
The Role of Immune Checkpoint Inhibitors in Metastatic Pancreatic Cancer: Current State and Outlook

Abstract
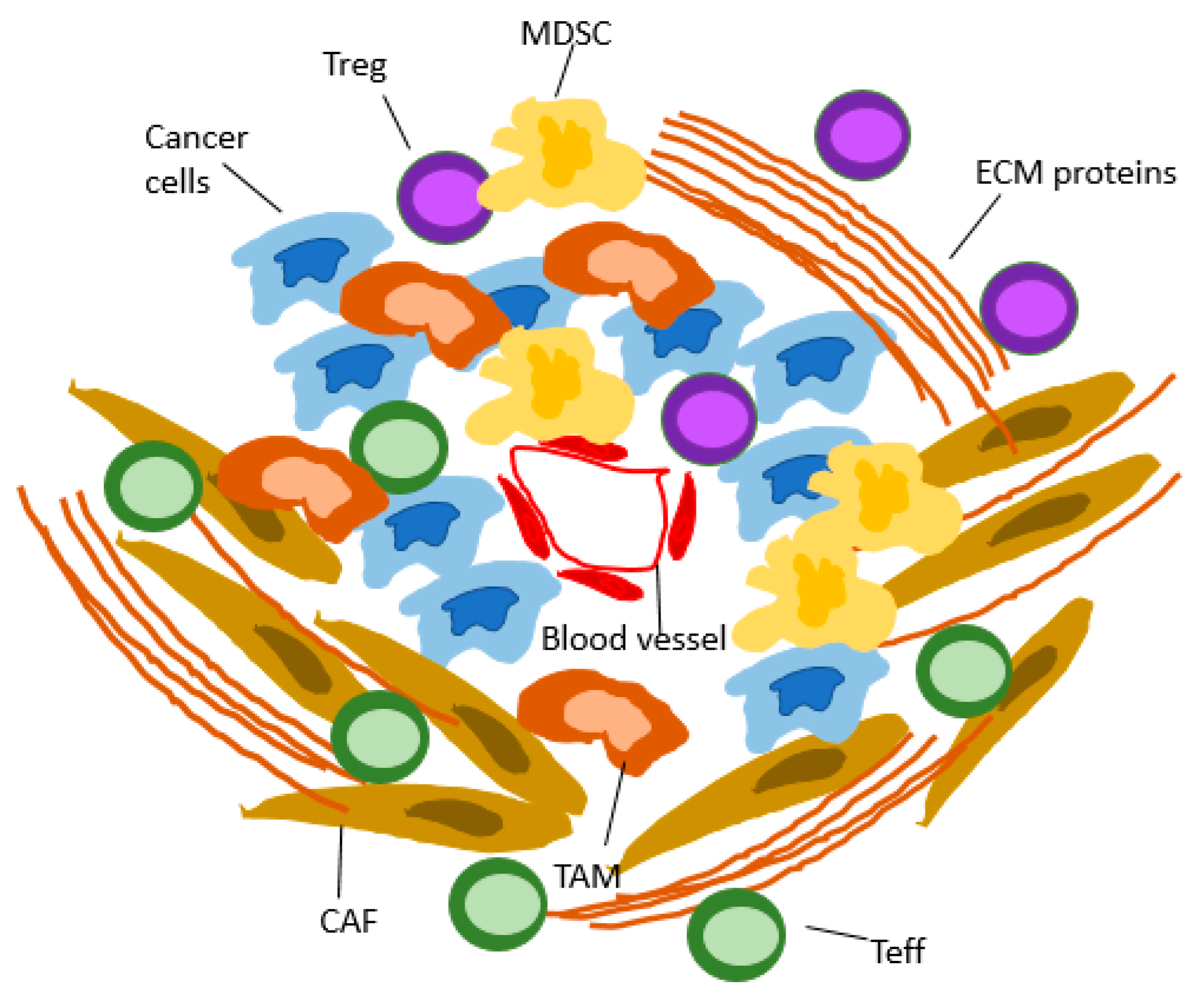
:1. Introduction
2. Rationale for Immune Checkpoint Inhibition in mPDAC
3. Current Role of Immune Checkpoint Inhibitors in mPDAC
3.1. Targeting PD-1/PD-L1 in mPDAC
3.2. Targeting CTLA-4 in mPDAC
3.3. Targeted Treatment in Combination with ICI in mPDAC
PARP Inhibitors
3.4. ICIs in Combination with TME-Modulating Agents in mPDAC
3.4.1. Immune Checkpoint Inhibition in Combination with Anti-CSF1R Ab
3.4.2. ICI in Combination with FAK Inhibition
3.4.3. ICIs in Combination with a CD40 Agonist
3.4.4. ICIs in Combination with an IDO Inhibitor
3.4.5. ICIs in Combination with an Anti-CCR4 Ab
3.4.6. Targeting the CXCL12/CXCR4 Axis in Combination with ICIs
3.4.7. Targeting CXCR2 in Combination with ICIs
3.4.8. ICIs Combined with Anti-Stromal Treatment
3.4.9. ICIs in Combination with Anti-TGFbeta
3.4.10. ICIs in Combination with Bruton Tyrosine Kinase Inhibitor
3.5. Combination of Immunomodulating Agents
3.5.1. Anti-PD-1 Ab in Combination with OX40 Agonists
3.5.2. Chemotherapy in Combination with an Anti-LAG-3 Ab
3.5.3. Targeting TIGIT
3.5.4. Targeting VISTA
3.5.5. ICIs in Combination with MET Kinase Inhibitors
3.5.6. ICIs Combined with a STING Agonist
3.5.7. Immunomodulating Triplet Treatment
3.6. Other Immunomodulating Therapeutic Approaches
3.6.1. Immune Checkpoint Inhibition in Combination with Oncolytic Viruses
3.6.2. Immune Checkpoint Inhibition in Combination with mRNA-Based Vaccines
3.7. Local Treatment Combined with Immune Checkpoint Inhibitors
3.7.1. Radiotherapy in Combination with ICIs
3.7.2. Radiofrequency Ablation (RFA) Combined with ICI
3.7.3. Irreversible Electroporation (IRE) Combined with ICI
4. Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Glatzer, M.; Horber, D.; Montemurro, M.; Winterhalder, R.; Inauen, R.; Berger, M.D.; Pestalozzi, B.; Pederiva, S.; Pless, M.; Putora, P.M. Choice of first line systemic treatment in pancreatic cancer among national experts. Pancreatology 2020, 20, 686–690. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R., Jr.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Philip, P.A.; Bahary, N.; Mahipal, A.; Kasi, A.; Rocha Lima, C.M.S.P.; Alistar, A.T.; Oberstein, P.E.; Golan, T.; Sahai, V.; Metges, J.P.; et al. Phase 3, multicenter, randomized study of CPI-613 with modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) as first-line therapy for patients with metastatic adenocarcinoma of the pancreas (AVENGER500). J. Clin. Oncol. 2022, 40 (Suppl. 16), 4023. [Google Scholar] [CrossRef]
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Schram, A.M.; Odintsov, I.; Espinosa-Cotton, M.; Khodos, I.; Sisso, W.J.; Mattar, M.S.; Lui, A.J.W.; Vojnic, M.; Shameem, S.H.; Chauhan, T.; et al. Zenocutuzumab, a HER2xHER3 Bispecific Antibody, Is Effective Therapy for Tumors Driven by NRG1 Gene Rearrangements. Cancer Discov. 2022, 12, 1233–1247. [Google Scholar] [CrossRef]
- Rhyner Agocs, G.; Assarzadegan, N.; Kirsch, R.; Dawson, H.; Galván, J.A.; Lugli, A.; Zlobec, I.; Berger, M.D. LAG-3 Expression Predicts Outcome in Stage II Colon Cancer. J. Pers. Med. 2021, 11, 749. [Google Scholar] [CrossRef]
- Puccini, A.; Seeber, A.; Berger, M.D. Biomarkers in Metastatic Colorectal Cancer: Status Quo and Future Perspective. Cancers 2022, 14, 4828. [Google Scholar] [CrossRef] [PubMed]
- Kole, C.; Charalampakis, N.; Tsakatikas, S.; Frountzas, M.; Apostolou, K.; Schizas, D. Immunotherapy in Combination with Well-Established Treatment Strategies in Pancreatic Cancer: Current Insights. Cancer Manag. Res. 2022, 14, 1043–1061. [Google Scholar] [CrossRef]
- Yeo, D.; Giardina, C.; Saxena, P.; Rasko, J.E.J. The next wave of cellular immunotherapies in pancreatic cancer. Mol. Ther. Oncolytics 2022, 24, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat. Rev. 2019, 78, 17–30. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Bang, Y.-J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated with Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Reyes, C.M.; Gärtner, P.; Rosenkranz, L.; Grippo, P.J.; Demir, I.E. In vivo Mouse Models of Pancreatic Ductal Adenocarcinoma. Pancreapedia Exocrine Pancreas Knowl. Base 2021. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Grünwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e18. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Q.; Wang, J.; Lou, Y.; Hong, Z.; Wie, S.; Sun, K.; Wang, J.; Chen, Y.; Sheng, J.; et al. Dynamic profiling of immune microenvironment during pancreatic cancer development suggests early intervention and combination strategy of immunotherapy. EBioMedicine 2022, 78, 103958. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell. 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-Induced Expression of ICAM-1 in Pancreatic Acinar Cells Causes Attraction of Macrophages to Expedite the Formation of Precancerous Lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Li, K.; Tandurella, J.A.; Gai, J.; Zhu, Q.; Lim, S.J.; Thomas, D.L., 2nd; Xia, T.; Mo, G.; Mitchell, J.T.; Montagne, J.; et al. Multi-omic analyses of changes in the tumor microenvironment of pancreatic adenocarcinoma following neoadjuvant treatment with anti-PD-1 therapy. Cancer Cell. 2022, 40, 1374–1391.e7. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, A.; Garasa, S.; Ochoa, M.D.C.; Cirella, A.; Olivera, I.; Glez-Vaz, J.; Andueza, M.P.; Migueliz, I.; Alvarez, M.; Rodríguez-Ruiz, M.E.; et al. Differential Interleukin-8 thresholds for chemotaxis and netosis in human neutrophils. Eur. J. Immunol. 2021, 51, 2274–2280. [Google Scholar] [CrossRef]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; Gögenur, I. The prognostic value of tumour-infiltrating lymphocytes in pancreatic cancer: A systematic review and meta-analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A., 3rd; Mulcahy, M. Ipilimumab and gemcitabine for advanced pancreatic cancer: A phase Ib study. Oncologist 2020, 25, e808–e815. [Google Scholar] [CrossRef] [PubMed]
- Renouf, D.J.; Loree, J.M.; Knox, J.J.; Topham, J.T.; Kavan, P.; Jonker, D.; Welch, S.; Couture, F.; Lemay, F.; Tehfe, M.; et al. The CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nat. Commun. 2022, 13, 5020. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Chen, Y.; Huang, D.; Guo, C.; Zhang, X.; Xiao, W.; Xue, X.; Zhang, Q.; Li, X.; Gao, S.; et al. Sintilimab Plus Modified FOLFIRINOX in Metastatic or Recurrent Pancreatic Cancer: The Randomized Phase II CISPD3 Trial. Ann. Surg. Oncol. 2023, 30, 5071–5080. [Google Scholar] [CrossRef]
- Padrón, L.J.; Maurer, D.M.; O’Hara, M.H.; O’Reilly, E.M.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: Clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat. Med. 2022, 28, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Morizane, C.; Ikeda, M.; Sudo, K.; Hirashima, Y.; Kuroda, M.; Fukuyama, Y.; Okusaka, T.; Furuse, J. A phase II study of nivolumab in combination with modified FOLFIRINOX for metastatic pancreatic cancer. J. Clin. Oncol. 2022, 40 (Suppl. 4), 553. [Google Scholar] [CrossRef]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF-Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: A phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Bahary, N.; Wang-Gillam, A.; Haraldsdottir, S.; Somer, B.G.; Lee, J.S.; O’Rourke, M.A.; Nayak-Kapoor, A.; Beatty, G.L.; Liu, M.; Delman, D.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus gemcitabine / nab-paclitaxel for the treatment of patients with metastatic pancreas cancer. J. Clin. Oncol. 2018, 36 (Suppl. 15), 4015. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. N. Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent ipilimumab (Anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Imamura, T.; Ashida, R.; Ohshima, K.; Uesaka, K.; Sugiura, T.; Ohgi, K.; Yamada, M.; Otsuka, S.; Hatakeyama, K.; Nagashima, T.; et al. Characterization of pancreatic cancer with ultra-low tumor mutational burden. Sci. Rep. 2023, 13, 4359. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Meng, Q.; Hua, J.; Yu, X.; Shi, S. The reciprocal regulation between host tissue and immune cells in pancreatic ductal adenocarcinoma: New insights and therapeutic implications. Mol. Cancer 2019, 18, 184. [Google Scholar] [CrossRef]
- Weiss, G.J.; Waypa, J.; Blaydorn, L.; Coats, J.; McGahey, K.; Sangal, A.; Niu, J.; Lynch, C.A.; Farley, J.H.; Khemka, V. A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br. J. Cancer 2017, 117, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.; Jain, R.; et al. Open-label, phase 1 study of Nivolumab combined with nab-paclitaxel plus gemcitabine in advanced pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822. [Google Scholar] [CrossRef]
- Borazanci, E.H.; Jameson, G.S.; Borad, M.J.; Ramanathan, R.K.; Korn, R.L.; Caldwell, L.; Ansaldo, K.; Hendrickson, K.; Marceau, K.; Von Hoff, D.D. A phase II pilot trial of nivolumab (N) + albumin bound paclitaxel (AP) + paricalcitol (P) + cisplatin (C) + gemcitabine (G) (NAPPCG) in patients with previously untreated metastatic pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2018, 36 (Suppl. 4), 358. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, U.K.; Goedegebuure, P.S.; Moore, T.T.; Viehl, C.T.; Moo-Young, T.A.; Larson, J.W.; Frey, D.M.; Ehlers, J.P.; Eberlein, T.J.; Linehan, D.C. Increased prevalence of regulatory T cells (Treg) is induced by pancreas adenocarcinoma. J. Immunother. 2006, 29, 416–424. [Google Scholar] [CrossRef]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A., Jr.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H., Jr.; Bagalà, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef]
- Bengsch, F.; Knoblock, D.M.; Liu, A.; McAllister, F.; Beatty, G.L. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017, 66, 1609–1617. [Google Scholar] [CrossRef]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Colombo, N.; Arrowsmith, E.; Narayan, V.; Yonemori, K.; Scambia, G.; Zelnak, A.; Bauer, T.M.; Jin, N.; Ulahannan, S.V.; et al. Avelumab Plus Talazoparib in Patients with BRCA1/2- or ATM-Altered Advanced Solid Tumors: Results from JAVELIN BRCA/ATM, an Open-Label, Multicenter, Phase 2b, Tumor-Agnostic Trial. JAMA Oncol. 2023, 9, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation. JCO Precis. Oncol. 2018, 2018, PO.17.00316. [Google Scholar] [CrossRef]
- Reiss, K.A.; Mick, R.; Teitelbaum, U.; O’Hara, M.; Schneider, C.; Massa, R.; Karasic, T.; Tondon, R.; Onyiah, C.; Gosselin, M.K.; et al. Niraparib plus nivolumab or niraparib plus ipilimumab in patients with platinum-sensitive advanced pancreatic cancer: A randomised, phase 1b/2 trial. Lancet Oncol. 2022, 23, 1009–1020. [Google Scholar] [CrossRef]
- Fumet, J.D.; Limagne, E.; Thibaudin, M.; Truntzer, C.; Bertaut, A.; Rederstorff, E.; Ghiringhelli, F. Precision medicine phase II study evaluating the efficacy of a double immunotherapy by durvalumab and tremelimumab combined with olaparib in patients with solid cancers and carriers of homologous recombination repair genes mutation in response or stable after olaparib treatment. BMC Cancer 2020, 20, 748. [Google Scholar]
- Chung, V.; Guthrie, K.A.; Pishvaian, M.J.; Lowy, A.M.; Chiorean, E.G.; Duong, M.T.; O’Reilly, E.M.; Philip, P.A. Randomized phase II trial of olaparib + pembrolizumab versus olaparib alone as maintenance therapy in metastatic pancreatic cancer patients with germline BRCA1 or BRCA2 (gBRCA1/2+) mutations: SWOG S2001. J. Clin. Oncol. 2021, 39 (Suppl. 3), TPS447. [Google Scholar] [CrossRef]
- Park, W.; O’Connor, C.; Chou, J.F.; Schwartz, C.; Varghese, A.M.; Larsen, M.; Balogun, F.; Brenner, R.; Yu, K.H.; Diguglielmo, E.; et al. Phase 2 trial of pembrolizumab and olaparib (POLAR) maintenance for patients (pts) with metastatic pancreatic cancer (mPDAC): Two cohorts B non-core homologous recombination deficiency (HRD) and C exceptional response to platinum-therapy. J. Clin. Oncol. 2023, 41 (Suppl 16), 4140. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014, 25, 846–859. [Google Scholar] [CrossRef]
- Razak, A.R.; Cleary, J.M.; Moreno, V.; Boyer, M.; Calvo Aller, E.; Edenfield, W.; Tie, J.; Harvey, R.D.; Rutten, A.; Shah, M.A.; et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J. Immunother. Cancer 2020, 8, e001006. [Google Scholar] [CrossRef]
- Johnson, M.; Dudek, A.Z.; Sukari, A.; Call, J.; Kunk, P.R.; Lewis, K.; Gainor, J.F.; Sarantopoulos, J.; Lee, P.; Golden, A.; et al. ARRY-382 in Combination with Pembrolizumab in Patients with Advanced Solid Tumors: Results from a Phase 1b/2 Study. Clin. Cancer Res. 2022, 28, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Piha-Paul, S.A.; Luke, J.; Kim, E.J.; Thompson, J.A.; Britten, C.D.; Johnson, J.M.; Pfanzelter, N.; Gordon, M.; Rasco, D.W.; et al. First-In-Human phase 1 dose escalation and expansion of a novel combination, anti–CSF-1 receptor (cabiralizumab) plus anti–PD-1 (nivolumab), in patients with advanced solid tumors. J. Immunother. Cancer 2017, 5 (Suppl. 3), 89. [Google Scholar]
- A Phase 2 Study of Cabiralizumab (BMS-986227, FPA008) Administered in Combination with Nivolumab (BMS-936558) with and without Chemotherapy in Patients with Advanced Pancreatic Cancer. Available online: https://clinicaltrials.gov/study/NCT03336216?term=NCT03336216&rank=1 (accessed on 24 March 2023).
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Lim, K.H.; McWilliams, R.; Suresh, R.; Lockhart, A.C.; Brown, A.; Breden, M.; Belle, J.I.; Herndon, J.; Bogner, S.J.; et al. Defactinib, Pembrolizumab, and Gemcitabine in Patients with Advanced Treatment Refractory Pancreatic Cancer: A Phase I Dose Escalation and Expansion Study. Clin. Cancer Res. 2022, 28, 5254–5262. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Bajor, D.L.; Winograd, R.; Evans, R.A.; Bayne, L.J.; Beatty, G.L. CD40 immunotherapy for pancreatic cancer. Cancer Immunol. Immunother. 2013, 62, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- Van Hooren, L.; Vaccaro, A.; Ramachandran, M.; Vazaios, K.; Libard, S.; van de Walle, T.; Georganaki, M.; Huang, H.; Pietilä, I.; Lau, J.; et al. Agonistic CD40 therapy induces tertiary lymphoid structures but impairs responses to checkpoint blockade in glioma. Nat. Commun. 2021, 12, 4127. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef]
- Zhang, T.; Tan, X.L.; Xu, Y.; Wang, Z.Z.; Xiao, C.H.; Liu, R. Expression and Prognostic Value of Indoleamine 2,3-dioxygenase in Pancreatic Cancer. Chin. Med. J. 2017, 130, 710–716. [Google Scholar] [CrossRef]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.J.; Hellmann, M.D.; Cervantes, A.; Ochoa de Olza, M.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Algazi, A.P.; Falchook, G.S.; Creelan, B.C.; Powderly, J.; Rosen, S.; Barve, M.; Mettu, N.B.; Triozzi, P.L.; Hamm, J.; et al. Phase 1/2 study of epacadostat in combination with durvalumab in patients with metastatic solid tumors. Cancer 2023, 129, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Yoshie, O.; Matsushima, K. CCR4 and its ligands: From bench to bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef] [PubMed]
- DeLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A phase I study of the Anti-CC chemokine receptor 4 antibody, mogamulizumab in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef]
- Zamarin, D.; Hamid, O.; Nayak-Kapoor, A.; Sahebjam, S.; Sznol, M.; Collaku, A.; Fox, F.E.; Marshall, M.A.; Hong, D.S. Mogamulizumab in Combination with Durvalumab or Tremelimumab in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2020, 26, 4531–4541. [Google Scholar] [CrossRef]
- Arora, S.; Majhail, N.S.; Liu, H. Hematopoietic Progenitor Cell Mobilization for Autologous Stem Cell Transplantation in Multiple Myeloma in Contemporary Era. Clin. Lymphoma Myeloma Leuk. 2019, 19, 200–205. [Google Scholar] [CrossRef]
- Brave, M.; Farrell, A.; Ching Lin, S.; Ocheltree, T.; Pope Miksinski, S.; Lee, S.L.; Saber, H.; Fourie, J.; Tornoe, C.; Booth, B.; et al. FDA review summary: Mozobil in combination with granulocyte colony-stimulating factor to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation. Oncology 2010, 78, 282–288. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8+ T cells via CXCR4 blockade facilitates PD-1 checkpoint therapy in human pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; Messersmith, W.; Kindler, H.; Zhang, W.; Pitou, C.; Szpurka, A.M.; Wang, D.; Peng, S.B.; Vangerow, B.; Khan, A.A.; et al. Safety and Pharmacokinetics of CXCR4 Peptide Antagonist, LY2510924, in Combination with Durvalumab in Advanced Refractory Solid Tumors. J. Pancreat. Cancer 2020, 6, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Williams, A.; Schreiber, J.; Hohmann, N.; Pruefer, U.; Krauss, J.; Jäger, D.; Frömming, A.; Beyer, D.; Eulberg, D.; et al. Combined inhibition of CXCL12 and PD-1 in MSS colorectal and pancreatic cancer: Modulation of the microenvironment and clinical effects. J. Immunother. Cancer 2021, 9, e002505. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Ullman, N.A.; Ruffolo, L.I.; Jackson, K.M.; Chacon, A.; Georger, M.; Jewell, R.; Belt, B.A.; Maeda, D.; Zebala, J.; Linehan, D. CXCR1/2 blockade to enhance response to immune checkpoint inhibition in an aggressive orthotopic pancreatic adenocarcinoma model. J. Clin. Oncol. 2020, 38 (Suppl. 5), 19. [Google Scholar] [CrossRef]
- Kudo, D.; Suto, A.; Hakamada, K. The Development of a Novel Therapeutic Strategy to Target Hyaluronan in the Extracellular Matrix of Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2017, 18, 600. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef]
- Rosengren, S.; Clift, R.; Zimmerman, S.J.; Souratha, J.; Thompson, B.J.; Blouw, B.; Li, X.; Zhao, Q.; Shepard, M.; Maneval, D.C.; et al. PEGylated recombinant hyaluronidase PH20 (PEGPH20) enhances checkpoint inhibitor efficacy in syngeneic mouse models of cancer. Cancer Res. 2016, 76 (Suppl. 14), 4886. [Google Scholar] [CrossRef]
- Zhen, D.B.; Whittle, M.; Ritch, P.S.; Hochster, H.S.; Coveler, A.L.; George, B.; Hendifar, A.E.; Dragovich, T.; Green, S.; Dion, B.; et al. Phase II study of PEGPH20 plus pembrolizumab for patients (pts) with hyaluronan (HA)-high refractory metastatic pancreatic ductal adenocarcinoma (mPC): PCRT16-001. J. Clin. Oncol. 2022, 40 (Suppl. 4), 576. [Google Scholar] [CrossRef]
- Principe, D.R.; DeCant, B.; Mascariñas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFβ blockade augments PD-1 inhibition to promote T-cell-mediated regression of pancreatic cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Sow, H.S.; Ren, J.; Camps, M.; Ossendorp, F.; Ten Dijke, P. Combined inhibition of TGF-β signaling and the PD-L1 immune checkpoint is differentially effective in tumor models. Cells 2019, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Heery, C.R.; Schlom, J.; Madan, R.A.; Cao, L.; Kang, Z.; Lamping, E.; Marté, J.L.; Donahue, R.N.; Grenga, I.; et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin. Cancer Res. 2018, 24, 1287–1295. [Google Scholar] [CrossRef]
- Liu, D.; Zhou, J.; Wang, Y.; Li, M.; Jiang, H.; Liu, Y.; Yin, X.; Ge, M.; Xiang, X.; Ying, J.; et al. Bifunctional anti-PD-L1/TGF-βRII agent SHR-1701 in advanced solid tumors: A dose-escalation, dose-expansion, and clinical-expansion phase 1 trial. BMC Med. 2022, 20, 408. [Google Scholar] [CrossRef]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef]
- Rule, S.; Dreyling, M.; Goy, A.; Hess, G.; Auer, R.; Kahl, B.; Hernández-Rivas, J.Á.; Qi, K.; Deshpande, S.; Parisi, L.; et al. Ibrutinib for the treatment of relapsed/refractory mantle cell lymphoma: Extended 3.5-year follow up from a pooled analysis. Haematologica 2019, 104, e211–e214. [Google Scholar] [CrossRef]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- Strouch, M.J.; Cheon, E.C.; Salabat, M.R.; Krantz, S.B.; Gounaris, E.; Melstrom, L.G.; Dangi-Garimella, S.; Wang, E.; Munshi, H.G.; Khazaie, K.; et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin. Cancer Res. 2010, 16, 2257–2265. [Google Scholar] [CrossRef]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef]
- Masso-Valles, D.; Jauset, T.; Serrano, E.; Sodir, N.M.; Pedersen, K.; Affara, N.I.; Whitfield, J.R.; Beaulieu, M.E.; Evan, G.I.; Elias, L.; et al. Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res. 2015, 75, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Rasco, D.; Veeder, M.; Luke, J.J.; Chandler, J.; Balmanoukian, A.; George, T.J.; Munster, P.; Berlin, J.D.; Gutierrez, M.; et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology 2019, 97, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.; Javle, M.; Davis, R.E.; Vats, P.; Kumar-Sinha, C.; Xiao, L.; Mettu, N.B.; Parra, E.R.; Benson, A.B.; Lopez, C.D.; et al. Randomized phase II study of the Bruton tyrosine kinase inhibitor acalabrutinib, alone or with pembrolizumab in patients with advanced pancreatic cancer. J. Immunother. Cancer 2020, 8, e000587. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.; Oh, D.Y.; Tabernero, J.; Reni, M.; Van Cutsem, E.; Hendifar, A.; Waldschmidt, D.T.; Starling, N.; Bachet, J.B.; Chang, H.M.; et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine for first-line treatment of patients with metastatic pancreatic adenocarcinoma: Phase III RESOLVE study. Ann. Oncol. 2021, 32, 600–608. [Google Scholar] [CrossRef]
- Ma, Y.; Li, J.; Wang, H.; Chiu, Y.; Kingsley, C.V.; Fry, D.; Delaney, S.N.; Wei, S.C.; Zhang, J.; Maitra, A.; et al. Combination of PD-1 inhibitor and OX40 agonist induces tumor rejection and immune memory in mouse models of pancreatic cancer. Gastroenterology 2020, 159, 306–319.e12. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; Plambeck-Suess, S.; Goedegebuure, P.; Simon, P.O.; Mitchem, J.B.; Hornick, J.R.; Sorscher, S.; Picus, J.; Suresh, R.; Lockhart, A.C.; et al. A phase I study of IMP321 and gemcitabine as the front-line therapy in patients with advanced pancreatic adenocarcinoma. Investig. N Drugs 2013, 31, 707–713. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Lambert, L.J.; Ely, Z.A.; Pattada, N.B.; Bhutkar, A.; Eng, G.; Mercer, K.L.; Garcia, A.P.; Lin, L.; Rideout, W.M., 3rd; et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell. 2021, 39, 1342–1360. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Yuan, L.; Tatineni, J.; Mahoney, K.M.; Freeman, G.J. VISTA: A Mediator of Quiescence and a Promising Target in Cancer Immunotherapy. Trends Immunol. 2021, 42, 209–227. [Google Scholar] [CrossRef]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef]
- Patnaik, A.; Yap, T.A.; Chung, H.C.; de Miguel, M.J.; Bang, Y.J.; Lin, C.C.; Su, W.C.; Italiano, A.; Chow, K.H.; Szpurka, A.M.; et al. Safety and Clinical Activity of a New Anti-PD-L1 Antibody as Monotherapy or Combined with Targeted Therapy in Advanced Solid Tumors: The PACT Phase Ia/Ib Trial. Cancer Res. 2021, 27, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Ager, C.R.; Boda, A.; Rajapakshe, K.; Lea, S.T.; Di Francesco, M.E.; Jayaprakash, P.; Slay, R.B.; Morrow, B.; Prasad, R.; Dean, M.A.; et al. High potency STING agonists engage unique myeloid pathways to reverse pancreatic cancer immune privilege. J. Immunother. Cancer 2021, 9, e003246. [Google Scholar] [CrossRef] [PubMed]
- Gulhati, P.; Schalck, A.; Jiang, S.; Shang, X.; Wu, C.J.; Hou, P.; Ruiz, S.H.; Soto, L.S.; Parra, E.; Ying, H.; et al. Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer. Nat. Cancer 2023, 4, 62–80. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilkinson, G.A.; Eng, K.H.; Fields, P.; Raber, P.; Moseley, J.L.; Cheetham, K.; Coffey, M.; Nuovo, G.; Kalinski, P.; et al. Pembrolizumab in combination with the oncolytic virus Pelareorep and chemotherapy in patients with advanced pancreatic adenocarcinoma: A phase Ib study. Clin. Cancer Res. 2020, 26, 71–81. [Google Scholar] [CrossRef]
- Collienne, M.; Loghmani, H.; Heineman, T.C.; Arnold, D. GOBLET: A phase I/II study of pelareorep and atezolizumab +/− chemo in advanced or metastatic gastrointestinal cancers. Future Oncol. 2022, 18, 2871–2878. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Herrera, F.G.; Bourhis, J.; Coukos, G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J. Clin. 2017, 67, 65–85. [Google Scholar] [CrossRef]
- Sharabi, A.B.; Lim, M.; DeWeese, T.L.; Drake, C.G. Radiation and checkpoint blockade immunotherapy: Radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015, 16, e498–e509. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.X. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef]
- Azad, A.; Yin Lim, S.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Chen, I.M.; Johansen, J.S.; Theile, S.; Hjaltelin, J.X.; Novitski, S.I.; Brunak, S.; Hasselby, J.P.; Willemoe, G.L.; Lorentzen, T.; Madsen, K.; et al. Randomized phase II study of nivolumab with or without ipilimumab combined with stereotactic body radiotherapy for refractory metastatic pancreatic cancer (CheckPAC). J. Clin. Oncol. 2022, 40, 3180–3189. [Google Scholar] [CrossRef]
- Parikh, A.R.; Szabolcs, A.; Allen, J.N.; Clark, J.W.; Wo, J.Y.; Raabe, M.; Thel, H.; Hoyos, D.; Mehta, A.; Arshad, S.; et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat. Cancer 2021, 2, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Duffy, A.G.; Brar, G.; Fioravanti, S.; Mabry-Hrones, D.; Walker, M.; Bonilla, C.M.; Wood, B.J.; Citrin, D.E.; Gil Ramirez, E.M.; et al. Immune Checkpoint Blockade in Combination with Stereotactic Body Radiotherapy in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2020, 26, 2318–2326. [Google Scholar] [CrossRef]
- Chen, I.M.; Donia, M.; Chamberlain, C.A.; Jensen, A.W.P.; Draghi, A.; Theile, S.; Madsen, K.; Hasselby, J.P.; Toxværd, A.; Høgdall, E.; et al. Phase 2 study of ipilimumab, nivolumab, and tocilizumab combined with stereotactic body radiotherapy in patients with refractory pancreatic cancer (TRIPLE-R). Eur. J. Cancer 2023, 180, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Dada, H.; Kotzin, J.J.; Henao-Mejia, J.; Minn, A.J.; Twyman-Saint Victor, C.; Vonderheide, R.H. Radiotherapy and CD40 activation separately augment immunity to checkpoint blockade in cancer. Cancer Res. 2018, 78, 4282–4291. [Google Scholar] [CrossRef]
- Fujiwara, K.; Saung, M.T.; Jing, H.; Herbst, B.; Zarecki, M.; Muth, S.; Wu, A.; Bigelow, E.; Chen, L.; Li, K.; et al. Interrogating the immune-modulating roles of radiation therapy for a rational combination with immune-checkpoint inhibitors in treating pancreatic cancer. J. Immunother. Cancer 2020, 8, e000351. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, L.; Wu, C.; Zhu, Y.; Xu, B.; Zheng, X.; Sun, M.; Wen, W.; Dai, X.; Yang, M.; et al. PD-1 blockade boosts radiofrequency ablation-elicited adaptive immune responses against tumor. Clin. Cancer Res. 2016, 22, 1173–1184. [Google Scholar] [CrossRef]
- Giardino, A.; Innamorati, G.; Ugel, S.; Perbellini, O.; Girelli, R.; Frigerio, I.; Regi, P.; Scopelliti, F.; Butturini, G.; Paiella, S.; et al. Immunomodulation after radiofrequency ablation of locally advanced pancreatic cancer by monitoring the immune response in 10 patients. Pancreatology 2017, 17, 962–966. [Google Scholar] [CrossRef]
- Martin, R.C., 2nd; Kwon, D.; Chalikonda, S.; Sellers, M.; Kotz, E.; Scoggins, C.; McMasters, K.M.; Watkins, K. Treatment of 200 Locally Advanced (Stage III) Pancreatic Adenocarcinoma Patients with Irreversible Electroporation: Safety and Efficacy. Ann. Surg. 2015, 262, 486–494. [Google Scholar] [CrossRef]
- Lin, M.; Alnaggar, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. An important discovery on combination of irreversible electroporation and allogeneic natural killer cell immunotherapy for unresectable pancreatic cancer. Oncotarget 2017, 8, 101795–101807. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. Short-term clinical efficacy of percutaneous irreversible electroporation combined with allogeneic natural killer cell for treating metastatic pancreatic cancer. Immunol. Lett. 2017, 186, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Bulvik, B.E.; Rozenblum, N.; Gourevich, S.; Ahmed, M.; Andriyanov, A.V.; Galun, E.; Goldberg, S.N. Irreversible Electroporation versus Radiofrequency Ablation: A Comparison of Local and Systemic Effects in a Small-Animal Model. Radiology 2016, 280, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Steger, A.; Lohr, H.; Bourhis, H.; Hoelz, H.; Kirchleitner, S.V.; Stieg, M.R.; Grassmann, S.; Kobold, S.; Siveke, J.T.; et al. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8+ T cells. Cell Death Differ. 2014, 21, 1825–1837. [Google Scholar] [CrossRef]
- Li, X.; Xu, K.; Li, W.; Qiu, X.; Ma, B.; Fan, Q.; Li, Z. Immunologic response to tumor ablation with irreversible electroporation. PLoS ONE 2012, 7, e48749. [Google Scholar] [CrossRef]
- Zhao, J.; Wen, X.; Tian, L.; Li, T.; Xu, C.; Wen, X.; Melancon, M.P.; Gupta, S.; Shen, B.; Peng, W.; et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat. Commun. 2019, 10, 899. [Google Scholar] [CrossRef]
- Narayanan, J.S.S.; Miller, A.; Hayashi, T.; Ray, P.; Schoenberger, S.; Carson, D.; White, R. Irreversible electroporation is an “in situ vaccine” and induces antitumor immune response in pancreatic cancer. Cancer Immunol. Res. 2020, 8 (Suppl. 4), A14. [Google Scholar] [CrossRef]
- Narayanan, J.S.S.; Ray, P.; Hayashi, T.; Whisenant, T.C.; Vicente, D.; Carson, D.A.; Miller, A.M.; Schoenberger, S.P.; White, R.R. Irreversible Electroporation Combined with Checkpoint Blockade and TLR7 Stimulation Induces Antitumor Immunity in a Murine Pancreatic Cancer Model. Cancer Immunol. Res. 2019, 7, 1714–1726. [Google Scholar] [CrossRef]
- Irreversible Electroporation (IRE) Followed by Nivolumab in Patients with Metastatic Pancreatic Cancer. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04212026 (accessed on 12 August 2023).
- Geboers, B.; Timmer, F.E.F.; Ruarus, A.H.; Pouw, J.E.E.; Schouten, E.A.C.; Bakker, J.; Puijk, R.S.; Nieuwenhuizen, S.; Dijkstra, M.; van den Tol, M.P.; et al. Irreversible Electroporation and Nivolumab Combined with Intratumoral Administration of a Toll-Like Receptor Ligand, as a Means of In Vivo Vaccination for Metastatic Pancreatic Ductal Adenocarcinoma (PANFIRE-III). A Phase-I Study Protocol. Cancers 2021, 13, 3902. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Trial | Phase | Treatment | Setting | N | Primary and Secondary Outcomes | Results | Ref. |
|---|---|---|---|---|---|---|---|
| CCTG PA.7 NCT02879318 | II | Gemcitabine + nab-paclitaxel + durvalumab + tremelimumab vs. Gemcitabine + nab-paclitaxel | First-line | 180 | 1. mOS 2. mPFS, ORR | mOS: 9.8 vs. 8.8 months (HR: 0.94, 90% CI, 0.71–1.25), p = 0.72 mPFS: 5.5 vs. 5.4 months (HR: 0.98, 90% CI, 0.75–1.29), p = 0.91 ORR: 30.3% vs. 23.0%, p = 0.28 | [41] |
| CISPD3 | II | Sintilimab + mFOLFIRINOX vs. mFOLFIRINOX | First- or second-line | 110 | 1. mOS 2. mPFS, ORR | mOS: 10.9 vs. 10.8 months (HR: 1.07, 95% CI, 0.69–1.68), p > 0.05 mPFS: 5.9 vs. 5.7 months (HR: 0.93, 95% CI, 0.62–1.40), p > 0.05 ORR: 50% vs. 23.9%, p < 0.05 | [42] |
| PRINCE | II | Arm 1: Gemcitabine + nab-paclitaxel + nivolumab Arm 2: Gemcitabine + nab-paclitaxel + sotigalimab Arm 3: Gemcitabine + nab-paclitaxel + nivolumab + sotigalimab | First-line | 105 | 1. 1-year OS vs. historical control rate of 35% 2. mPFS, ORR, DCR, DOR | Arm 1: 1-year OS: 57.7%, p = 0.006 mOS: 16.7 months mPFS: 6.4 months Arm 2: 1-year OS: 48.1%, p = 0.062 Arm 3: 1-year OS: 41.3%, p = 0.223 | [43] |
| JapicCTI-184230 | II | mFOLFIRINOX + nivolumab | First-line | 31 | 1. ORR 2. mOS, mPFS | ORR: 32.3% (CR: 0.%, PR: 32.3%) mOS: 13.4 months mPFS: 7.4 months | [44] |
| NCT01896869 | II | FOLFIRINOX followed by ipilimumab + GVAX vs. FOLFIRINOX | Maintenance after 8–12 cycles first-line FOLFIRINOX | 82 | 1. mOS 2. mPFS | mOS: 9.4 vs. 14.7 months (HR: 1.75, 95% CI, 1.09–2.79), p = 0.019 mPFS: 2.4 vs. 5.6 months (HR: 2.92, 95% CI, 1.70–5.02), p < 0.001 | [45] |
| NCT02558894 | II | 4 cycles durvalumab + tremelimumab, followed by durvalumab vs. durvalumab monotherapy, up to 12 months | Second-line | 65 | 1. ORR 2. mPFS, mOS | ORR: 3.1% vs. 0% mPFS: 1.5 vs. 1.5 months mOS: 3.1 vs. 3.6 months | [46] |
| NCT02077881 | II | Gemcitabine + nab-paclitaxel + indoximod | First- or second-line | 104 | 1. mOS 2. ORR | mOS: 10.9 months ORR: 46.2% | [47] |
| NCT02331251 | Ib/II | Gemcitabine + nab-paclitaxel + pembrolizumab | First-or second line | 17 | 1. >15% CR 2. mOS, mPFS | ORR: 17.6% (0 CR + 3 PR) mOS: 15.0 months mPFS: 9.1 months | [48] |
| NCT00112580 | II | Ipilimumab monotherapy | First-/second-/or further-line | 27 | ORR | ORR: 0% | [49] |
| Combination Strategy | Phase | Setting | N | Drugs/Intervention | Efficacy and Survival Data | Potential Mechanism |
|---|---|---|---|---|---|---|
| ICI + PARP inhibitor [65] | I/II | Maintenance after 4 months of platin-based treatment in LAPC and mPDAC | 84 | Anti-PD1 Ab nivolumab + niraparib Anti-CTLA4 Ab ipilimumab + niraparib | mPFS at 6 months: 20.6% vs. 59.6% mOS: 13.2 vs. 17.3 months ORR: 7.7% vs. 15.4% | Increased intratumoral CD8+ activity following treatment with anti-CTLA4 Ab and PARP inhibitor |
| ICI + anti-CSF1R inhibitor [71] | I/II | Second or further-line | 27 | Anti-PD-1 Ab pembrolizumab + CSF1R inhibitor | ORR: 3.7% mPFS: 1.4 months mOS: 2.2 months | Inhibition of TAMs and MDSCs |
| ICI + anti-CCR4 Ab [88] | I | Second or further-line | 24 | Anti-PD-L1 Ab durvalumab + anti-CCR4 Ab mogamulizumab vs. Anti-CTLA4 Ab tremelimumab + anti-CCR4 Ab mogamulizumab | ORR: 0% vs. 0% | Targeting CCR4 in combination with ICI results in a decrease in Tregs and an increased amount of CD8+ T cells |
| ICI + CXCR4 antagonist [94] | II | Second or further- line | 29 | CXCR4 antagonist motixafortide + anti-PD-1 Ab pembrolizumab | ORR: 3.4% DCR: 34.5% mOS: 3.3 months | Increase in intratumoral CD8+ T cells and a reduction in MDSCs |
| ICI + TGFbeta inhibitor [107] | I | Second or further-line | 32 | Anti-PD-L1 Ab durvalumab + TGFbeta receptor I kinase inhibitor galunisertib | mPFS: 1.9 months mOS: 5.7 months ORR: 3.1% | ICI and anti-TGFbeta enhance effector T cell activity |
| ICI + Bruton tyrosine kinase inhibitor [115] | II | Second or further-line | 73 | Acalabrutinib vs. Anti-PD-1 Ab pembrolizumab + acalabrutinib | ORR: 0% vs. 7.9% DCR: 14.3% vs. 21.1% mPFS: 1.4 vs. 1.4 months mOS: 3.6 vs. 3.8 months | Combined treatment decreases MDSCs and activates CD4+ and CD8+ T cells |
| ICI + MET kinase inhibitor [123] | I | Further-line | 17 | PD-L1 inhibitor + MET kinase inhibitor merestinib | ORR: 20% (dose-escalation cohort) 0% (expansion cohort) | ICI + MET kinase inhibitor enhances T cell response and decreases immunosuppressive effects of MDSCs |
| ICI + oncolytic viruses [126] | I | Further-line | 11 | Anti-PD-1 Ab pembrolizumab + pelareorep + chemotherapy | ORR: 9% mPFS: 2.0 months mOS: 3.1 months | Enhances T cell migration to the TME |
| Radiotherapy + ICI [134] | II | Further-line | 84 | SBRT + nivolumab vs. SBRT + nivolumab + ipilimumab | DCR: 17.1% vs. 37.2% mPFS: 1.7 vs. 1.6 months mOS: 3.8 vs. 3.8 months | Radiotherapy + ICI induce an antitumoral effect by increasing CD8+ T cells and CD8+/Treg ratio while decreasing MDSCs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, L.C.; Özdemir, B.C.; Berger, M.D. The Role of Immune Checkpoint Inhibitors in Metastatic Pancreatic Cancer: Current State and Outlook. Pharmaceuticals 2023, 16, 1411. https://doi.org/10.3390/ph16101411
Tran LC, Özdemir BC, Berger MD. The Role of Immune Checkpoint Inhibitors in Metastatic Pancreatic Cancer: Current State and Outlook. Pharmaceuticals. 2023; 16(10):1411. https://doi.org/10.3390/ph16101411
Chicago/Turabian StyleTran, Linh Chi, Berna C. Özdemir, and Martin D. Berger. 2023. "The Role of Immune Checkpoint Inhibitors in Metastatic Pancreatic Cancer: Current State and Outlook" Pharmaceuticals 16, no. 10: 1411. https://doi.org/10.3390/ph16101411
APA StyleTran, L. C., Özdemir, B. C., & Berger, M. D. (2023). The Role of Immune Checkpoint Inhibitors in Metastatic Pancreatic Cancer: Current State and Outlook. Pharmaceuticals, 16(10), 1411. https://doi.org/10.3390/ph16101411