
A Study on Pharmacokinetic Functionalities and Safety Margins of an Optimized Simvastatin Nanoformulation


 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. In Vitro Absorption
2.2. In Vitro Plasma Protein Binding (Distribution)
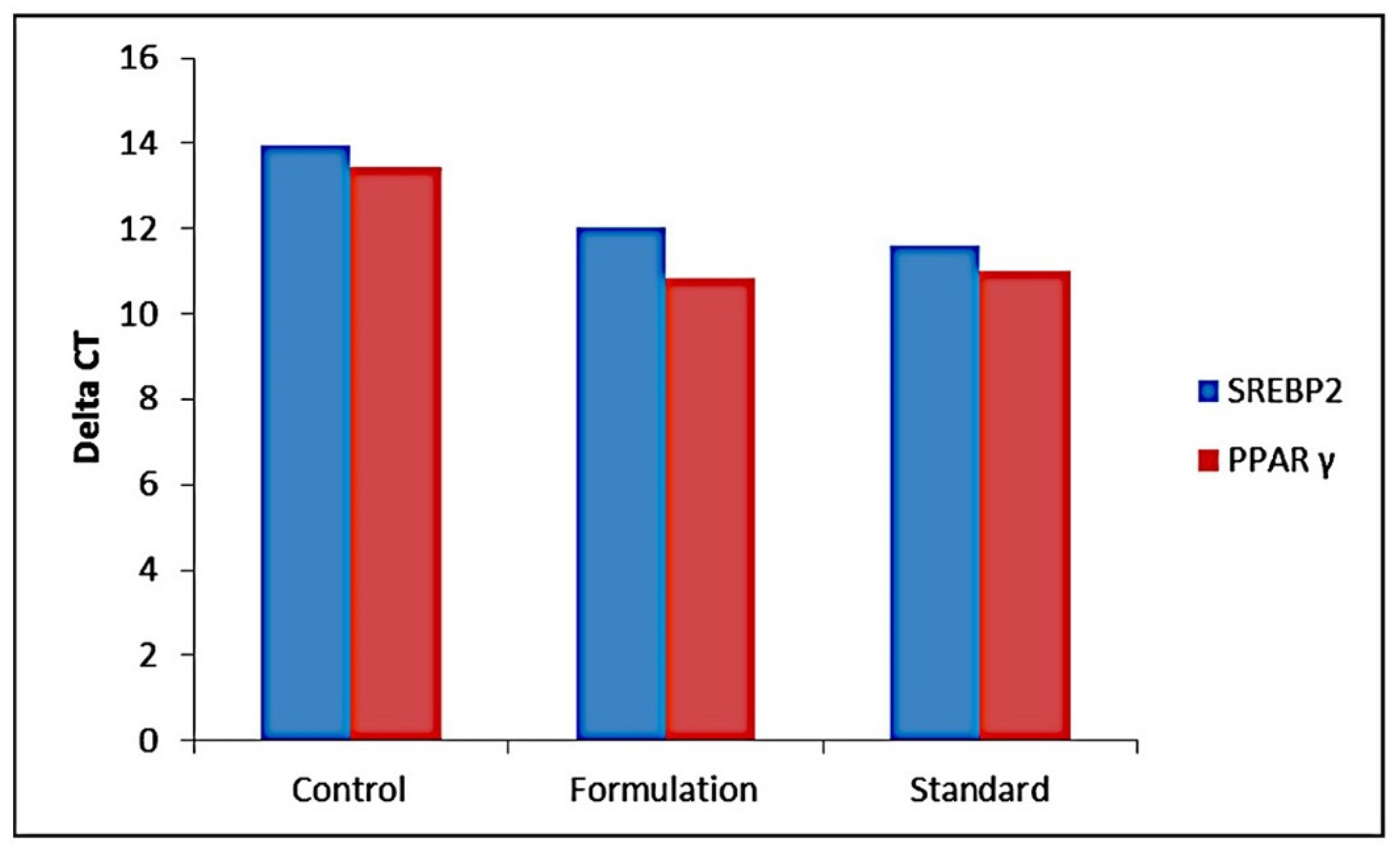
2.3. Metabolism
2.3.1. CYP3A4 Activity of Standard Simvastatin and Formulation F40
2.3.2. Metabolic Pathway of Standard Simvastatin and the Formulation
2.4. Excretion
2.4.1. Food Intake and Body Weight
2.4.2. Fecal Dry Weight
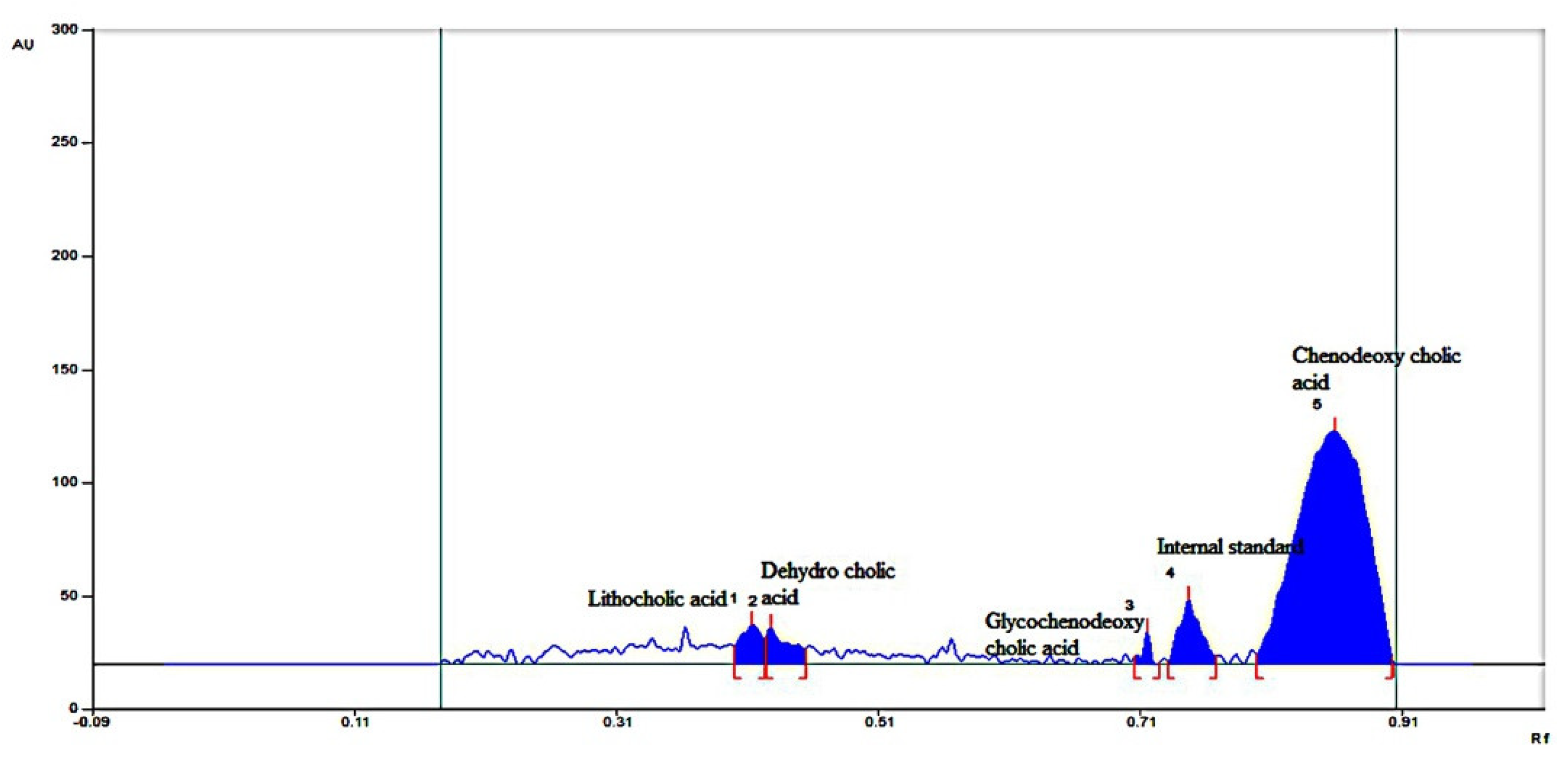
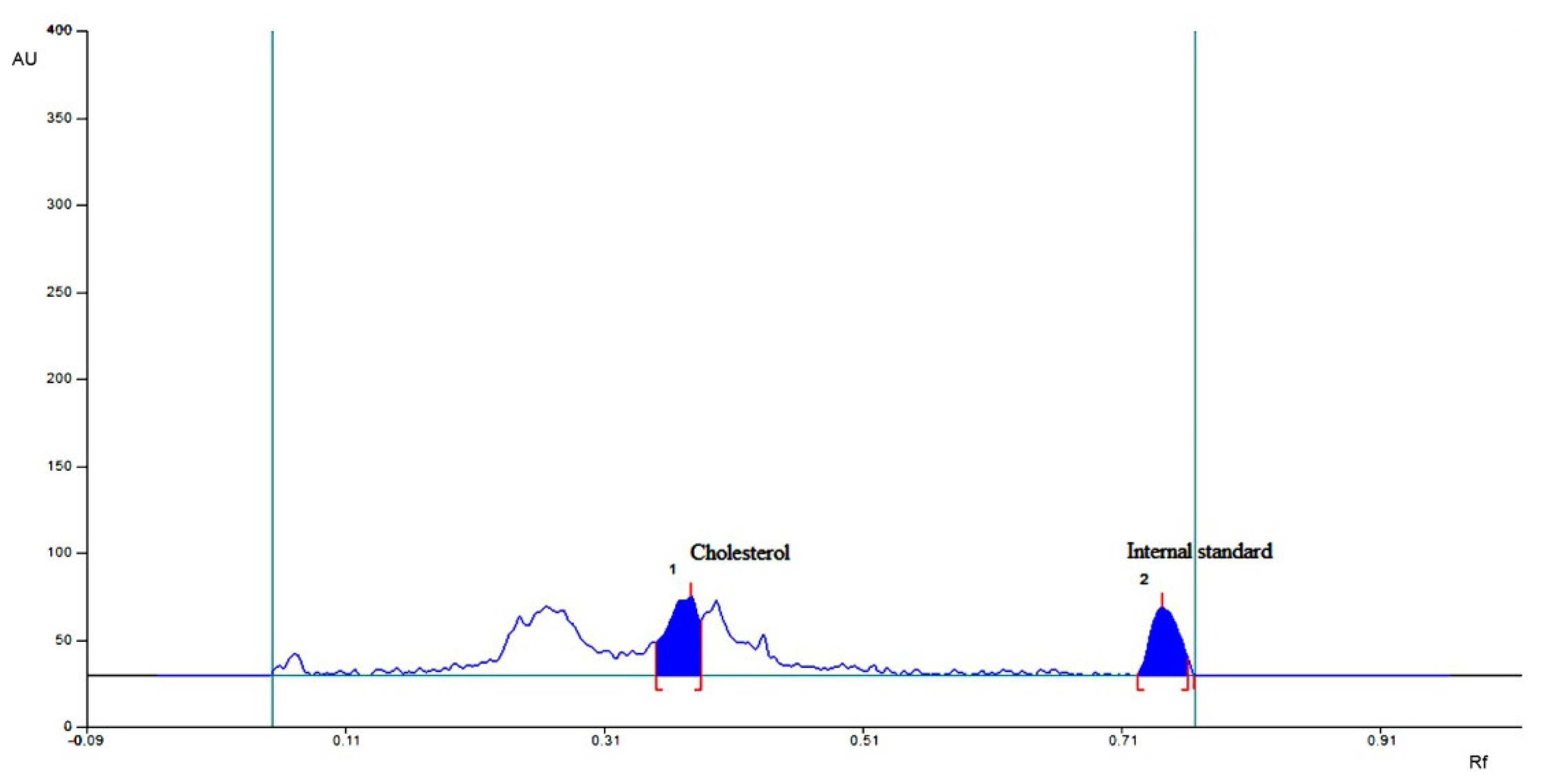
2.4.3. Neutral Sterol and Bile Acids
2.4.4. HPTLC for Individual Bile Acid and Sterols
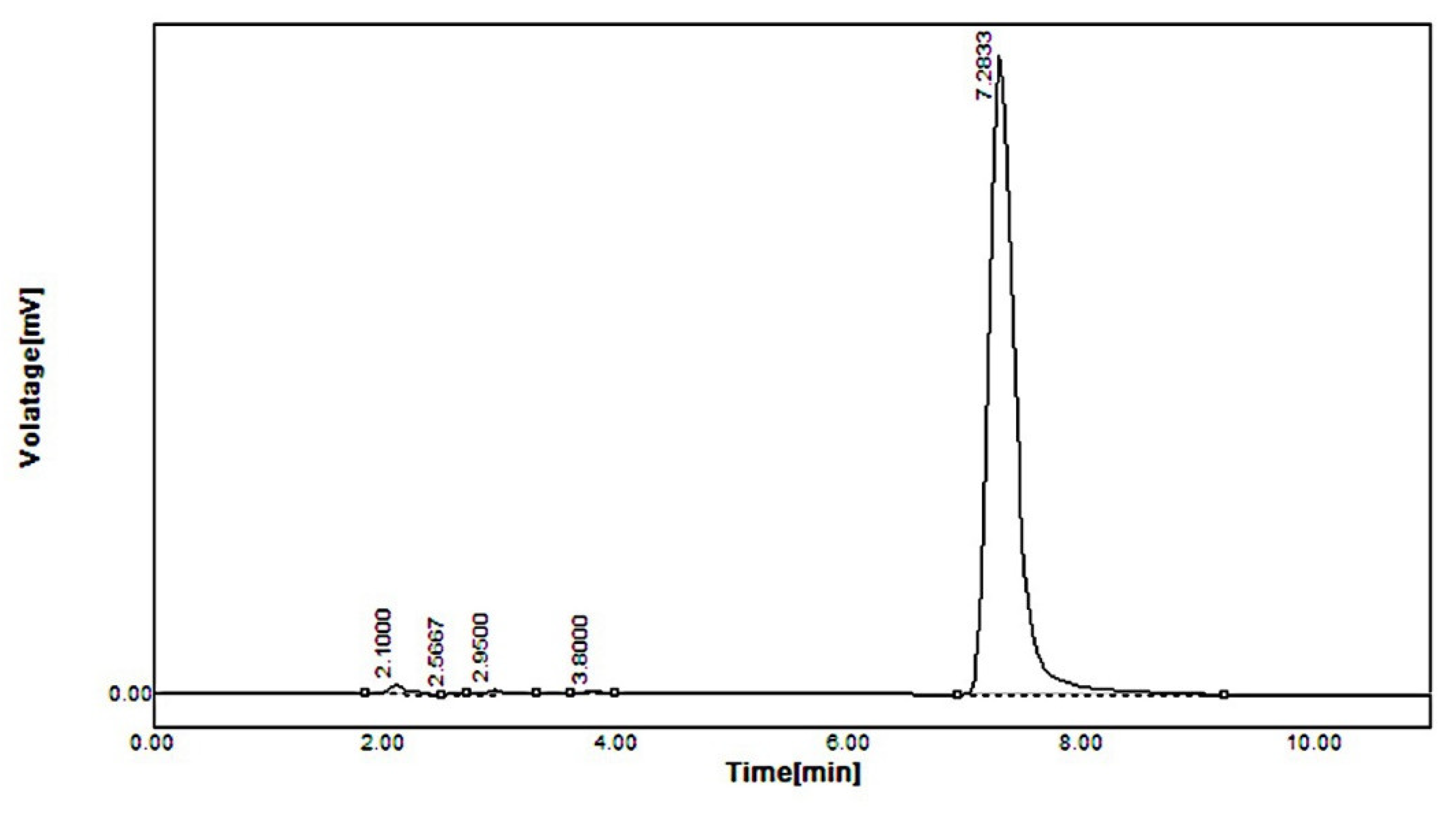
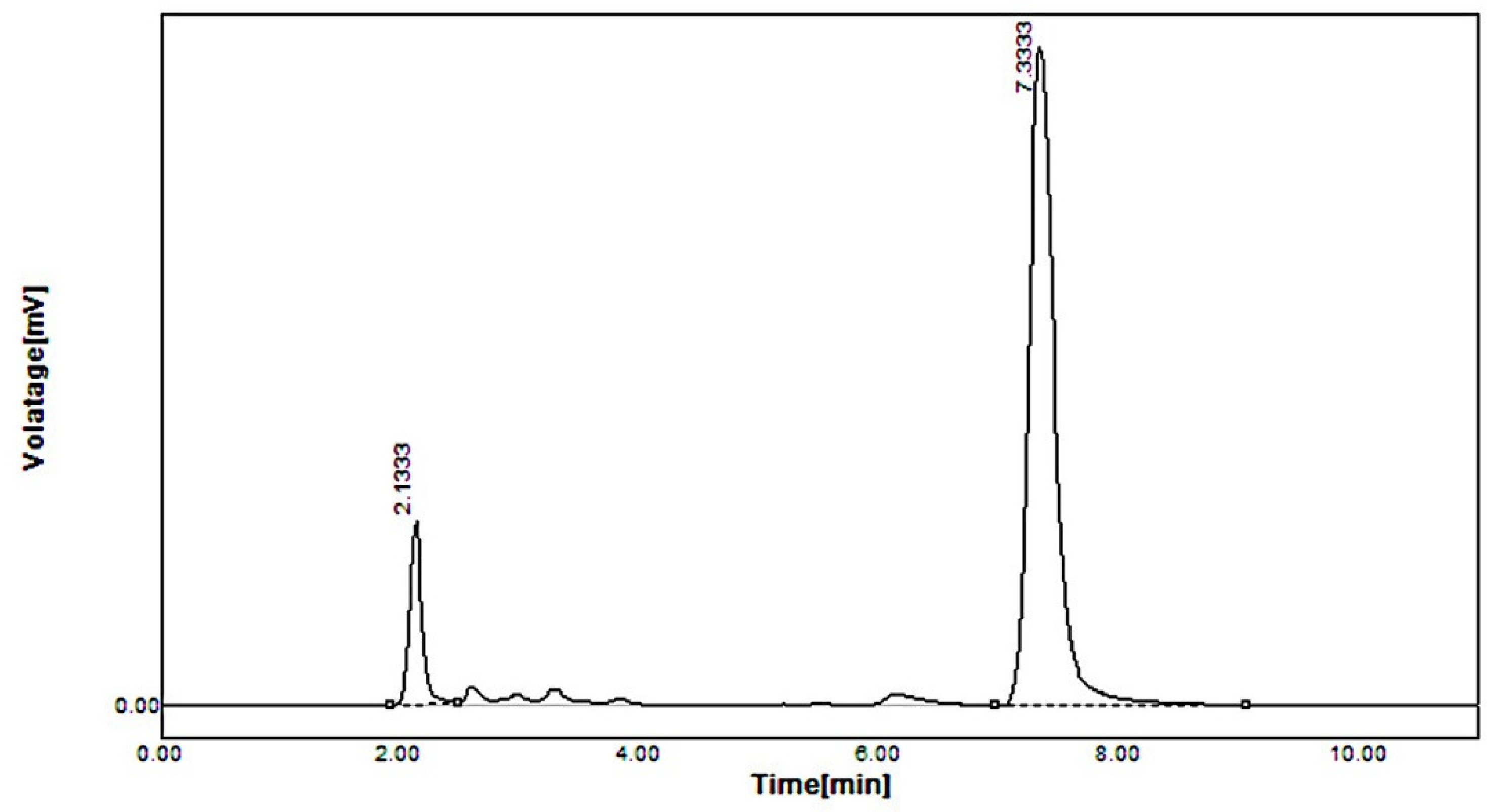
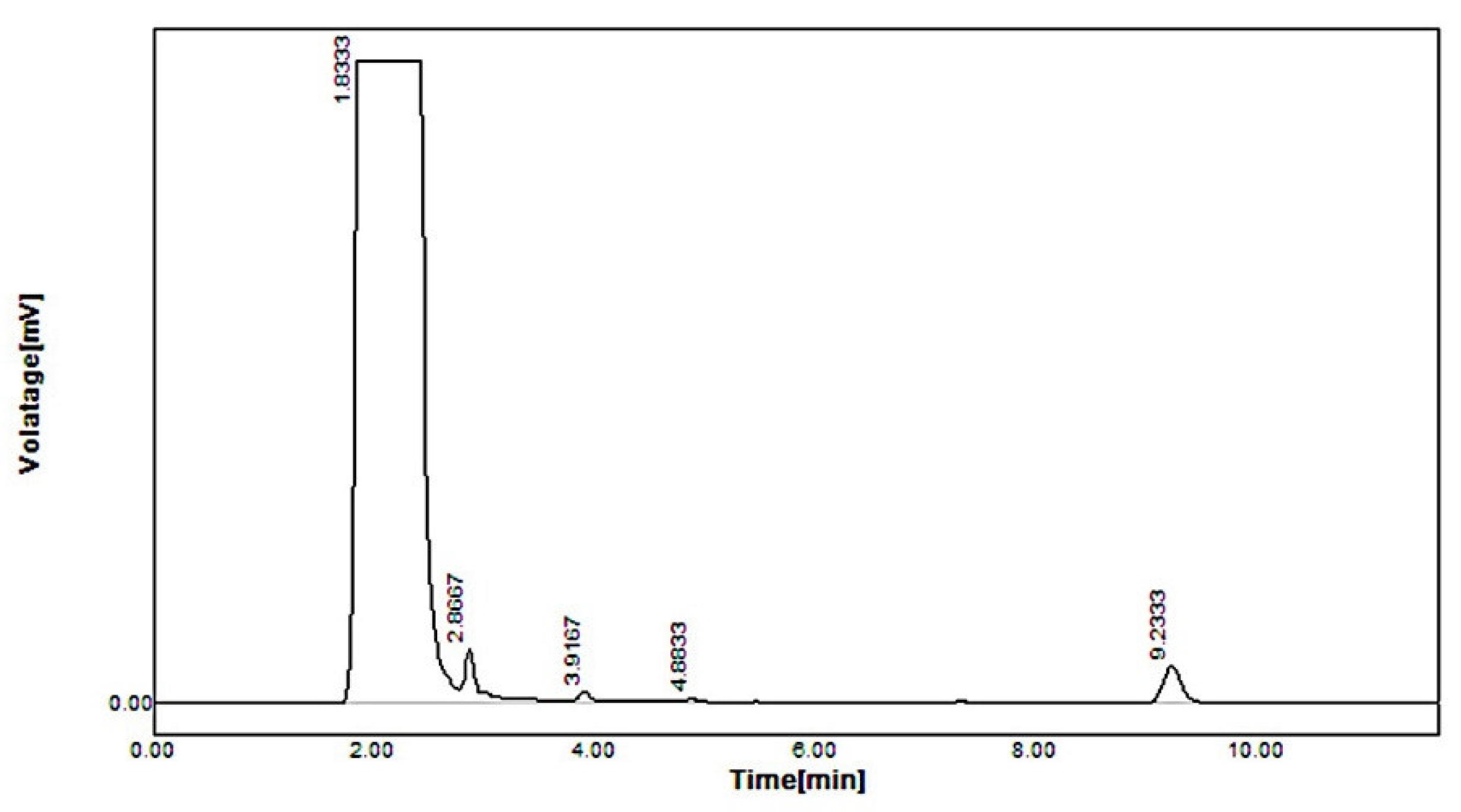
2.5. In Vivo Pharmacokinetic Study
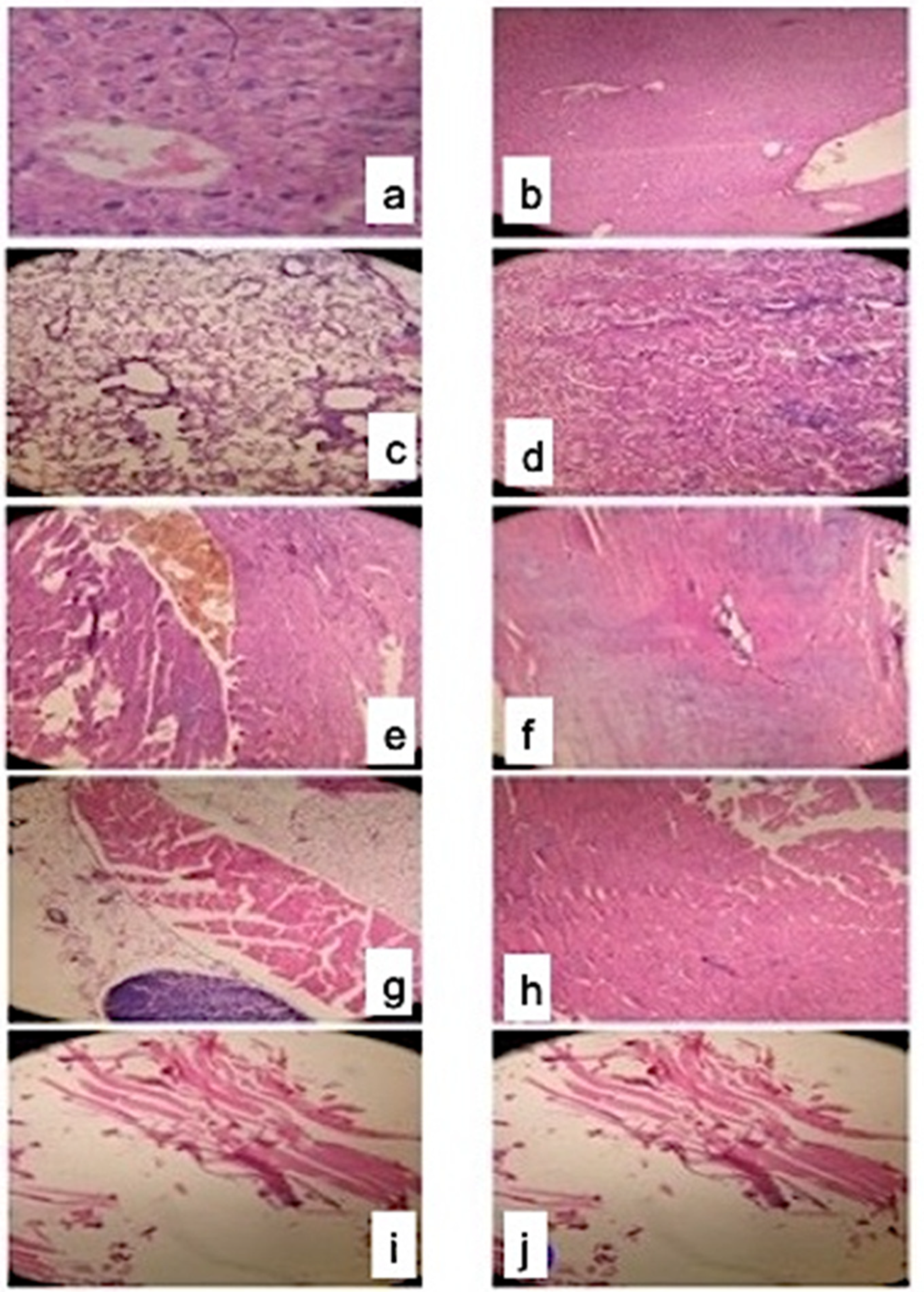
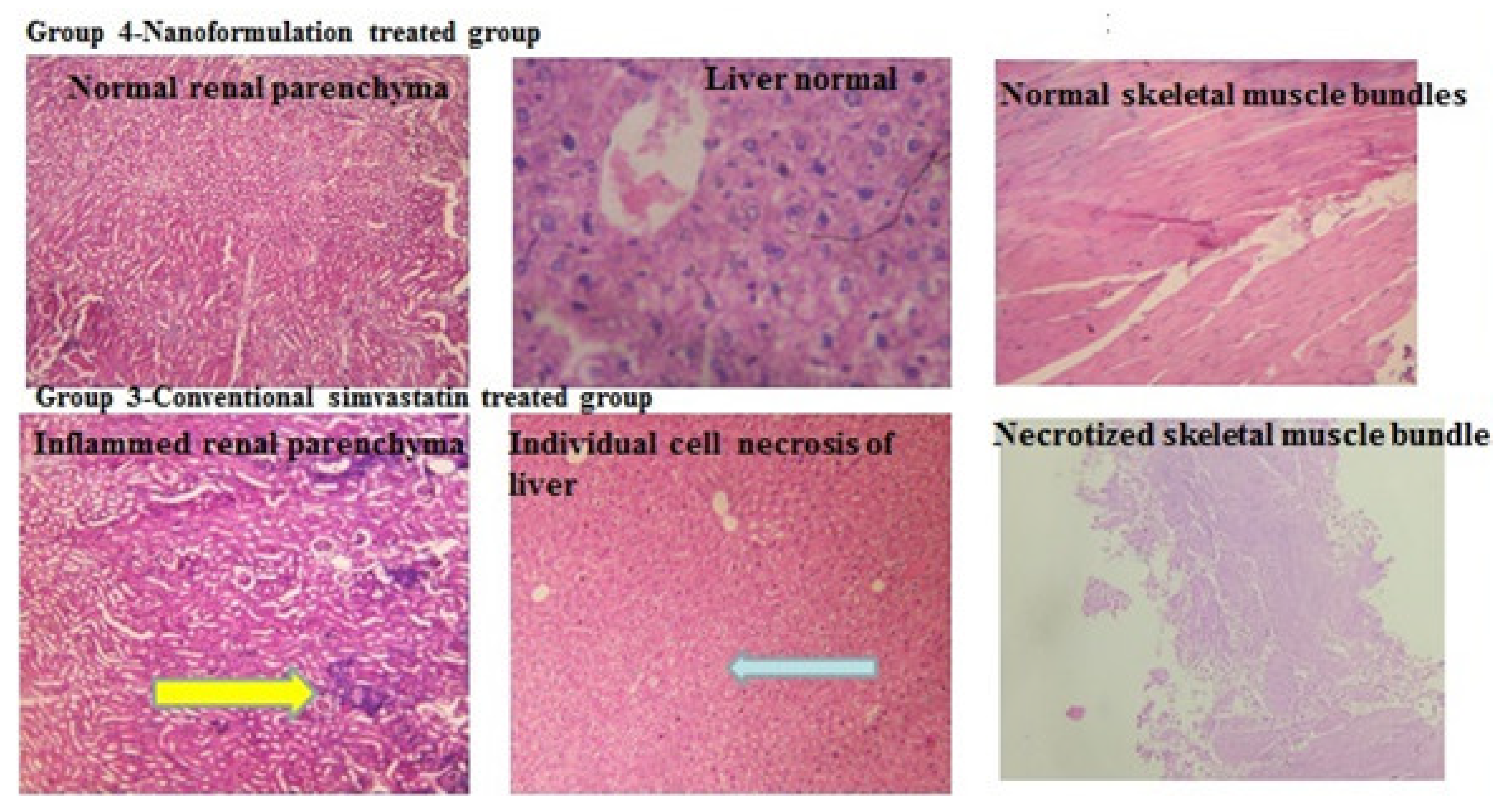
2.6. Histopathology
2.6.1. Observation
2.6.2. Fiber Typing and Necrosis in Standard Simvastatin and Nanoformulation F40
2.7. Hemolysis Assay for Biocompatibility
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methodology
4.2.1. The Everted Sac Technique: In Vitro Absorption Study
4.2.2. In Vitro Plasma Protein Binding: Distribution Studies
4.2.3. qRT-PCR: Metabolic Pathway Determination
Separation of m-RNA
RNA Isolation by Trizol Method
qRT-PCR (Quantitative Reverse Transcriptase Polymerase Chain Reaction) for CYP3A4 Microenzyme Analysis
Determination of Metabolic Pathway of Simvastatin and Chitosan in the Standard Simvastatin and Formulation F40-Treated Groups—qRT-PCR
4.2.4. Fecal Matter Evaluation—In Vitro Excretion
Determination of Fecal Cholesterol and Bile Acid Contents
4.2.5. In Vivo Pharmacokinetic Study
4.2.6. Histopathological Analysis (Toxicity)
4.2.7. Biocompatibility Analysis: Hemolytic Activity on Human Blood Agar Plate
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korani, S.; Bahrami, S.; Korani, M.; Banach, M.; Johnston, T.P.; Sahebkar, A. Parenteral systems for statin delivery: A review. Lipids Health Dis. 2019, 18, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filppula, A.M.; Hirvensalo, P.; Parviainen, H.; Ivaska, V.E.; Lönnberg, K.I.; Deng, F.; Viinamäki, J.; Kurkela, M.; Neuvonen, M.; Niemi, M. Comparative Hepatic and Intestinal Metabolism and Pharmacodynamics of Statins. Drug Metab. Dispos. Biol. Fate Chem. 2021, 49, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.Y.; Okochi, H.; Huang, Y.; Benet, L.Z. Pharmacokinetics of atorvastatin and its hydroxy metabolites in rats and the effects of concomitant rifampicin single doses: Relevance of first-pass effect from hepatic uptake transporters, and intestinal and hepatic metabolism. Drug Metab. Dispos. Biol. Fate Chem. 2006, 34, 1175–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenmann, E.D.; Talebi, Z.; Sparreboom, A.; Baker, S.D. Boosting the oral bioavailability of anticancer drugs through intentional drug-drug interactions. Basic Clin. Pharmacol. Toxicol. 2022, 130 (Suppl. S1), 23–35. [Google Scholar] [CrossRef]
- Hirota, T.; Fujita, Y.; Ieiri, I. An updated review of pharmacokinetic drug interactions and pharmacogenetics of statins. Expert Opin. Drug Metab. Toxicol. 2020, 16, 809–822. [Google Scholar] [CrossRef]
- Selvasudha, N.; Koumaravelou, K. The multifunctional synergistic effect of chitosan on simvastatin loaded nanoparticulate drug delivery system. Carbohydr. Polym. 2017, 163, 70–80. [Google Scholar] [CrossRef]
- Baldwin, K.M.; Haddad, F. Effects of different activity and inactivity paradigms on myosin heavy chain gene expression in striated muscle. J. Appl. Physiol. 2001, 90, 345–357. [Google Scholar] [CrossRef]
- Larson, L.; Lioy, J.; Johnson, J.; Medler, S. Transitional Hybrid Skeletal Muscle Fibers in Rat Soleus Development. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2019, 67, 891–900. [Google Scholar] [CrossRef]
- Zendehdel Baher, S.; Yaqoubi, S.; Asare-Addo, K.; Hamishehkar, H.; Nokhodchi, A. Dry Powder Formulation of Simvastatin Nanoparticles for Potential Application in Pulmonary Arterial Hypertension. Pharmaceutics 2022, 14, 895. [Google Scholar] [CrossRef]
- Rodrigues, A.C.; Curi, R.; Genvigir, F.D.; Hirata, M.H.; Hirata, R.D. The expression of efflux and uptake transporters are regulated by statins in Caco-2 and HepG2 cells. Acta Pharmacol. Sin. 2009, 30, 956–964. [Google Scholar] [CrossRef]
- Choo, E.F.; Kurnik, D.; Muszkat, M.; Ohkubo, T.; Shay, S.D.; Higginbotham, J.N.; Glaeser, H.; Kim, R.B.; Wood, A.J.; Wilkinson, G.R. Differential in vivo sensitivity to inhibition of P-glycoprotein located in lymphocytes, testes, and the blood-brain barrier. J. Pharmacol. Exp. Ther. 2006, 317, 1012–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werle, M.; Bernkop-Schnürch, A. Thiolated chitosans: Useful excipients for oral drug delivery. J. Pharm. Pharmacol. 2008, 60, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Maezaki, Y.; Tsuji, K.; Nakagawa, Y.; Kawai, Y.; Akimoto, M.; Tsugita, T.; Takekawa, W.; Terada, A.; Hara, H.; Mitsuoka, T. Hypocholesterolemic effect of chitosan in adult males. Biosci. Biotechnol. Biochem. 1993, 57, 1439–1444. [Google Scholar] [CrossRef]
- Al-Mohizea, A.M. Influence of intestinal efflux pumps on the absorption and transport of furosemide. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2010, 18, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Si, L.; Pan, H.; Rabba, A.K.; Yan, F.; Qiu, J.; Li, G. Excipients enhance intestinal absorption of ganciclovir by P-gp inhibition: Assessed in vitro by everted gut sac and in situ by improved intestinal perfusion. Int. J. Pharm. 2011, 403, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Abbas, K.; Amin, M.; Hussain, M.A.; Sher, M.; Abbas Bukhari, S.N.; Edgar, K.J. Design, characterization and pharmaceutical/pharmacological applications of ibuprofen conjugates based on hydroxyethylcellulose. RSC Adv. 2017, 7, 50672–50679. [Google Scholar] [CrossRef] [Green Version]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J.P. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials 2006, 27, 4356–4373. [Google Scholar] [CrossRef]
- Saptarshi, S.R.; Duschl, A.; Lopata, A.L. Interaction of nanoparticles with proteins: Relation to bio-reactivity of the nanoparticle. J. Nanobiotechnology 2013, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Dhana Lekshmi, U.M.; Poovi, G.; Kishore, N.; Reddy, P.N. In vitro characterization and invivo toxicity study of repaglinide loaded poly (methyl methacrylate) nanoparticles. Int. J. Pharm. 2010, 396, 194–203. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Preiss, D.; Kuchenbaecker, K.B.; Holmes, M.V.; Engmann, J.E.; Shah, T.; Sofat, R.; Stender, S.; Johnson, P.C.; Scott, R.A.; et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: Evidence from genetic analysis and randomised trials. Lancet 2015, 385, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Defour, M.; Dijk, W.; Ruppert, P.; Nascimento, E.B.M.; Schrauwen, P.; Kersten, S. The Peroxisome Proliferator-Activated Receptor α is dispensable for cold-induced adipose tissue browning in mice. Mol. Metab. 2018, 10, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Muzio, G.; Barrera, G.; Pizzimenti, S. Peroxisome Proliferator-Activated Receptors (PPARs) and Oxidative Stress in Physiological Conditions and in Cancer. Antioxidants 2021, 10, 1734. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriamornsak, P.; Dass, C.R. Chitosan Nanoparticles in Atherosclerosis-Development to Preclinical Testing. Pharmaceutics 2022, 14, 935. [Google Scholar] [CrossRef]
- Preta, G. Role of Lactone and Acid Forms in the Pleiotropic Effects of Statins. Pharmaceutics 2022, 14, 1899. [Google Scholar] [CrossRef]
- Shiozawa, A.; Yamaori, S.; Kamijo, S.; Ohmori, S. Effects of acid and lactone forms of statins on S-warfarin 7-hydroxylation catalyzed by human liver microsomes and recombinant CYP2C9 variants (CYP2C9.1 and CYP2C9.3). Drug Metab. Pharmacokinet. 2021, 36, 100364. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Alam, M.A.; Al-Jenoobi, F.I.; Al-Mohizea, A.M. Everted gut sac model as a tool in pharmaceutical research: Limitations and applications. J. Pharm. Pharmacol. 2012, 64, 326–336. [Google Scholar] [CrossRef]
- Barthe, L.; Woodley, J.F.; Kenworthy, S.; Houin, G. An improved everted gut sac as a simple and accurate technique to measure paracellular transport across the small intestine. Eur. J. Drug Metab. Pharmacokinet. 1998, 23, 313–323. [Google Scholar] [CrossRef]
- Stolnik, S.; Daudali, B.; Arien, A.; Whetstone, J.; Heald, C.R.; Garnett, M.C.; Davis, S.S.; Illum, L. The effect of surface coverage and conformation of poly(ethylene oxide) (PEO) chains of poloxamer 407 on the biological fate of model colloidal drug carriers. Biochim. Et Biophys. Acta 2001, 1514, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Pinto, F.L.; Thapper, A.; Sontheim, W.; Lindblad, P. Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol. Biol. 2009, 10, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, S.; Jahreis, G. Determination of underivatised sterols and bile acid trimethyl silyl ether methyl esters by gas chromatography-mass spectrometry-single ion monitoring in faeces. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 813, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Karthikayalu, S.; Rama, V.; Kirubagaran, R.; Venkatesan, R. Hemolytic toxin from the soft coral Sarcophyton trocheliophorum: Isolation and physiological characterization. J. Venom. Anim. Toxins Incl. Trop. Dis. 2010, 16, 107–120. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time in Min | Concentration μg/mL in Mucosal Side | |
|---|---|---|
| Standard | CS-SS Nanoformulation | |
| 0 | 25 ± 1.21 | 27 ± 0.90 |
| 15 | 20 ± 1.01 | 26 ± 0.70 |
| 30 | 22 ± 1.90 | 26 ± 0.90 |
| 45 | 19 ± 1.30 | 22 ± 0.56 |
| 60 | 20 ± 1.00 | 18 ± 0.43 |
| 75 | 23 ± 2.10 | 16 ± 0.41 |
| Binding Type | Pure Simvastatin (%) | CS-SS Nanoformulation (%) |
|---|---|---|
| Binding with bovine serum albumin | 95.00 ± 3.11 | 77.02 ± 4.58 |
| Binding with human plasma | 95.70 ± 1.00 | 81.80 ± 1.90 |
| Binding with mice plasma | 96.00 ± 0.73 | 83.10 ± 2.22 |
| Parameters | Time | Control Group | HFD Group | Simvastatin-Treated Group | CS-SS Nanoformulation-Treated Group |
|---|---|---|---|---|---|
| Bodyweight in mg | Initial | 24.50 ± 2.01 | 24.00 ± 2.30 | 24.01 ± 3.30 | 24.50 ± 3.10 |
| 8 weeks | 26.30 ± 1.80 | 30.70 ± 3.22 | 30.20 ± 1.90 | 29.04 ± 3.04 | |
| 16 weeks | 28.90 ± 0.90 | 34.00 ± 5.00 | 32.90 ± 2.40 | 29.20 ± 2.10 | |
| Food intake in mg | Initial | 1.50 ± 0.20 | 1.50 ± 0.20 | 1.50 ± 0.21 | 1.50 ± 0.20 |
| 8 weeks | 1.62 ± 0.41 | 1.89 ± 0.67 | 1.52 ± 0.30 | 1.48 ± 0.11 | |
| 16 weeks | 1.69 ± 0.54 | 1.96 ± 0.34 | 1.52 ± 0.81 | 1.46 ± 0.13 | |
| Fecal dry weight | Initial | 0.12 ± 0.003 | 0.10 ± 0.004 | 0.12 ± 0.003 | 0.10 ± 0.002 |
| 8 weeks | 0.13 ± 0.007 | 0.18 ± 0.001 | 0.13 ± 0.00 | 0.32 ± 0.002 | |
| 16 weeks | 0.12 ± 0.007 | 0.27 ± 0.020 | 0.13 ± 0.008 | 0.49 ± 0.005 | |
| Total cholesterol concentration in feces mg/day/animal | Initial | Traces | Traces | Traces | Traces |
| 8 weeks | Traces | Traces | Traces | 2.8 ± 1.2 | |
| 16 weeks | Traces | Traces | Traces | 3.4 ± 0.9 | |
| Total bile acids in feces mg/day/animal | Initial | Traces | Traces | Traces | Traces |
| 8 weeks | Traces | Traces | Traces | 5.9 ± 0.87 | |
| 16 weeks | Traces | Traces | Traces | 8.2 ± 1.43 | |
| Simvastatin concentration in ng/mL | 16 weeks | NA | NA | 18.98 ± 0.20 | NF |
| Simvastatin metabolite in ng/mL | 16 weeks | NA | NA | 23.12 ± 1.3 | Traces |
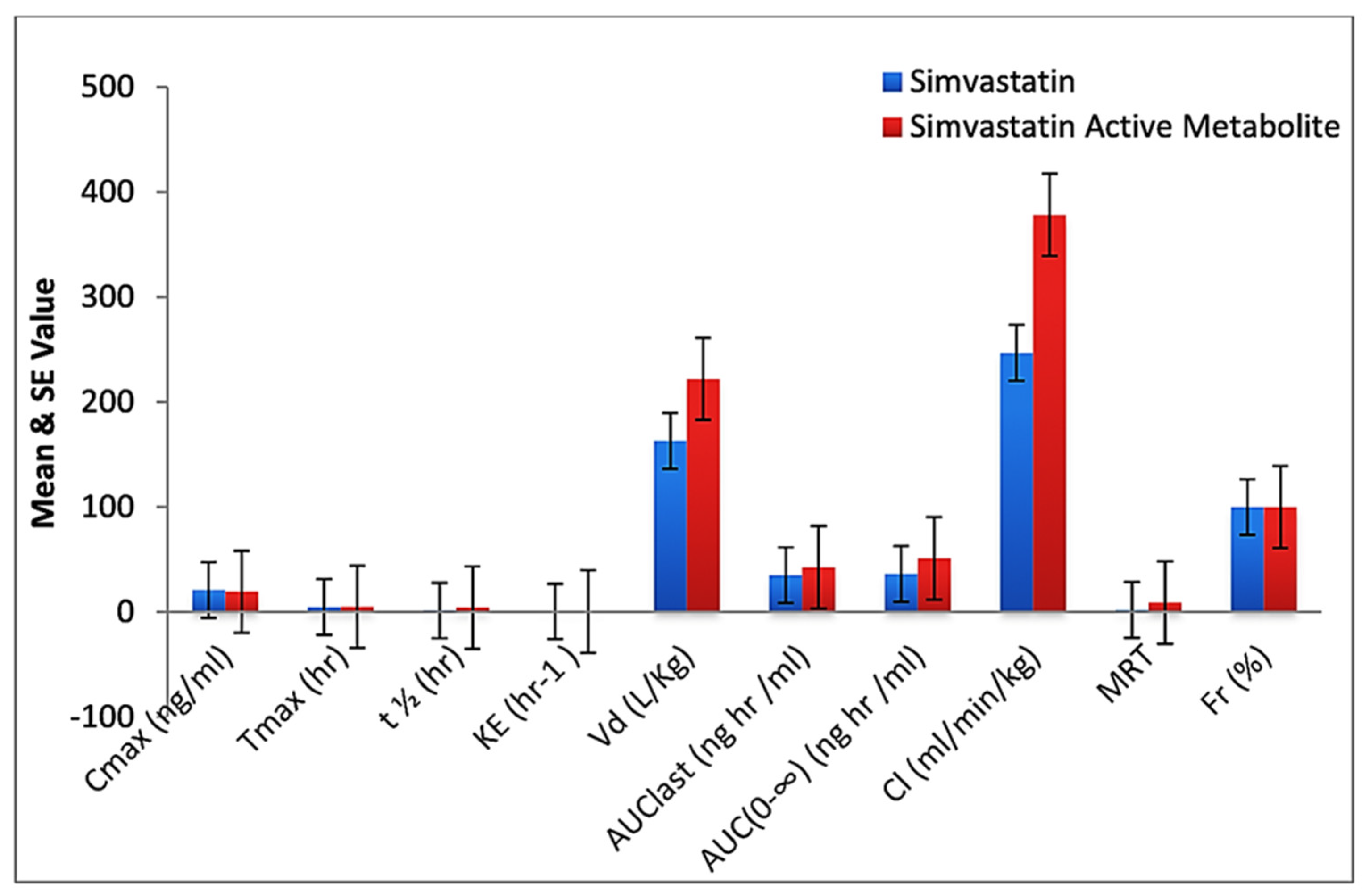
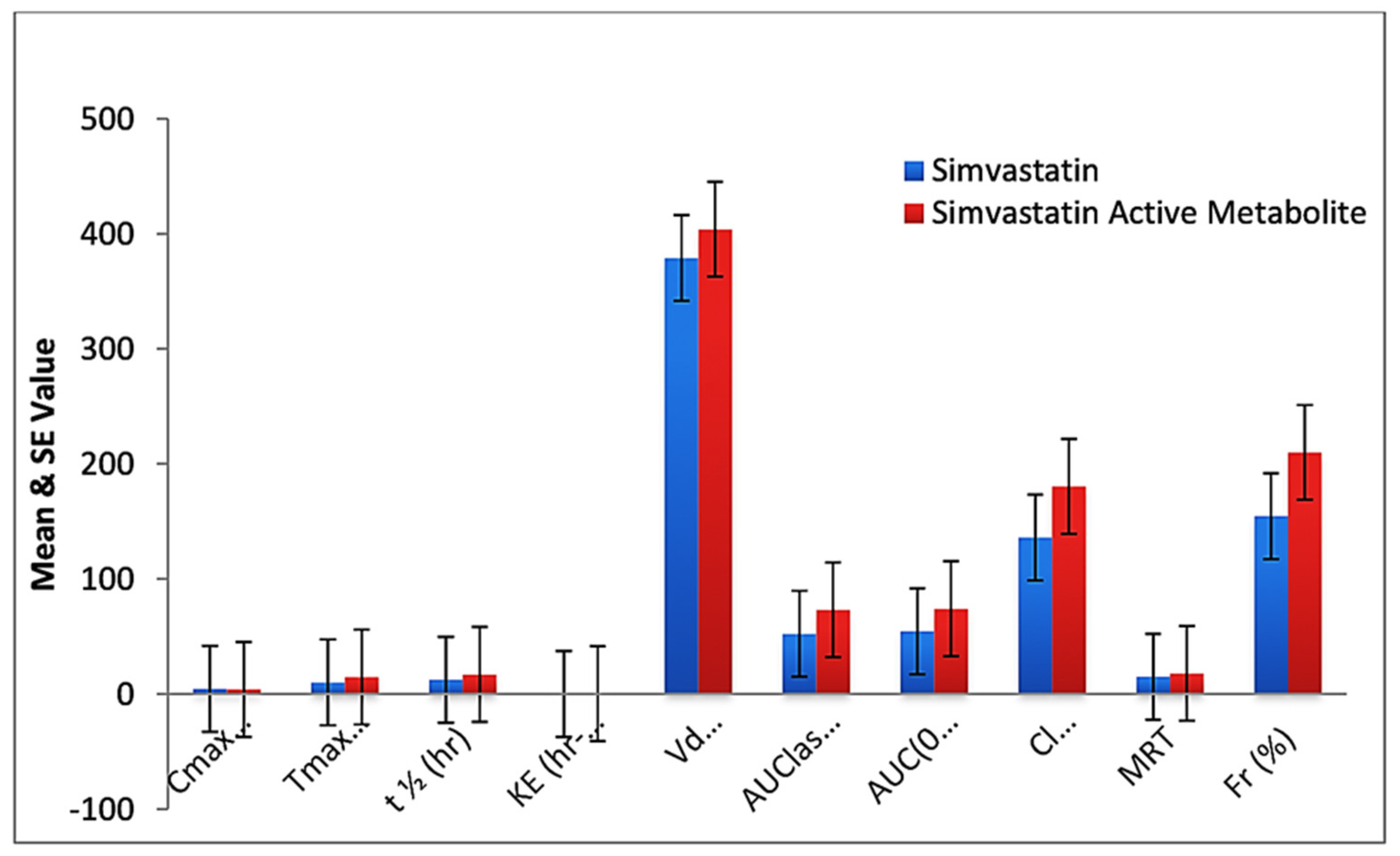
| Parameters | Standard Simvastatin * | Standard Simvastatin Active Metabolite * | Simvastatin in CS-SS Nanoformulation * | Simvastatin Active Metabolite in CS-SS Nanoformulation * | p Value |
|---|---|---|---|---|---|
| Cmax (ng/mL) | 21.12 ± 7.24 | 19.42 ± 6.90 | 4.33 ± 1.70 | 3.98 ± 1.60 | 0.005 * |
| Tmax (h) | 04.72 ± 1.20 | 5.00 ± 0.31 | 10.00 ± 2.78 | 14.56 ± 2.19 | 0.001 * |
| t½ (h) | 1.343 ± 0.689 | 4.20 ± 2.20 | 12.29 ± 4.57 | 16.87 ± 3.91 | 0.001 * |
| KE (h–1) | 0.57 ± 0.140 | 0.420 ± 0.060 | 0.0445 ± 0.008 | 0.0195 ± 0.0147 | 0.001 * |
| Vd (L/Kg) | 163.20 ± 79.00 | 222.15 ± 69.35 | 378.90 ± 112.32 | 404.00 ± 134.98 | 0.050 * |
| AUC last (nghr/mL) | 35.09 ± 12.23 | 42.72 ± 10.00 | 52.17 ± 9.86 | 73.11 ± 12.56 | 0.016 * |
| AUC(0–∞) (nghr/mL) | 36.38 ± 10.90 | 51.11 ± 14.00 | 54.35 ± 8.31 | 73.98 ± 11.90 | 0.024 * |
| Cl (mL/min/kg) | 246.88 ± 121.62 | 378.30 ± 96.22 | 135.78 ± 77.06 | 180.34 ± 90.00 | 0.049 * |
| MRT | 2.00 ± 1.10 | 9.00 ± 2.30 | 14.98 ± 3.40 | 17.85 ± 2.87 | 0.001 * |
| Fr (%) | 100.00 ± 0.0 | 100.00 ± 0.0 | 154.46 ± 23.41 | 209.66 ± 31.53 | 0.001 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A.; Dhanalekshmi, U.M.; Koumaravelu, K.; Francis, A.P.; Khan, S.A.; Abuzinadah, M.F.; Selvasudha, N. A Study on Pharmacokinetic Functionalities and Safety Margins of an Optimized Simvastatin Nanoformulation. Pharmaceuticals 2023, 16, 380. https://doi.org/10.3390/ph16030380
Ahmad A, Dhanalekshmi UM, Koumaravelu K, Francis AP, Khan SA, Abuzinadah MF, Selvasudha N. A Study on Pharmacokinetic Functionalities and Safety Margins of an Optimized Simvastatin Nanoformulation. Pharmaceuticals. 2023; 16(3):380. https://doi.org/10.3390/ph16030380
Chicago/Turabian StyleAhmad, Aftab, Unnikrishnan Meenakshi Dhanalekshmi, Kailasam Koumaravelu, Arul Prakash Francis, Shah Alam Khan, Mohammed F. Abuzinadah, and Nandakumar Selvasudha. 2023. "A Study on Pharmacokinetic Functionalities and Safety Margins of an Optimized Simvastatin Nanoformulation" Pharmaceuticals 16, no. 3: 380. https://doi.org/10.3390/ph16030380
APA StyleAhmad, A., Dhanalekshmi, U. M., Koumaravelu, K., Francis, A. P., Khan, S. A., Abuzinadah, M. F., & Selvasudha, N. (2023). A Study on Pharmacokinetic Functionalities and Safety Margins of an Optimized Simvastatin Nanoformulation. Pharmaceuticals, 16(3), 380. https://doi.org/10.3390/ph16030380