
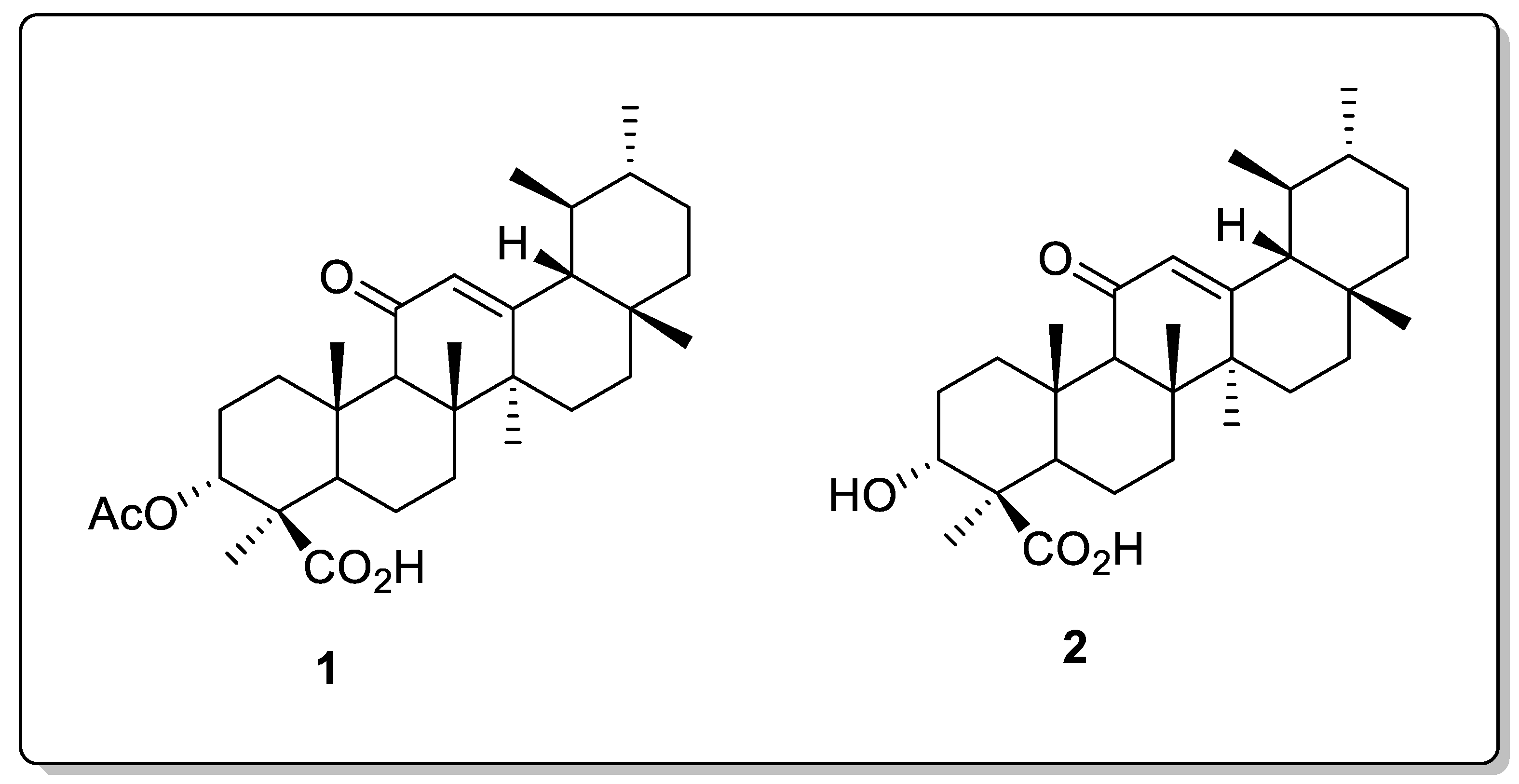
Discovery of New Boswellic Acid Hybrid 1H-1,2,3-Triazoles for Diabetic Management: In Vitro and In Silico Studies

 , ,
, ,  ,
,  ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
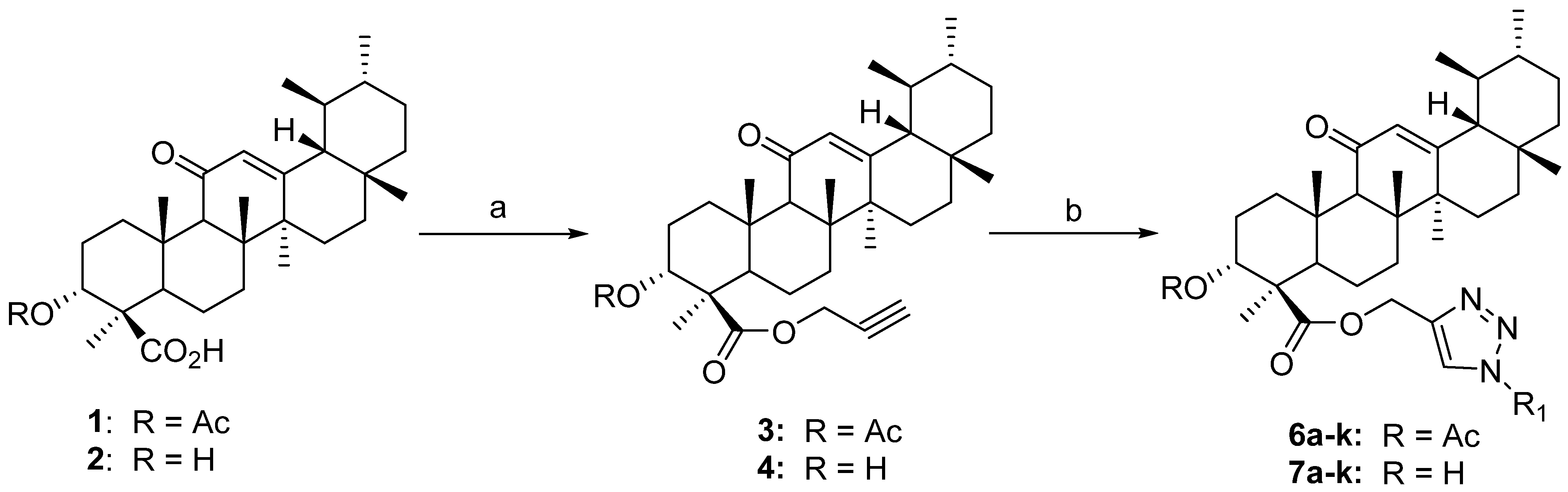
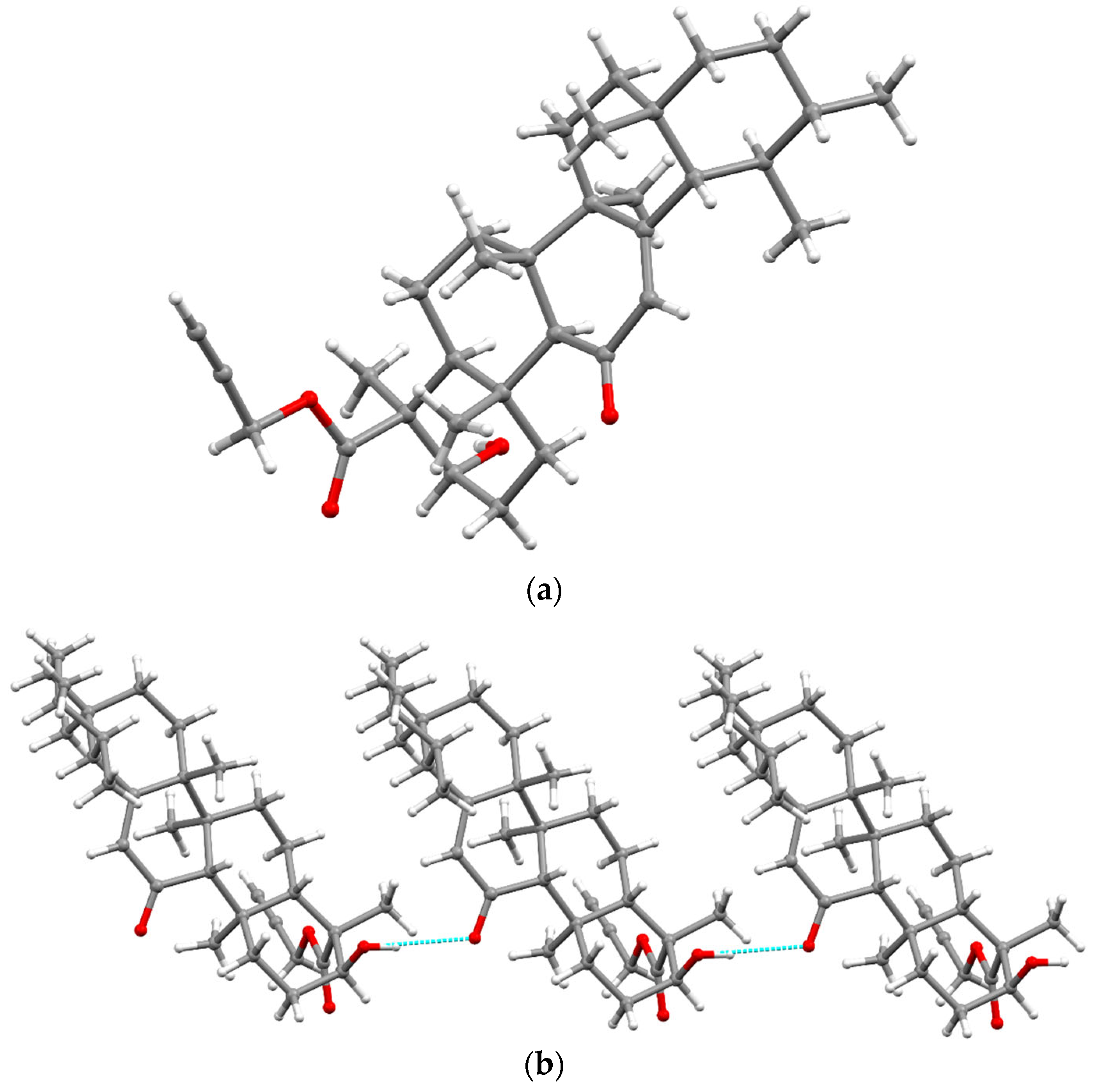
2.1. Chemistry
2.2. α-Glucosidase Activity
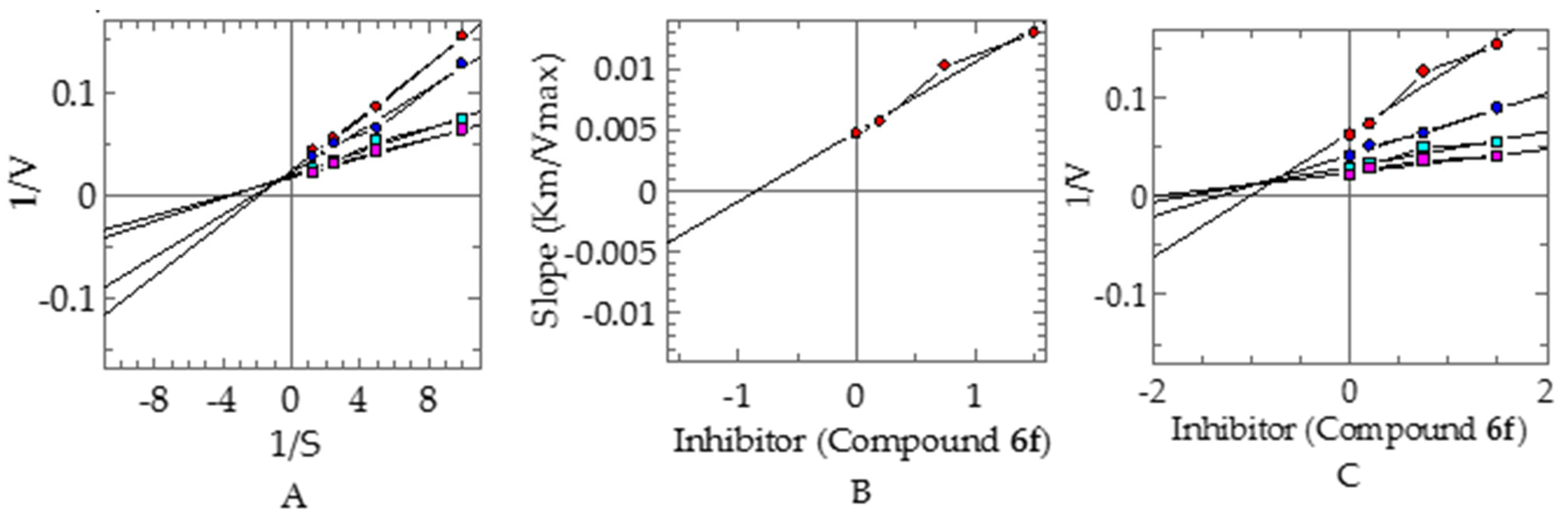
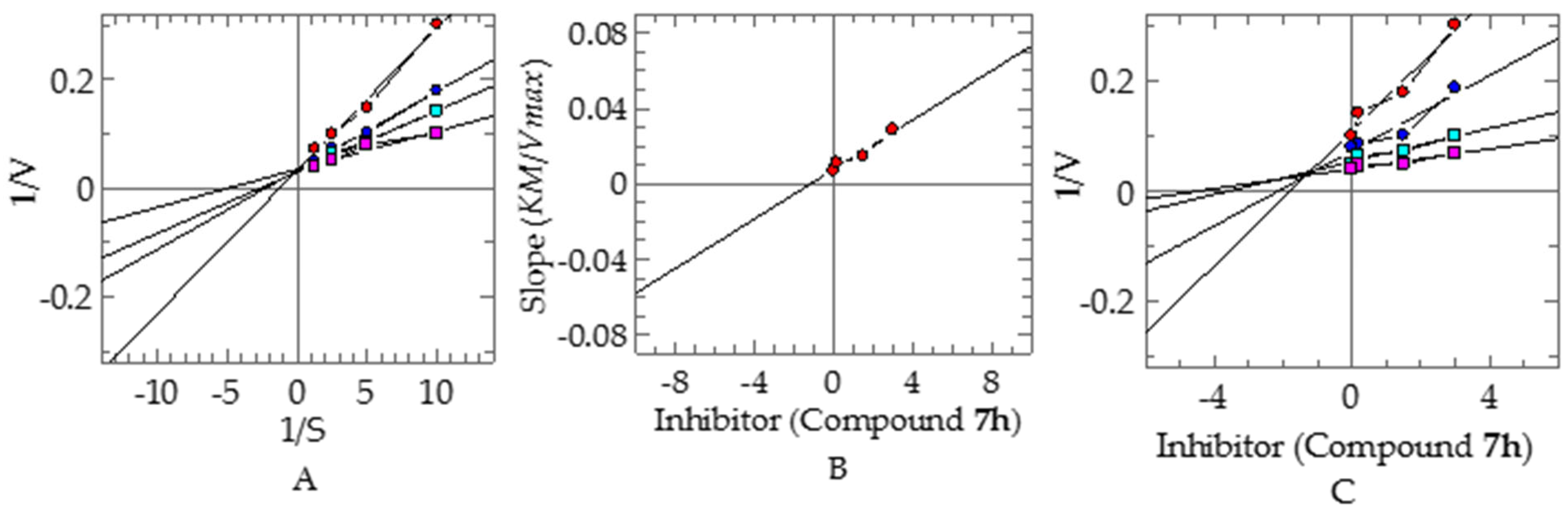
Kinetic Studies
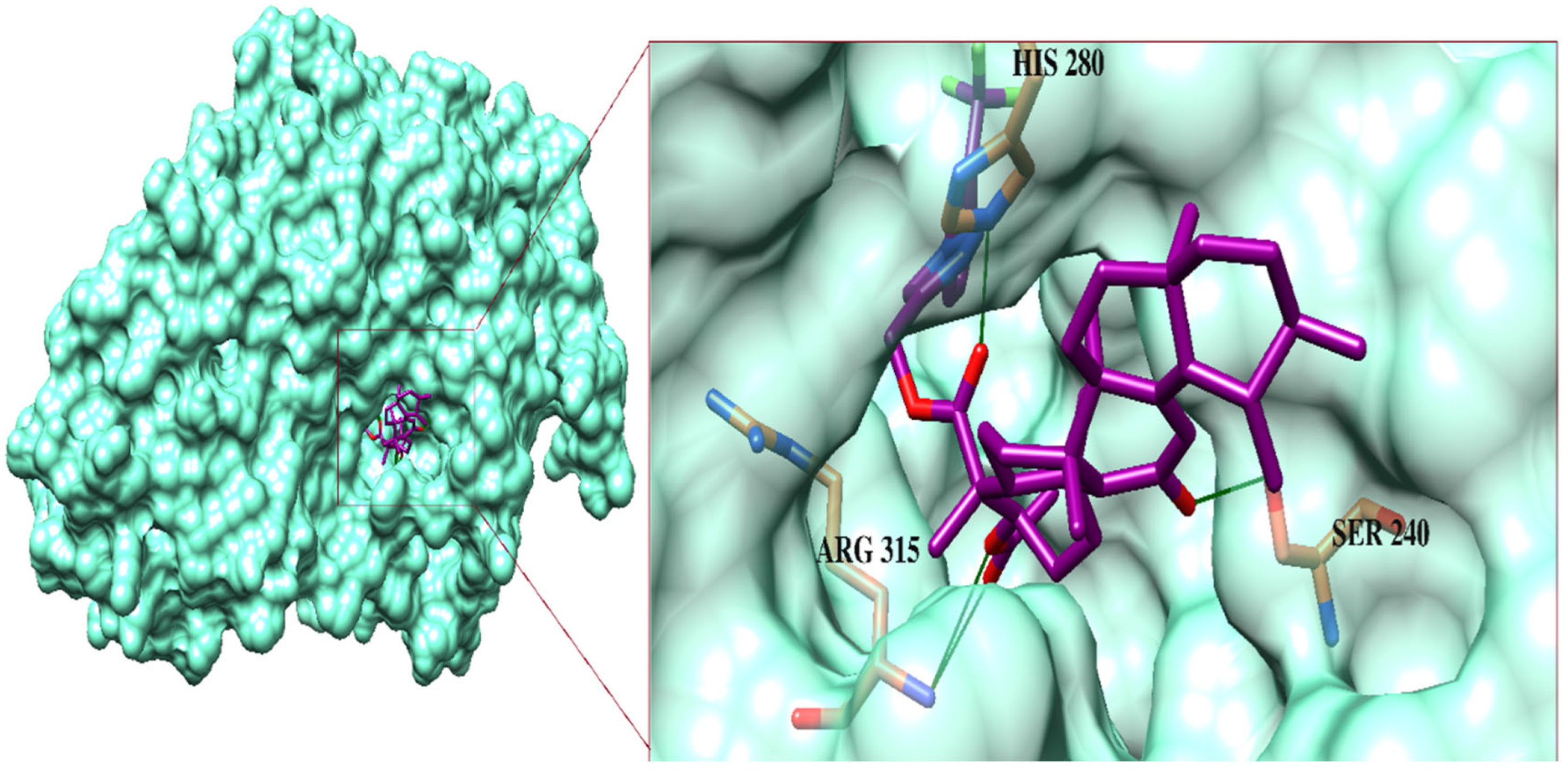
2.3. Molecular Docking Studies
3. Experimental Section
3.1. General
3.2. Preparation of Boswellic Acids (BAs) Cluster
3.3. General Procedure for Synthesis of 3 and 4
3.3.1. (Prop-2-yne-1-yl) 3α-acetyloxy-11-oxo-urs-12-en-24-oate (3)
3.3.2. (Prop-2-yne-1-yl) 3α-hydroxy-11-oxo-urs-12-en-24-oate (4)
3.4. General Procedure for Synthesis of 1H-1,2,3-Triazol Hybrids of 3-Acetyl-11-keto-β-boswellic Acid (6a–6k) and 11-Keto-β-boswellic Acid (7a–7k) Derivatives
3.4.1. (4-Phenyl-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6a)
3.4.2. (4-(O-tolyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6b)
3.4.3. (4-(Ortho-methoxyphenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6c)
3.4.4. (4-(Ortho-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6d)
3.4.5. (4-(Para-methoxyphenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6e)
3.4.6. (4-(Meta-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6f)
3.4.7. (4-(Meta-bromo) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-Acetyloxy-11-oxo-urs-12-en-24-oate (6g)
3.4.8. (4-(Para-bromo) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6h)
3.4.9. 4-(Para-chloro) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6i)
3.4.10. 4-(Para-fluoro) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6j)
3.4.11. 4-(Para-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (6k)
3.4.12. (4-Phenyl-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7a)
3.4.13. (4-(Ortho-tolyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7b)
3.4.14. (4-(Ortho-methoxyphenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7c)
3.4.15. (4-(Ortho-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (7d)
3.4.16. (4-(Para-methoxyphenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7e)
3.4.17. (4-(Meta-bromo) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7g)
3.4.18. (4-(Para-bromo) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-hydroxy-11-oxo-urs-12-en-24-oate (7h)
3.4.19. 4-(Para-chloro) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (7i)
3.4.20. 4-(Para-fluoro) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (7j)
3.4.21. 4-(Para-trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl) Methyl 3α-acetyloxy-11-oxo-urs-12-en-24-oate (7k)
3.5. α-Glucosidase-Inhibitory Assay
3.6. In Silico Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Harrasi, A.; Csuk, R.; Khan, A.; Hussain, J. Distribution of the Anti-Inflammatory and Anti-Depressant Compounds: Incensole and Incensole Acetate in Genus Boswellia. Phytochemistry 2019, 161, 28–40. [Google Scholar] [CrossRef]
- Asteggiano, A.; Curatolo, L.; Schiavo, V.; Occhipinti, A.; Medana, C. Development, Validation, and Application of a Simple and Rugged HPLC Method for Boswellic Acids for a Comparative Study of Their Abundance in Different Species of Boswellia Gum Resins. Appl. Sci. 2023, 13, 1254. [Google Scholar] [CrossRef]
- Liu, Y.M. Harmonograph of Uighur, Part One. Xinjiang Sci. Technol. Hyg. Publ. House, Urumqi 1999, 222. [Google Scholar]
- The Chinese Pharmacopeia Pharmacopia; CotC 1; China Medical Sciences Press: Beijing, China, 2015; p. 223.
- Singh, S.; Khajuria, A.; Taneja, S.C.; Johri, R.K.; Singh, J.; Qazi, G.N. Boswellic Acids: A Leukotriene Inhibitor Also Effective through Topical Application in Inflammatory Disorders. Phytomedicine 2008, 15, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Bertocchi, M.; Isani, G.; Medici, F.; Andreani, G.; Tubon Usca, I.; Roncada, P.; Forni, M.; Bernardini, C. Anti-inflammatory activity of Boswellia serrata extracts: An in vitro study on porcine aortic endothelial cells. Oxid. Med. Cell. Longev. 2018, 2018, 2504305. [Google Scholar] [CrossRef] [PubMed]
- Mehrzadi, S.; Tavakolifar, B.; Huseini, H.F.; Mosavat, S.H.; Heydari, M. The effects of Boswellia serrata gum resin on the blood glucose and lipid profile of diabetic patients: A double-blind randomized placebo-controlled clinical trial. J. Evid. Based Integr. Med. 2018, 13, 2515690X18772728. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, Q.; Sun, L. Five Terpenoids from the Gum Resin of Boswellia Carterii and Their Cytotoxicity. Fitoterapia 2021, 154, 105017. [Google Scholar] [CrossRef] [PubMed]
- Barakat, B.M.; Ahmed, H.I.; Bahr, H.I.; Elbahaie, A.M. Protective effect of boswellic acids against doxorubicin-induced hepatotoxicity: Impact on Nrf2/HO-1 defense pathway. Oxid. Med. Cell. Longev. 2018, 2018, 8296451. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.G.; Ren, J.; Wang, A.G.; Yang, J.B.; Ji, T.F.; Ma, Q.G.; Tian, J.; Su, Y.L. Hepatoprotective prenylaromadendrane-type diterpenes from the gum resin of Boswellia carterii. J. Nat. Prod. 2013, 76, 2074–2079. [Google Scholar] [CrossRef]
- Mahesh, B.U.; Shrivastava, S.; Pragada, R.R.; Naidu, V.G.; Sistla, R. Antioxidant and hepatoprotective effects of Boswellia ovalifoliolata bark extracts. Chin. J. Nat. Med. 2014, 12, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Safayhi, H.; Mack, T.; Sabieraj, J.; Anazodo, M.I.; Subramanian, L.R.; Ammon, H.P. Boswellic Acids: Novel, Specific, Nonredox Inhibitors of 5-Lipoxygenase. J. Pharmacol. Exp. Ther. 1992, 261, 1143–1146. [Google Scholar]
- Moussaieff, A.; Shein, N.A.; Tsenter, J.; Grigoriadis, S.; Simeonidou, C.; Alexandrovich, A.G.; Trembovler, V.; Ben-Neriah, Y.; Schmitz, M.L.; Fiebich, B.L.; et al. Incensole Acetate: A Novel Neuroprotective Agent Isolated from Boswellia Carterii. J. Cereb. Blood Flow Metab. 2008, 28, 1341–1352. [Google Scholar] [CrossRef]
- Liu, J.J.; Nilsson, Å.; Oredsson, S.; Badmaev, V.; Zhao, W.Z.; Duan, R.D. Boswellic Acids Trigger Apoptosis via a Pathway Dependent on Caspase-8 Activation but Independent on Fas/Fas Ligand Interaction in Colon Cancer HT-29 Cells. Carcinogenesis 2002, 23, 2087–2093. [Google Scholar] [CrossRef]
- Solanki, N.; Mehta, M.; Chellappan, D.K.; Gupta, G.; Hansbro, N.G.; Tambuwala, M.M.; AA Aljabali, A.; Paudel, K.R.; Liu, G.; Satija, S.; et al. Antiproliferative Effects of Boswellic Acid-Loaded Chitosan Nanoparticles on Human Lung Cancer Cell Line A549. Future Med. Chem. 2020, 12, 2019–2034. [Google Scholar] [CrossRef]
- Shah, B.A.; Qazi, G.N.; Taneja, S.C. Boswellic Acids: A Group of Medicinally Important Compounds. Nat. Prod. Rep. 2009, 26, 72–89. [Google Scholar] [CrossRef]
- Hussain, H.; Ali, I.; Wang, D.; Hakkim, F.L.; Westermann, B.; Rashan, L.; Ahmed, I.; Green, I.R. Boswellic Acids: Privileged Structures to Develop Lead Compounds for Anticancer Drug Discovery. Expert Opin. Drug Discov. 2021, 16, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Iram, F.; Khan, S.A.; Husain, A. Phytochemistry and Potential Therapeutic Actions of Boswellic Acids: A Mini-Review. Asian Pac. J. Trop. Biomed. 2017, 7, 513–523. [Google Scholar] [CrossRef]
- Li, C.; He, Q.; Xu, Y.; Lou, H.; Fan, P. Synthesis of 3-O-Acetyl-11-Keto-β-Boswellic Acid (AKBA)-Derived Amides and Their Mitochondria-Targeted Antitumor Activities. ACS Omega 2022, 7, 9853–9866. [Google Scholar] [CrossRef]
- Verma, K.N.; Mondal, D.; Bera, S. Pharmacological and Cellular Significance of Triazole-Surrogated Compounds. Curr. Org. Chem. 2019, 23, 2505–2572. [Google Scholar] [CrossRef]
- Lauria, A.; Delisi, R.; Mingoia, F.; Terenzi, A.; Martorana, A.; Barone, G.; Almerico, A.M. 1,2,3-Triazole in Heterocyclic Compounds, Endowed with Biological Activity, through 1,3-Dipolar Cycloadditions. Eur. J. Org. Chem. 2014, 2014, 3289–3306. [Google Scholar] [CrossRef]
- Nehra, N.; Tittal, R.K.; Ghule, V.D. 1,2,3-Triazoles of 8-Hydroxyquinoline and HBT: Synthesis and Studies (DNA Binding, Antimicrobial, Molecular Docking, ADME, and DFT). ACS Omega 2021, 6, 27089–27100. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.L.; Hoover, J.R.; Berges, D.A.; Taggart, J.J.; Davis, L.D.; Dietz, E.M.; Jakas, D.R.; Yim, N.; Actor, P.; Uri, J.V.; et al. Orally active 7-phenylglycyl cephalosporins. Structure-activity studies related to cefatrizine (SK&F 60771). J. Antibiotics 1976, 29, 65–80. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; Khalaf, H.S.; Mohamed, S.F.; Hussien, H.A.; Kutkat, O.M.; Amr, A.E. Synthesis and Antiviral Activity of 1,2,3-Triazole Glycosides Based Substituted Pyridine via Click Cycloaddition. Russ. J. Gen. Chem. 2017, 87, 2444–2453. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, Z.; Liu, J.; Huang, B.; Kang, D.; Zhang, H.; De Clercq, E.; Daelemans, D.; Pannecouque, C.; Lee, K.-H.; et al. Targeting the Entrance Channel of NNIBP: Discovery of Diarylnicotinamide 1,4-Disubstituted 1,2,3-Triazoles as Novel HIV-1 NNRTIs with High Potency against Wild-Type and E138K Mutant Virus. Eur. J. Med. Chem. 2018, 151, 339–350. [Google Scholar] [CrossRef]
- Angajala, K.K.; Vianala, S.; Macha, R.; Raghavender, M.; Thupurani, M.K.; Pathi, P.J. Synthesis, Anti-Inflammatory, Bactericidal Activities and Docking Studies of Novel 1,2,3-Triazoles Derived from Ibuprofen Using Click Chemistry. Springerplus 2016, 5, 423. [Google Scholar] [CrossRef]
- El Bourakadi, K.; Mekhzoum, M.E.M.; Saby, C.; Morjani, H.; Chakchak, H.; Merghoub, N.; el kacem Qaiss, A.; Bouhfid, R. Synthesis, Characterization and in Vitro Anticancer Activity of Thiabendazole-Derived 1,2,3-Triazole Derivatives. New J. Chem. 2020, 44, 12099–12106. [Google Scholar] [CrossRef]
- Avula, S.K.; Rehman, N.U.; Khan, M.; Halim, S.A.; Khan, A.; Rafiq, K.; Csuk, R.; Das, B.; Al-Harrasi, A. New Synthetic 1H-1,2,3-Triazole Derivatives of 3-O-Acetyl-β-Boswellic Acid and 3-O-Acetyl-11-Keto-β-Boswellic Acid from Boswellia Sacra Inhibit Carbonic Anhydrase II in Vitro. Med. Chem. Res. 2021, 30, 1185–1198. [Google Scholar] [CrossRef]
- Kausar, N.; Ullah, S.; Khan, M.A.; Zafar, H.; Atia-tul-Wahab; Choudhary, M.I.; Yousuf, S. Celebrex Derivatives: Synthesis, α-Glucosidase Inhibition, Crystal Structures and Molecular Docking Studies. Bioorg. Chem. 2021, 106, 104499. [Google Scholar] [CrossRef]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global Estimates of the Prevalence of Diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef]
- McCulloch, D.K.; Kurtz, A.B.; Tattersall, R.B. A new approach to the treatment of nocturnal hypoglycemia using alpha-glucosidase inhibition. Diabetes Care. 1983, 6, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Umm-E-Farwa; Ullah, S.; Khan, M.A.; Zafar, H.; Atia-tul-Wahab; Younus, M.; Choudhary, M.I.; Basha, F.Z. Dibenzazepine-Linked Isoxazoles: New and Potent Class of α-Glucosidase Inhibitors. Bioorg. Med. Chem. Lett. 2021, 40, 127979. [Google Scholar] [CrossRef] [PubMed]
- Akhter, S.; Ullah, S.; Yousuf, S.; Atia-tul-Wahab; Siddiqui, H.; Choudhary, M.I. Synthesis, Crystal Structure and Hirshfeld Surface Analysis of Benzamide Derivatives of Thiourea as Potent Inhibitors of α-Glucosidase in-Vitro. Bioorg. Chem. 2021, 107, 104531. [Google Scholar] [CrossRef] [PubMed]
- Maurya, A.K.; Mulpuru, V.; Mishra, N. Discovery of Novel Coumarin Analogs against the α-Glucosidase Protein Target of Diabetes Mellitus: Pharmacophore-Based QSAR, Docking, and Molecular Dynamics Simulation Studies. ACS Omega 2020, 5, 32234–32249. [Google Scholar] [CrossRef]
- Rehman, N.U.; Khan, A.; Al-Harrasi, A.; Hussain, H.; Wadood, A.; Riaz, M.; Al-Abri, Z. New α-Glucosidase Inhibitors from the Resins of Boswellia Species with Structure–Glucosidase Activity and Molecular Docking Studies. Bioorg. Chem. 2018, 79, 27–33. [Google Scholar] [CrossRef]
- Ye, G.-J.; Lan, T.; Huang, Z.-X.; Cheng, X.-N.; Cai, C.-Y.; Ding, S.-M.; Xie, M.-L.; Wang, B. Design and Synthesis of Novel Xanthone-Triazole Derivatives as Potential Antidiabetic Agents: α-Glucosidase Inhibition and Glucose Uptake Promotion. Eur. J. Med. Chem. 2019, 177, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Jauch, J.; Bergmann, J. An Efficient Method for the Large-Scale Preparation of 3-O-Acetyl-11-Oxo-β-Boswellic Acid and Other Boswellic Acids. Eur. J. Org. Chem. 2003, 2003, 4752–4756. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Gubaidullin, R.R.; Galimshina, Z.R.; Nedopekina, D.A.; Odinokov, V.N. Effective Synthesis of Novel C(2)-Propargyl Derivatives of Betulinic and Ursolic Acids and Their Conjugation with β-d-Glucopyranoside Azides via Click Chemistry. Tetrahedron 2016, 72, 1249–1256. [Google Scholar] [CrossRef]
- Carvalho, I.; Andrade, P.; Campo, V.L.; Guedes, P.M.M.; Sesti-Costa, R.; Silva, J.S.; Schenkman, S.; Dedola, S.; Hill, L.; Rejzek, M.; et al. ‘Click Chemistry’ Synthesis of a Library of 1,2,3-Triazole-Substituted Galactose Derivatives and Their Evaluation against Trypanosoma Cruzi and Its Cell Surface Trans-Sialidase. Bioorg. Med. Chem. 2010, 18, 2412–2427. [Google Scholar] [CrossRef]
- Das, R.; Mukhopadhyay, B. Use of ‘Click Chemistry’ for the Synthesis of Carbohydrate-Porphyrin Dendrimers and Their Multivalent Approach toward Lectin Sensing. Tetrahedron Lett. 2016, 57, 1775–1781. [Google Scholar] [CrossRef]
- Artyushin, O.I.; Sharova, E.V.; Vinogradova, N.M.; Genkina, G.K.; Moiseeva, A.A.; Klemenkova, Z.S.; Orshanskaya, I.R.; Shtro, A.A.; Kadyrova, R.A.; Zarubaev, V.V.; et al. Synthesis of Camphecene Derivatives Using Click Chemistry Methodology and Study of Their Antiviral Activity. Bioorg. Med. Chem. Lett. 2017, 27, 2181–2184. [Google Scholar] [CrossRef]
- APEX-II, Bruker AXS, Madison, WI, USA.
- SADABS, Bruker AXS, Madison, WI, USA.
- Bruker SAINT. AXS, Madison, WI, USA.
- Sheldrick, G.M. Acta Crystallographica Section A. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Chemical Computing Group ULC Molecular Operating Environment, 1010 Sherbooke St. West, Suite 910, Montr. QC, Canada, 2014.
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal Structures of Isomaltase from Saccharomyces Cerevisiae and in Complex with Its Competitive Inhibitor Maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Azides (5a-k) R1-Group | 1H-1,2,3-Triazole Products (6a–k and 7a–k) | Yields of Products % a |
|---|---|---|---|
| 3 | Ph | 6a | 90 |
| 3 | 2-MeC6H4 | 6b | 87 |
| 3 | 2-MeOC6H4 | 6c | 89 |
| 3 | 2-F3CC6H4 | 6d | 96 |
| 3 | 4-MeOC6H4 | 6e | 88 |
| 3 | 3-F3CC6H4 | 6f | 95 |
| 3 | 3-BrC6H4 | 6g | 92 |
| 3 | 4-BrC6H4 | 6h | 91 |
| 3 | 4-ClC6H4 | 6i | 93 |
| 3 | 4-FC6H4 | 6j | 94 |
| 3 | 4-F3CC6H4 | 6k | 96 |
| 4 | Ph | 7a | 89 |
| 4 | 2-MeC6H4 | 7b | 86 |
| 4 | 2-MeOC6H4 | 7c | 88 |
| 4 | 2-F3CC6H4 | 7d | 96 |
| 4 | 4-MeOC6H4 | 7e | 87 |
| 4 | 3-F3CC6H4 | 7f | 94 |
| 4 | 3-BrC6H4 | 7g | 91 |
| 4 | 4-BrC6H4 | 7h | 93 |
| 4 | 4-ClC6H4 | 7i | 92 |
| 4 | 4-FC6H4 | 7j | 94 |
| 4 | 4-F3CC6H4 | 7k | 96 |
| Compounds | IC50 ± µM | Compounds | IC50 ± µM | R1-Group |
|---|---|---|---|---|
| 6a | 2.02 ± 0.03 | 7a | 2.31 ± 0.17 | 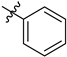 |
| 6b | 2.25 ± 0.10 | 7b | 2.11 ± 0.10 | 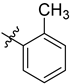 |
| 6c | 1.23 ± 0.08 | 7c | 2.18 ± 0.01 | 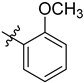 |
| 6d | 2.37 ± 0.11 | 7d | 2.62 ± 0.06 |  |
| 6e | 2.01 ± 0.09 | 7e | 2.37 ± 0.07 | 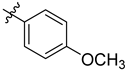 |
| 6f | 0.22 ± 0.08 | 7f | 2.52 ± 0.12 | 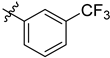 |
| 6g | 0.78 ± 0.01 | 7g | 1.34 ± 0.02 | 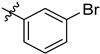 |
| 6h | 0.69 ± 0.04 | 7h | 0.43 ± 0.05 | 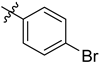 |
| 6i | 3.06 ± 0.22 | 7i | 3.18 ± 0.69 | 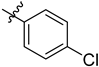 |
| 6j | 0.58 ± 0.04 | 7j | 2.64 ± 0.03 |  |
| 6k | 1.26 ± 0.05 | 7k | 1.39 ± 0.11 | 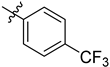 |
| Acarbose (Standard) | 942 ± 0.74 |  | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, N.U.; Ullah, S.; Alam, T.; Halim, S.A.; Mohanta, T.K.; Khan, A.; Anwar, M.U.; Csuk, R.; Avula, S.K.; Al-Harrasi, A. Discovery of New Boswellic Acid Hybrid 1H-1,2,3-Triazoles for Diabetic Management: In Vitro and In Silico Studies. Pharmaceuticals 2023, 16, 229. https://doi.org/10.3390/ph16020229
Rehman NU, Ullah S, Alam T, Halim SA, Mohanta TK, Khan A, Anwar MU, Csuk R, Avula SK, Al-Harrasi A. Discovery of New Boswellic Acid Hybrid 1H-1,2,3-Triazoles for Diabetic Management: In Vitro and In Silico Studies. Pharmaceuticals. 2023; 16(2):229. https://doi.org/10.3390/ph16020229
Chicago/Turabian StyleRehman, Najeeb Ur, Saeed Ullah, Tanveer Alam, Sobia Ahsan Halim, Tapan Kumar Mohanta, Ajmal Khan, Muhammad U. Anwar, René Csuk, Satya Kumar Avula, and Ahmed Al-Harrasi. 2023. "Discovery of New Boswellic Acid Hybrid 1H-1,2,3-Triazoles for Diabetic Management: In Vitro and In Silico Studies" Pharmaceuticals 16, no. 2: 229. https://doi.org/10.3390/ph16020229
APA StyleRehman, N. U., Ullah, S., Alam, T., Halim, S. A., Mohanta, T. K., Khan, A., Anwar, M. U., Csuk, R., Avula, S. K., & Al-Harrasi, A. (2023). Discovery of New Boswellic Acid Hybrid 1H-1,2,3-Triazoles for Diabetic Management: In Vitro and In Silico Studies. Pharmaceuticals, 16(2), 229. https://doi.org/10.3390/ph16020229