
Photocaging of Pyridinylimidazole-Based Covalent JNK3 Inhibitors Affords Spatiotemporal Control of the Binding Affinity in Live Cells

Abstract
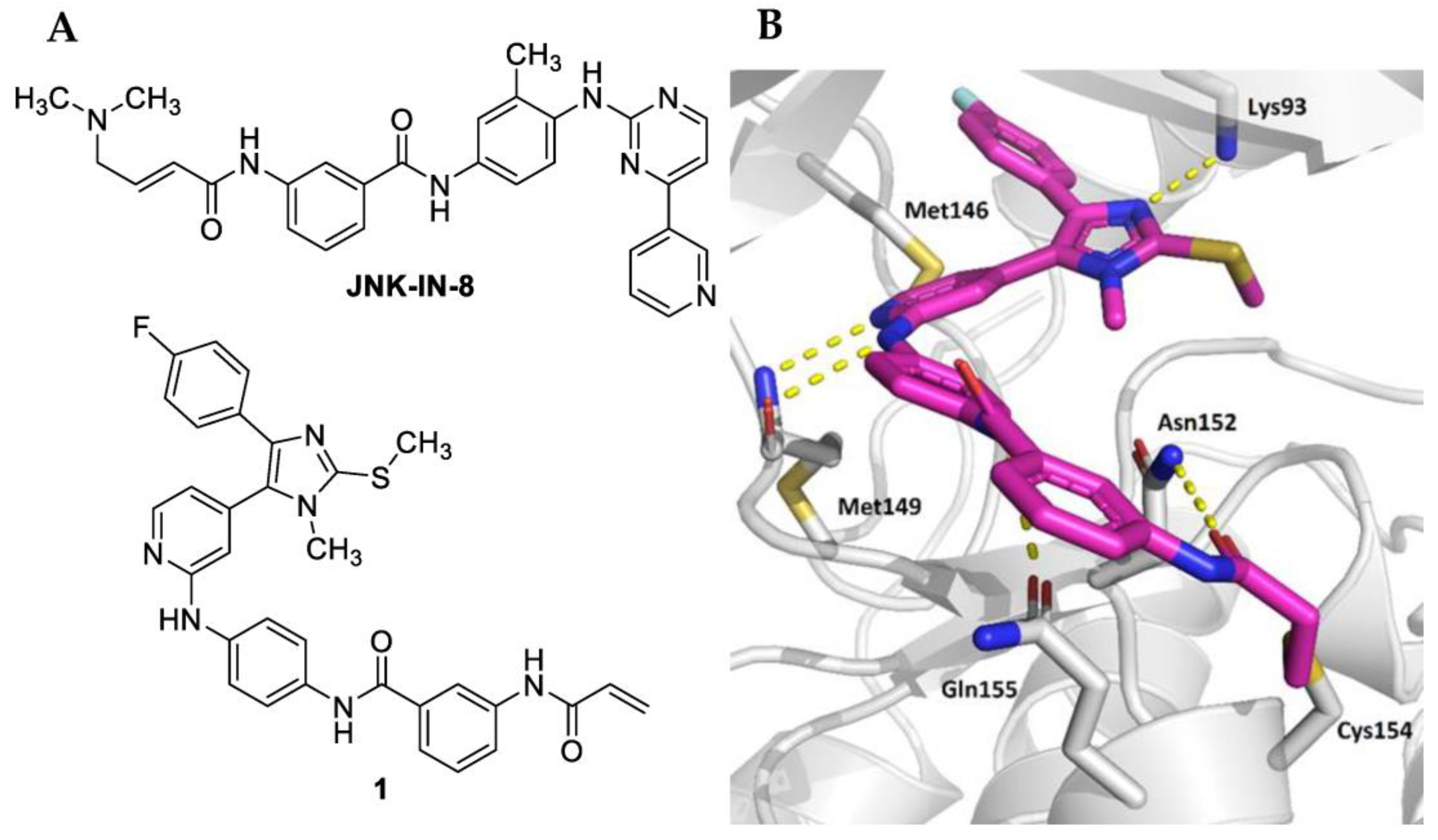
:1. Introduction
2. Results and Discussion
2.1. Biological Evaluation
2.2. Chemistry
2.3. Conclusions
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Procedures
- (1)
- General Procedure A (Buchwald-Hartwig Arylamination)
- (2)
- General Procedure B (HATU-mediated Amide Coupling)
3.1.3. Detailed Procedures
- N-(4-Bromophenyl)-N-methyl-3-nitrobenzamide (4)
- 3-Amino-N-(4-bromophenyl)-N-methylbenzamide (5)
- 3-Acrylamido-N-(4-bromophenyl)-N-methylbenzamide (6)
- 3-Acrylamido-N-(4-((4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)phenyl)-N-methylbenzamide hydrotrifluoroacetate (8)
- Synthesis of N-(4-Bromophenyl)-N-methyl-3-propionamidobenzamide (9)
- N-(4-((4-(4-(4-Fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)phenyl)-N-methyl-3-propionamidobenzamide (10)
- Methyl 4-((4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)benzoate (11)
- 4-((4-(4-(4-Fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)benzoic acid (12)
- N-(3-Acrylamidophenyl)-4-((4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)benzamide hydrotrifluoroacetate (13)
- 4-((4-(4-(4-Fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)-N-(3-propionamidophenyl)benzamide (14)
- Methyl 4-((4,5-dimethoxy-2-nitrobenzyl)(4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridine-2-yl)amino)benzoate (15)
- 4-((4,5-Dimethoxy-2-nitrobenzyl)(4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)benzoic acid (16)
- N-(3-Acrylamidophenyl)-4-((4,5-dimethoxy-2-nitrobenzyl)(4-(4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)amino)benzamide hydrotrifluoroacetate (17)
3.2. Biological Assays
3.2.1. NanoBRET™ Assay
3.2.2. Point Mutation of Cys154 in the JNK3-NanoLuc Fusion Vector
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barr, R.K.; Bogoyevitch, M.A. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int. J. Biochem. Cell Biol. 2001, 33, 1047–1063. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A. The isoform-specific functions of the c-Jun N-terminal kinases (JNKs): Differences revealed by gene targeting. Bioessays 2006, 28, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Wityak, J.; McGee, K.F.; Conlon, M.P.; Song, R.H.; Duffy, B.C.; Clayton, B.; Lynch, M.; Wang, G.; Freeman, E.; Haber, J.; et al. Lead Optimization toward Proof-of-Concept Tools for Huntington’s Disease within a 4-(1H-Pyrazol-4-yl)pyrimidine Class of Pan-JNK Inhibitors. J. Med. Chem. 2015, 58, 2967–2987. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Vila, M.; Teismann, P.; Davis, R.J.; Hirsch, E.C.; Przedborski, S.; Rakic, P.; Flavell, R.A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, S.P.; Schmid, R.S.; He, D.N.; Sung, M.L.; Cho, S.; Resnick, L.; Monaghan, M.M.; Hirst, W.D.; Essrich, C.; Reinhart, P.H.; et al. Inhibition of c-Jun kinase provides neuroprotection in a model of Alzheimer’s disease. Neurobiol. Dis. 2010, 39, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Koch, P.; Laufer, S.A.; Knapp, S. The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem. Int. Edit. 2018, 57, 4372–4385. [Google Scholar] [CrossRef]
- Zhang, T.; Inesta-Vaquera, F.; Niepel, M.; Zhang, J.M.; Ficarro, S.B.; Machleidt, T.; Xie, T.; Marto, J.A.; Kim, N.; Sim, T.; et al. Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. [Google Scholar] [CrossRef]
- Muth, F.; El-Gokha, A.; Ansideri, F.; Eitel, M.; Döring, E.; Sievers-Engler, A.; Lange, A.; Boeckler, F.M.; Lämmerhofer, M.; Koch, P.; et al. Tri- and Tetrasubstituted Pyridinylimidazoles as Covalent Inhibitors of c-Jun N-Terminal Kinase 3. J. Med. Chem. 2017, 60, 594–607. [Google Scholar] [CrossRef]
- Reynders, M.; Chaikuad, A.; Berger, B.T.; Bauer, K.; Koch, P.; Laufer, S.; Knapp, S.; Trauner, D. Controlling the Covalent Reactivity of a Kinase Inhibitor with Light. Angew. Chem. Int. Edit. 2021, 60, 20178–20183. [Google Scholar] [CrossRef]
- Wu, G.C.; Zhao, T.; Kang, D.W.; Zhang, J.; Song, Y.N.; Namasivayam, V.; Kongsted, J.; Pannecouque, C.; De Clercq, E.; Poongavanam, V.; et al. Overview of Recent Strategic Advances in Medicinal Chemistry. J. Med. Chem. 2019, 62, 9375–9414. [Google Scholar] [CrossRef] [PubMed]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G.; Heckel, A. Biologically active molecules with a “light switch”. Angew. Chem. Int. Edit. 2006, 45, 4900–4921. [Google Scholar] [CrossRef]
- Yu, H.T.; Li, J.B.; Wu, D.D.; Qiu, Z.J.; Zhang, Y. Chemistry and biological applications of photo-labile organic molecules. Chem. Soc. Rev. 2010, 39, 464–473. [Google Scholar] [CrossRef]
- Kaplan, J.H.; Forbush, B.; Hoffman, J.F. Rapid Photolytic Release of Adenosine 5’-Triphosphate from a Protected Analog—Utilization by Na-K Pump of Human Red Blood-Cell Ghosts. Biochemistry 1978, 17, 1929–1935. [Google Scholar] [CrossRef]
- Chen, R.; Wang, Z.Y.; Liu, L.H.; Pan, Z.Y. Discovery of novel photocaged ERK1/2 inhibitors as light-controlled anticancer agents. Chem. Commun. 2022, 58, 4901–4904. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Ke, R.; Song, Z.Q.; Zhou, Y.; Ren, X.M.; Huang, W.X.; Wang, Z.; Ding, K. A novel photocaged B-Raf(V600E) inhibitor toward precise melanoma treatment. Bioorg. Med. Chem. Lett. 2022, 64, 128683. [Google Scholar] [CrossRef] [PubMed]
- Fleming, C.L.; Grotli, M.; Andreasson, J. On-Command Regulation of Kinase Activity using Photonic Stimuli. ChemPhotoChem 2019, 3, 318–326. [Google Scholar] [CrossRef]
- Zhang, K.H.; Ji, M.; Lin, S.W.; Peng, S.G.; Zhang, Z.H.; Zhang, M.Y.; Zhang, J.B.; Zhang, Y.; Wu, D.Y.; Tian, H.; et al. Design, Synthesis, and Biological Evaluation of a Novel Photocaged PI3K Inhibitor toward Precise Cancer Treatment. J. Med. Chem. 2021, 64, 7331–7340. [Google Scholar] [CrossRef]
- Silva, J.M.; Silva, E.; Reis, R.L. Light-triggered release of photocaged therapeutics—Where are we now? J. Control. Release 2019, 298, 154–176. [Google Scholar] [CrossRef]
- Robers, M.B.; Vasta, J.D.; Corona, C.R.; Ohana, R.F.; Hurst, R.; Jhala, M.A.; Comess, K.M.; Wood, K.V. Quantitative, Real-Time Measurements of Intracellular Target Engagement Using Energy Transfer. In Systems Chemical Biology: Methods and Protocols; Ziegler, S., Waldmann, H., Eds.; Springer: New York, NY, USA, 2019; pp. 45–71. [Google Scholar]
- Forster, M.; Liang, X.J.; Schröder, M.; Gerstenecker, S.; Chaikuad, A.; Knapp, S.; Laufer, S.; Gehringer, M. Discovery of a Novel Class of Covalent Dual Inhibitors Targeting the Protein Kinases BMX and BTK. Int. J. Mol. Sci. 2020, 21, 9269. [Google Scholar] [CrossRef] [PubMed]
- Laufer, S.A.; Zimmermann, W.; Ruff, K.J. Tetrasubstituted imidazole inhibitors of cytokine release: Probing substituents in the N-1 position. J. Med. Chem. 2004, 47, 6311–6325. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.K.; Kotschenreuther, D.; Zimmermann, W.; Laufer, S.A. Identification of regioisomers in a series of N-substituted pyridin-4-yl imidazole derivatives by regiospecific synthesis, GC/MS, and H-1 NMR. J. Org. Chem. 2003, 68, 4527–4530. [Google Scholar] [CrossRef]
- Jiang, X.-Y.; Chen, T.-K.; Zhou, J.-T.; He, S.-Y.; Yang, H.-Y.; Chen, Y.; Qu, W.; Feng, F.; Sun, H.-P. Dual GSK-3beta/AChE Inhibitors as a New Strategy for Multitargeting Anti-Alzheimer’s Disease Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 171–176. [Google Scholar] [CrossRef]
- Kirschner, S.; Dobber, A.; Krebs, M.; Witt, C.; Hartke, B.; Peifer, C. The Impact of Electronic Effects on Photolysis: A Model Study on the 4,5-Dimethoxy-2-nitrobenzyl Caged N-Phenylpyrimidine-2-amine Scaffold. ChemPhotoChem 2020, 4, 638–643. [Google Scholar] [CrossRef]
- Andreev, S.; Pantsar, T.; Tesch, R.; Kahlke, N.; El-Gokha, A.; Ansideri, F.; Gratz, L.; Romasco, J.; Sita, G.; Geibel, C.; et al. Addressing a Trapped High-Energy Water: Design and Synthesis of Highly Potent Pyrimidoindole-Based Glycogen Synthase Kinase-3 beta Inhibitors. J. Med. Chem. 2022, 65, 1283–1301. [Google Scholar] [CrossRef]
- Bernhardt, G.; Reile, H.; Birnbock, H.; Spruss, T.; Schoenenberger, H. Standardized Kinetic Microassay to Quantify Differential Chemosensitivity on the Basis of Proliferative Activity. J. Cancer Res. Clin. 1992, 118, 35–43. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Cpd. | R | JNK3 IC50 [nM] a | JNK3 IC50 ± SEM [nM] b |
| 1 |  | 6 | 243 ± 75 |
| 8 |  | 14 | 1360 ± 342 |
| 10 |  | 24 | 5015 ± 97 |
| 13 |  | 13 | 1064 ± 252 |
| 14 |  | 22 | >10,000 |
| 17 |  | n.d. c | 9487 ± 538 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffelner, B.S.; Andreev, S.; Plank, N.; Koch, P. Photocaging of Pyridinylimidazole-Based Covalent JNK3 Inhibitors Affords Spatiotemporal Control of the Binding Affinity in Live Cells. Pharmaceuticals 2023, 16, 264. https://doi.org/10.3390/ph16020264
Hoffelner BS, Andreev S, Plank N, Koch P. Photocaging of Pyridinylimidazole-Based Covalent JNK3 Inhibitors Affords Spatiotemporal Control of the Binding Affinity in Live Cells. Pharmaceuticals. 2023; 16(2):264. https://doi.org/10.3390/ph16020264
Chicago/Turabian StyleHoffelner, Beate Sandra, Stanislav Andreev, Nicole Plank, and Pierre Koch. 2023. "Photocaging of Pyridinylimidazole-Based Covalent JNK3 Inhibitors Affords Spatiotemporal Control of the Binding Affinity in Live Cells" Pharmaceuticals 16, no. 2: 264. https://doi.org/10.3390/ph16020264