
Synthesis and Evaluation of Antiproliferative Activity, Topoisomerase IIα Inhibition, DNA Binding and Non-Clinical Toxicity of New Acridine–Thiosemicarbazone Derivatives

 ,
,  ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Cytotoxicity Assay
2.3. Human Topoisomerase IIα Inhibition Assay
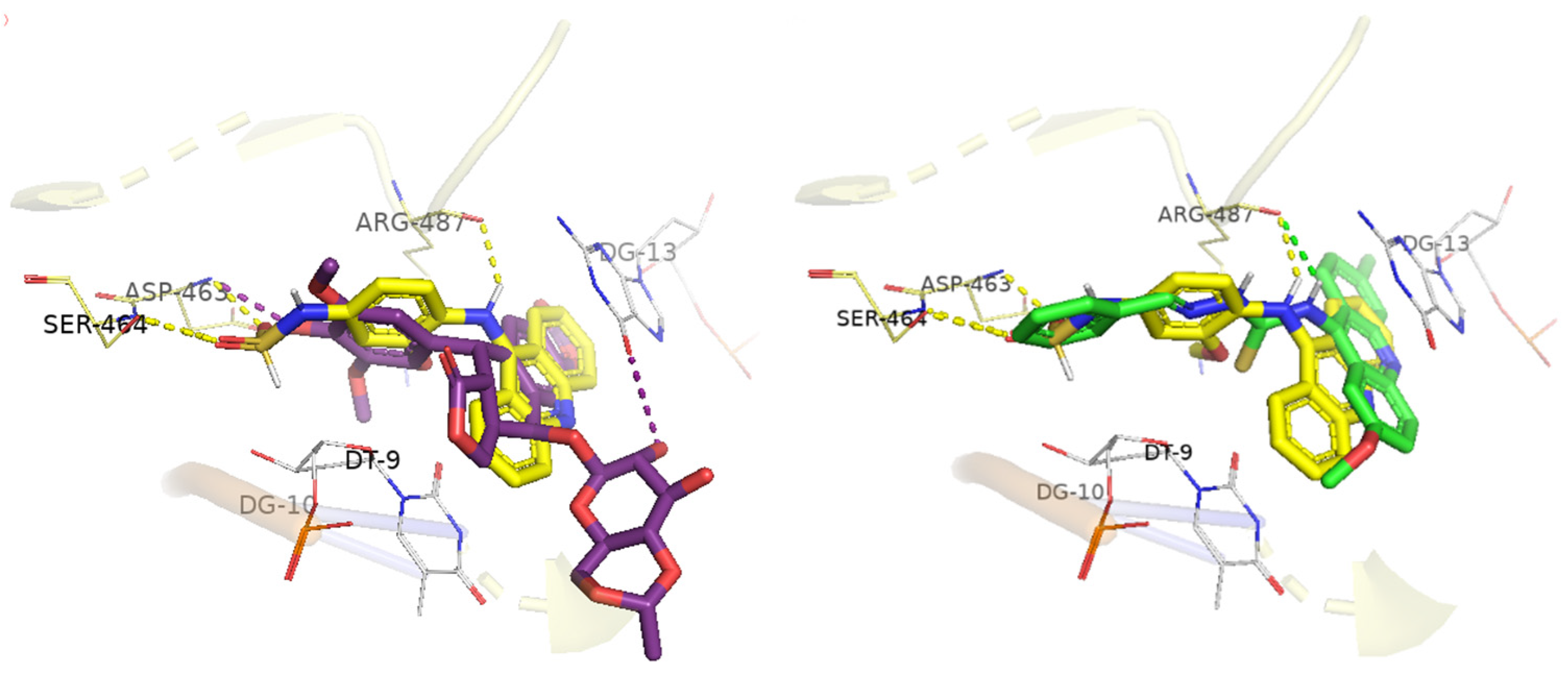
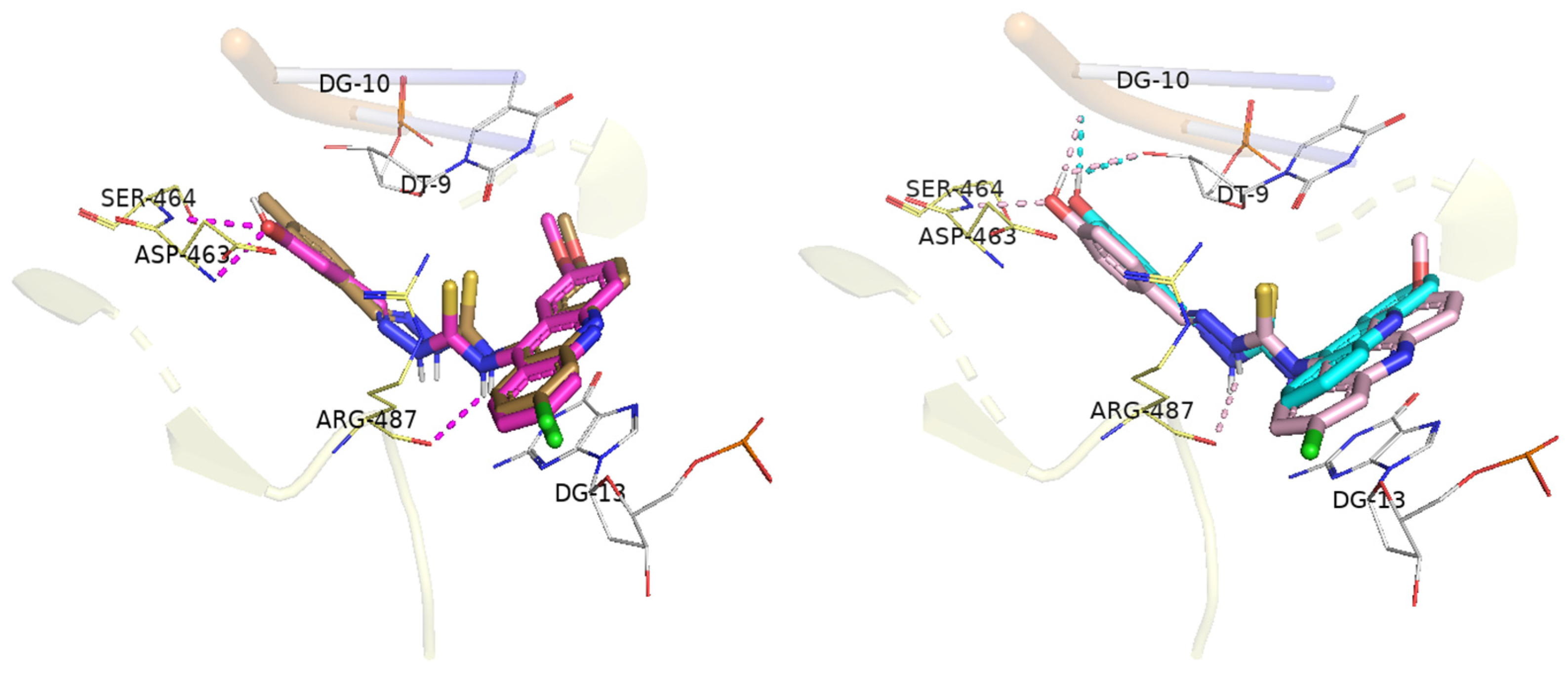
2.4. Molecular Docking
2.5. DNA Interaction Studies
2.5.1. CT DNA Binding by UV–Vis Absorption Spectroscopy
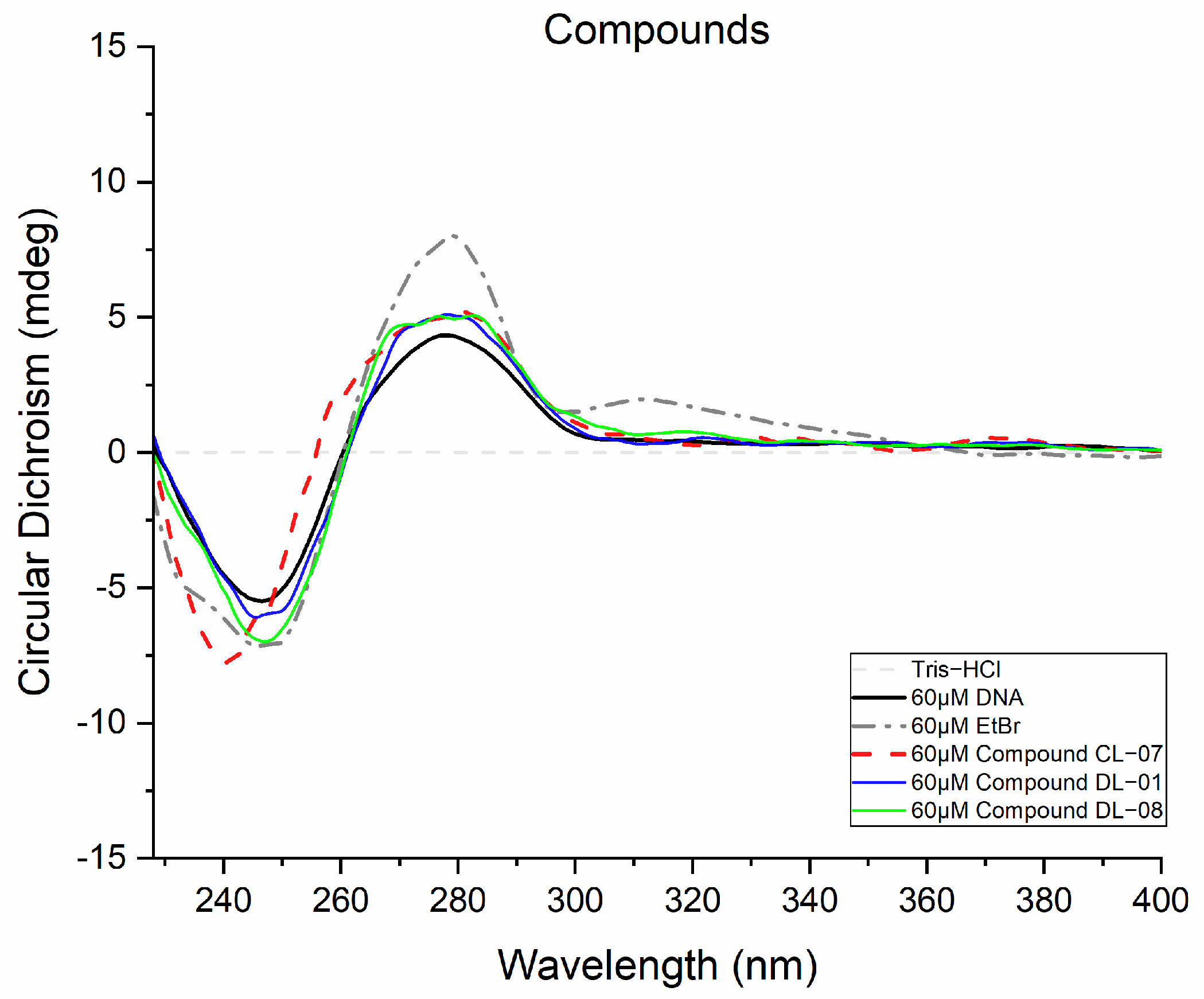
2.5.2. Circular Dichroism
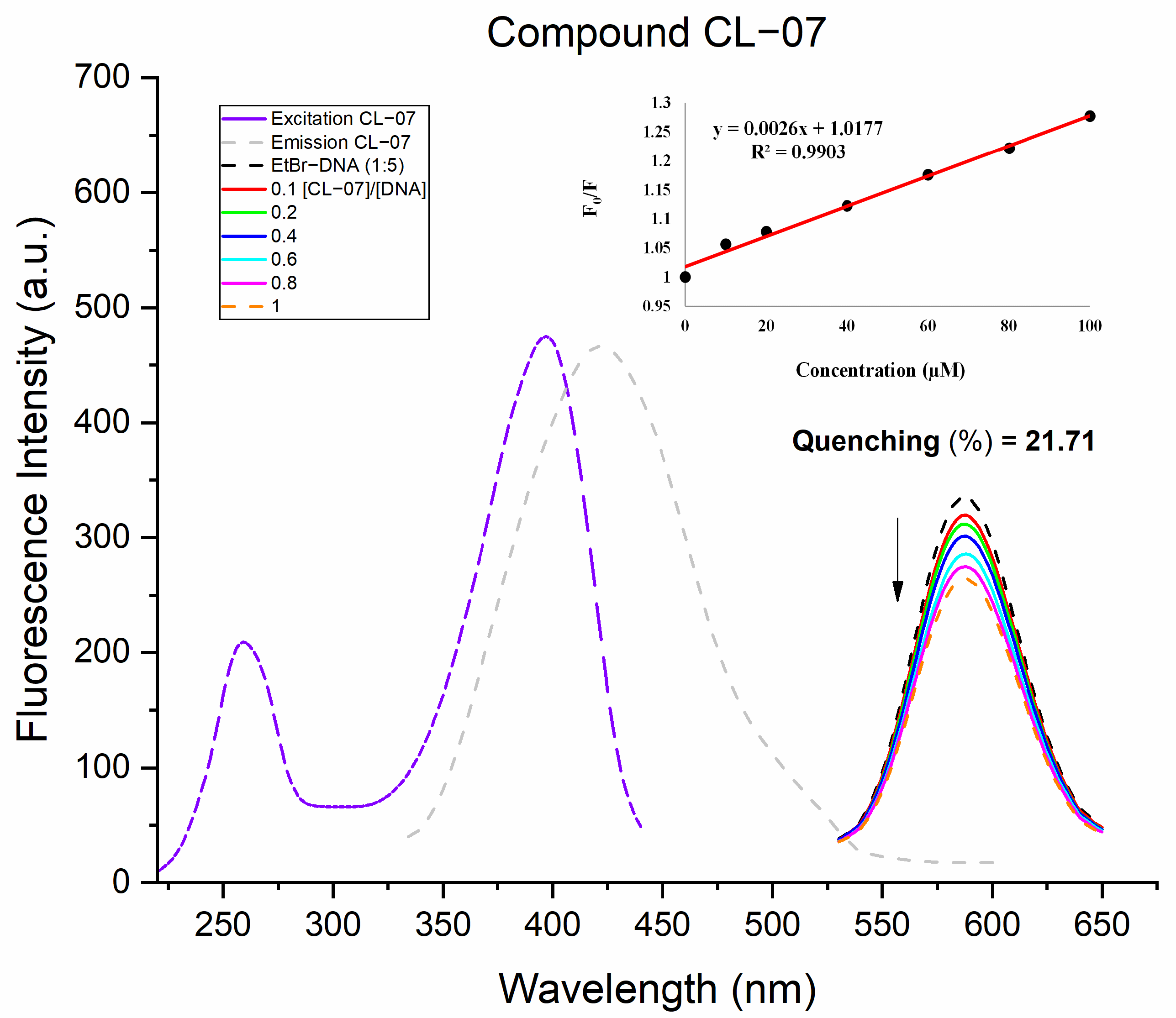
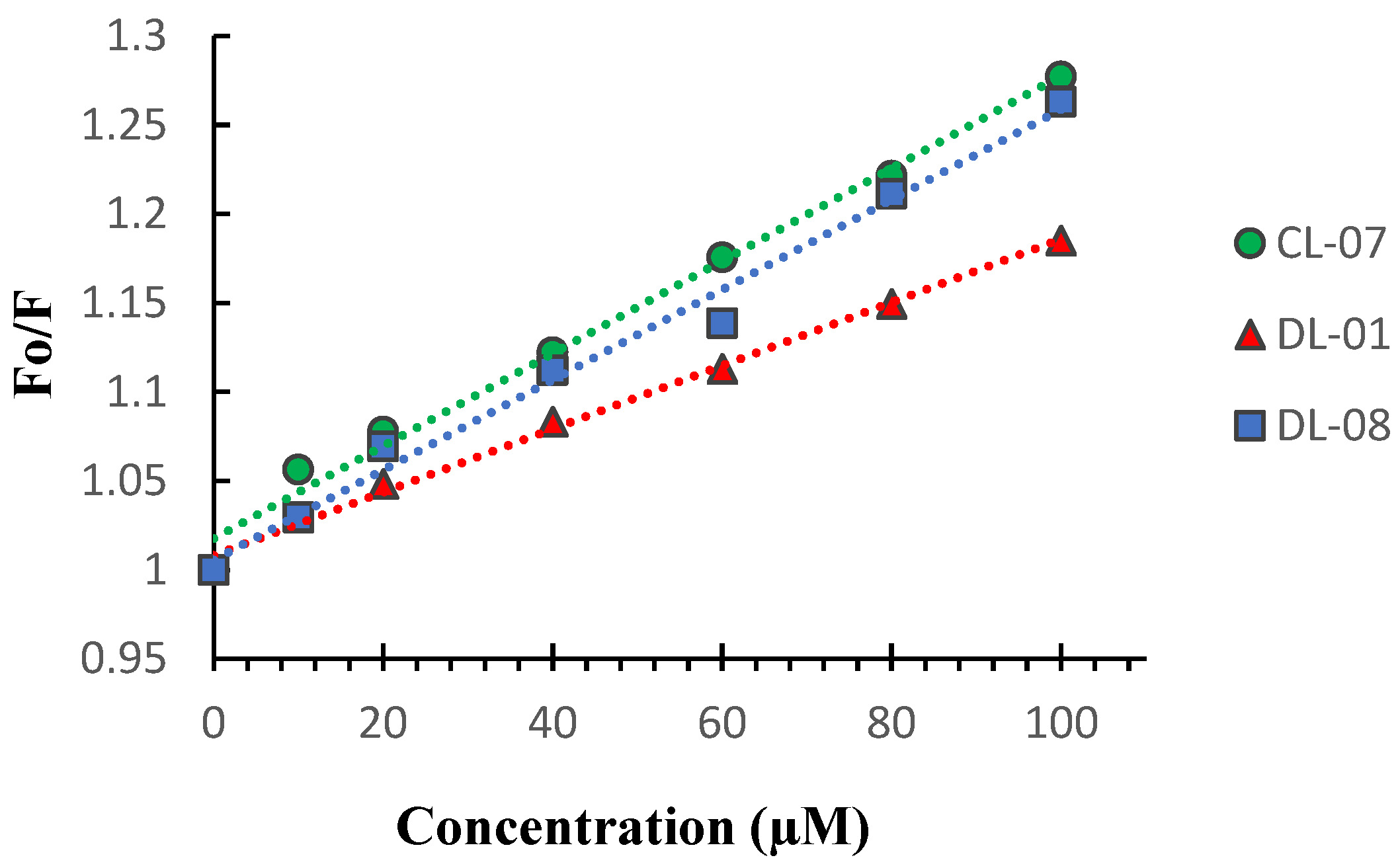
2.5.3. Fluorescent Studies with EtBr
2.6. Evaluation of the Non-Clinical Toxicity
2.7. Cytotoxicity Assay on Leukemic Normal and Resistant Cells
3. Materials and Methods
3.1. Chemicals, Reagents, and Equipment
3.2. Synthesis of the Novel Derivatives
3.2.1. Synthesis of 9-Chloroacridine Cores
- Route A:
- Route B:
3.2.2. Obtaining Thiosemicarbazone Intermediates
3.2.3. Obtaining Acridine–Thiosemicarbazone Derivatives (CL 01-10 and DL 01-10)
3.2.4. (E)-N-(acridin-9-yl)-2-benzylidenehydrazine-1-carbothioamide (CL-01)
3.2.5. (E)-N-(acridin-9-yl)-2-(4-chlorobenzylidene)hydrazine-1-carbothioamide (CL-02)
3.2.6. (E)-N-(acridin-9-yl)-2-(4-methoxybenzylidene)hydrazine-1-carbothioamide (CL-03)
3.2.7. (E)-N-(acridin-9-yl)-2-(2,4-dichlorobenzylidene)hydrazine-1-carbothioamide (CL-04)
3.2.8. (E)-N-(acridin-9-yl)-2-(4-nitrobenzylidene)hydrazine-1-carbothioamide (CL-05)
3.2.9. (E)-N-(acridin-9-yl)-2-(4-methylbenzylidene)hydrazine-1-carbothioamide (CL-06)
3.2.10. (E)-N-(acridin-9-yl)-2-(4-hydroxybenzylidene)hydrazine-1-carbothioamide (CL-07)
3.2.11. (E)-N-(acridin-9-yl)-2-(3-hydroxybenzylidene)hydrazine-1-carbothioamide (CL-08)
3.2.12. (E)-N-(acridin-9-yl)-2-(4-(dimethylamino)benzylidene)hydrazine-1-carbothioamide (CL-09)
3.2.13. (E)-N-(acridin-9-yl)-2-(4-bromobenzylidene)hydrazine-1-carbothioamide (CL-10)
3.2.14. (E)-2-benzylidene-N-(6-chloro-2-methoxyacridin-9-yl)hydrazine-1-carbothioamide (DL-01)
3.2.15. (E)-N-(6-chloro-2-methoxyacridin-9-yl)-2-(4-chlorobenzylidene)hydrazine-1-carbothioamide (DL-02)
3.2.16. (E)-N-(6-chloro-2-methoxyacridin-9-yl)-2-(4-methoxybenzylidene)hydrazine-1-carbothioamide (DL-03)
3.2.17. (E)-N-(6-chloro-2-methoxyacridin-9-yl)-2-(2,4-dichlorobenzylidene)hydrazine-1-carbothioamide (DL-04)
3.2.18. (E)-N-(6-chloro-2-methoxyacridin-9-yl)-2-(4-nitrobenzylidene)hydrazine-1-carbothioamide (DL-05)
3.2.19. (E)-N-(6-chloro-2-methoxyacridin-9-yl)-2-(4-methylbenzylidene)hydrazine-1-carbothioamide (DL-06)
3.2.20. (E)-N-(6-chloro-2-methoxyacridin-9-yl)-2-(4-hydroxybenzylidene)hydrazine-1-carbothioamide (DL-07)
3.2.21. (E)-N-(6-chloro-2-methoxyacridin-9-yl)-2-(3-hydroxybenzylidene)hydrazine-1-carbothioamide (DL-08)
3.2.22. (E)-N-(6-chloro-2-methoxyacridin-9-yl)-2-(4-(dimethylamino)benzylidene)hydrazine-1-carbothioamide (DL-09)
3.2.23. (E)-2-(4-bromobenzylidene)-N-(6-chloro-2-methoxyacridin-9-yl)hydrazine-1-carbothioamide (DL-10)
3.3. Cytotoxic Activity
3.3.1. Cells
3.3.2. Cytotoxicity Assay
3.4. Interaction with DNA
3.4.1. UV–Visible Spectroscopy
3.4.2. Circular Dichroism
3.4.3. Fluorescence Competition with EtBr
3.5. Human Topoisomerase IIα Inhibition Assay
3.6. Molecular Docking
3.7. Evaluation of the Non-Clinical Toxicity
3.7.1. Acute Toxicity Study (14 Days)
3.7.2. Necropsy, Macroscopic Analysis and Organ Weight
3.8. Cytotoxicity Assay on Leukemic Cells
3.8.1. Cells
3.8.2. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Markham, M.J.; Wachter, K.; Agarwal, N.; Bertagnolli, M.M.; Chang, S.M.; Dale, W.; Diefenbach, C.S.M.; Rodriguez-Galindo, C.; George, D.J.; Gilligan, T.D.; et al. Clinical Cancer Advances 2020: Annual Report on Progress Against Cancer From the American Society of Clinical Oncology. J. Clin. Oncol. 2020, 38, 1081. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, W.; Thompson, P.; Hannun, Y.A. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat. Commun. 2018, 9, 3490. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.S.; Silva, D.K.F.; Lisboa, T.M.H.; Gouveia, R.G.; Ferreira, R.C.; De Moura, R.O.; Da Silva, J.M.; De Almeida Lima, É.; Rodrigues-Mascarenhas, S.; Da Silva, P.M.; et al. Anticancer Effect of a Spiro-acridine Compound Involves Immunomodulatory and Anti-angiogenic Actions. Anticancer Res. 2020, 40, 5049–5057. [Google Scholar] [CrossRef]
- Salem, O.M.; Vilková, M.; JanoĿková, J.; Jendželovský, R.; FedoroĿko, P.; Žilecká, E.; Kašpárková, J.; Brabec, V.; Imrich, J.; Kožurková, M. New spiro tria(thia)zolidine acridines as topoisomerase inhibitors, DNA binders and cytostatic compounds. Int. J. Biol. Macromol. 2016, 86, 690–700. [Google Scholar] [CrossRef]
- Haider, M.R.; Ahmad, K.; Siddiqui, N.; Ali, Z.; Akhtar, M.J.; Fuloria, N.; Fuloria, S.; Ravichandran, M.; Yar, M.S. Novel 9-(2-(1-arylethylidene)hydrazinyl)acridine derivatives: Target Topoisomerase 1 and growth inhibition of HeLa cancer cells. Bioorg. Chem. 2019, 88, 102962. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- McKie, S.J.; Neuman, K.C.; Maxwell, A. DNA topoisomerases: Advances in understanding of cellular roles and multi-protein complexes via structure-function analysis. BioEssays 2021, 43, 2000286. [Google Scholar] [CrossRef]
- Riccio, A.A.; Schellenberg, M.J.; Williams, R.S. Molecular mechanisms of topoisomerase 2 DNA–protein crosslink resolution. Cell. Mol. Life Sci. 2020, 77, 81–91. [Google Scholar] [CrossRef]
- Kitdumrongthum, S.; Reabroi, S.; Suksen, K.; Tuchinda, P.; Munyoo, B.; Mahalapbutr, P.; Rungrotmongkol, T.; Ounjai, P.; Chairoungdua, A. Inhibition of topoisomerase IIα and induction of DNA damage in cholangiocarcinoma cells by altholactone and its halogenated benzoate derivatives. Biomed. Pharmacother. 2020, 127, 110149. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Berger, J.M. Cell Cycle-Dependent Control and Roles of DNA Topoisomerase II. Genes 2019, 10, 859. [Google Scholar] [CrossRef] [PubMed]
- Matias-Barrios, V.M.; Radaeva, M.; Song, Y.; Alperstein, Z.; Lee, A.R.; Schmitt, V.; Lee, J.; Ban, F.; Xie, N.; Qi, J.; et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Front. Oncol. 2021, 10, 633142. [Google Scholar] [CrossRef]
- Coss, A.; Tosetto, M.; Fox, E.J.; Sapetto-Rebow, B.; Gorman, S.; Kennedy, B.N.; Lloyd, A.T.; Hyland, J.M.; O’Donoghue, D.P.; Sheahan, K.; et al. Increased topoisomerase IIα expression in colorectal cancer is associated with advanced disease and chemotherapeutic resistance via inhibition of apoptosis. Cancer Lett. 2009, 276, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Rody, A.; Karn, T.; Ruckhäberle, E.; Müller, V.; Gehrmann, M.; Solbach, C.; Ahr, A.; Gätje, R.; Holtrich, U.; Kaufmann, M. Gene expression of topoisomerase II alpha (TOP2A) by microarray analysis is highly prognostic in estrogen receptor (ER) positive breast cancer. Breast Cancer Res. Treat. 2009, 113, 457–466. [Google Scholar] [CrossRef]
- An, X.; Xu, F.; Luo, R.; Zheng, Q.; Lu, J.; Yang, Y.; Qin, T.; Yuan, Z.; Shi, Y.; Jiang, W.; et al. The prognostic significance of topoisomerase II alpha protein in early stage luminal breast cancer. BMC Cancer 2018, 18, 331. [Google Scholar] [CrossRef]
- Vann, K.R.; Oviatt, A.A.; Osheroff, N. Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry 2021, 60, 1630–1641. [Google Scholar] [CrossRef]
- Ketron, A.C.; Denny, W.A.; Graves, D.E.; Osheroff, N. Amsacrine as a Topoisomerase II Poison: Importance of Drug–DNA Interactions. Biochemistry 2012, 51, 1730–1739. [Google Scholar] [CrossRef]
- Loboda, K.B.; Valjavec, K.; Štampar, M.; Wolber, G.; Žegura, B.; Filipič, M.; Dolenc, M.S.; Perdih, A. Design and synthesis of 3,5-substituted 1,2,4-oxadiazoles as catalytic inhibitors of human DNA topoisomerase IIα. Bioorg. Chem. 2020, 99, 103828. [Google Scholar] [CrossRef]
- Skok, Ž.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2020, 63, 884–904. [Google Scholar] [CrossRef]
- Li, D.; Yuan, Z.; Chen, S.; Zhang, C.; Song, L.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Synthesis and biological research of novel azaacridine derivatives as potent DNA-binding ligands and topoisomerase II inhibitors. Bioorg. Med. Chem. 2017, 25, 3437–3446. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Pyronaridine: An update of its pharmacological activities and mechanisms of action. Biopolymers 2021, 112, e23398. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Huang, X.-S.; Wu, J.-F.; Yang, L.; Zheng, Y.-T.; Shen, Y.-M.; Li, Z.-Y.; Li, X. Discovery of Novel Topoisomerase II Inhibitors by Medicinal Chemistry Approaches. J. Med. Chem. 2018, 61, 8947–8980. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-M.; Chen, X.; Deng, Y.; Lu, J. Recent advances of N-heterocyclic carbenes in the applications of constructing carbo- and heterocyclic frameworks with potential biological activity. RSC Adv. 2021, 11, 38060–38078. [Google Scholar] [CrossRef]
- Silva, C.F.M.; Pinto, D.C.G.A.; Fernandes, P.A.; Silva, A.M.S. Evolution of Acridines and Xanthenes as a Core Structure for the Development of Antileishmanial Agents. Pharmaceuticals 2022, 15, 148. [Google Scholar] [CrossRef]
- Gabriel, I. ‘Acridines’ as New Horizons in Antifungal Treatment. Molecules 2020, 25, 1480. [Google Scholar] [CrossRef]
- Kozurkova, M.; Sabolova, D.; Kristian, P. A new look at 9-substituted acridines with various biological activities. J. Appl. Toxicol. 2021, 41, 175–189. [Google Scholar] [CrossRef]
- Albino, S.L.; da Silva, J.M.; de C. Nobre, M.S.; de M. e Silva, Y.M.S.; Santos, M.B.; de Araújo, R.S.A.; do C. A. de Lima, M.; Schmitt, M.; de Moura, R.O. Bioprospecting of Nitrogenous Heterocyclic Scaffolds with Potential Action for Neglected Parasitosis: A Review. Curr. Pharm. Des. 2020, 26, 4112–4150. [Google Scholar] [CrossRef]
- Chen, G.; Seukep, A.J.; Guo, M. Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs. Mar. Drugs 2020, 18, 545. [Google Scholar] [CrossRef]
- Czarnecka, K.; Lisiecki, P.; Szewczyk, E.; Chufarova, N.; Wójtowicz, P.; Kręcisz, P.; Szymański, P. New acridine derivatives as promising agents against methicillin-resistant staphylococci—From tests to in silico analysis. Comput. Biol. Chem. 2020, 88, 107321. [Google Scholar] [CrossRef]
- Medapi, B.; Meda, N.; Kulkarni, P.; Yogeeswari, P.; Sriram, D. Development of acridine derivatives as selective Mycobacterium tuberculosis DNA gyrase inhibitors. Bioorg. Med. Chem. 2016, 24, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Gündüz, M.G.; Tahir, M.N.; Armaković, S.; Özkul Koçak, C.; Armaković, S.J. Design, synthesis and computational analysis of novel acridine-(sulfadiazine/sulfathiazole) hybrids as antibacterial agents. J. Mol. Struct. 2019, 1186, 39–49. [Google Scholar] [CrossRef]
- Silva, M.d.M.; Macedo, T.S.; Teixeira, H.M.P.; Moreira, D.R.M.; Soares, M.B.P.; Pereira, A.L.d.C.; Serafim, V.d.L.; Mendonça-Júnior, F.J.B.; do Carmo, A.; de Lima, M.; et al. Correlation between DNA/HSA-interactions and antimalarial activity of acridine derivatives: Proposing a possible mechanism of action. J. Photochem. Photobiol. B Biol. 2018, 189, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Fonte, M.; Tassi, N.; Fontinha, D.; Bouzón-Arnáiz, I.; Ferraz, R.; Araújo, M.J.; Fernàndez-Busquets, X.; Prudêncio, M.; Gomes, P.; Teixeira, C. 4,9-Diaminoacridines and 4-Aminoacridines as Dual-Stage Antiplasmodial Hits. ChemMedChem 2021, 16, 788–792. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, K.; Raja Kumar, S.; Puri, S.K.; Chauhan, P.M.S. Synthesis of new 4-aminoquinolines and quinoline–acridine hybrids as antimalarial agents. Bioorg. Med. Chem. Lett. 2010, 20, 7059–7063. [Google Scholar] [CrossRef]
- Gao, C.; Li, B.; Zhang, B.; Sun, Q.; Li, L.; Li, X.; Chen, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and biological evaluation of benzimidazole acridine derivatives as potential DNA-binding and apoptosis-inducing agents. Bioorg. Med. Chem. 2015, 23, 1800–1807. [Google Scholar] [CrossRef]
- Cisáriková, A.; Barbieriková, Z.; Janovec, L.; Imrich, J.; Hunáková, L.; Bačová, Z.; Paulíková, H. Acridin-3,6-dialkyldithiourea hydrochlorides as new photosensitizers for photodynamic therapy of mouse leukemia cells. Bioorg. Med. Chem. 2016, 24, 2011–2022. [Google Scholar] [CrossRef]
- de Almeida, S.M.V.; Lafayette, E.A.; Silva, W.L.; de Lima Serafim, V.; Menezes, T.M.; Neves, J.L.; Ruiz, A.L.T.G.; de Carvalho, J.E.; de Moura, R.O.; Beltrão, E.I.C.; et al. New spiro-acridines: DNA interaction, antiproliferative activity and inhibition of human DNA topoisomerases. Int. J. Biol. Macromol. 2016, 92, 467–475. [Google Scholar] [CrossRef]
- Dai, Q.; Chen, J.; Gao, C.; Sun, Q.; Yuan, Z.; Jiang, Y. Design, synthesis and biological evaluation of novel phthalazinone acridine derivatives as dual PARP and Topo inhibitors for potential anticancer agents. Chin. Chem. Lett. 2020, 31, 404–408. [Google Scholar] [CrossRef]
- Rupar, J.; Dobričić, V.; Grahovac, J.; Radulović, S.; Skok, Ž.; Ilaš, J.; Aleksić, M.; Brborić, J.; Čudina, O. Synthesis and evaluation of anticancer activity of new 9-acridinyl amino acid derivatives. RSC Med. Chem. 2020, 11, 378–386. [Google Scholar] [CrossRef]
- Heffeter, P.; Pape, V.F.S.; Enyedy, É.A.; Keppler, B.K.; Szakacs, G.; Kowol, C.R. Anticancer Thiosemicarbazones: Chemical Properties, Interaction with Iron Metabolism, and Resistance Development. Antioxid. Redox Signal. 2019, 30, 1062–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalowich, J.C.; Wu, X.; Zhang, R.; Kanagasabai, R.; Hornbaker, M.; Hasinoff, B.B. The anticancer thiosemicarbazones Dp44mT and triapine lack inhibitory effects as catalytic inhibitors or poisons of DNA topoisomerase IIα. Biochem. Pharmacol. 2012, 84, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.M.; Nascimento, G.P.; Magalhães, K.G.; Iglesias, B.A.; Gatto, C.C. Crystal structures, DNA-binding ability and influence on cellular viability of gold(I) complexes of thiosemicarbazones. J. Coord. Chem. 2018, 71, 502–519. [Google Scholar] [CrossRef]
- Queiroz, C.M.; de Oliveira Filho, G.B.; Espíndola, J.W.P.; do Nascimento, A.V.; Aliança, A.S.D.S.; de Lorena, V.M.B.; Feitosa, A.P.S.; da Silva, P.R.; Alves, L.C.; Leite, A.C.L.; et al. Thiosemicarbazone and thiazole: In vitro evaluation of leishmanicidal and ultrastructural activity on Leishmania infantum. Med. Chem. Res. 2020, 29, 2050–2065. [Google Scholar] [CrossRef]
- Zeglis, B.M.; Divilov, V.; Lewis, J.S. Role of Metalation in the Topoisomerase IIα Inhibition and Antiproliferation Activity of a Series of α-Heterocyclic-N 4 -Substituted Thiosemicarbazones and Their Cu(II) Complexes. J. Med. Chem. 2011, 54, 2391–2398. [Google Scholar] [CrossRef]
- Govender, H.; Mocktar, C.; Kumalo, H.M.; Koorbanally, N.A. Synthesis, antibacterial activity and docking studies of substituted quinolone thiosemicarbazones. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 1074–1081. [Google Scholar] [CrossRef]
- Matsa, R.; Makam, P.; Kaushik, M.; Hoti, S.L.; Kannan, T. Thiosemicarbazone derivatives: Design, synthesis and in vitro antimalarial activity studies. Eur. J. Pharm. Sci. 2019, 137, 104986. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Khaleefathullah, S.; Kaveri, K.; Palani, G.; Ramanathan, G.; Thennarasu, S.; Tirichurapalli Sivagnanam, U. Antiviral activity of Thiosemicarbazones derived from α-amino acids against Dengue virus: Anti Viralactivity of Thiosemicarbazones Derivatives. J. Med. Virol. 2017, 89, 546–552. [Google Scholar] [CrossRef]
- Haribabu, J.; Jeyalakshmi, K.; Arun, Y.; Bhuvanesh, N.S.P.; Perumal, P.T.; Karvembu, R. Synthesis, DNA/protein binding, molecular docking, DNA cleavage and in vitro anticancer activity of nickel(II) bis(thiosemicarbazone) complexes. RSC Adv. 2015, 5, 46031–46049. [Google Scholar] [CrossRef]
- De Almeida, S.; Lafayette, E.; da Silva, L.; Amorim, C.; de Oliveira, T.; Ruiz, A.; de Carvalho, J.; de Moura, R.; Beltrão, E.; de Lima, M.; et al. Synthesis, DNA Binding, and Antiproliferative Activity of Novel Acridine-Thiosemicarbazone Derivatives. Int. J. Mol. Sci. 2015, 16, 13023–13042. [Google Scholar] [CrossRef]
- Ribeiro, A.G.; de Almeida, S.M.V.; de Oliveira, J.F.; de Lima Souza, T.R.C.; Dos Santos, K.L.; de Barros Albuquerque, A.P.; Nogueira, M.C.D.B.L.; de Carvalho Junior, L.B.; de Moura, R.O.; da Silva, A.C.; et al. Novel 4-quinoline-thiosemicarbazone derivatives: Synthesis, antiproliferative activity, in vitro and in silico biomacromolecule interaction studies and topoisomerase inhibition. Eur. J. Med. Chem. 2019, 182, 111592. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Filho, F.A.; de Freitas Souza, T.; Ribeiro, A.G.; Alves, J.E.F.; de Oliveira, J.F.; de Lima Souza, T.R.C.; de Moura, R.O.; do Carmo Alves de Lima, M.; de Carvalho Junior, L.B.; de Almeida, S.M.V. Topoisomerase inhibition and albumin interaction studies of acridine-thiosemicarbazone derivatives. Int. J. Biol. Macromol. 2019, 138, 582–589. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, J.F.; da Silva, A.L.; Vendramini-Costa, D.B.; da Cruz Amorim, C.A.; Campos, J.F.; Ribeiro, A.G.; Olímpio de Moura, R.; Neves, J.L.; Ruiz, A.L.T.G.; Ernesto de Carvalho, J.; et al. Synthesis of thiophene-thiosemicarbazone derivatives and evaluation of their in vitro and in vivo antitumor activities. Eur. J. Med. Chem. 2015, 104, 148–156. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, J.F.; Lima, T.S.; Vendramini-Costa, D.B.; de Lacerda Pedrosa, S.C.B.; Lafayette, E.A.; da Silva, R.M.F.; de Almeida, S.M.V.; de Moura, R.O.; Ruiz, A.L.T.G.; de Carvalho, J.E.; et al. Thiosemicarbazones and 4-thiazolidinones indole-based derivatives: Synthesis, evaluation of antiproliferative activity, cell death mechanisms and topoisomerase inhibition assay. Eur. J. Med. Chem. 2017, 136, 305–314. [Google Scholar] [CrossRef]
- Lima, L.; Barreiro, E. Bioisosterism: A Useful Strategy for Molecular Modification and Drug Design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Richter, L. Topliss Batchwise Schemes Reviewed in the Era of Open Data Reveal Significant Differences between Enzymes and Membrane Receptors. J. Chem. Inf. Model. 2017, 57, 2575–2583. [Google Scholar] [CrossRef]
- Topliss, J.G. Utilization of Operational Schemes for Analog Synthesis in Drug Design? J. Med. Chem. 1972, 15, 1006–1011. [Google Scholar] [CrossRef]
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA binding—A review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal. 2011, 9, 13. [Google Scholar] [CrossRef]
- Aapro, M.; Bossi, P.; Dasari, A.; Fallowfield, L.; Gascón, P.; Geller, M.; Jordan, K.; Kim, J.; Martin, K.; Porzig, S. Digital health for optimal supportive care in oncology: Benefits, limits, and future perspectives. Support Care Cancer 2020, 28, 4589–4612. [Google Scholar] [CrossRef]
- Pearce, A.; Haas, M.; Viney, R.; Pearson, S.-A.; Haywood, P.; Brown, C.; Ward, R. Incidence and severity of self-reported chemotherapy side effects in routine care: A prospective cohort study. PLoS ONE 2017, 12, e0184360. [Google Scholar] [CrossRef]
- Lisboa, T.; Silva, D.; Duarte, S.; Ferreira, R.; Andrade, C.; Lopes, A.L.; Ribeiro, J.; Farias, D.; Moura, R.; Reis, M.; et al. Toxicity and Antitumor Activity of a Thiophene–Acridine Hybrid. Molecules 2019, 25, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Kusuzaki, K.; Matsubara, T.; Matsumine, A.; Asanuma, K.; Satonaka, H.; Uchida, A.; Sudo, A. Determination of the LD50 of Acridine Orange via Intravenous Administration in Mice in Preparation for Clinical Application to Cancer Therapy. In Vivo 2014, 28, 523–527. [Google Scholar]
- Kumar, P.; Kumar, R.; Prasad, D.N. Synthesis and anticancer study of 9-aminoacridine derivatives. Arab. J. Chem. 2013, 6, 79–85. [Google Scholar] [CrossRef]
- Hosseinpoor, H.; Iraji, A.; Edraki, N.; Pirhadi, S.; Attarroshan, M.; Khoshneviszadeh, M.; Khoshneviszadeh, M. A Series of Benzylidenes Linked to Hydrazine-1-carbothioamide as Tyrosinase Inhibitors: Synthesis, Biological Evaluation and Structure−Activity Relationship. Chem. Biodiver. 2020, 17, e2000285. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, S.; Wang, Y.; Li, R.; Wang, J.; Wang, L.; Zhao, Y.; Gong, P. Design, synthesis and biological evaluation of novel thieno[3,2-d]pyrimidine derivatives possessing diaryl semicarbazone scaffolds as potent antitumor agents. Eur. J. Med. Chem. 2014, 87, 782–793. [Google Scholar] [CrossRef]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618. [Google Scholar] [CrossRef]
- Silva, V.R.; Corrêa, R.S.; Santos, L.d.S.; Soares, M.B.P.; Batista, A.A.; Bezerra, D.P. A ruthenium-based 5-fluorouracil complex with enhanced cytotoxicity and apoptosis induction action in HCT116 cells. Sci. Rep. 2018, 8, 288. [Google Scholar] [CrossRef]
- Gouveia, R.G.; Ribeiro, A.G.; Segundo, M.Â.S.P.; de Oliveira, J.F.; de Lima, M.d.C.A.; de Lima Souza, T.R.C.; de Almeida, S.M.V.; de Moura, R.O. Synthesis, DNA and protein interactions and human topoisomerase inhibition of novel Spiroacridine derivatives. Bioorg. Med. Chem. 2018, 26, 5911–5921. [Google Scholar] [CrossRef]
- Wolf, S.J.; Wakelin, L.P.G.; He, Z.; Stewart, B.W.; Catchpoole, D.R. In vitro assessment of novel transcription inhibitors and topoisomerase poisons in rhabdomyosarcoma cell lines. Cancer Chemother. Pharmacol. 2009, 64, 1059–1069. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Chen, S.-F.; Wu, C.-C.; Liao, Y.-W.; Lin, T.-S.; Liu, K.-T.; Chen, Y.-S.; Li, T.-K.; Chien, T.-C.; Chan, N.-L. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt(II)-methionine coordination chemistry. Nucleic Acids Res. 2017, 45, 10861–10871. [Google Scholar] [CrossRef]
- Gilad, Y.; Senderowitz, H. Docking Studies on DNA Intercalators. J. Chem. Inf. Model. 2014, 54, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Marinello, J.; Manzo, S.G.; Wakelin, L.P.G.; Capranico, G.; Griffith, R. Novel DNA Topoisomerase IIα Inhibitors from Combined Ligand- and Structure-Based Virtual Screening. PLoS ONE 2014, 9, e114904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sader, S.; Wu, C. Computational analysis of Amsacrine resistance in human topoisomerase II alpha mutants (R487K and E571K) using homology modeling, docking and all-atom molecular dynamics simulation in explicit solvent. J. Mol. Graph. Model. 2017, 72, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef]
- Buzun, K.; Bielawska, A.; Bielawski, K.; Gornowicz, A. DNA topoisomerases as molecular targets for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2020, 35, 1781–1799. [Google Scholar] [CrossRef]
- Yang, J.; Zhen, X.; Wang, B.; Gao, X.; Ren, Z.; Wang, J.; Xie, Y.; Li, J.; Peng, Q.; Pu, K.; et al. The influence of the molecular packing on the room temperature phosphorescence of purely organic luminogens. Nat. Commun. 2018, 9, 840. [Google Scholar] [CrossRef]
- Alves, J.E.F.; de Oliveira, J.F.; de Lima Souza, T.R.C.; de Moura, R.O.; de Carvalho Júnior, L.B.; de Lima, M.d.C.A.; de Almeida, S.M.V. Novel indole-thiazole and indole-thiazolidinone derivatives as DNA groove binders. Int. J. Biol. Macromol. 2021, 170, 622–635. [Google Scholar] [CrossRef]
- Kumar, S.; Nair, M.S. Deciphering the interaction of flavones with calf thymus DNA and octamer DNA sequence (CCAATTGG)2. RSC Adv. 2021, 11, 29354–29371. [Google Scholar] [CrossRef]
- Barra, C.V.; Rocha, F.V.; Morel, L.; Gautier, A.; Garrido, S.S.; Mauro, A.E.; Frem, R.C.G.; Netto, A.V.G. DNA binding, topoisomerase inhibition and cytotoxicity of palladium(II) complexes with 1,10-phenanthroline and thioureas. Inorg. Chim. Acta 2016, 446, 54–60. [Google Scholar] [CrossRef]
- Alsaedi, S.; Babgi, B.A.; Abdellattif, M.H.; Arshad, M.N.; Emwas, A.-H.M.; Jaremko, M.; Humphrey, M.G.; Asiri, A.M.; Hussien, M.A. DNA-Binding and Cytotoxicity of Copper(I) Complexes Containing Functionalized Dipyridylphenazine Ligands. Pharmaceutics 2021, 13, 764. [Google Scholar] [CrossRef]
- Alagesan, M.; Sathyadevi, P.; Krishnamoorthy, P.; Bhuvanesh, N.S.P.; Dharmaraj, N. DMSO containing ruthenium(II) hydrazone complexes: In vitro evaluation of biomolecular interaction and anticancer activity. Dalton Trans. 2014, 43, 15829–15840. [Google Scholar] [CrossRef] [PubMed]
- Aleksi, M.M.; Kapetanovi, V. An Overview of the Optical and Electrochemical Methods for Detection of DNA—Drug Interactions. Acta Chim. Slov. 2014, 61, 555–573. [Google Scholar]
- Jangir, D.K.; Dey, S.K.; Kundu, S.; Mehrotra, R. Assessment of amsacrine binding with DNA using UV–visible, circular dichroism and Raman spectroscopic techniques. J. Photochem. Photobiol. B Biol. 2012, 114, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Satpathi, S.; Sengupta, A.; Hridya, V.M.; Gavvala, K.; Koninti, R.K.; Roy, B.; Hazra, P. A Green Solvent Induced DNA Package. Sci. Rep. 2015, 5, srep09137. [Google Scholar] [CrossRef]
- Chatterjee, S.; Mallick, S.; Buzzetti, F.; Fiorillo, G.; Syeda, T.M.; Lombardi, P.; Saha, K.D.; Kumar, G.S. New 13-pyridinealkyl berberine analogues intercalate to DNA and induce apoptosis in HepG2 and MCF-7 cells through ROS mediated p53 dependent pathway: Biophysical, biochemical and molecular modeling studies. RSC Adv. 2015, 5, 90632–90644. [Google Scholar] [CrossRef]
- de Almeida, P.S.V.B.; de Arruda, H.J.; Sousa, G.L.S.; Ribeiro, F.V.; de Azevedo-França, J.A.; Ferreira, L.A.; Guedes, G.P.; Silva, H.; Kummerle, A.E.; Neves, A.P. Cytotoxicity evaluation and DNA interaction of Ru II -bipy complexes containing coumarin-based ligands. Dalton Trans. 2021, 50, 14908–14919. [Google Scholar] [CrossRef]
- Wang, G.; Wu, H.; Wang, D.; Yan, C.; Lu, Y. Exploring the binding mechanism of phosphoramidate derivative with DNA: Spectroscopy, calorimetry and modeling. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 104, 492–496. [Google Scholar] [CrossRef]
- Da Silva Oliveira, G.L.; Machado, K.C.; Machado, K.C.; da Silva, A.P.d.S.C.L.; Feitosa, C.M.; de Castro Almeida, F.R. Non-clinical toxicity of β-caryophyllene, a dietary cannabinoid: Absence of adverse effects in female Swiss mice. Regul. Toxicol. Pharmacol. 2018, 92, 338–346. [Google Scholar] [CrossRef]
- da Silva Oliveira, G.L.; da Silva, A.P.d.S.C.L. Evaluation of the non-clinical toxicity of an antiparasitic agent: Diminazene aceturate. Drug Chem. Toxicol. 2021, 1–11. [Google Scholar] [CrossRef]
- Tang, G.; Nierath, W.-F.; Palme, R.; Vollmar, B.; Zechner, D. Analysis of Animal Well-Being When Supplementing Drinking Water with Tramadol or Metamizole during Chronic Pancreatitis. Animals 2020, 10, 2306. [Google Scholar] [CrossRef]
- Kifayatullah, M.; Mustafa, M.S.; Sengupta, P.; Sarker, M.d.M.R.; Das, A.; Das, S.K. Evaluation of the acute and sub-acute toxicity of the ethanolic extract of Pericampylus glaucus (Lam.) Merr. in BALB/c mice. J. Acute Dis. 2015, 4, 309–315. [Google Scholar] [CrossRef]
- Laroche-Clary, A.; Larrue, A.; Robert, J. Down-regulation of bcr-abl and bcl-xL expression in a leukemia cell line and its doxorubicin-resistant variant by topoisomerase II inhibitors. Biochem. Pharmacol. 2000, 60, 1823–1828. [Google Scholar] [CrossRef]
- Quarti, J.; Torres, D.N.M.; Ferreira, E.; Vidal, R.S.; Casanova, F.; Chiarini, L.B.; Fialho, E.; Rumjanek, V.M. Selective Cytotoxicity of Piperine over Multidrug Resistance Leukemic Cells. Molecules 2021, 26, 934. [Google Scholar] [CrossRef]
- de Sousa Portilho, A.J.; da Silva, E.L.; Bezerra, E.C.A.; Moraes Rego Gomes, C.B.d.S.; Ferreira, V.; de Moraes, M.E.A.; da Rocha, D.R.; Burbano, R.M.R.; Moreira-Nunes, C.A.; Montenegro, R.C. 1,4-Naphthoquinone (CNN1) Induces Apoptosis through DNA Damage and Promotes Upregulation of H2AFX in Leukemia Multidrug Resistant Cell Line. Int. J. Mol. Sci. 2022, 23, 8105. [Google Scholar] [CrossRef]
- Mohammadi-Khanaposhtani, M.; Shabani, M.; Faizi, M.; Aghaei, I.; Jahani, R.; Sharafi, Z.; Shamsaei Zafarghandi, N.; Mahdavi, M.; Akbarzadeh, T.; Emami, S.; et al. Design, synthesis, pharmacological evaluation, and docking study of new acridone-based 1,2,4-oxadiazoles as potential anticonvulsant agents. Eur. J. Med. Chem. 2016, 112, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.d.S.; Silva, V.R.; Menezes, L.R.A.; Soares, M.B.P.; Costa, E.V.; Bezerra, D.P. Xylopine Induces Oxidative Stress and Causes G 2 /M Phase Arrest, Triggering Caspase-Mediated Apoptosis by p53-Independent Pathway in HCT116 Cells. Oxidative Med. Cell. Longev. 2017, 2017, 7126872. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.V.; Soares, L.d.N.; Chaar, J.d.S.; Silva, V.R.; Santos, L.d.S.; Koolen, H.H.F.; Silva, F.M.A.d.; Tavares, J.F.; Zengin, G.; Soares, M.B.P.; et al. Benzylated Dihydroflavones and Isoquinoline-Derived Alkaloids from the Bark of Diclinanona calycina (Annonaceae) and Their Cytotoxicities. Molecules 2021, 26, 3714. [Google Scholar] [CrossRef]
- Pakravan, P.; Kashanian, S.; Khodaei, M.M.; Harding, F.J. Biochemical and pharmacological characterization of isatin and its derivatives: From structure to activity. Pharmacol. Rep. 2013, 65, 313–335. [Google Scholar] [CrossRef]
- Wolfe, A.; Shimer, G.H.; Meehan, T. Polycyclic aromatic hydrocarbons physically intercalate into duplex regions of denatured DNA. Biochemistry 1987, 26, 6392–6396. [Google Scholar] [CrossRef]
- Miles, A.J.; Janes, R.W.; Wallace, B.A. Tools and methods for circular dichroism spectroscopy of proteins: A tutorial review. Chem. Soc. Rev. 2021, 50, 8400–8413. [Google Scholar] [CrossRef]
- Keizer, J. Nonlinear fluorescence quenching and the origin of positive curvature in Stern-Volmer plots. J. Am. Chem. Soc. 1983, 105, 1494–1498. [Google Scholar] [CrossRef]
- OECD. Test No. 423: Acute Oral Toxicity—Acute Toxic Class Method; OECD Guidelines for the Testing of Chemicals, Section 4; OECD Publishing: Paris, France, 2002. [Google Scholar] [CrossRef]
- Barbano, D.B.A. Guia para a Condução de Estudos não Clínicos de Toxicologia e Segurança Farmacológica Necessários ao Desenvolvimento de Medicamentos; Agência Nacional de Vigilância Sanitária (Anvisa): Brasilia, Brazil, 2013; p. 48.
- Saleem, U.; Amin, S.; Ahmad, B.; Azeem, H.; Anwar, F.; Mary, S. Acute oral toxicity evaluation of aqueous ethanolic extract of Saccharum munja Roxb. roots in albino mice as per OECD 425 TG. Toxicol. Rep. 2017, 4, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Flecknell, P.A. Laboratory Animal Anaesthesia, 3rd ed.; Elsevier/Academic Press: Amsterdam, The Netherlands; Boston, MA, USA; London, UK, 2009; ISBN 978-0-12-369376-1. [Google Scholar]
- Rumjanek, V.M.; Trindade, G.S.; Wagner-Souza, K.; Meletti-De-Oliveira, M.C.; Marques-Santos, L.F.; Maia, R.C.; Capella, M.A.M. Multidrug resistance in tumour cells: Characterisation of the multidrug resistant cell line K562-Lucena 1. An. Acad. Bras. Ciênc. 2001, 73, 57–69. [Google Scholar] [CrossRef]
- Daflon-Yunes, N.; Pinto-Silva, F.E.; Vidal, R.S.; Novis, B.F.; Berguetti, T.; Lopes, R.R.S.; Polycarpo, C.; Rumjanek, V.M. Characterization of a multidrug-resistant chronic myeloid leukemia cell line presenting multiple resistance mechanisms. Mol. Cell. Biochem. 2013, 383, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Dung, D.T.M.; Park, E.J.; Anh, D.T.; Phan, D.T.P.; Na, I.H.; Kwon, J.H.; Kang, J.S.; Tung, T.T.; Han, S.; Nam, N. De-sign, Synthesis and Evaluation of Novel 2-oxoindoline-based Acetohydrazides as Antitumor Agents. Sci. Rep. 2022, 12, 2886. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| R | Comp | IC50 (µM) 1 | |||
| HCT116 | HepG2 | B16-F10 | MRC-5 | ||
| H | CL-01 | >70.00 | >70.00 | ND | >70.00 |
| 4-Cl | CL-02 | >70.00 | >70.00 | ND | >70.00 |
| 4-OCH3 | CL-03 | >70.00 | >70.00 | >70.00 | >70.00 |
| 2,4-Cl | CL-04 | >60.00 | >60.00 | ND | >60.00 |
| 4-NO2 | CL-05 | >60.00 | >60.00 | 22.71 | 26.78 |
| 16.60–31.09 | 22.26–32.19 | ||||
| 4-CH3 | CL-06 | 64.76 | 58.06 | 23.54 | 28.02 |
| 44.07–95.16 | 38.93–86.62 | 15.94–28.50 | 21.59–36.31 | ||
| 4-OH | CL-07 | 39.67 | 50.66 | 19.83 | 43.40 |
| 19.13–63.40 | 43.24–59.31 | 18.73–29.61 | 36.28–51.89 | ||
| 3-OH | CL-08 | 39.16 | 49.42 | 21.32 | 33.38 |
| 17.76–59.53 | 43.99–55.52 | 14.56–27.00 | 27.79–40.07 | ||
| 4-N(CH3)2 | CL-09 | >60.00 | >60.00 | ND | >60.00 |
| 4-Br | CL-10 | >60.00 | >60.00 | ND | >60.00 |
| H | DL-01 | >60.00 | 21.87 | 39.56 | 36.97 |
| 15.97–29.97 | 24.11–64.89 | 25.35–53.97 | |||
| 4-Cl | DL-02 | >60.00 | >60.00 | 30.57 | >60.00 |
| 21.74–42.99 | |||||
| 4-OCH3 | DL-03 | >60.00 | >60.00 | >60.00 | >60.00 |
| 2,4-Cl | DL-04 | >60.00 | >60.00 | >60.00 | >60.00 |
| 4-NO2 | DL-05 | >60.00 | >60.00 | >60.00 | >60.00 |
| 4-CH3 | DL-06 | >60.00 | 33.38 | 26.26 | >60.00 |
| 23.64–47.13 | 14.81–46.53 | ||||
| 4-OH | DL-07 | 28.46 | 24.84 | 21.12 | 23.64 |
| 25.13–32.24 | 16.21–38.04 | 16.39–26.72 | 17.06–32.79 | ||
| 3-OH | DL-08 | 24.22 | 21.28 | 14.79 | 14.36 |
| 21.32–27.49 | 11.17–40.50 | 11.51–18.99 | 9.61–21.44 | ||
| 4-N(CH3)2 | DL-09 | >60.00 | >60.00 | 46.04 | >60.00 |
| 27.68–50.85 | |||||
| 4-Br | DL-10 | >50.00 | 20.88 | 34.62 | >50.00 |
| 11.22–38.83 | 25.74–53.55 | ||||
| DOX 2 | 0.15 | 0.15 | 0.03 | 2.22 | |
| 5-FU 3 | 4.1 | 1.3 | 3.5 | 57.9 | |
| Compounds | mAMSA | DL01 | DL06 | DL07 | DL08 | CL07 |
|---|---|---|---|---|---|---|
| % of enzyme inhibition | 100 | 77 | - | 74 | 79 | - |
| Compounds | Docking Score | Hydrogen Bond | Hydrophobic Interactions | |
|---|---|---|---|---|
| π-Alkyl | Stacked π–π | |||
| CL-07 | 82.02 | T9a G10 * | Arg487 | A12 G13 |
| DL-01 | 93.88 | Arg487 | T9 Arg487 | G13 |
| DL-06 | 84.01 | - | T9 Arg487 | A12 G13 |
| DL-07 | 97.98 | Arg487 T9a Ser464 G10 * | T9 Arg487 | A12 G13 |
| DL-08 | 98.06 | Arg487 Asp463 Ser464 | T9 Arg487 | T9 G13 |
| mAMSA | 95.21 | Asp463 Ser464 Arg487 | Arg487 | C8 T9 G13 |
| Etoposide | 98.35 | Asp463 | T9 Arg487 | G13 Arg487 |
| Compounds | Absorptive Properties | ||||
|---|---|---|---|---|---|
| λ Kb (nm) | ∆λ | R2 | Hypochromic (%) | Kb (104) (M−1) | |
| CL-07 | 325 | 3 1 | 0.9586 | 25.52 | 4.51 ± 0.14 |
| DL-01 | 320 | 2 2 | 0.9626 | 12.89 | 0.75 ± 0.07 |
| DL-08 | 305 | 3 2 | 0.9708 | 14.79 | 1.73 ± 0.23 |
| EtBr | 489 | 6 2 | 0.9652 | 35.15 | 5.56 ± 0.19 |
| Compounds | Emissive Properties | ||
|---|---|---|---|
| R2 | Quenching (%) | Ksv (103) (M−1) | |
| CL-07 | 0.9903 | 21.71 | 2.6 |
| DL-01 | 0.9954 | 15.61 | 1.8 |
| DL-08 | 0.9886 | 20.83 | 2.5 |
| Toxicity Study | DA/TA | Mortality (%) | Signs of Toxicity |
|---|---|---|---|
| CL-07 | 0/3 | 0 | None |
| DL-01 | 0/3 | 0 | Reduced feed consumption |
| DL-08 | 0/3 | 0 | Hepatic steatosis |
| NC | 0/3 | 0 | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, G.; de Almeida, M.C.F.; Lócio, L.L.; dos Santos, V.L.; Bezerra, D.P.; Silva, V.R.; de Almeida, S.M.V.; Simon, A.; Honório, T.d.S.; Cabral, L.M.; et al. Synthesis and Evaluation of Antiproliferative Activity, Topoisomerase IIα Inhibition, DNA Binding and Non-Clinical Toxicity of New Acridine–Thiosemicarbazone Derivatives. Pharmaceuticals 2022, 15, 1098. https://doi.org/10.3390/ph15091098
Sousa G, de Almeida MCF, Lócio LL, dos Santos VL, Bezerra DP, Silva VR, de Almeida SMV, Simon A, Honório TdS, Cabral LM, et al. Synthesis and Evaluation of Antiproliferative Activity, Topoisomerase IIα Inhibition, DNA Binding and Non-Clinical Toxicity of New Acridine–Thiosemicarbazone Derivatives. Pharmaceuticals. 2022; 15(9):1098. https://doi.org/10.3390/ph15091098
Chicago/Turabian StyleSousa, Gleyton, Maria C. F. de Almeida, Lucas L. Lócio, Vanda L. dos Santos, Daniel P. Bezerra, Valdenizia R. Silva, Sinara M. V. de Almeida, Alice Simon, Thiago da S. Honório, Lucio M. Cabral, and et al. 2022. "Synthesis and Evaluation of Antiproliferative Activity, Topoisomerase IIα Inhibition, DNA Binding and Non-Clinical Toxicity of New Acridine–Thiosemicarbazone Derivatives" Pharmaceuticals 15, no. 9: 1098. https://doi.org/10.3390/ph15091098
APA StyleSousa, G., de Almeida, M. C. F., Lócio, L. L., dos Santos, V. L., Bezerra, D. P., Silva, V. R., de Almeida, S. M. V., Simon, A., Honório, T. d. S., Cabral, L. M., Castro, R. N., de Moura, R. O., & Kümmerle, A. E. (2022). Synthesis and Evaluation of Antiproliferative Activity, Topoisomerase IIα Inhibition, DNA Binding and Non-Clinical Toxicity of New Acridine–Thiosemicarbazone Derivatives. Pharmaceuticals, 15(9), 1098. https://doi.org/10.3390/ph15091098