
Discovery of Polyphenolic Natural Products as SARS-CoV-2 Mpro Inhibitors for COVID-19

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
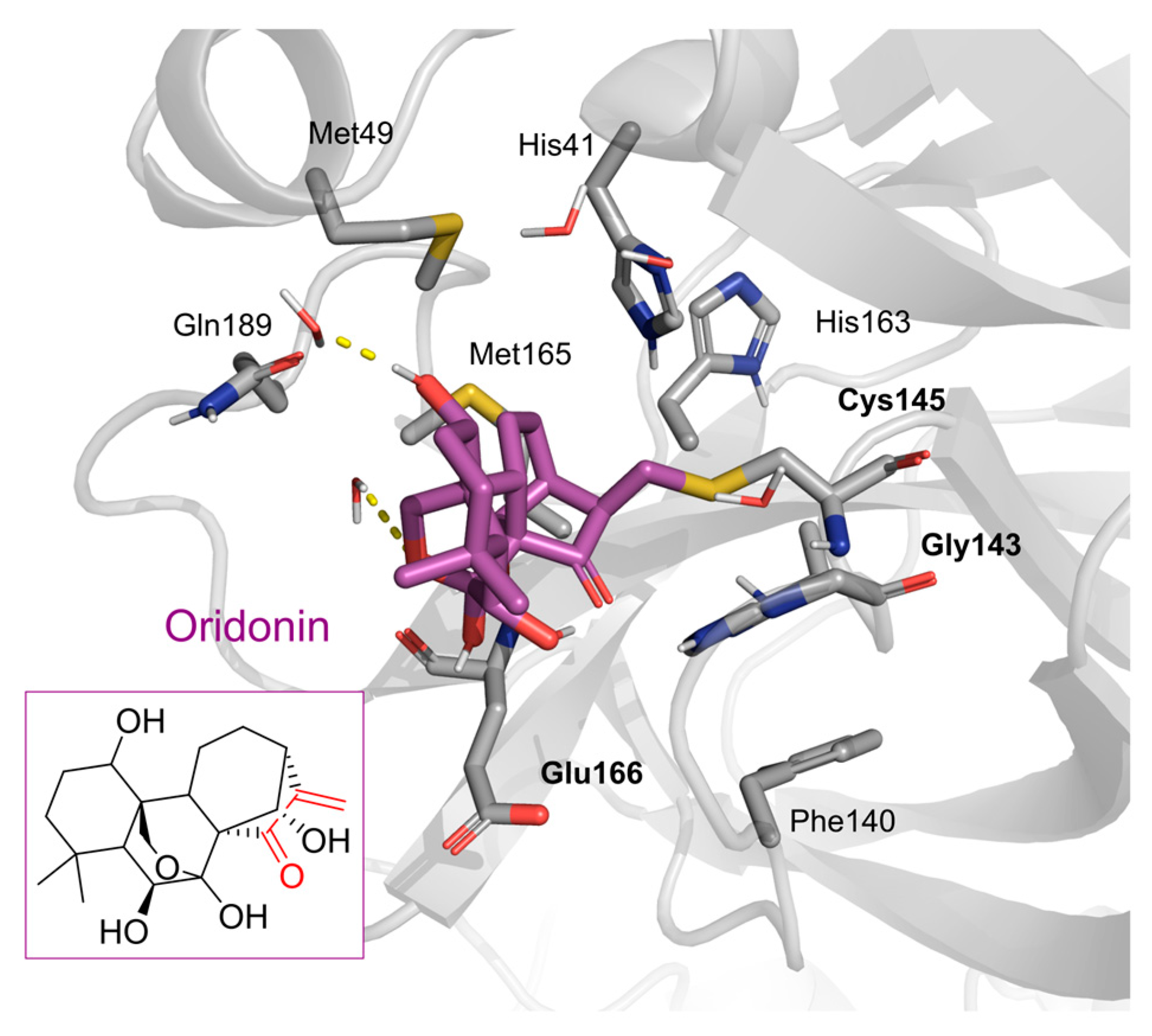
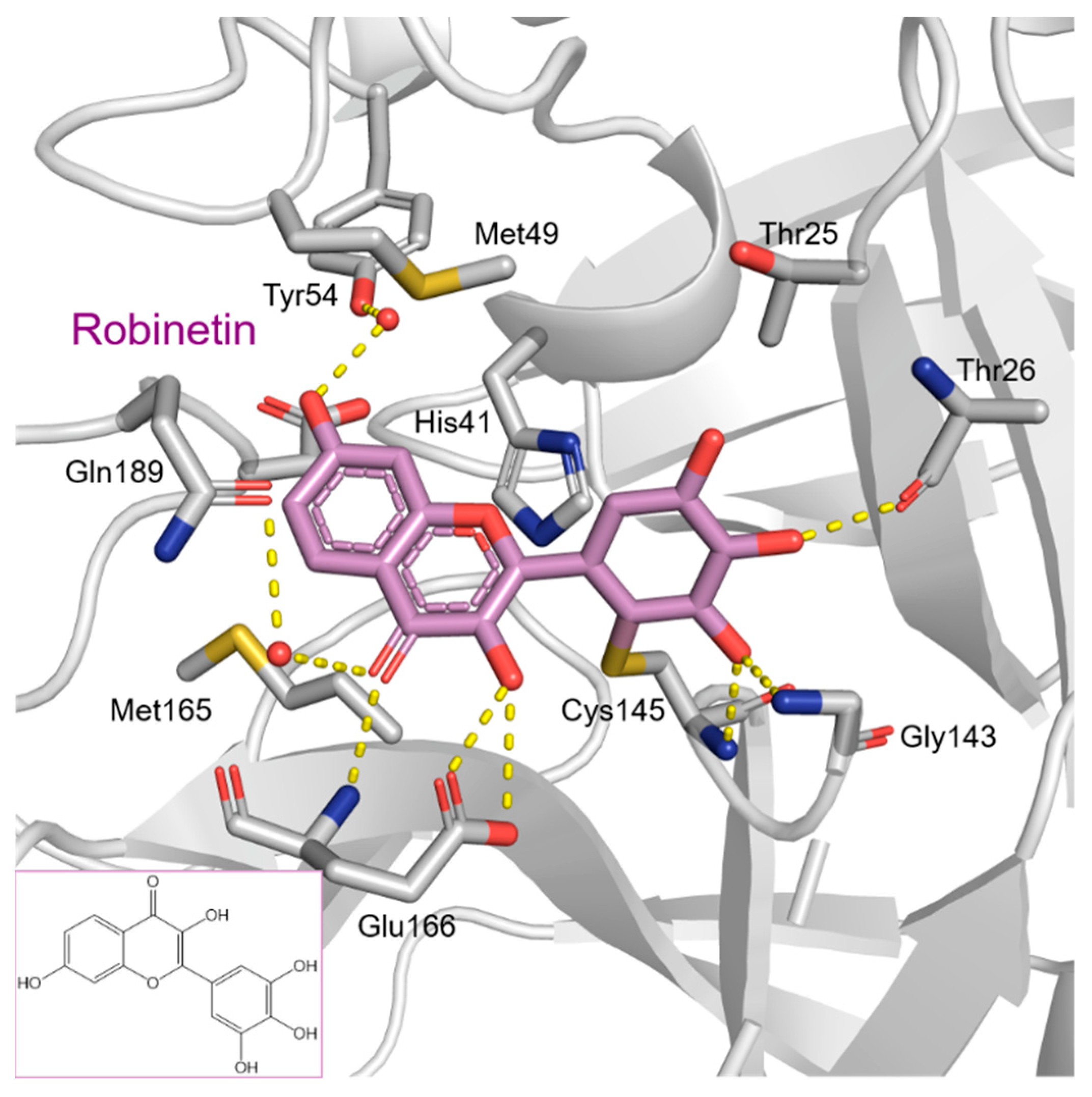
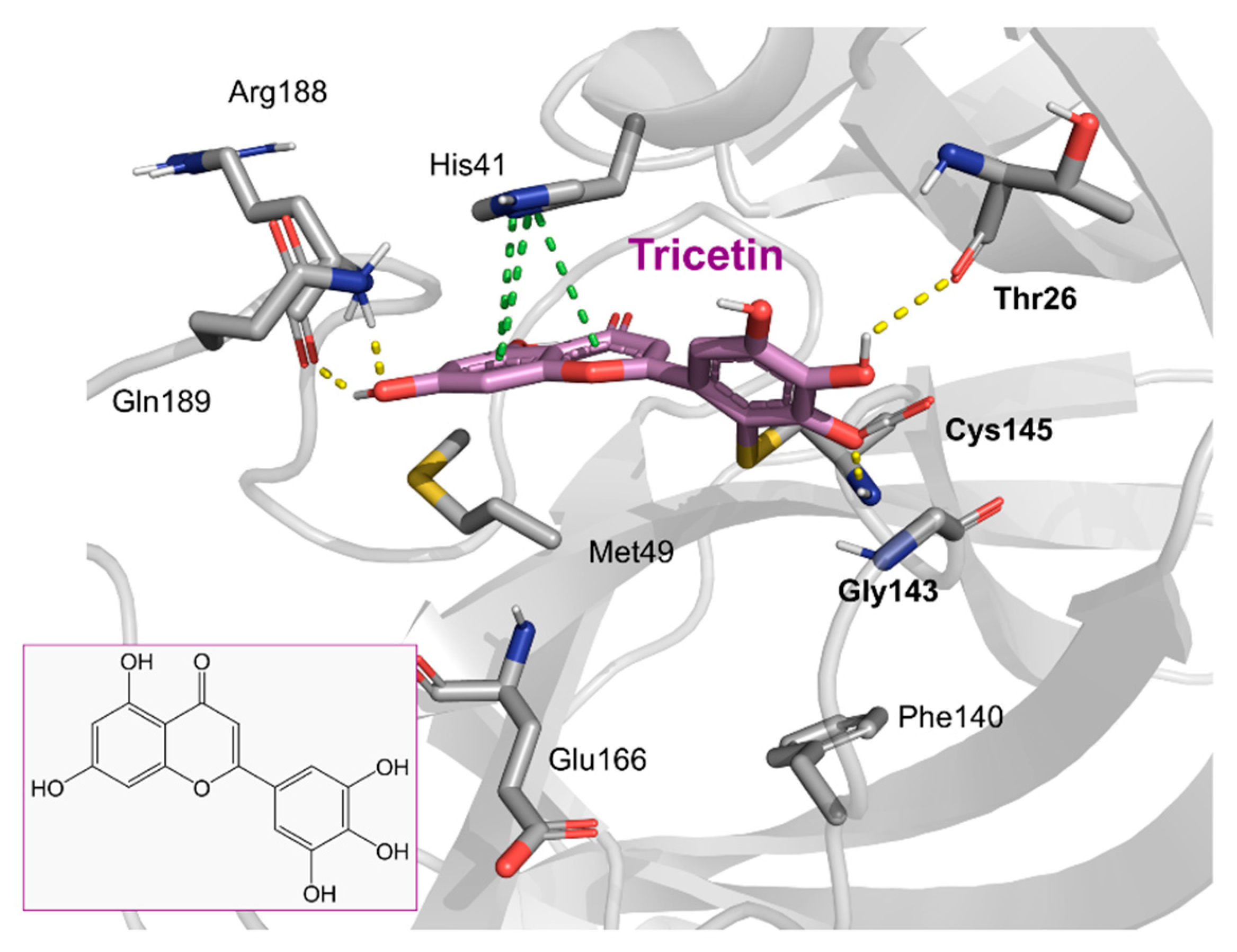
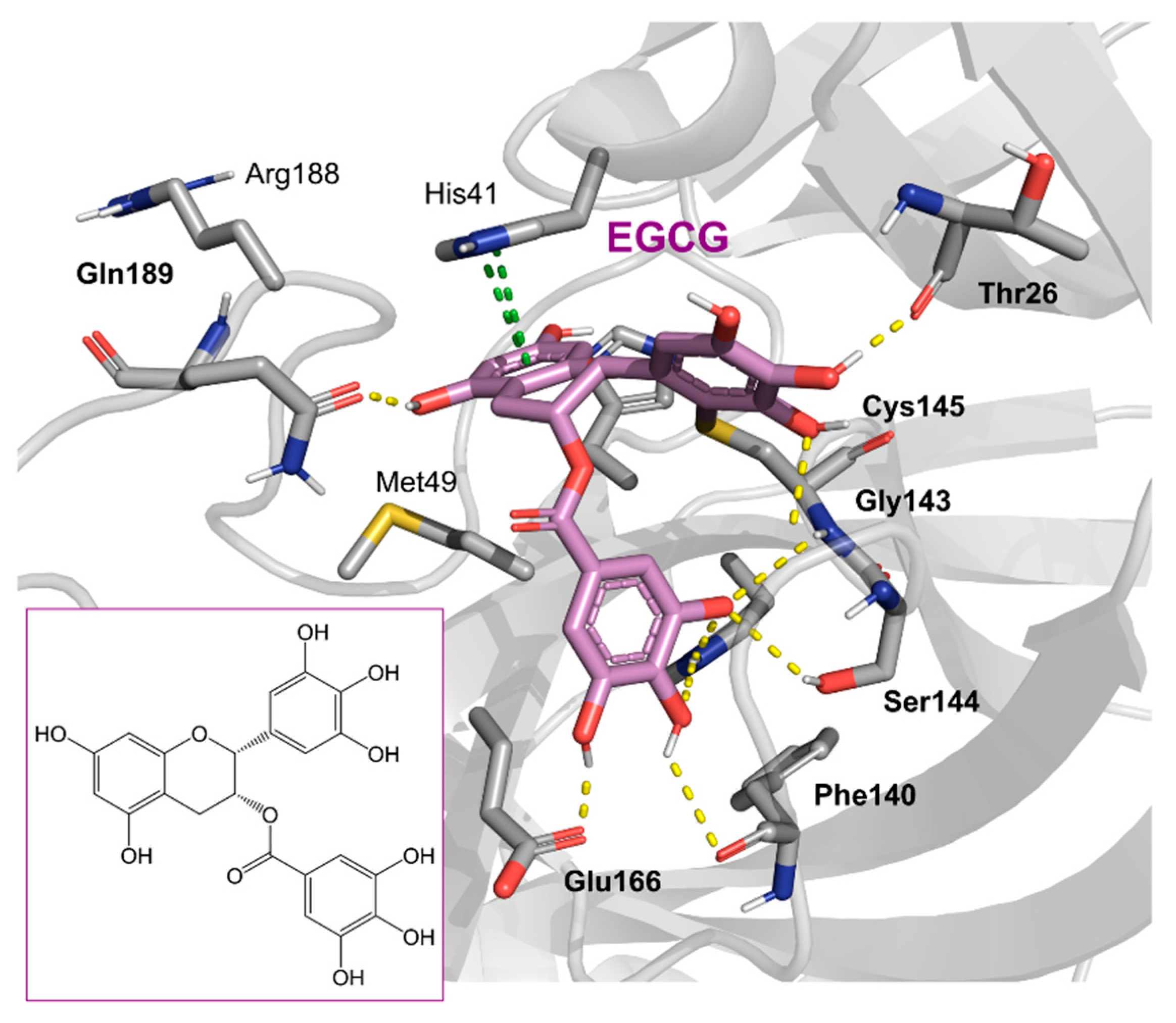
2. Results and Discussion
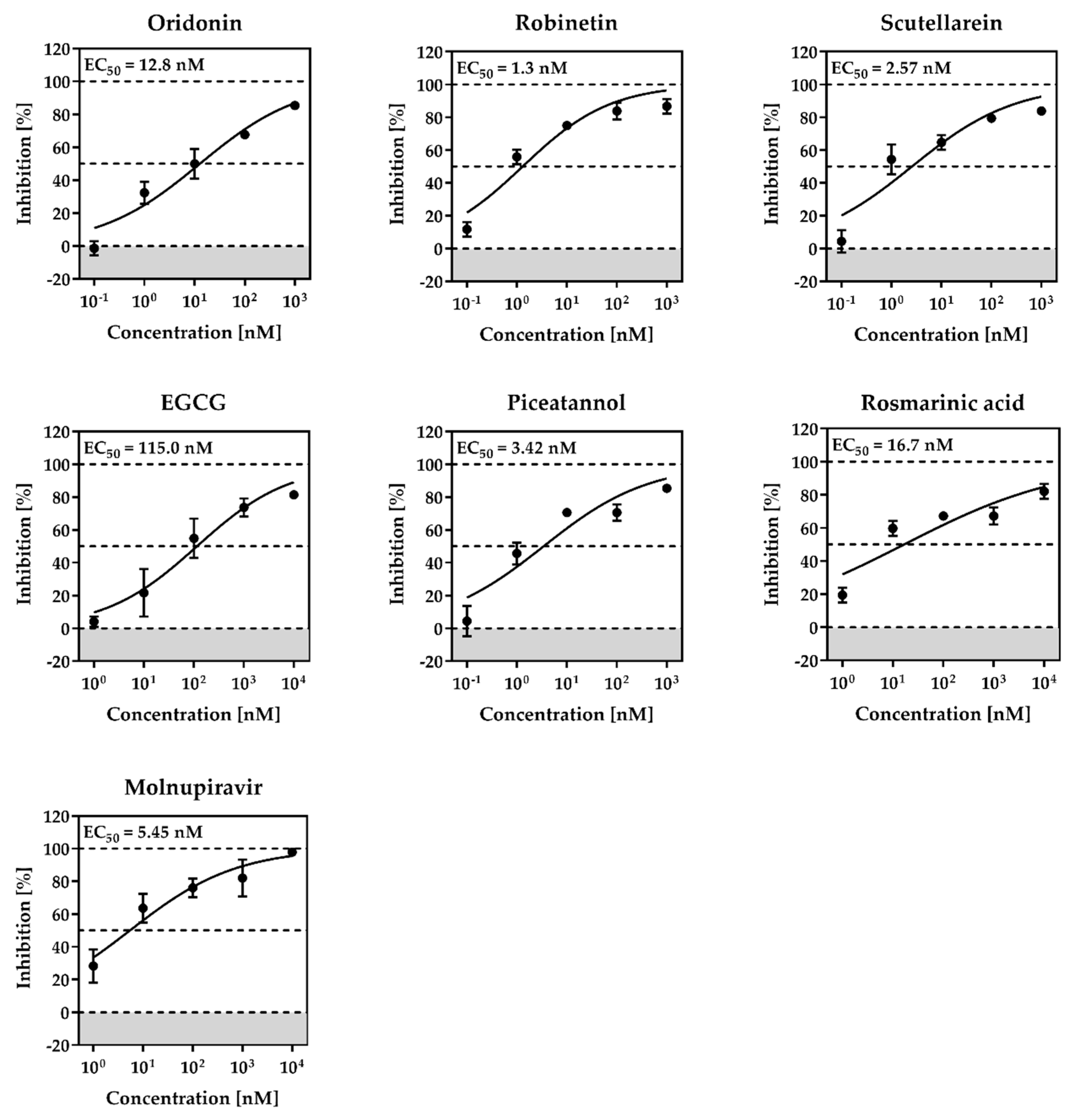
2.1. The Mpro Inhibition Assay
2.2. Cytotoxicity and Anti-SARS-CoV-2 Activity
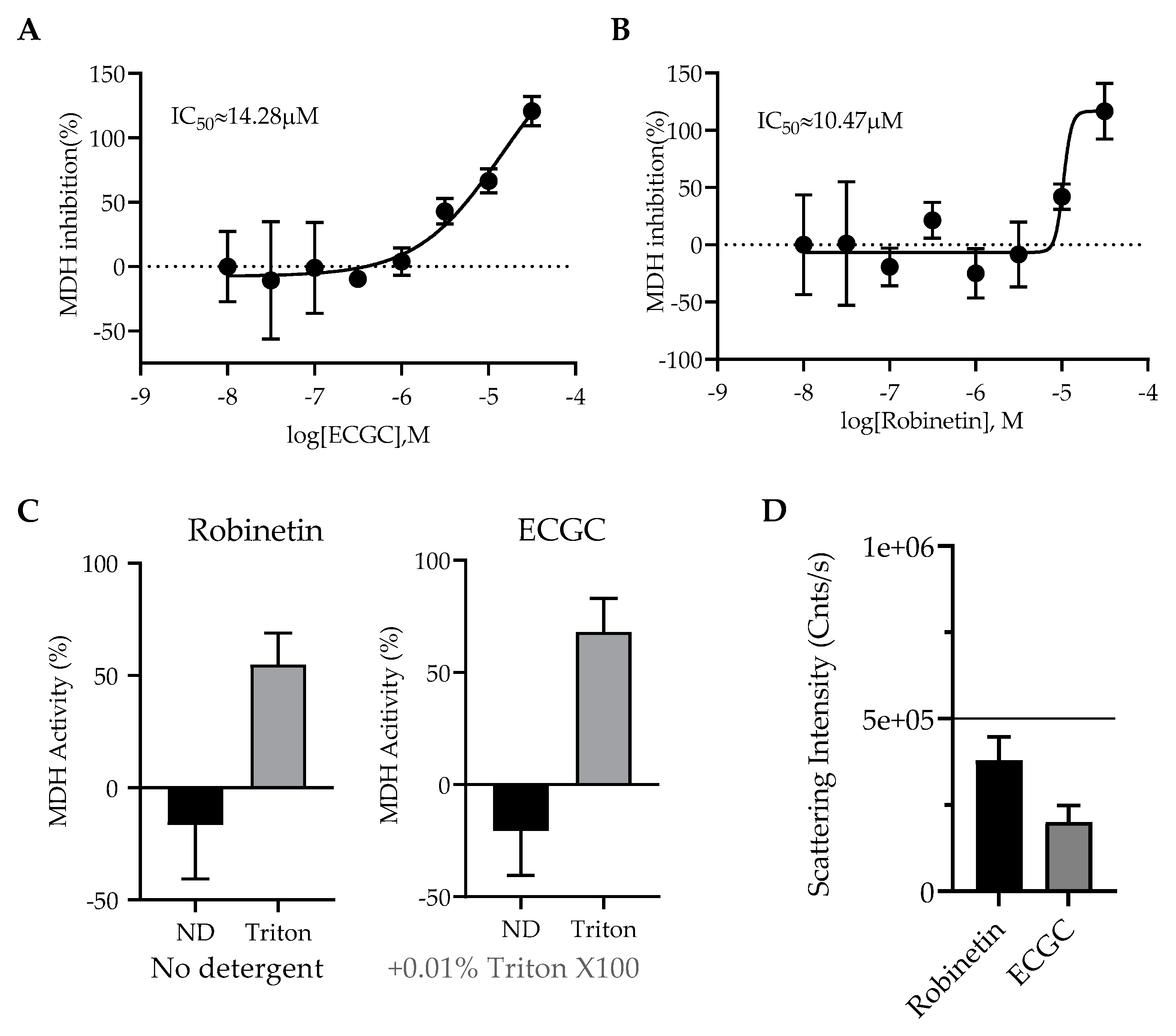
2.3. Colloidal Aggregation Assays
3. Materials and Methods
3.1. Compounds
3.2. The Inhibition Assay of SARS-CoV-2 Mpro
3.3. Dynamic Light Scattering (DLS)
3.4. Malate Dehydrogenase Inhibition Assays
3.5. Cytotoxicity and Antiviral Assays
3.6. Molecular Modelling
3.7. X-ray Protein Crystallography Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pillaiyar, T.; Wendt, L.L.; Manickam, M.; Easwaran, M. The Recent Outbreaks of Human Coronaviruses: A Medicinal Chemistry Perspective. Med. Res. Rev. 2021, 41, 72–135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. Addendum: A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 588, E6. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A Familial Cluster of Pneumonia Associated with the 2019 Novel Coronavirus Indicating Person-to-Person Transmission: A Study of a Family Cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Mellott, D.M.; Tseng, C.-T.; Drelich, A.; Fajtová, P.; Chenna, B.C.; Kostomiris, D.H.; Hsu, J.; Zhu, J.; Taylor, Z.W.; Kocurek, K.I.; et al. A Clinical-Stage Cysteine Protease Inhibitor Blocks SARS-CoV-2 Infection of Human and Monkey Cells. ACS Chem. Biol. 2021, 16, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Konno, S.; Kobayashi, K.; Senda, M.; Funai, Y.; Seki, Y.; Tamai, I.; Schäkel, L.; Sakata, K.; Pillaiyar, T.; Taguchi, A.; et al. 3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents. J. Med. Chem. 2022, 65, 2926–2939. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Kovela, S.; Osswald, H.L.; Amano, M.; Aoki, M.; Agniswamy, J.; Wang, Y.-F.; Weber, I.T.; Mitsuya, H. Structure-Based Design of Highly Potent HIV-1 Protease Inhibitors Containing New Tricyclic Ring P2-Ligands: Design, Synthesis, Biological, and X-ray Structural Studies. J. Med. Chem. 2020, 63, 4867–4879. [Google Scholar] [CrossRef]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Preclinical Characterization of an Intravenous Coronavirus 3CL Protease Inhibitor for the Potential Treatment of COVID-19. Nat. Commun. 2021, 12, 6055. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent Discovery and Development of Inhibitors Targeting Coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.-H. An Overview of Severe Acute Respiratory Syndrome–Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef] [PubMed]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent Insights into Emerging Coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T.; Takemoto, C.; Kim, Y.-T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL Protease Cleaves Its C-Terminal Autoprocessing Site by Novel Subsite Cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of Curcumin: Problems and Promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases. PLoS Biol. 2005, 3, e324. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug Discovery and Therapeutic Options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X.; et al. Structural Basis for the Inhibition of SARS-CoV-2 Main Protease by Antineoplastic Drug Carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar] [CrossRef]
- Al-Harrasi, A.; Behl, T.; Upadhyay, T.; Chigurupati, S.; Bhatt, S.; Sehgal, A.; Bhatia, S.; Singh, S.; Sharma, N.; Vijayabalan, S.; et al. Targeting Natural Products against SARS-CoV-2. Environ. Sci. Pollut. Res. 2022, 29, 42404–42432. [Google Scholar] [CrossRef]
- Li, S.-Y.; Chen, C.; Zhang, H.-Q.; Guo, H.-Y.; Wang, H.; Wang, L.; Zhang, X.; Hua, S.-N.; Yu, J.; Xiao, P.-G.; et al. Identification of Natural Compounds with Antiviral Activities against SARS-Associated Coronavirus. Antivir. Res. 2005, 67, 18–23. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of Pyrogallol as a Warhead in Design of Covalent Inhibitors for the SARS-CoV-2 3CL Protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-X.; Yao, S.; Zhao, W.-F.; Li, M.-J.; Liu, J.; Shang, W.-J.; Xie, H.; Ke, C.-Q.; Hu, H.-C.; Gao, M.-N.; et al. Anti-SARS-CoV-2 Activities in Vitro of Shuanghuanglian Preparations and Bioactive Ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef]
- Miyazawa, M.; Nakamura, Y.; Ishikawa, Y. Insecticidal Sesquiterpene from Alpinia Oxyphylla against Drosophila Melanogaster. J. Agric. Food Chem. 2000, 48, 3639–3641. [Google Scholar] [CrossRef] [PubMed]
- Biolatto, A.; Sancho, A.M.; Cantet, R.J.C.; Güemes, D.R.; Pensel, N.A. Use of Nootkatone as a Senescence Indicator for Rouge La Toma Cv. Grapefruit (Citrus paradisi Macf.). J. Agric. Food Chem. 2002, 50, 4816–4819. [Google Scholar] [CrossRef] [PubMed]
- Kadota, S.; Basnet, P.; Ishii, E.; Tamura, T.; Namba, T. Antibacterial Activity of Trichorabdal A from Rabdosia trichocarpa against Helicobacter pylori. Zentralblatt Bakteriol. Int. J. Med. Microbiol. 1997, 286, 63–67. [Google Scholar] [CrossRef]
- Kuo, L.-M.; Kuo, C.-Y.; Lin, C.-Y.; Hung, M.-F.; Shen, J.-J.; Hwang, T.-L. Intracellular Glutathione Depletion by Oridonin Leads to Apoptosis in Hepatic Stellate Cells. Molecules 2014, 19, 3327–3344. [Google Scholar] [CrossRef]
- Huang, J.; Wu, L.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. A Comparison of the Signal Pathways between the TNF Alpha- and Oridonin-Induced Murine L929 Fibrosarcoma Cell Death. Acta Med. Okayama 2005, 59, 261–270. [Google Scholar] [CrossRef]
- Xu, Y.; Xue, Y.; Wang, Y.; Feng, D.; Lin, S.; Xu, L. Multiple-Modulation Effects of Oridonin on the Production of Proinflammatory Cytokines and Neurotrophic Factors in LPS-Activated Microglia. Int. Immunopharmacol. 2009, 9, 360–365. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, T.; Ma, X.; Jiang, K.; Wu, H.; Qiu, C.; Guo, M.; Deng, G. Oridonin Attenuates the Release of Pro-Inflammatory Cytokines in Lipopolysaccharide-Induced RAW264.7 Cells and Acute Lung Injury. Oncotarget 2017, 8, 68153–68164. [Google Scholar] [CrossRef]
- Song, M.; Liu, X.; Liu, K.; Zhao, R.; Huang, H.; Shi, Y.; Zhang, M.; Zhou, S.; Xie, H.; Chen, H.; et al. Targeting AKT with Oridonin Inhibits Growth of Esophageal Squamous Cell Carcinoma In Vitro and Patient-Derived Xenografts In Vivo. Mol. Cancer Ther. 2018, 17, 1540–1553. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin Is a Covalent NLRP3 Inhibitor with Strong Anti-Inflammasome Activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Peng, W.; Du, S.; Chen, B.; Feng, Y.; Hu, X.; Lai, Q.; Liu, S.; Zhou, Z.-W.; Fang, P.; et al. Oridonin Inhibits SARS-CoV-2 by Targeting Its 3C-Like Protease. Small Sci. 2022, 2, 2270012. [Google Scholar] [CrossRef] [PubMed]
- Birt, D.F.; Walker, B.; Tibbels, M.G.; Bresnick, E. Anti-Mutagenesis and Anti-Promotion by Apigenin, Robinetin and Indole-3-Carbinol. Carcinogenesis 1986, 7, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.L.; Huang, M.T.; Wood, A.W.; Wong, C.Q.; Newmark, H.L.; Yagi, H.; Sayer, J.M.; Jerina, D.M.; Conney, A.H. Effect of Ellagic Acid and Hydroxylated Flavonoids on the Tumorigenicity of Benzo[a]Pyrene and (±)-7β, 8α-Dihydroxy-9α, 10α-Epoxy-7,8,9,10-Tetrahydrobenzo[a]Pyrene on Mouse Skin and in the Newborn Mouse. Carcinogenesis 1985, 6, 1127–1133. [Google Scholar] [CrossRef]
- Kyaw, M.; Yoshizumi, M.; Tsuchiya, K.; Izawa, Y.; Kanematsu, Y.; Tamaki, T. Atheroprotective Effects of Antioxidants through Inhibition of Mitogen-Activated Protein Kinases. Acta Pharmacol. Sin. 2004, 25, 977–985. [Google Scholar]
- Mahmud, S.; Uddin, M.A.R.; Paul, G.K.; Shimu, M.S.S.; Islam, S.; Rahman, E.; Islam, A.; Islam, M.S.; Promi, M.M.; Emran, T.B.; et al. Virtual Screening and Molecular Dynamics Simulation Study of Plant-Derived Compounds to Identify Potential Inhibitors of Main Protease from SARS-CoV-2. Brief. Bioinform. 2021, 22, 1402–1414. [Google Scholar] [CrossRef]
- Ren, J.; Yuan, L.; Wang, W.; Zhang, M.; Wang, Q.; Li, S.; Zhang, L.; Hu, K. Tricetin Protects against 6-OHDA-Induced Neurotoxicity in Parkinson’s Disease Model by Activating Nrf2/HO-1 Signaling Pathway and Preventing Mitochondria-Dependent Apoptosis Pathway. Toxicol. Appl. Pharmacol. 2019, 378, 114617. [Google Scholar] [CrossRef]
- Wu, Q.; Yan, S.; Wang, Y.; Li, M.; Xiao, Y.; Li, Y. Discovery of 4′-O-Methylscutellarein as a Potent SARS-CoV-2 Main Protease Inhibitor. Biochem. Biophys. Res. Commun. 2022, 604, 76–82. [Google Scholar] [CrossRef]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria Baicalensis Extract and Baicalein Inhibit Replication of SARS-CoV-2 and Its 3C-like Protease in Vitro. J. Enzyme Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef]
- Xing, L.; Zhang, H.; Qi, R.; Tsao, R.; Mine, Y. Recent Advances in the Understanding of the Health Benefits and Molecular Mechanisms Associated with Green Tea Polyphenols. J. Agric. Food Chem. 2019, 67, 1029–1043. [Google Scholar] [CrossRef]
- Jang, M.; Park, Y.-I.; Cha, Y.-E.; Park, R.; Namkoong, S.; Lee, J.I.; Park, J. Tea Polyphenols EGCG and Theaflavin Inhibit the Activity of SARS-CoV-2 3CL-Protease In Vitro. Evid.-Based Complement. Altern. Med. ECAM 2020, 2020, 5630838. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.; Li, Z.; Chen, L.; Xu, X. Activity of Compounds from Chinese Herbal Medicine Rhodiola kirilowii (Regel) Maxim against HCV NS3 Serine Protease. Antivir. Res. 2007, 76, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.H.; Woo, H.-J.; Kang, H.-K.; Nguyen, V.D.; Kim, Y.-M.; Kim, D.-W.; Ahn, S.-A.; Xia, Y.; Kim, D. Flavonoid-Mediated Inhibition of SARS Coronavirus 3C-like Protease Expressed in Pichia Pastoris. Biotechnol. Lett. 2012, 34, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Du, A.; Zheng, R.; Disoma, C.; Li, S.; Chen, Z.; Li, S.; Liu, P.; Zhou, Y.; Shen, Y.; Liu, S.; et al. Epigallocatechin-3-Gallate, an Active Ingredient of Traditional Chinese Medicines, Inhibits the 3CLpro Activity of SARS-CoV-2. Int. J. Biol. Macromol. 2021, 176, 1–12. [Google Scholar] [CrossRef]
- Chiou, W.-C.; Chen, J.-C.; Chen, Y.-T.; Yang, J.-M.; Hwang, L.-H.; Lyu, Y.-S.; Yang, H.-Y.; Huang, C. The Inhibitory Effects of PGG and EGCG against the SARS-CoV-2 3C-like Protease. Biochem. Biophys. Res. Commun. 2022, 591, 130–136. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Laufer, S. Kinases as Potential Therapeutic Targets for Anti-Coronaviral Therapy. J. Med. Chem. 2022, 65, 955–982. [Google Scholar] [CrossRef]
- Menegazzi, M.; Mariotto, S.; Dal Bosco, M.; Darra, E.; Vaiana, N.; Shoji, K.; Safwat, A.-A.; Marechal, J.D.; Perahia, D.; Suzuki, H.; et al. Direct Interaction of Natural and Synthetic Catechins with Signal Transducer Activator of Transcription 1 Affects Both Its Phosphorylation and Activity. FEBS J. 2014, 281, 724–738. [Google Scholar] [CrossRef]
- Menegazzi, M.; Campagnari, R.; Bertoldi, M.; Crupi, R.; Di Paola, R.; Cuzzocrea, S. Protective Effect of Epigallocatechin-3-Gallate (EGCG) in Diseases with Uncontrolled Immune Activation: Could Such a Scenario Be Helpful to Counteract COVID-19? Int. J. Mol. Sci. 2020, 21, 5171. [Google Scholar] [CrossRef]
- Joo, S.-Y.; Song, Y.-A.; Park, Y.-L.; Myung, E.; Chung, C.-Y.; Park, K.-J.; Cho, S.-B.; Lee, W.-S.; Kim, H.-S.; Rew, J.-S.; et al. Epigallocatechin-3-Gallate Inhibits LPS-Induced NF-ΚB and MAPK Signaling Pathways in Bone Marrow-Derived Macrophages. Gut Liver 2012, 6, 188–196. [Google Scholar] [CrossRef]
- Yang, F.; Oz, H.S.; Barve, S.; de Villiers, W.J.S.; McClain, C.J.; Varilek, G.W. The Green Tea Polyphenol (−)-Epigallocatechin-3-Gallate Blocks Nuclear Factor-ΚB Activation by Inhibiting IκB Kinase Activity in the Intestinal Epithelial Cell Line IEC-6. Mol. Pharmacol. 2001, 60, 528–533. [Google Scholar]
- Almatroodi, S.A.; Almatroudi, A.; Khan, A.A.; Alhumaydhi, F.A.; Alsahli, M.A.; Rahmani, A.H. Potential Therapeutic Targets of Epigallocatechin Gallate (EGCG), the Most Abundant Catechin in Green Tea, and Its Role in the Therapy of Various Types of Cancer. Molecules 2020, 25, 3146. [Google Scholar] [CrossRef] [PubMed]
- Münzenberger, B.; Heilemann, J.; Strack, D.; Kottke, I.; Oberwinkler, F. Phenolics of Mycorrhizas and Non-Mycorrhizal Roots of Norway Spruce. Planta 1990, 182, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Geahlen, R.L.; McLaughlin, J.L. Piceatannol (3,4,3′,5′-Tetrahydroxy-Trans-Stilbene) Is a Naturally Occurring Protein-Tyrosine Kinase Inhibitor. Biochem. Biophys. Res. Commun. 1989, 165, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Scarpati, M.L.; Oriente, G. Isolamento e Costituzione Dell’acido Rosmarinico (Dal Rosmarinus Off.). Ric. Sci. 1958, 28, 2329–2333. [Google Scholar]
- Al-Dhabi, N.A.; Arasu, M.V.; Park, C.H.; Park, S.U. Recent Studies on Rosmarinic Acid and Its Biological and Pharmacological Activities. EXCLI J. 2014, 13, 1192–1195. [Google Scholar]
- Guan, H.; Luo, W.; Bao, B.; Cao, Y.; Cheng, F.; Yu, S.; Fan, Q.; Zhang, L.; Wu, Q.; Shan, M. A Comprehensive Review of Rosmarinic Acid: From Phytochemistry to Pharmacology and Its New Insight. Molecules 2022, 27, 3292. [Google Scholar] [CrossRef]
- May, G.; Willuhn, G. Antiviral effect of aqueous plant extracts in tissue culture. Arzneimittel-Forschung 1978, 28, 1–7. [Google Scholar]
- Häusler, E.; Petersen, M.; Alfermann, A.W. Isolation of Protoplasts and Vacuoles from Cell Suspension Cultures of Coleus Blumei Benth. Plant Cell Rep. 1993, 12, 510–512. [Google Scholar] [CrossRef]
- McGovern, S.L.; Shoichet, B.K. Kinase Inhibitors: Not Just for Kinases Anymore. J. Med. Chem. 2003, 46, 1478–1483. [Google Scholar] [CrossRef]
- Doak, A.K.; Wille, H.; Prusiner, S.B.; Shoichet, B.K. Colloid Formation by Drugs in Simulated Intestinal Fluid. J. Med. Chem. 2010, 53, 4259–4265. [Google Scholar] [CrossRef]
- Seidler, J.; McGovern, S.L.; Doman, T.N.; Shoichet, B.K. Identification and Prediction of Promiscuous Aggregating Inhibitors among Known Drugs. J. Med. Chem. 2003, 46, 4477–4486. [Google Scholar] [CrossRef]
- Ganesh, A.N.; Donders, E.N.; Shoichet, B.K.; Shoichet, M.S. Colloidal Aggregation: From Screening Nuisance to Formulation Nuance. Nano Today 2018, 19, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Coan, K.E.D.; Maltby, D.A.; Burlingame, A.L.; Shoichet, B.K. Promiscuous Aggregate-Based Inhibitors Promote Enzyme Unfolding. J. Med. Chem. 2009, 52, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Blevitt, J.M.; Hack, M.D.; Herman, K.L.; Jackson, P.F.; Krawczuk, P.J.; Lebsack, A.D.; Liu, A.X.; Mirzadegan, T.; Nelen, M.I.; Patrick, A.N.; et al. Structural Basis of Small-Molecule Aggregate Induced Inhibition of a Protein-Protein Interaction. J. Med. Chem. 2017, 60, 3511–3517. [Google Scholar] [CrossRef] [PubMed]
- McGovern, S.L.; Helfand, B.T.; Feng, B.; Shoichet, B.K. A Specific Mechanism of Nonspecific Inhibition. J. Med. Chem. 2003, 46, 4265–4272. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Toyama, B.H.; Wille, H.; Colby, D.W.; Collins, S.R.; May, B.C.H.; Prusiner, S.B.; Weissman, J.; Shoichet, B.K. Small-Molecule Aggregates Inhibit Amyloid Polymerization. Nat. Chem. Biol. 2008, 4, 197–199. [Google Scholar] [CrossRef] [PubMed]
- LaPlante, S.R.; Aubry, N.; Bolger, G.; Bonneau, P.; Carson, R.; Coulombe, R.; Sturino, C.; Beaulieu, P.L. Monitoring Drug Self-Aggregation and Potential for Promiscuity in off-Target In Vitro Pharmacology Screens by a Practical NMR Strategy. J. Med. Chem. 2013, 56, 7073–7083. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PKaprediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach to Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Wang, Q.-S.; Zhang, K.-H.; Cui, Y.; Wang, Z.-J.; Pan, Q.-Y.; Liu, K.; Sun, B.; Zhou, H.; Li, M.-J.; Xu, Q.; et al. Upgrade of Macromolecular Crystallography Beamline BL17U1 at SSRF. Nucl. Sci. Tech. 2018, 29, 68. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1948–1954. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
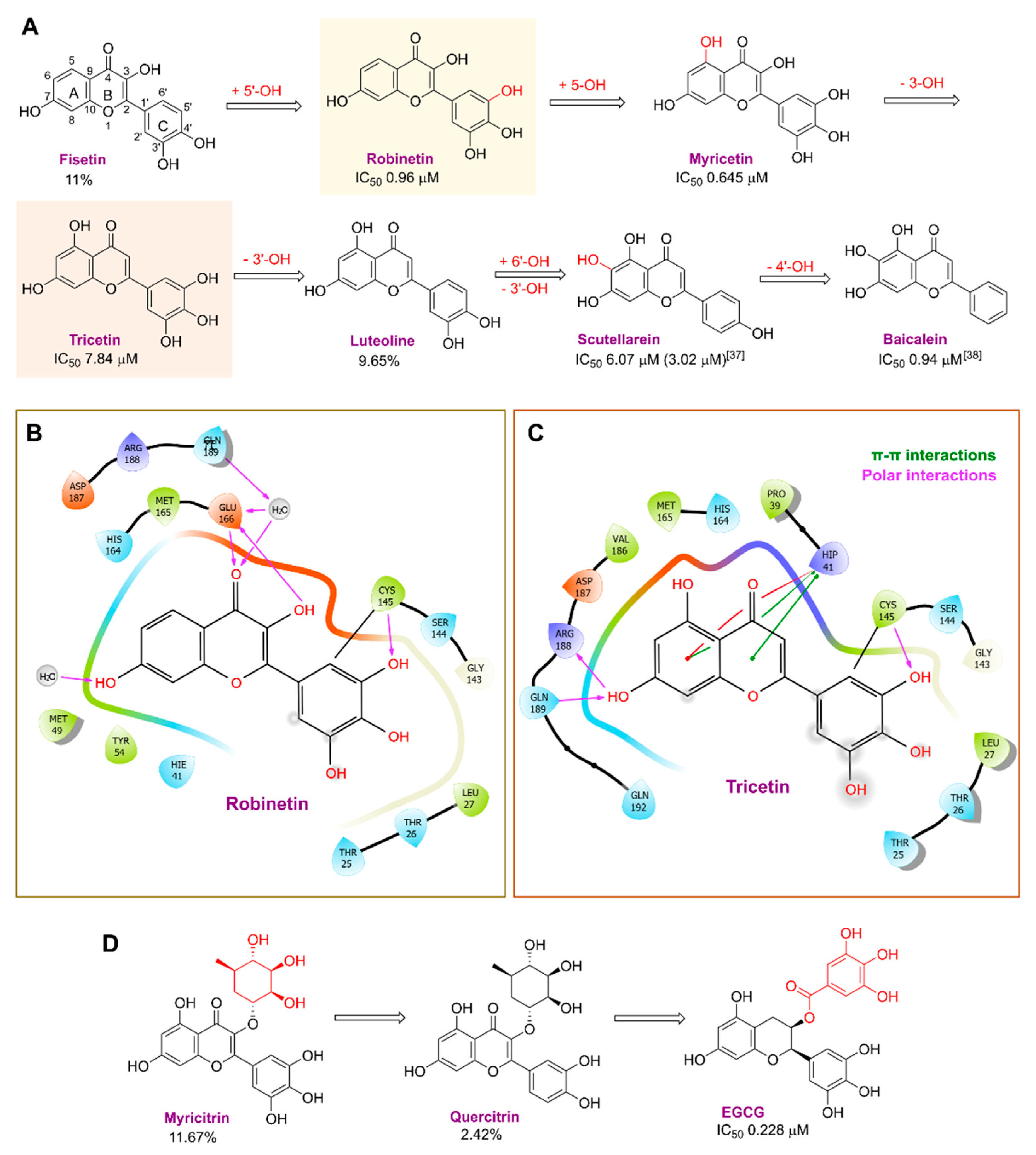
| Chemical Structure | SARS-CoV-2 Mpro | |
|---|---|---|
| % Inhibition at 10 μM | IC50 (μM) | |
 (+)-Nootkaton | 32.04 | >10 |
 Costunolide | 27.96 | >10 |
 Alantolacton | 27.55 | >10 |
 (−)-Parthenolide | 20.06 | >10 |
 (−)-α-Santonin | 24.13 | >10 |
 Dehydrocostus lactone | 13.39 | >10 |
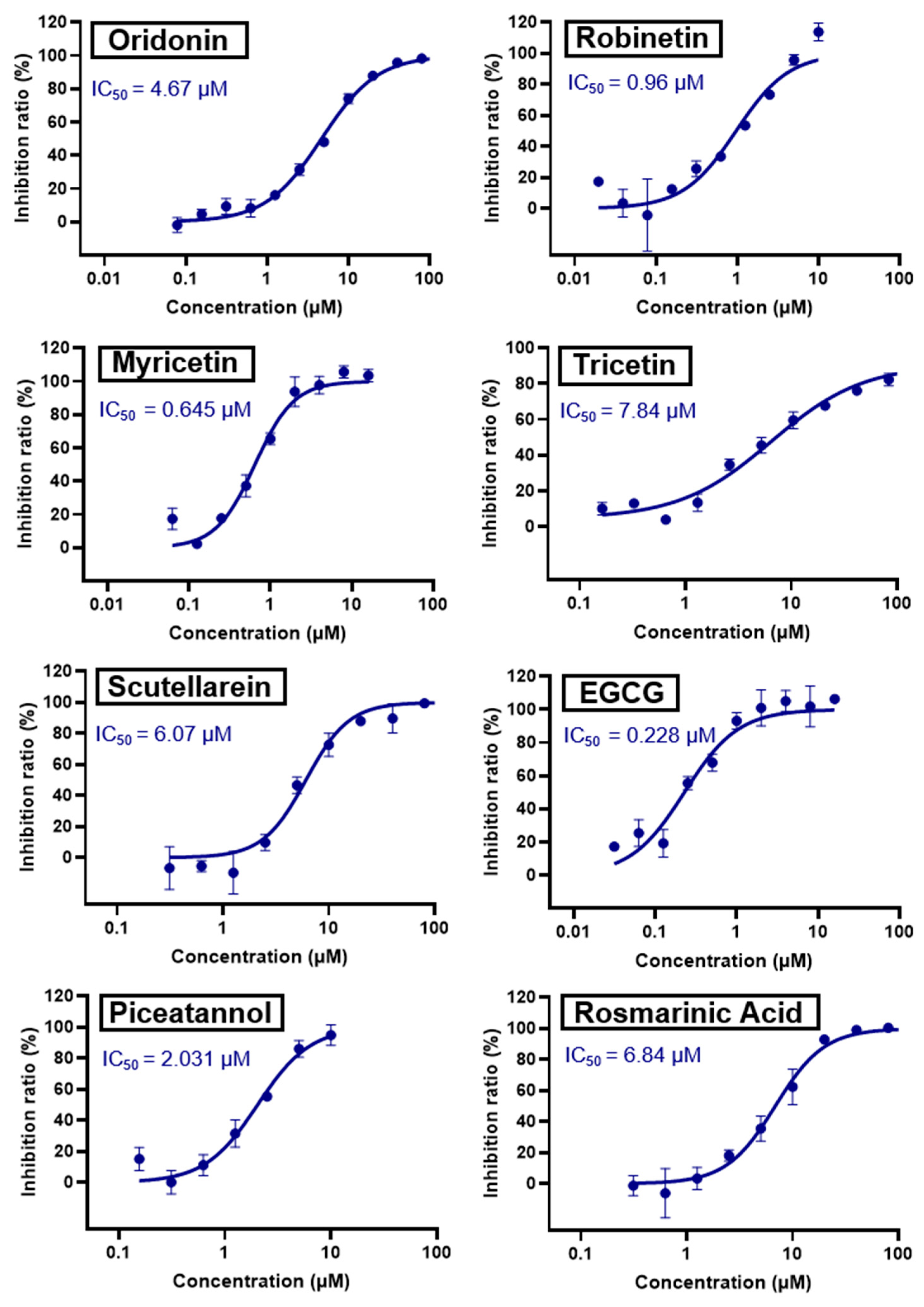
 Oridonin | 81.87 | 4.67 |
 Osthole | 4.98 | >10 |
 Tanshinone I | 7.03 | >10 |
 Sinomenine | 4.80 | >10 |
 Resibufogenin | 6.32 | >10 |
 Cinobufagin | 0.53 | >10 |
 α-Mangostin | 38.56 | 25.12 |
 Gambogic acid | 22.75 | >10 |
 Loganin | 29.43 | >10 |
 l-Ascorbic acid | 22.25 | >10 |
 Fisetin | −11.21 | >10 |
 Robinetin | 86.13 | 0.96 |
 Myricetin | 104.00 | 0.645 |
 Tricetin | 72.28 | 7.84 |
 Luteoline | 9.65 | >10 |
 Scutellarein | 76.07 | 6.07 |
 Myricitrin | 11.67 | >10 |
 Quercitrin | 2.42 | >10 |
 l-Epigallocatechin gallate | 105.40 | 0.228 |
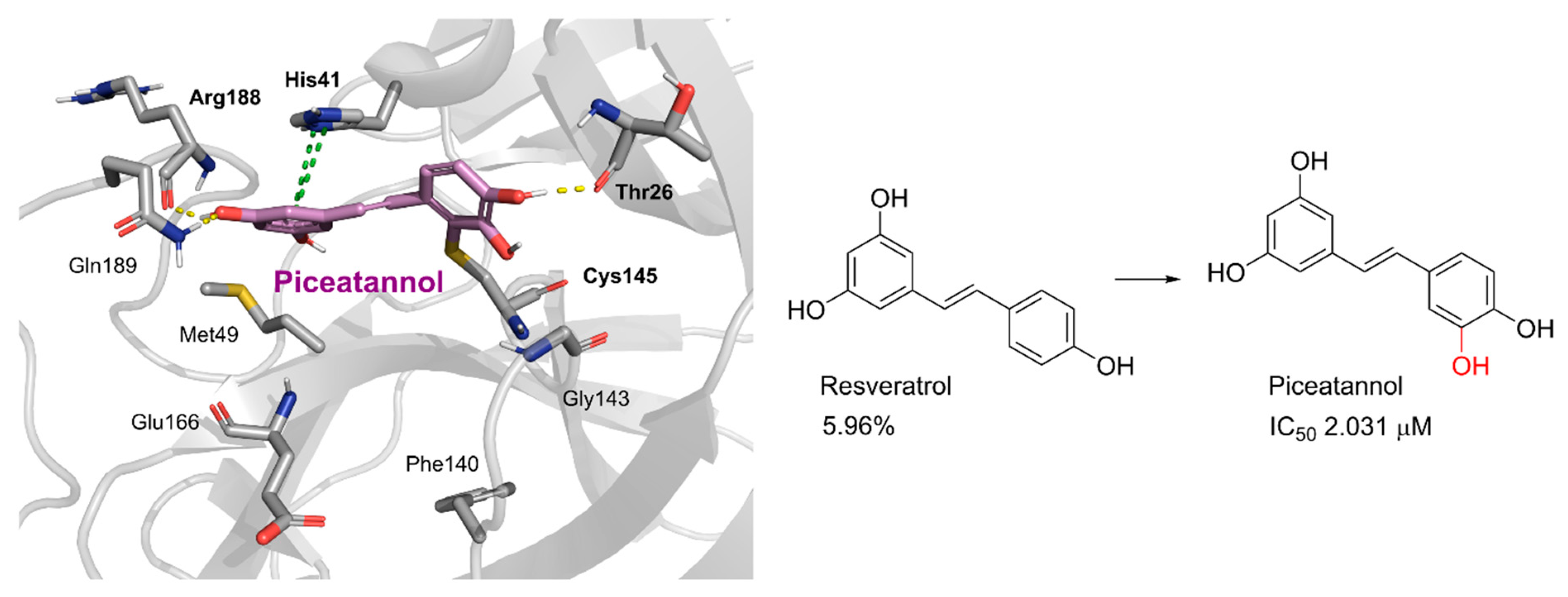
 Piceatannol | 76.90 | 2.031 |
 Resveratrol | 5.96 | >10 |
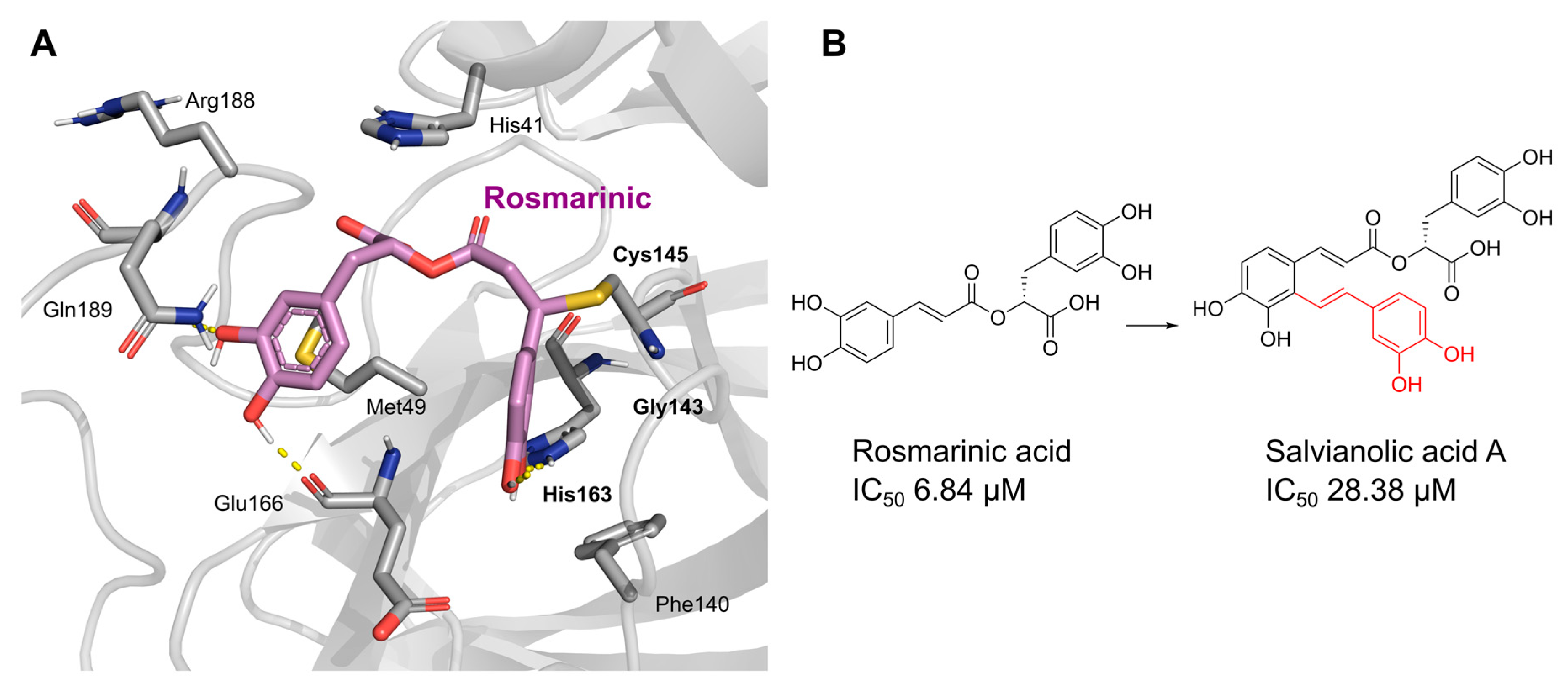
 Rosmarinic acid | 50.56 | 6.84 |
 Salvianolic acid A | 50.84 | 28.38 |
 Orlistat | 20.99 | >10 |
 3-O-Ethyl-l-ascorbic acid | 24.94 | >10 |
| SARS-CoV-2 Mpro–Robinetin | |
|---|---|
| PDB ID | 8HI9 |
| Space Group | P 21 |
| Cell Dimension: a (Å) | 44.156 |
| b (Å) | 54.182 |
| c (Å) | 115.416 |
| Wavelength (Å) | 0.979 |
| Reflections (unique) | 24333 |
| Resolution Range (Å) | 2.28-37.78 |
| Highest-Resolution Shell (Å) | 2.28-2.39 |
| Redundancy | 6.1(6.2) |
| I/σ (I) | 14.8(6.9) |
| Completeness (%) | 99.0(99.0) |
| Rwork/Rfree | 0.2457/0.2553 |
| Clashscore | 1.72 |
| MolProbity Score | 0.93 |
| RMS Values | |
| Bond Length (Å) | 0.003 |
| Bond Angle (°) | 0.638 |
| Number of Non-hydrogen Atoms | |
| Protein | 4449 |
| Inhibitor | 44 |
| Water Oxygen | 108 |
| Others | 0 |
| B-factor (Å2) | |
| Protein | 33.33 |
| Inhibitor | 35.13 |
| Water Oxygen | 29.42 |
| Ramachandran Plot | |
| Favored (%) | 98.32 |
| Allowed (%) | 1.52 |
| Outliers (%) | 0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krüger, N.; Kronenberger, T.; Xie, H.; Rocha, C.; Pöhlmann, S.; Su, H.; Xu, Y.; Laufer, S.A.; Pillaiyar, T. Discovery of Polyphenolic Natural Products as SARS-CoV-2 Mpro Inhibitors for COVID-19. Pharmaceuticals 2023, 16, 190. https://doi.org/10.3390/ph16020190
Krüger N, Kronenberger T, Xie H, Rocha C, Pöhlmann S, Su H, Xu Y, Laufer SA, Pillaiyar T. Discovery of Polyphenolic Natural Products as SARS-CoV-2 Mpro Inhibitors for COVID-19. Pharmaceuticals. 2023; 16(2):190. https://doi.org/10.3390/ph16020190
Chicago/Turabian StyleKrüger, Nadine, Thales Kronenberger, Hang Xie, Cheila Rocha, Stefan Pöhlmann, Haixia Su, Yechun Xu, Stefan A. Laufer, and Thanigaimalai Pillaiyar. 2023. "Discovery of Polyphenolic Natural Products as SARS-CoV-2 Mpro Inhibitors for COVID-19" Pharmaceuticals 16, no. 2: 190. https://doi.org/10.3390/ph16020190
APA StyleKrüger, N., Kronenberger, T., Xie, H., Rocha, C., Pöhlmann, S., Su, H., Xu, Y., Laufer, S. A., & Pillaiyar, T. (2023). Discovery of Polyphenolic Natural Products as SARS-CoV-2 Mpro Inhibitors for COVID-19. Pharmaceuticals, 16(2), 190. https://doi.org/10.3390/ph16020190