
Towards Radiolabeled EGFR-Specific Peptides: Alternatives to GE11


 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. General Radiotracer Design and Synthesis of the Peptidic Labeling Precursors 1–11
2.2. 68Ga-Radiolabeling of 1–11 and Investigation of [68Ga]Ga-1–[68Ga]Ga-11 with Regard to logD(7.4) and Stability towards Serum Peptidase Degradation
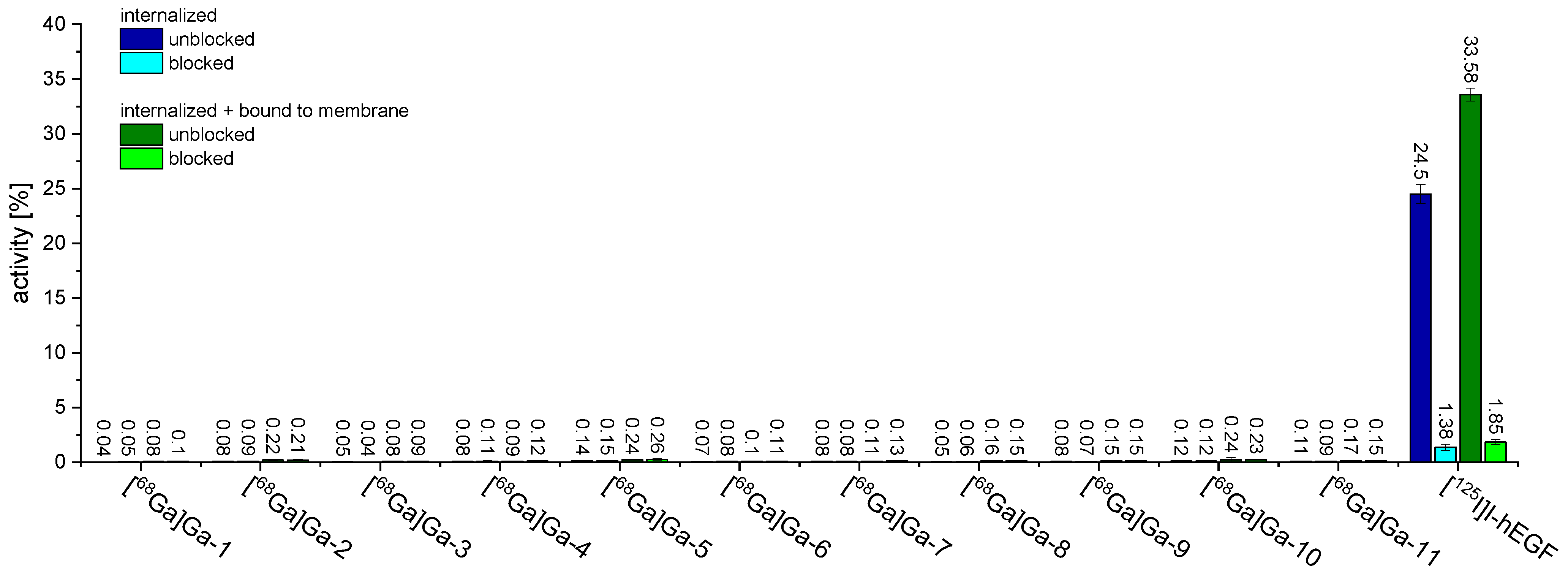
2.3. In Vitro Evaluation of [68Ga]Ga-1–[68Ga]Ga-11 Regarding Cell Uptake into A431 Cells and Receptor Affinity to the EGFR by Competitive Displacement Assays on the Same Cell Line
3. Materials and Methods
Syntheses of the Peptidic Labeling Precursors 1–11
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z.X. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, N.E.M.; Dhingra, S.; Jois, S.D.; Vicente, M.D.H. Molecular Targeting of Epidermal Growth Factor Receptor (EGFR) and Vascular Endothelial Growth Factor Receptor (VEGFR). Molecules 2021, 26, 1076. [Google Scholar] [CrossRef] [PubMed]
- Genta, I.; Chiesa, E.; Colzani, B.; Modena, T.; Conti, B.; Dorati, R. GE11 Peptide as an Active Targeting Agent in Antitumor Therapy: A Minireview. Pharmaceutics 2018, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Rinne, S.S.; Orlova, A.; Tolmachev, V. PET and SPECT Imaging of the EGFR Family (RTK Class I) in Oncology. Int. J. Mol. Sci. 2021, 22, 3663. [Google Scholar] [CrossRef]
- Santarius, T.; Shipley, J.; Brewer, D.; Stratton, M.R.; Cooper, C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 2010, 10, 59–64. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Thomas, R.; Zhang, W.H. Rethink of EGFR in Cancer With Its Kinase Independent Function on Board. Front. Oncol. 2019, 9, 800. [Google Scholar] [CrossRef]
- Chen, C.J.; Chan, C.H.; Lin, K.L.; Chen, J.H.; Tseng, C.H.; Wang, P.Y.; Chien, C.Y.; Yu, H.M.; Lin, W.J. 68Ga-labelled NOTA-RGD-GE11 peptide for dual integrin and EGFR-targeted tumour imaging. Nucl. Med. Biol. 2019, 68–69, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.M.; Chen, J.H.; Lin, K.L.; Lin, W.J. Synthesis of 68Ga-labeled NOTA-RGD-GE11 heterodimeric peptide for dual integrin and epidermal growth factor receptor-targeted tumor imaging. J. Labelled Comp. Radiopharm. 2015, 58, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Striese, F.; Sihver, W.; Gao, F.; Bergmann, R.; Walther, M.; Pietzsch, J.; Steinbach, J.; Pietzsch, H.J. Exploring pitfalls of 64Cu-labeled EGFR-targeting peptide GE11 as a potential PET tracer. Amino Acids 2018, 50, 1415–1431. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Hu, K.Z.; Liu, W.F.; Wei, Y.F.; Sha, R.H.; Long, Y.X.; Han, Y.J.; Sun, P.H.; Wu, H.B.; Li, G.P.; et al. Synthesis and evaluation of [18F]FP-Lys-GE11 as a new radiolabeled peptide probe for epidermal growth factor receptor (EGFR) imaging. Nucl. Med. Biol. 2020, 90–91, 84–92. [Google Scholar] [CrossRef]
- Dissoki, S.; Hagooly, A.; Elmachily, S.; Mishani, E. Labeling approaches for the GE11 peptide, an epidermal growth factor receptor biomarker. J. Labelled Comp. Radiopharm. 2011, 54, 693–701. [Google Scholar] [CrossRef]
- Judmann, B.; Braun, D.; Schirrmacher, R.; Wängler, B.; Fricker, G.; Wängler, C. Toward the Development of GE11-Based Radioligands for Imaging of Epidermal Growth Factor Receptor-Positive Tumors. ACS Omega 2022, 7, 27690–27702. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Zhao, R.J.; Wu, X.H.; Sun, Y.; Yao, M.; Li, J.J.; Xu, Y.H.; Gu, J.R. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005, 19, 1978–1985. [Google Scholar] [CrossRef]
- Song, S.X.; Liu, D.; Peng, J.L.; Deng, H.W.; Guo, Y.; Xu, L.X.; Miller, A.D.; Xu, Y.H. Novel peptide ligand directs liposomes toward EGF-R high-expressing cancer cells in vitro and in vivo. FASEB J. 2009, 23, 1396–1404. [Google Scholar] [CrossRef]
- Tesauro, D.; Mastro, R.; Cusimano, A.; Emma, M.R.; Cervello, M. Synthetic peptide-labelled micelles for active targeting of cells overexpressing EGF receptors. Amino Acids 2019, 51, 1177–1185. [Google Scholar] [CrossRef]
- Williams, T.M.; Sable, R.; Singh, S.; Vicente, M.G.H.; Jois, S.D. Peptide ligands for targeting the extracellular domain of EGFR: Comparison between linear and cyclic peptides. Chem. Biol. Drug Des. 2018, 91, 605–619. [Google Scholar] [CrossRef]
- Zahmatkesh, M.H.; Abedi, S.M.; Hosseinimehr, S.J. Tc-99m-HYNIC-D4 Peptide: A New Small Radiolabeled Peptide for Non Small Cell Lung Tumor Targeting. Anticancer Agents Med. Chem. 2017, 17, 734–740. [Google Scholar] [CrossRef]
- Kazemi, Z.; Zahmatkesh, M.H.; Abedi, S.M.; Hosseinimehr, S.J. Biological Evaluation of 99mTc-HYNIC-EDDA/tricine-(Ser)-D4 Peptide for Tumor Targeting. Curr. Radiopharm. 2017, 10, 123–130. [Google Scholar] [CrossRef]
- Zahmatkesh, M.H.; Abedi, S.M.; Hosseinimehr, S.J. Preparation and biological evaluation of Tc-99m-HYNIC-(Ser)3-D4 peptide for targeting and imaging of non-small-cell lung cancer. Future Oncol. 2017, 13, 893–905. [Google Scholar] [CrossRef]
- Gan, B.K.; Yong, C.Y.; Ho, K.L.; Omar, A.R.; Alitheen, N.B.; Tan, W.S. Targeted Delivery of Cell Penetrating Peptide Virus-like Nanoparticles to Skin Cancer Cells. Sci. Rep. 2018, 8, 8499. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh-Mivehroud, M.; Mahmoudpour, A.; Dastmalchi, S. Identification of New Peptide Ligands for Epidermal Growth Factor Receptor Using Phage Display and Computationally Modeling their Mode of Binding. Chem. Biol. Drug Des. 2012, 79, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, S.G.; Kim, D.W. A novel dual-labeled small peptide as a multimodal imaging agent for targeting wild-type EGFR in tumors. PLoS ONE 2022, 17, e0263474. [Google Scholar] [CrossRef] [PubMed]
- Xue, E.Y.; Wong, R.C.H.; Wong, C.T.T.; Fong, W.P.; Ng, D.K.P. Synthesis and biological evaluation of an epidermal growth factor receptor-targeted peptide-conjugated phthalocyanine-based photosensitiser. RSC Adv. 2019, 9, 20652–20662. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, S.; Joo, H.; Tsai, J.; Jasti, B.; Li, X.L. A Rational Approach for Creating Peptides Mimicking Antibody Binding. Sci. Rep. 2019, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Joshi, B.P.; Duan, X.Y.; Pant, A.; Qiu, Z.; Kuick, R.; Owens, S.R.; Wang, T.D. EGFR Overexpressed in Colonic Neoplasia Can be Detected on Wide-Field Endoscopic Imaging. Clin. Transl. Gastroenterol. 2015, 6, e101. [Google Scholar] [CrossRef]
- Jeong, M.H.; Kim, K.; Kim, E.M.; Cheong, S.J.; Lee, C.M.; Jeong, H.J.; Kim, D.W.; Lim, S.T.; Sohn, M.H.; Chung, J. In vivo and in vitro evaluation of Cy5.5 conjugated epidermal growth factor receptor binding peptide. Nucl. Med. Biol. 2012, 39, 805–812. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Brown, P.M.; Debanne, M.T.; Grothe, S.; Bergsma, D.; Caron, M.; Kay, C.; Oconnormccourt, M.D. The Extracellular Domain of the Epidermal Growth-Factor Receptor—Studies on the Affinity and Stoichiometry of Binding, Receptor Dimerization and a Binding-Domain Mutant. Eur. J. Biochem. 1994, 225, 223–233. [Google Scholar] [CrossRef]
- Komoriya, A.; Hortsch, M.; Meyers, C.; Smith, M.; Kanety, H.; Schlessinger, J. Biologically-Active Synthetic Fragments of Epidermal Growth-Factor—Localization of a Major Receptor-Binding Region. Proc. Natl. Acad. Sci. USA 1984, 81, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Charron, C.L.; Hickey, J.L.; Nsiama, T.K.; Cruickshank, D.R.; Turnbull, W.L.; Luyt, L.G. Molecular imaging probes derived from natural peptides. Nat. Prod. Rep. 2016, 33, 761–800. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Pomper, M.G. Clinical applications of Gallium-68. Appl. Radiat. Isot. 2013, 76, 2–13. [Google Scholar] [CrossRef]
- Martiniova, L.; Palatis, L.; Etchebehere, E.; Ravizzini, G. Gallium-68 in Medical Imaging. Curr. Radiopharm. 2016, 9, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.K.; Rullah, K.; Yong, C.Y.; Ho, K.L.; Omar, A.R.; Alitheen, N.B.; Tan, W.S. Targeted delivery of 5-fluorouracil-1-acetic acid (5-FA) to cancer cells overexpressing epithelial growth factor receptor (EGFR) using virus-like nanoparticles. Sci. Rep. 2020, 10, 16867. [Google Scholar] [CrossRef]
- Wellings, D.A.; Atherton, E. Standard Fmoc protocols. Methods Enzymol. 1997, 289, 44–67. [Google Scholar]
- Merlino, F.; Tomassi, S.; Yousif, A.M.; Messere, A.; Marinelli, L.; Grieco, P.; Novellino, E.; Cosconati, S.; Di Maro, S. Boosting Fmoc Solid-Phase Peptide Synthesis by Ultrasonication. Org. Lett. 2019, 21, 6378–6382. [Google Scholar] [CrossRef]
- Wolczanski, G.; Plociennik, H.; Lisowski, M.; Stefanowicz, P. A faster solid phase peptide synthesis method using ultrasonic agitation. Tet. Lett. 2019, 60, 1814–1818. [Google Scholar] [CrossRef]
- Lindner, S.; Wängler, C.; Bailey, J.J.; Jurkschat, K.; Bartenstein, P.; Wängler, B.; Schirrmacher, R. Radiosynthesis of [F-18]SiFAlin-TATE for clinical neuroendocrine tumor positron emission tomography. Nat. Protoc. 2020, 15, 3827–3843. [Google Scholar] [CrossRef]
- Postma, T.M.; Albericio, F. N-chlorosuccinimide, an efficient peptide disulfide bond-forming reagent in aqueous solution. RSC Adv. 2013, 3, 14277–14280. [Google Scholar] [CrossRef]
- Postma, T.M.; Albericio, F. N-Chlorosuccinimide, an Efficient Reagent for On-Resin Disulfide Formation in Solid-Phase Peptide Synthesis. Org. Lett. 2013, 15, 616–619. [Google Scholar] [CrossRef]
- Reilly, R.M.; Chen, P.; Wang, J.; Scollard, D.; Cameron, R.; Vallis, K.A. Preclinical pharmacokinetic, biodistribution, toxicology, and dosimetry studies of In-111-DTPA-human epidermal growth factor: An Auger electron-emitting radiotherapeutic agent for epidermal growth factor receptor-positive breast cancer. J. Nucl. Med. 2006, 47, 1023–1031. [Google Scholar] [PubMed]
- Garayoa, E.G.; Schweinsberg, C.; Maes, V.; Brans, L.; Blauenstein, P.; Tourwe, D.A.; Schibli, R.; Schubiger, P.A. Influence of the Molecular Charge on the Biodistribution of Bombesin Analogues Labeled with the [Tc-99m(CO)(3)]-Core. Bioconjug. Chem. 2008, 19, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- Glaser, M.; Morrison, M.; Solbakken, M.; Arukwe, J.; Karlsen, H.; Wiggen, U.; Champion, S.; Kindberg, G.M.; Cuthbertson, A. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with novel [18F]fluorinated aldehyde-containing prosthetic groups. Bioconjug. Chem. 2008, 19, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.J.; Baik, I.H.; Ye, S.K.; Lee, Y.H. Molecular Targeted Therapy for Hepatocellular Carcinoma: Present Status and Future Directions. Biol. Pharm. Bull. 2015, 38, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.Y.; Wei, Y.; Chen, J.W.; Chang, W.J.; Ye, L.C.; Zhu, D.X.; Xu, J.M. Anti-EGFR and anti-VEGF agents: Important targeted therapies of colorectal liver metastases. World J. Gastroenterol. 2014, 20, 4263–4275. [Google Scholar] [CrossRef]
- Oroujeni, M.; Xu, T.Q.; Gagnon, K.; Rinne, S.S.; Weis, J.; Garousi, J.; Andersson, K.G.; Lofblom, J.; Orlova, A.; Tolmachev, V. The Use of a Non-Conventional Long-Lived Gallium Radioisotope Ga-66 Improves Imaging Contrast of EGFR Expression in Malignant Tumours Using DFO-ZEGFR:2377 Affibody Molecule. Pharmaceutics 2021, 13, 292. [Google Scholar] [CrossRef]
- Chang, A.J.; De Silva, R.A.; Lapi, S.E. Development and Characterization of Zr-89-Labeled Panitumumab for Immuno-Positron Emission Tomographic Imaging of the Epidermal Growth Factor Receptor. Mol. Imaging 2013, 12, 17–27. [Google Scholar]
- Benedetto, S.; Pulito, R.; Crich, S.G.; Tarone, G.; Aime, S.; Silengo, L.; Hamm, J. Quantification of the expression level of integrin receptor alpha(V)beta(3) in cell lines and MR imaging with antibody-coated iron oxide particles. Magn. Reson. Med. 2006, 56, 711–716. [Google Scholar] [CrossRef]
- Abourbeh, G.; Shir, A.; Mishani, E.; Ogris, M.; Rodl, W.; Wagner, E.; Levitzki, A. PolyIC GE11 polyplex inhibits EGFR-overexpressing tumors. IUBMB Life 2012, 64, 324–330. [Google Scholar] [CrossRef]
- Storch, D.; Behe, M.; Walter, M.A.; Chen, J.H.; Powell, P.; Mikolajczak, R.; Macke, H.R. Evaluation of [Tc-99m/EDDA/HYNIC0]octreotide derivatives compared with [In-111-DOTA(0),Tyr(3), Thr(8)]octreotide and [In-111-DTPA(0)]octreotide: Does tumor or pancreas uptake correlate with the rate of internalization? J. Nucl. Med. 2005, 46, 1561–1569. [Google Scholar] [PubMed]
- Fani, M.; Braun, F.; Waser, B.; Beetschen, K.; Cescato, R.; Erchegyi, J.; Rivier, J.E.; Weber, W.A.; Maecke, H.R.; Reubi, J.C. Unexpected Sensitivity of sst(2) Antagonists to N-Terminal Radiometal Modifications. J. Nucl. Med. 2012, 53, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Judmann, B.; Cheng, X.; Wängler, B.; Schirrmacher, R.; Fricker, G.; Wängler, C. Synthesis, Radiolabeling, and In Vitro and In Vivo Characterization of Heterobivalent Peptidic Agents for Bispecific EGFR and Integrin alpha v beta 3 Targeting. ACS Omega 2023, 8, 2793–2807. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | logD(7.4) | Calculated Half-Life in Human Serum [min] |
|---|---|---|
| [68Ga]Ga-1 | −3.44 ± 0.08 | 295.7 ± 2.7 |
| [68Ga]Ga-2 | −3.51 ± 0.22 | 2089.2 ± 52.3 |
| [68Ga]Ga-3 | −3.92 ± 0.09 | 8077.7 ± 307.2 |
| [68Ga]Ga-4 | −4.01 ± 0.13 | 2427.2 ± 38.7 |
| [68Ga]Ga-5 | −3.91 ± 0.06 | – 1 |
| [68Ga]Ga-6 | −3.86 ± 0.11 | 1064.1 ± 43.0 |
| [68Ga]Ga-7 | −3.03 ± 0.20 | 66.4 ± 3.0 |
| [68Ga]Ga-8 | −3.16 ± 0.11 | – |
| [68Ga]Ga-9 | −2.93 ± 0.08 | – |
| [68Ga]Ga-10 | −3.92 ± 0.09 | 702.2 ± 42.2 |
| [68Ga]Ga-11 | −3.98 ± 0.06 | 562.0 ± 6.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Judmann, B.; Wängler, B.; Schirrmacher, R.; Fricker, G.; Wängler, C. Towards Radiolabeled EGFR-Specific Peptides: Alternatives to GE11. Pharmaceuticals 2023, 16, 273. https://doi.org/10.3390/ph16020273
Judmann B, Wängler B, Schirrmacher R, Fricker G, Wängler C. Towards Radiolabeled EGFR-Specific Peptides: Alternatives to GE11. Pharmaceuticals. 2023; 16(2):273. https://doi.org/10.3390/ph16020273
Chicago/Turabian StyleJudmann, Benedikt, Björn Wängler, Ralf Schirrmacher, Gert Fricker, and Carmen Wängler. 2023. "Towards Radiolabeled EGFR-Specific Peptides: Alternatives to GE11" Pharmaceuticals 16, no. 2: 273. https://doi.org/10.3390/ph16020273
APA StyleJudmann, B., Wängler, B., Schirrmacher, R., Fricker, G., & Wängler, C. (2023). Towards Radiolabeled EGFR-Specific Peptides: Alternatives to GE11. Pharmaceuticals, 16(2), 273. https://doi.org/10.3390/ph16020273