, But Do Not Inhibit P-Glycoprotein in Caco-2 Intestinal Epithelial Cells. J. Pharm. Sci. 2014, 103, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Trevaskis, N.; Charman, W. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Zughaid, H.; Forbes, B.; Martin, G.P.; Patel, N. Bile salt composition is secondary to bile salt concentration in determining hydrocortisone and progesterone solubility in intestinal mimetic fluids. Int. J. Pharm. 2012, 422, 295–301. [Google Scholar] [CrossRef]
- Kotze, A.; Luessen, H.; Thanou, M.; Verhoef, J.; De Boer, A.; Junginger, H. Chitosan and Chitosan Derivatives as Absorption Enhancers for Peptide Drugs Across Mucosal Epithelia. Drugs Pharm. Sci. 1999, 98, 341–386. [Google Scholar] [CrossRef]
- Borchard, G.; Lueβen, H.L.; de Boer, A.G.; Verhoef, J.; Lehr, C.-M.; Junginger, H.E. The potential of mucoadhesive polymers in enhancing intestinal peptide drug absorption. III: Effects of chitosan-glutamate and carbomer on epithelial tight junctions in vitro. J. Control. Release 1996, 39, 131–138. [Google Scholar] [CrossRef]
- Fan, D.; Wu, X.; Dong, W.; Sun, W.; Li, J.; Tang, X. Enhancement by sodium caprate and sodium deoxycholate of the gastrointestinal absorption of berberine chloride in rats. Drug Dev. Ind. Pharm. 2012, 39, 1447–1456. [Google Scholar] [CrossRef]
- Mathavan, S.; Chen-Tan, N.; Arfuso, F.; Al-Salami, H. A comprehensive study of novel microcapsules incorporating gliclazide and a permeation enhancing bile acid: Hypoglycemic effect in an animal model of Type-1 diabetes. Drug Deliv. 2015, 23, 2869–2880. [Google Scholar] [CrossRef]
- Yu, Q.; Cao, Y.; Liu, L.-F.; Jiang, S.-X.; Yang, Q. Enhancement of sodium taurocholate to the absorption of cefquinome. Pak. J. Pharm. Sci. 2016, 29, 139–143. [Google Scholar]
- Handali, S.; Moghimipour, E.; Tabassi, S.A.S.; Ramezani, M.; Lobenberg, R. Brush border membrane vesicle and Caco-2 cell line: Two experimental models for evaluation of absorption enhancing effects of saponins, bile salts, and some synthetic surfactants. J. Adv. Pharm. Technol. Res. 2016, 7, 75–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaowa, A.; Horibe, T.; Kohno, M.; Kawakami, K. Bile Acid as an Effective Absorption Enhancer for Oral Delivery of Epidermal Growth Factor Receptor–Targeted Hybrid Peptide. J. Pharm. Sci. 2018, 107, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Tajdini, F.; Amini, M.A.; Nafissi-Varcheh, N.; Faramarzi, M.A. Production, physiochemical and antimicrobial properties of fungal chitosan from Rhizomucor miehei and Mucor racemosus. Int. J. Biol. Macromol. 2010, 47, 180–183. [Google Scholar] [CrossRef] [PubMed]
- He, Z.G.; Lian, H.; Sun, J.; Yu, Y.P.; Liu, Y.H.; Wang, Y.J. Supramolecular micellar nanoaggregates based on a novel chitosan/vitamin E succinate copolymer for paclitaxel selective delivery. Int. J. Nanomed. 2011, 6, 3323–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadai, M.; Tajiri, C.; Yoshizumi, H.; Suzuki, Y.; Zhao, Y.L.; Kimura, M.; Tsunekawa, Y.; Hasegawa, T. Effect of Chitosan on Gastrointestinal Absorption of Water-Insoluble Drugs Following Oral Administration in Rats. Biol. Pharm. Bull. 2006, 29, 1941–1946. [Google Scholar] [CrossRef] [Green Version]
- Dodane, V. Effect of chitosan on epithelial permeability and structure. Int. J. Pharm. 1999, 182, 21–32. [Google Scholar] [CrossRef]
- Liang, K.; Jia, Z.-Y.; Jin, X.; Zhang, S.-B.; Li, S.-M. Influence of chitosan nanoparticles as the absorption enhancers on salvianolic acid B In vitro and In vivo evaluation. Pharmacogn. Mag. 2016, 12, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.; Wood, E.; Dornish, M. Effect of Chitosan on Epithelial Cell Tight Junctions. Pharm. Res. 2004, 21, 43–49. [Google Scholar] [CrossRef]
- Schipper, N.G.M.; Vårum, K.M.; Artursson, P. Chitosans as Absorption Enhancers for Poorly Absorbable Drugs. 1: Influence of Molecular Weight and Degree of Acetylation on Drug Transport Across Human Intestinal Epithelial (Caco-2) Cells. Pharm. Res. 1996, 13, 1686–1692. [Google Scholar] [CrossRef]
- Thanou, M.; Verhoef, J.; Junginger, H. Oral drug absorption enhancement by chitosan and its derivatives. Adv. Drug Deliv. Rev. 2001, 52, 117–126. [Google Scholar] [CrossRef]
- Kubbinga, M.; Augustijns, P.; García, M.A.; Heinen, C.; Wortelboer, H.M.; Verwei, M.; Langguth, P. The effect of chitosan on the bioaccessibility and intestinal permeability of acyclovir. Eur. J. Pharm. Biopharm. 2019, 136, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.R. The Interesting Case of Acyclovir Delivered Using Chitosan in Humans: Is it a Drug Issue or Formulation Issue? Pharm. Res. 2015, 33, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Valdes, S.A.; Alzhrani, R.F.; Lansakara-P, D.S.P.; Cui, Z. Effect of a Solid Lipid Nanoparticle Formulation on the Bioavailability of 4-(N)-Docosahexaenoyl 2′, 2′-Difluorodeoxycytidine After Oral Administration. AAPS PharmSciTech 2020, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Fan, D.; Meng, L.; Miao, Y.; Yang, S.; Weng, Y.; He, H.; Tang, X. Enhancing effects of chitosan and chitosan hydrochloride on intestinal absorption of berberine in rats. Drug Dev. Ind. Pharm. 2011, 38, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Kontogiannidou, E.; Meikopoulos, T.; Virgiliou, C.; Bouropoulos, N.; Gika, H.; Vizirianakis, I.; Müllertz, A.; Fatouros, D.G. Towards the development of Self-Nano-Emulsifying Drug Delivery Systems (SNEDDS) containing trimethyl chitosan for the oral delivery of amphotericin B: In vitro assessment and cytocompatibility studies. J. Drug Deliv. Sci. Technol. 2020, 56, 101524. [Google Scholar] [CrossRef]
- Qian, S.; Zhang, Q.; Wang, Y.; Lee, B.; Betageri, G.V.; Chow, M.S.; Huang, M.; Zuo, Z. Bioavailability enhancement of glucosamine hydrochloride by chitosan. Int. J. Pharm. 2013, 455, 365–373. [Google Scholar] [CrossRef]
- Teixeira, M.; Carbone, C.; Souto, E. Beyond liposomes: Recent advances on lipid based nanostructures for poorly soluble/poorly permeable drug delivery. Prog. Lipid Res. 2017, 68, 1–11. [Google Scholar] [CrossRef]
- Baek, J.-S.; Cho, C.-W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140. [Google Scholar] [CrossRef]
- Porter, C.; Charman, W.N. Uptake of drugs into the intestinal lymphatics after oral administration. Adv. Drug Deliv. Rev. 1997, 25, 71–89. [Google Scholar] [CrossRef]
- Garg, A.; Bhalala, K.; Tomar, D.S.; Wahajuddin. In-situ single pass intestinal permeability and pharmacokinetic study of developed Lumefantrine loaded solid lipid nanoparticles. Int. J. Pharm. 2017, 516, 120–130. [Google Scholar] [CrossRef]
- Ansari, M.J.; Anwer, K.; Jamil, S.; Al-Shdefat, R.; Ali, B.E.; Ahmad, M.M. Enhanced oral bioavailability of insulin-loaded solid lipid nanoparticles: Pharmacokinetic bioavailability of insulin-loaded solid lipid nanoparticles in diabetic rats. Drug Deliv. 2015, 23, 1972–1979. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.; Mundada, V.; Sawant, K. Enhanced intestinal absorption of asenapine maleate by fabricating solid lipid nanoparticles using TPGS: Elucidation of transport mechanism, permeability across Caco-2 cell line and in vivo pharmacokinetic studies. Artif. Cells Nanomed. Biotechnol. 2019, 47, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Quach, T.; Han, S.; Lim, S.F.; Yadav, P.; Senyschyn, D.; Trevaskis, N.L.; Simpson, J.S.; Porter, C.J.H. Glyceride-Mimetic Prodrugs Incorporating Self-Immolative Spacers Promote Lymphatic Transport, Avoid First-Pass Metabolism, and Enhance Oral Bioavailability. Angew. Chem. Int. Ed. 2016, 55, 13700–13705. [Google Scholar] [CrossRef]
- Palmerston Mendes, L.; Pan, J.; Torchilin, V.P. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef] [Green Version]
- Beezer, A.; King, A.; Martin, I.; Mitchel, J.; Twyman, L.; Wain, C. Dendrimers as potential drug carriers; encapsulation of acidic hydrophobes within water soluble PAMAM derivatives. Tetrahedron 2003, 59, 3873–3880. [Google Scholar] [CrossRef]
- Yan, C.; Gu, J.; Lv, Y.; Shi, W.; Jing, H. Improved intestinal absorption of water-soluble drugs by acetylation of G2 PAMAM dendrimer nanocomplexes in rat. Drug Deliv. Transl. Res. 2017, 11, 408–415. [Google Scholar] [CrossRef]
- Qi, R.; Zhang, H.; Xu, L.; Shen, W.; Chen, C.; Wang, C.; Cao, Y.; Wang, Y.; van Dongen, M.A.; He, B.; et al. G5 PAMAM dendrimer versus liposome: A comparison study on the in vitro transepithelial transport and in vivo oral absorption of simvastatin. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1141–1151. [Google Scholar] [CrossRef]
- Sadekar, S.; Thiagarajan, G.; Bartlett, K.; Hubbard, D.; Ray, A.; McGill, L.; Ghandehari, H. Poly(amido amine) dendrimers as absorption enhancers for oral delivery of camptothecin. Int. J. Pharm. 2013, 456, 175–185. [Google Scholar] [CrossRef] [Green Version]
- D’Emanuele, A.; Jevprasesphant, R.; Penny, J.; Attwood, D. The use of a dendrimer-propranolol prodrug to bypass efflux transporters and enhance oral bioavailability. J. Control. Release 2004, 95, 447–453. [Google Scholar] [CrossRef]
- Yiyun, C.; Tongwen, X. Dendrimers as Potential Drug Carriers. Part I. Solubilization of Non-Steroidal Anti-Inflammatory Drugs in the Presence of Polyamidoamine Dendrimers. Eur. J. Med. Chem. 2005, 40, 1188–1192. [Google Scholar] [CrossRef]
- Choudhury, H.; Pandey, M.; Gorain, B.; Chatterjee, B.; Madheswaran, T.; Shadab; Mak, K.-K.; Tambuwala, M.; Chourasia, M.K.; Kesharwani, P. Nanoemulsions as Effective Carriers for the Treatment of Lung Cancer. In Nanotechnology-Based Targeted Drug Delivery Systems for Lung Cancer; Academic Press: Cambridge, MA, USA, 2019; pp. 217–247. [Google Scholar] [CrossRef]
- Kawakami, K.; Yoshikawa, T.; Moroto, Y.; Kanaoka, E.; Takahashi, K.; Nishihara, Y.; Masuda, K. Microemulsion formulation for enhanced absorption of poorly soluble drugs: I. Prescription design. J. Control. Release 2002, 81, 65–74. [Google Scholar] [CrossRef]
- Aboofazeli, R. Nanometric-Scaled Emulsions (Nanoemulsions). Iran. J. Pharm. Res. 2010, 9, 325–326. [Google Scholar] [CrossRef] [PubMed]
- Isailović, T.M.; Todosijević, M.N.; Dordević, S.M.; Savić, S.D. Natural Surfactants-Based Micro/Nanoemulsion Systems for NSAIDs—Practical Formulation Approach, Physicochemical and Biopharmaceutical Characteristics/Performances. In Microsized and Nanosized Carriers for Nonsteroidal Anti-Inflammatory Drugs; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Baral, K.; Song, J.-G.; Lee, S.; Bajracharya, R.; Sreenivasulu, G.; Kim, M.; Lee, K.; Han, H.-K. Enhanced Bioavailability of AC1497, a Novel Anticancer Drug Candidate, via a Self-Nanoemulsifying Drug Delivery System. Pharmaceutics 2021, 13, 1142. [Google Scholar] [CrossRef] [PubMed]
- Kuncahyo, I.; Choiri, S.; Fudholi, A.; Martien, R.; Rohman, A. Development of pitavastatin-loaded super-saturable self-nano emulsion: A continues screening and optimization approach using statistical technique. J. Dispers. Sci. Technol. 2021, 1–10. [Google Scholar] [CrossRef]
- Li, M.; Cui, J.; Ngadi, M.O.; Ma, Y. Absorption mechanism of whey-protein-delivered curcumin using Caco-2 cell monolayers. Food Chem. 2015, 180, 48–54. [Google Scholar] [CrossRef]
- Anuar, N.; Sabri, A.H.; Effendi, T.J.B.; Hamid, K.A. Development and characterisation of ibuprofen-loaded nanoemulsion with enhanced oral bioavailability. Heliyon 2020, 6, e04570. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Z.; Bu, H.; Huang, Y.; Gao, Z.; Shen, J.; Zhao, C.; Li, Y. Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: Performance and mechanism. J. Control. Release 2011, 149, 168–174. [Google Scholar] [CrossRef]
- Date, A.A.; Desai, N.; Dixit, R.; Nagarsenker, M. Self-nanoemulsifying drug delivery systems: Formulation insights, applications and advances. Nanomedicine 2010, 5, 1595–1616. [Google Scholar] [CrossRef]
- Thakkar, H.P.; Khunt, A.; Dhande, R.D.; Patel, A.A. Formulation and evaluation of Itraconazole nanoemulsion for enhanced oral bioavailability. J. Microencapsul. 2015, 32, 559–569. [Google Scholar] [CrossRef]
- Li, Y.-J.; Hu, X.-B.; Lu, X.-L.; Liao, D.-H.; Tang, T.-T.; Wu, J.-Y.; Xiang, D.-X. Nanoemulsion-based delivery system for enhanced oral bioavailability and Caco-2 cell monolayers permeability of berberine hydrochloride. Drug Deliv. 2017, 24, 1868–1873. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhang, J.; Wu, L.; Wu, H.; Dai, M. Paeonol nanoemulsion for enhanced oral bioavailability: Optimization and mechanism. Nanomedicine 2018, 13, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Talekar, M.; Singh, A.; Coleman, T.P.; Amiji, M.M. Nanoemulsions in Translational Research—Opportunities and Challenges in Targeted Cancer Therapy. AAPS PharmSciTech 2014, 15, 694–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Drug (s) | Absorption Enhancer | Model | Results | Ref. |
|---|---|---|---|---|
| [14C]-mannitol | Sucrose laurate | In vitro: Caco-2 cell | Papp: 9-fold increase | [57] |
| Sucrose laurate | In vitro: Ussing chamber | Papp: 2.6-fold increase | ||
| Insulin | Sucrose laurate | In situ: rat jejunum and colon | Relative bioavailability (F, %): 8.9% increase | [57] |
| Fluorescein, atenolol, rhodamine 123, and vinblastine | Sucrose laurate | In vitro: Caco-2 cell | Papp: several folds increase for all drugs. | [65] |
| Carbamazepine | Cyclodextrins | In vivo: dogs | Tmax: 0.6-fold decrease Cmax: 0.004-fold increase | [63] |
| Erlotinib | Cyclodextrins | In vivo: rats | Tmax: 5.4-fold decrease Cmax: 3.2-fold increase AUC: 3.6-fold increase | [59] |
| Drug (s) | Model | Results | Ref. |
|---|---|---|---|
| Lumefantrine | In situ: single pass intestinal permeability study | Cellular uptake: 3-fold increase Ka: 2.96-fold increase | [92] |
| In vivo: rat intestine | AUC and Cmax: 2.7-fold increase Tmax: no change | ||
| Curcumin | In vivo: rat intestine | Lymphatic uptake: 6.3-fold increase Oral bioavailability: 9.5-fold increase Cmax: several folds increase Tmax: 2-fold increase AUC: increased | [90] |
| Asenapine maleate | In vitro: Caco-2 cell | Papp: increased | |
| In vivo: rat intestine | Bioavailability: 50.19-fold increase AUC: increased Cmax: 20.78-fold increase Tmax: 8-fold increase | [94] | |
| 4-(N)-docosahexaenoyl 2′, 2′-difluorodeoxycytidine (DHA-dFdC) | In vitro: simulated gastrointestinal fluids | Cmax: increased Tmax: decreased AUC: increased | [95] |
| Insulin | Ex vivo: rat everted intestinal sac | Papp: 2-fold increase Cmax: increased AUC: increased | [93] |
| Drug (s) | Model | Results | Ref. |
|---|---|---|---|
| Paeonol | In situ: single-pass intestine perfusion | Papp: 1.64-fold increase Ka: 0.65-fold increase | [115] |
| In vitro: everted gut sacs | Papp: increased (p < 0.01) | ||
| In vitro: Caco-2 cell | Papp: increased | ||
| In vivo: rat intestinal uptake | AUC0→t: 4.27-fold increase Cmax: 4.02-fold increase Tmax: 40-min increase | ||
| Berberine hydrochloride | In vivo: rat intestinal uptake | AUC: 4.4-fold increase Cmax: 1.6-fold increase Tmax: 4.3-fold increase | [114] |
| In vitro: Caco-2 cell | Papp: increased to 0.574 ± 0.18 × 10−8 cm/s | ||
| Curcumin | In vitro: Caco-2 cell | The digested nanoemulsion had the highest permeation rate (7.07 × 105 cm/s) | [109] |
| Candesartan cilexetil | In situ single-pass intestinal perfusion | Cellular uptake: 1.75-, 1.93-, and 1.84-fold increase in the duodenum, jejunum, and ileum, respectively. | [111] |
| In vitro: Caco-2 cell | The cellular uptake of CCN at 4 °C reduced 92% compared with that at 37 °C (p < 0.01) | ||
| In vivo: rat intestinal uptake | AUC: 10-fold increase Cmax: 27-fold increase Tmax: no change | ||
| Ibuprofen | In vitro diffusion chamber: rat intestinal membrane | Papp: 10.6-fold | [110] |
| In vivo: rat intestinal uptake | AUC 0–6h: 2.2-fold increase Cmax: 27-fold increase Tmax: no change |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals 2022, 15, 975. https://doi.org/10.3390/ph15080975
Azman M, Sabri AH, Anjani QK, Mustaffa MF, Hamid KA. Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals. 2022; 15(8):975. https://doi.org/10.3390/ph15080975
Chicago/Turabian StyleAzman, Maisarah, Akmal H. Sabri, Qonita Kurnia Anjani, Mohd Faiz Mustaffa, and Khuriah Abdul Hamid. 2022. "Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery" Pharmaceuticals 15, no. 8: 975. https://doi.org/10.3390/ph15080975
APA StyleAzman, M., Sabri, A. H., Anjani, Q. K., Mustaffa, M. F., & Hamid, K. A. (2022). Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals, 15(8), 975. https://doi.org/10.3390/ph15080975