
Identification of Potential Allosteric Site Binders of Indoleamine 2,3-Dioxygenase 1 from Plants: A Virtual and Molecular Dynamics Investigation



Abstract
1. Introduction
1.1. Tryptophan Metabolism and the Kynurenine Pathway
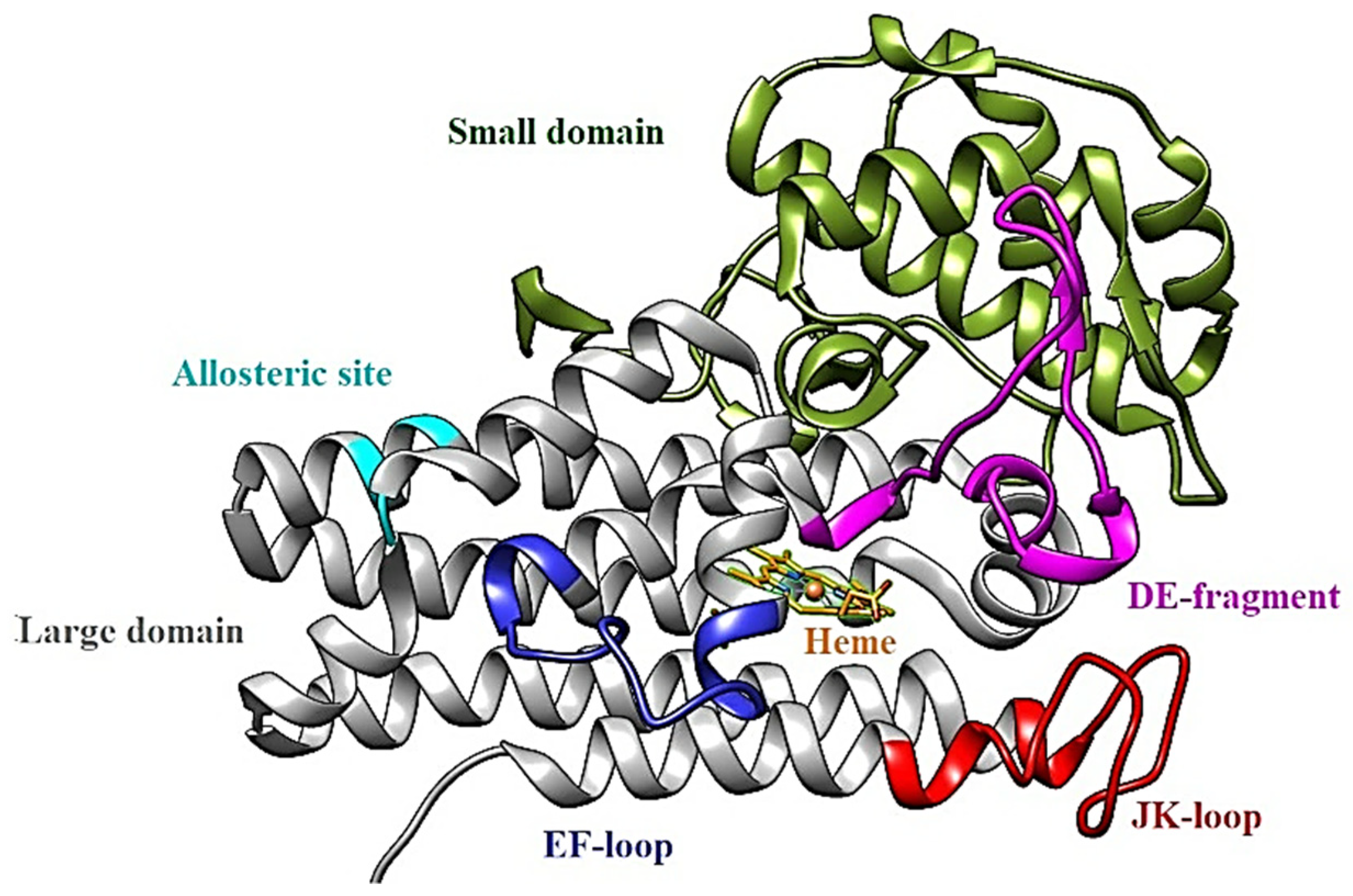
1.2. IDO1: Structure
- apo-IDO1: without the heme cofactor;
- holo-IDO1: containing the iron atom of the heme group in the ferric state (Fe3+);
- holo-IDO1 with the iron atom of the heme group in the ferrous state (Fe2+);
- O2-linked holo-IDO1 complex.
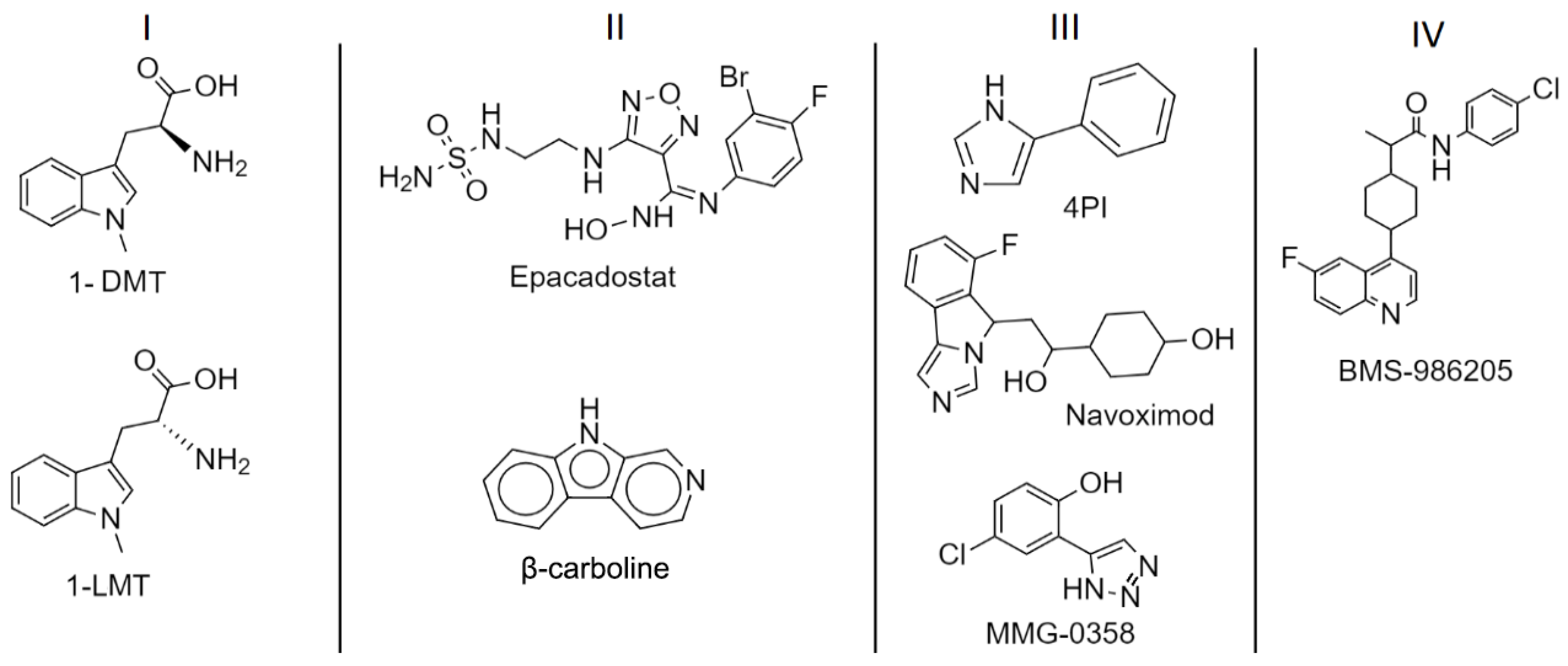
1.3. IDO1: Inhibitors
2. Results and Discussion
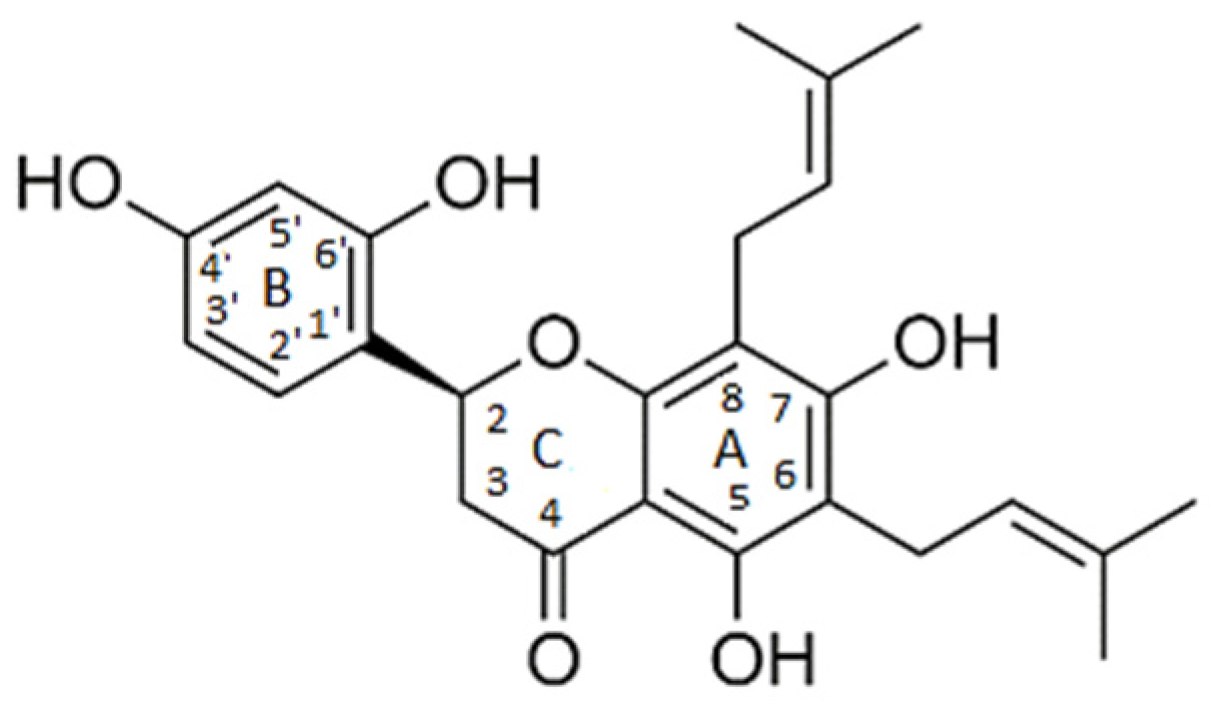
2.1. Ligand-Based Virtual Screening
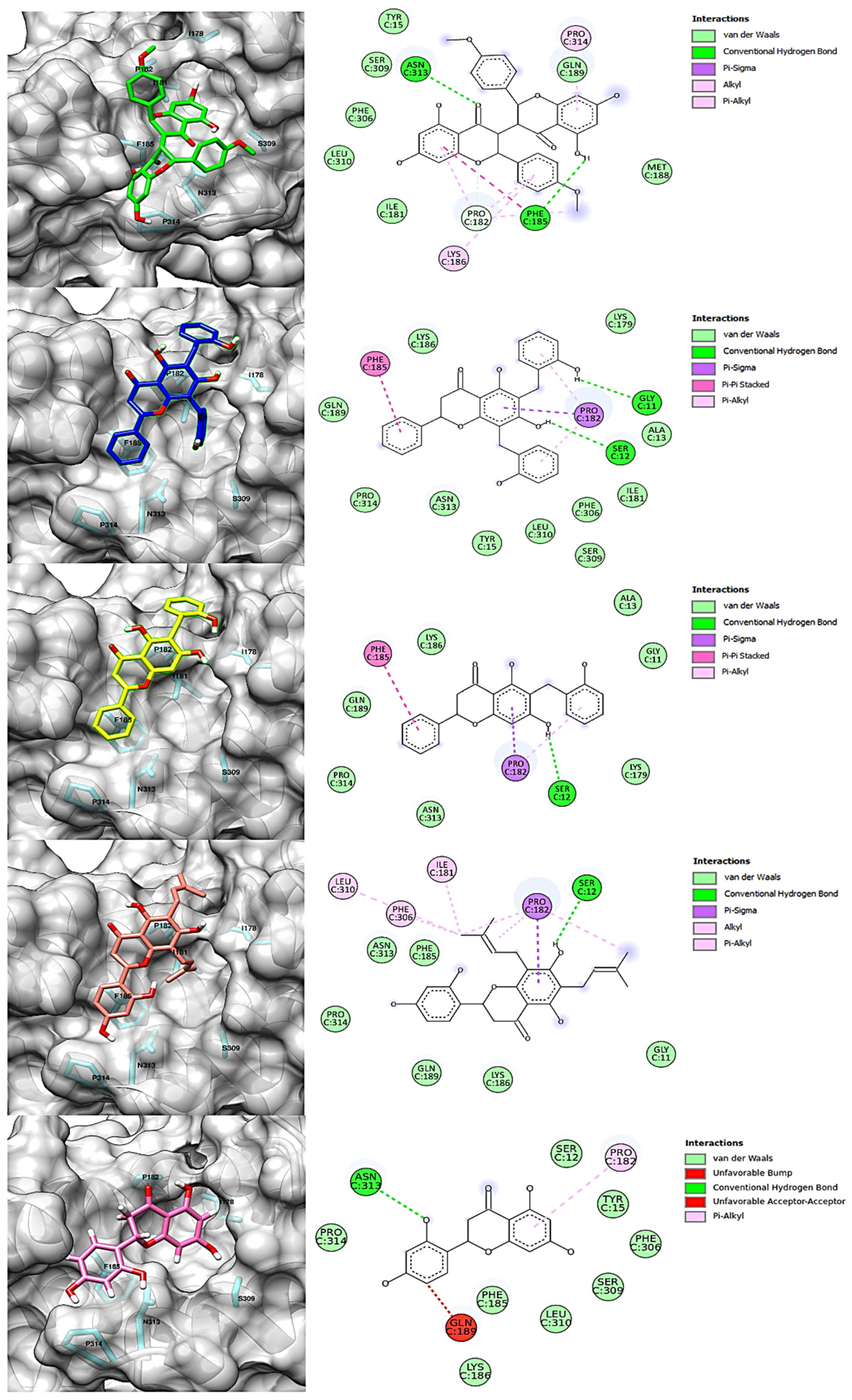
2.2. Structure-Based Virtual Screening and Molecular Docking
2.3. Molecular Dynamics Simulations and Free Energy Calculation
3. Materials and Methods
3.1. IDO1
3.2. Virtual Screening
3.3. Molecular Docking
3.4. Molecular Dynamics Simulations and Free Energy Calculation
3.5. Visualization Tools and Plots
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’h, N.; Otten, W.; Merlot, E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids 2011, 41, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Lanser, L.; Kink, P.; Egger, E.M.; Willenbacher, W.; Fuchs, D.; Weiss, G.; Kurz, K. Inflammation-Induced Tryptophan Breakdown is Related with Anemia, Fatigue, and Depression in Cancer. Front. Immunol. 2020, 11, 249. [Google Scholar] [CrossRef]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef]
- Pallotta, M.T.; Fallarino, F.; Matino, D.; Macchiarulo, A.; Orabona, C. AhR-mediated, non-genomic modulation of IDO1 function. Front. Immunol. 2014, 5, 497. [Google Scholar] [CrossRef][Green Version]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef]
- Hascitha, J.; Priya, R.; Jayavelu, S.; Dhandapani, H.; Selvaluxmy, G.; Sunder Singh, S.; Rajkumar, T. Analysis of Kynurenine/Tryptophan ratio and expression of IDO1 and 2 mRNA in tumour tissue of cervical cancer patients. Clin. Biochem. 2016, 49, 919–924. [Google Scholar] [CrossRef]
- Löb, S.; Königsrainer, A.; Zieker, D.; Brücher, B.L.D.M.; Rammensee, H.G.; Opelz, G.; Terness, P. IDO1 and IDO2 are expressed in human tumors: Levo-but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol. Immunother. 2009, 58, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Theate, I.; Van Baren, N.; Pilotte, L.; Moulin, P.; Larrieu, P.; Renauld, J.C.; Herve, C.; Gutierrez-Roelens, I.; Marbaix, E.; Sempoux, C.; et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol. Res. 2015, 3, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Sook, Y.; Hamdy, H. Involvement of Two Regulatory Elements in Interferon-7-Regulated Expression of Human Indoleamine 2,3-Dioxygenase Gene. J. Interferon Cytokine Res. 1995, 15, 517–526. [Google Scholar]
- Prendergast, G.C.; Jaffee, E.M. Cancer Immunotherapy: Immune Suppression and Tumor Growth, 2nd ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Mirgaux, M.; Leherte, L.; Wouters, J. Influence of the presence of the heme cofactor on the JK-loop structure in indoleamine 2,3-dioxygenase 1. Acta Crystallogr. Sect. D: Struct. Biol. 2020, 76, 1211–1221. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Duan, H.; Luo, Q.; Liu, W.; Liang, L.; Wan, H.; Chang, S.; Hu, J.; Shi, H. Inhibition Mechanism of Indoleamine 2, 3-Dioxygenase 1 (IDO1) by Amidoxime Derivatives and Its Revelation in Drug Design: Comparative Molecular Dynamics Simulations. Front. Mol. Biosci. 2020, 6, 164. [Google Scholar] [CrossRef]
- Lewis-Ballester, A.; Pham, K.N.; Batabyal, D.; Karkashon, S.; Bonanno, J.B.; Poulos, T.L.; Yeh, S.R. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Nelp, M.T.; Kates, P.A.; Hunt, J.T.; Newitt, J.A.; Balog, A.; Maley, D.; Zhu, X.; Abell, L.; Allentoff, A.; Borzilleri, R.; et al. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. USA 2018, 115, 3249–3254. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Reynaud, A.; Majjigapu, S.R.; Vogel, P.; Pojer, F.; Zoete, V. Inhibition Mechanisms of Indoleamine 2,3-Dioxygenase 1 (IDO1). J. Med. Chem. 2019, 62, 8784–8795. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Opitz, U.; Sahm, F.; Ochs, K.; Lutz, C.; Wick, W.; Platten, M. The indoleamine-2,3-dioxygenase (IDO) inhibitor 1-methyl-d-tryptophan upregulates IDO1 in human cancer cells. PLoS ONE 2011, 6, e19823. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, J.; Li, Q.; Xu, H.; Chen, G.; Li, Z.; Liu, D.; Yang, X. Discovery of potent indoleamine 2,3-dioxygenase (IDO) inhibitor from alkaloids in Picrasma quassioides by virtual screening and in vitro evaluation. Fitoterapia 2019, 133, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Yue, E.W.; Sparks, R.; Polam, P.; Modi, D.; Douty, B.; Wayland, B.; Glass, B.; Takvorian, A.; Glenn, J.; Zhu, W.; et al. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Meoz, R.F.; Wang, L.; Matico, R.; Rutkowska-Klute, A.; De la Rosa, M.; Bedard, S.; Midgett, R.; Strohmer, K.; Thomson, D.; Zhang, C.; et al. Characterization of Apo-Form Selective Inhibition of Indoleamine 2,3-Dioxygenase. ChemBioChem 2021, 22, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Ko, S.K.; Jang, M.; Kim, G.H.; Ryoo, I.J.; Son, S.; Ryu, H.W.; Oh, S.R.; Lee, W.K.; Kim, B.Y.; et al. Inhibitory effects of flavonoids isolated from Sophora flavescens on indoleamine 2,3-dioxygenase 1 activity. J. Enzym. Inhib. Med. Chem. 2019, 34, 1481–1488. [Google Scholar] [CrossRef]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand Discovery for Everyone. J. Chem. Inf. Modeling 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Tanimoto, T.T. Elementary Mathematical Theory of Classification and Prediction, 1st ed.; International Business Machines Corporation: New York, NY, USA, 1958. [Google Scholar]
- Chokchaisiri, R.; Kunkaewom, S.; Chokchaisiri, S.; Ganranoo, L.; Chalermglin, R.; Suksamrarn, A. Potent cytotoxicity against human small cell lung cancer cells of the heptenes from the stem bark of Xylopia pierrei Hance. Med. Chem. Res. 2017, 26, 1291–1296. [Google Scholar] [CrossRef]
- Quimque, M.T.; Notarte, K.I.; Letada, A.; Fernandez, R.A.; Pilapil, D.Y.; Pueblos, K.R.; Agbay, J.C.; Dahse, H.M.; Wenzel-Storjohann, A.; Tasdemir, D.; et al. Potential Cancer- And Alzheimer’s Disease-Targeting Phosphodiesterase Inhibitors from Uvaria alba: Insights from in Vitro and Consensus Virtual Screening. ACS Omega 2021, 6, 8403–8417. [Google Scholar] [CrossRef]
- Yong, Y.; Matthew, S.; Wittwer, J.; Pan, L.; Shen, Q.; Kinghorn, A.D.; Swanson, S.M.; Blanco, E.J.C.D. Dichamanetin Inhibits Cancer Cell Growth by Affecting ROS-related Signaling Components through Mitochondrial-mediated Apoptosis. Anticancer. Res. 2013, 33, 5349–5355. [Google Scholar]
- Swarnalatha, Y.; Vidhya, V.G.; Murugan, A. Isochamanetin is a Selective Inhibitor for CyclinD1 in SKOV3 Cell Lines. Nutr. Cancer 2019, 71, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Lasswell, W.L., Jr.; Hufford, C.D. Cytotoxic C-Benzylated Flavonoids from Uvaria chamae. J. Org. Chem. 1976, 42, 4–11. [Google Scholar]
- Zhang, C.; Zhou, S.S.; Feng, L.Y.; Zhang, D.Y.; Lin, N.M.; Zhang, L.H.; Pan, J.P.; Wang, J.B.; Li, J. In vitro anti-cancer activity of chamaejasmenin B and neochamaejasmin C isolated from the root of Stellera chamaejasme L. Acta Pharmacol. Sin. 2013, 34, 262–270. [Google Scholar] [CrossRef]
- Zondlo, N.J. Aromatic-Proline Interactions: Electronically Tunable CH/π Interactions. Acc. Chem. Res. 2012, 46, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes; 21.1.0; Discovery Studio Visualizer; Dassault Systèmes: San Diego, CA, USA, 2021.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Grace. Available online: https://plasma-gate.weizmann.ac.il/Grace/ (accessed on 12 July 2022).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Docking (kcal/mol) | Compound | Docking (kcal/mol) |
|---|---|---|---|
| chamuvaritin | −7.9 | Kushenol C | −6.5 |
| chamaejasmin B | −7.9 | butin | −6.5 |
| dichamanetin | −7.8 | estrobopinin | −6.4 |
| chamaejasmin | −7.7 | rhamnocitrin | −6.4 |
| neochamaejasmin A | −7.6 | 7-benziloxicumarin | −6.3 |
| obovatine | −7.5 | naringenin | −6.3 |
| isochamanetin | −7.5 | uvaretin | −6.2 |
| β-naftoflavone | −6.9 | pinocembrin | −6.1 |
| pinobanksine | −6.9 | genkwanin | −6.1 |
| tectocrisina | −6.8 | glabranin | −6.0 |
| soforaflavanone B | −6.7 | 7-hidroxiflavanone | −6.0 |
| strobopinin-7-methyl-ether | −6.7 | apigenin-4’,7-dimethyl-ether | −6.0 |
| diuvaretin | −6.6 | 2-hidroxiflavanone | −6.0 |
| izalpinin | −6.6 | asebogenine | −5.9 |
| Kushenol E | −6.6 | steppogenin | −5.8 |
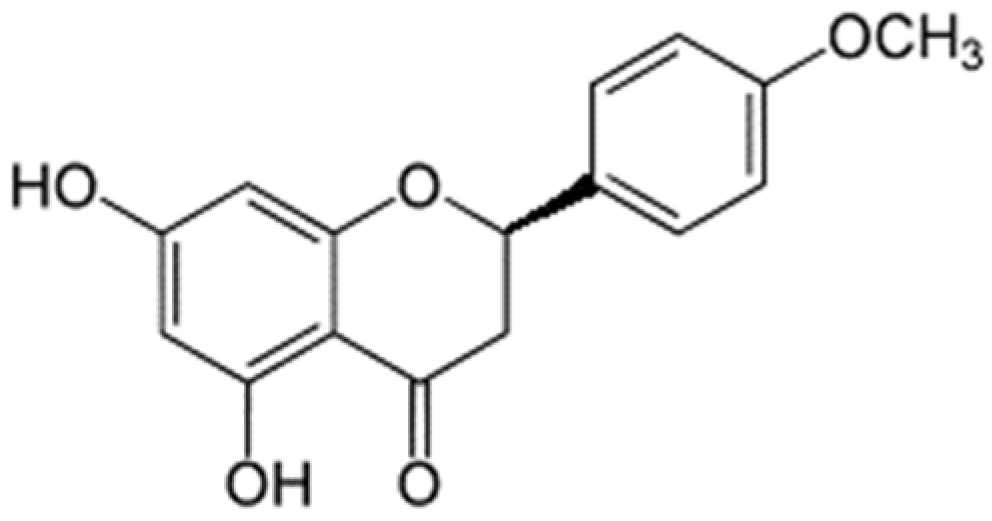
| Compound | Molecular Structure | H Bond | van der Waals | Pi-Alkyl | Pi-Sigma | Pi-Pi | Alkyl | Unfavorable |
|---|---|---|---|---|---|---|---|---|
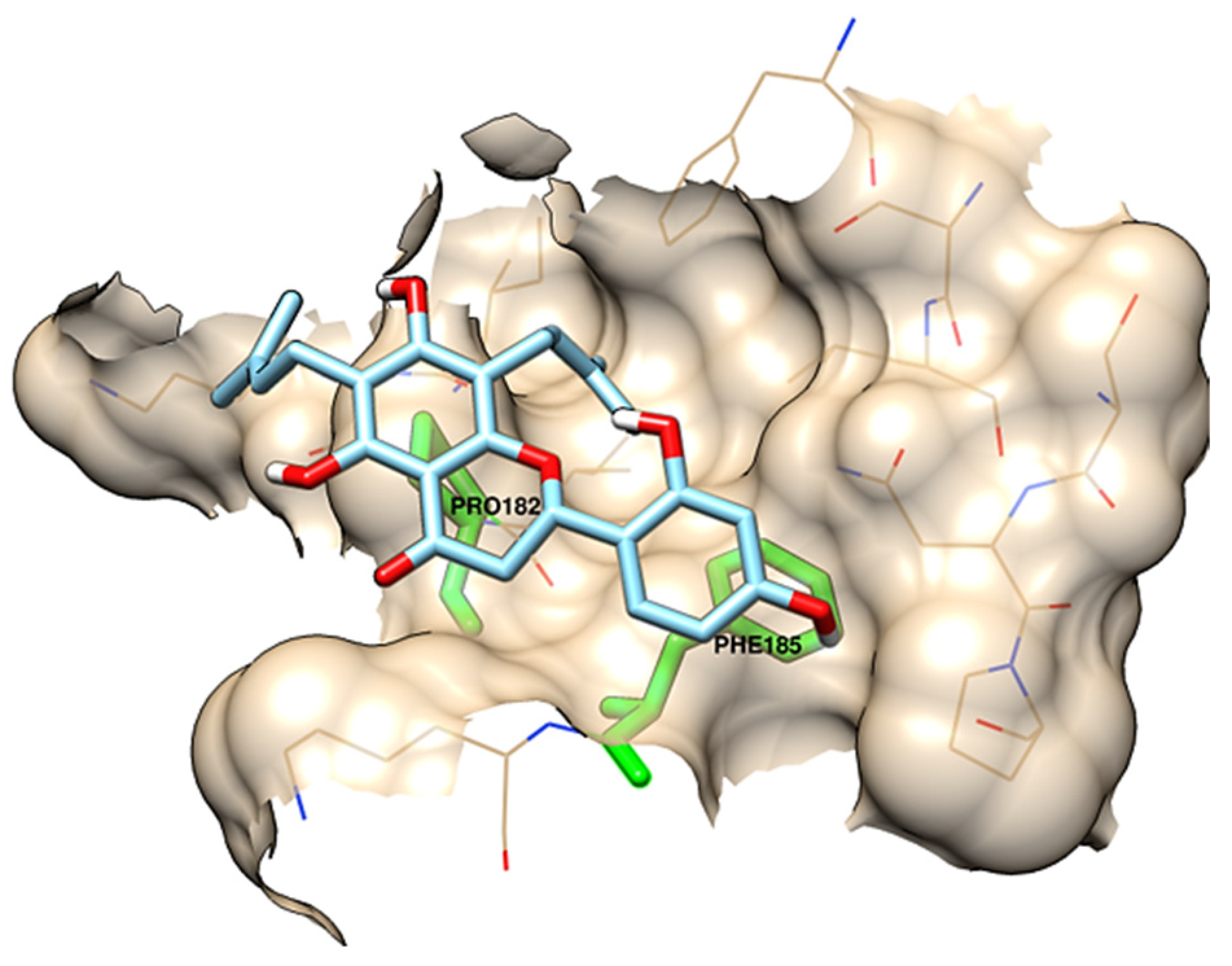
| chamaejasmin B |  | Phe185, Asn313 | Tyr15, Ile181, Gln189, Met188, Phe306, Ser309, Leu310 | Pro182, Pro314 | - | Phe185 | Lys186, Pro182 | - |
| dichamanetin |  | Ser12, Gly11 | Ala13, Tyr15, Lys179, Ile181, Lys186, Gln189, Phe306, Ser309, Leu310, Asn313, Pro314 | Pro182 | Pro182 | Phe185 | - | - |
| isochamanetin |  | Ser12, | Gly11, Ala13, Lys179, Lys186, Gln189, Asn313, Pro314 | Pro182 | Pro182 | Phe185 | - | - |
| Kushenol E |  | Ser12 | Gly11, Lys179, Phe185, Lys186, Gln189, Asn313, Pro314 | Pro182 | Pro182 | - | Ile181, Pro182, Phe306, Leu310 | - |
| steppogenin |  | Asn313 | Ser12, Tyr15, Phe185, Lys186, Phe306, Ser309, Leu310, Pro314 | Pro182 | - | - | - | Gln189 |
| Energy Component | Kushenol E ΔG (kcal/mol) | Steppogenin ΔG (kcal/mol) | Dichamanetin ΔG (kcal/mol) | Isochamanetin ΔG (kcal/mol) |
|---|---|---|---|---|
| van der Waals | −33.4464 +/− 2.7222 | −12.6589 +/− 5.4591 | −38.7571 +/− 2.8011 | −33.1692 +/− 2.6773 |
| Electrostatic | −0.4995 +/− 2.3494 | −9.5176 +/− 5.6553 | −8.1238 +/− 3.5253 | −2.7772 +/− 2.1816 |
| Polar Solvation | 16.2786 +/− 3.5898 | 18.1821 +/− 8.5497 | 25.4326 +/− 4.2624 | 17.7325 +/− 2.3312 |
| SASA | −3.9866 +/− 0.2390 | −1.7428 +/− 0.7148 | −4.4861 +/− 0.2483 | −3.8790 +/− 0.3585 |
| Free energy of interaction | −21.6563 +/− 3.1142 | −5.7373 +/− 5.2005 | −25.9345 +/− 3.1214 | −22.0960 +/− 3.1859 |
| Alloesteric Site | Kushenol E ΔG (kcal/mol) | Steppogenin ΔG (kcal/mol) | Dichamanetin ΔG (kcal/mol) | Isochamanetin ΔG (kcal/mol) |
|---|---|---|---|---|
| Lys179 | −5.6572 +/− 0.009 | 0.0522 +/− 0.0146 | −10.2480 +/− 0.0138 | −0.1242 +/− 0.0023 |
| Ile181 | −5.5043 +/− 0.003 | −0.0478 +/− 0.0006 | −7.1328 +/− 0.0045 | −5.9646 +/− 0.0042 |
| Pro182 | −2.7174 +/− 0.009 | −0.6856 +/− 0.0075 | −2.9304 +/− 0.0094 | −0.9383 +/− 0.0058 |
| Phe185 | −9.7036 +/− 0.0064 | −0.1037 +/− 0.0017 | −10.9729 +/− 0.0063 | −2.6943 +/− 0.0152 |
| Lys186 | −0.0069 +/− 0.0271 | −0.6023 +/− 0.0201 | −12.8116 +/− 0.0194 | −3.0506 +/− 0.0093 |
| Met188 | −0.1075 +/− 0.0007 | −0.0451 +/− 0.0006 | −0.1199 +/− 0.0006 | −6.0922 +/− 0.0049 |
| Gln189 | −4.2292 +/− 0.0045 | −0.0770 +/− 0.0020 | −4.7260 +/− 0.0061 | −7.7712 +/− 0.0108 |
| Phe306 | −13.3145 +/− 0.0084 | −0.0055 +/− 0.0003 | −14.2844 +/− 0.0076 | −7.4103 +/− 0.0070 |
| Ser309 | 0.0639 +/− 0.0090 | 0.0004 +/− 0.0012 | 0.1190 +/− 0.0077 | 2.6300 +/− 0.0092 |
| Leu310 | −6.0265 +/− 0.0047 | −0.0027 +/− 0.0004 | −6.1063 +/− 0.0043 | −6.0442 +/− 0.0048 |
| Asn313 | 3.2576 +/− 0.0086 | −0.0099 +/− 0.0015 | −0.0466 +/− 0.0065 | −0.0041 +/− 0.0150 |
| Pro314 | −0.0440 +/− 0.0013 | −0.0007 +/− 0.0007 | −0.0231 +/− 0.0009 | −5.5250 +/− 0.0143 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Almeida, V.M.; Santos-Filho, O.A. Identification of Potential Allosteric Site Binders of Indoleamine 2,3-Dioxygenase 1 from Plants: A Virtual and Molecular Dynamics Investigation. Pharmaceuticals 2022, 15, 1099. https://doi.org/10.3390/ph15091099
de Almeida VM, Santos-Filho OA. Identification of Potential Allosteric Site Binders of Indoleamine 2,3-Dioxygenase 1 from Plants: A Virtual and Molecular Dynamics Investigation. Pharmaceuticals. 2022; 15(9):1099. https://doi.org/10.3390/ph15091099
Chicago/Turabian Stylede Almeida, Vitor Martins, and Osvaldo Andrade Santos-Filho. 2022. "Identification of Potential Allosteric Site Binders of Indoleamine 2,3-Dioxygenase 1 from Plants: A Virtual and Molecular Dynamics Investigation" Pharmaceuticals 15, no. 9: 1099. https://doi.org/10.3390/ph15091099
APA Stylede Almeida, V. M., & Santos-Filho, O. A. (2022). Identification of Potential Allosteric Site Binders of Indoleamine 2,3-Dioxygenase 1 from Plants: A Virtual and Molecular Dynamics Investigation. Pharmaceuticals, 15(9), 1099. https://doi.org/10.3390/ph15091099