
Glycoconjugates of Mucochloric Acid—Synthesis and Biological Activity

 ,
,  , and
, and
Abstract
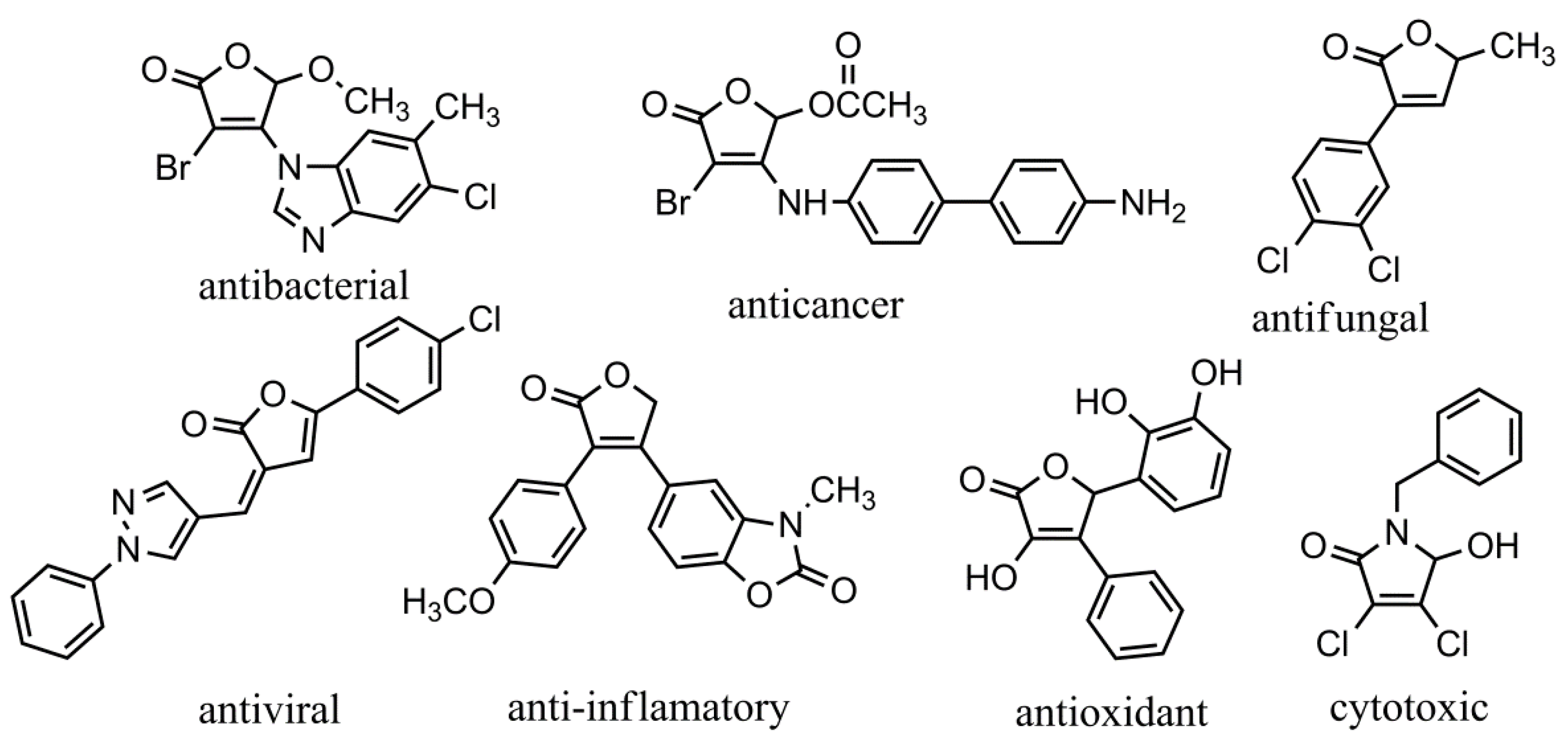
1. Introduction
2. Results and Discussion
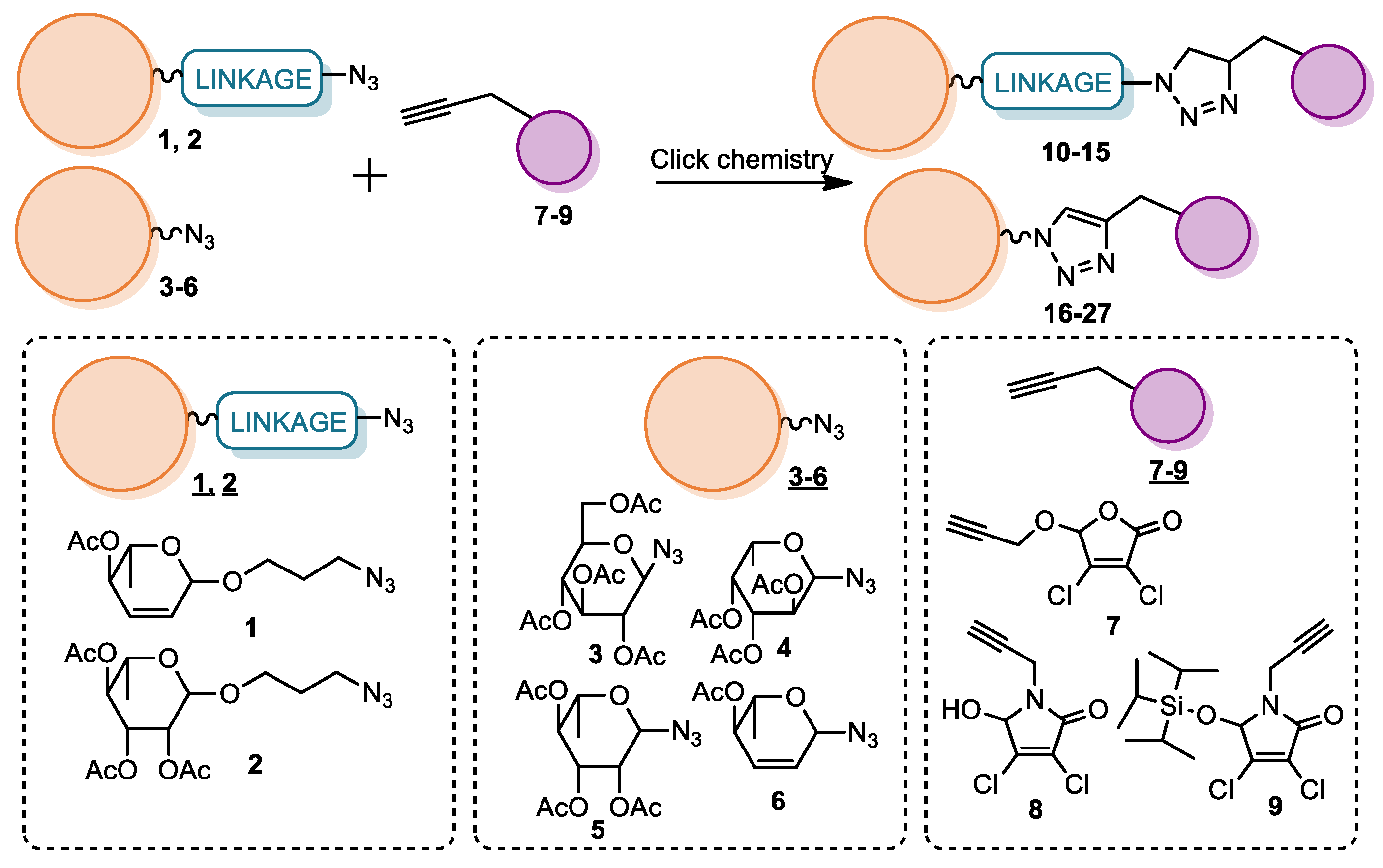
2.1. Synthesis
2.2. Anticancer Screening
2.2.1. Cytotoxicity and Anticancer Activities
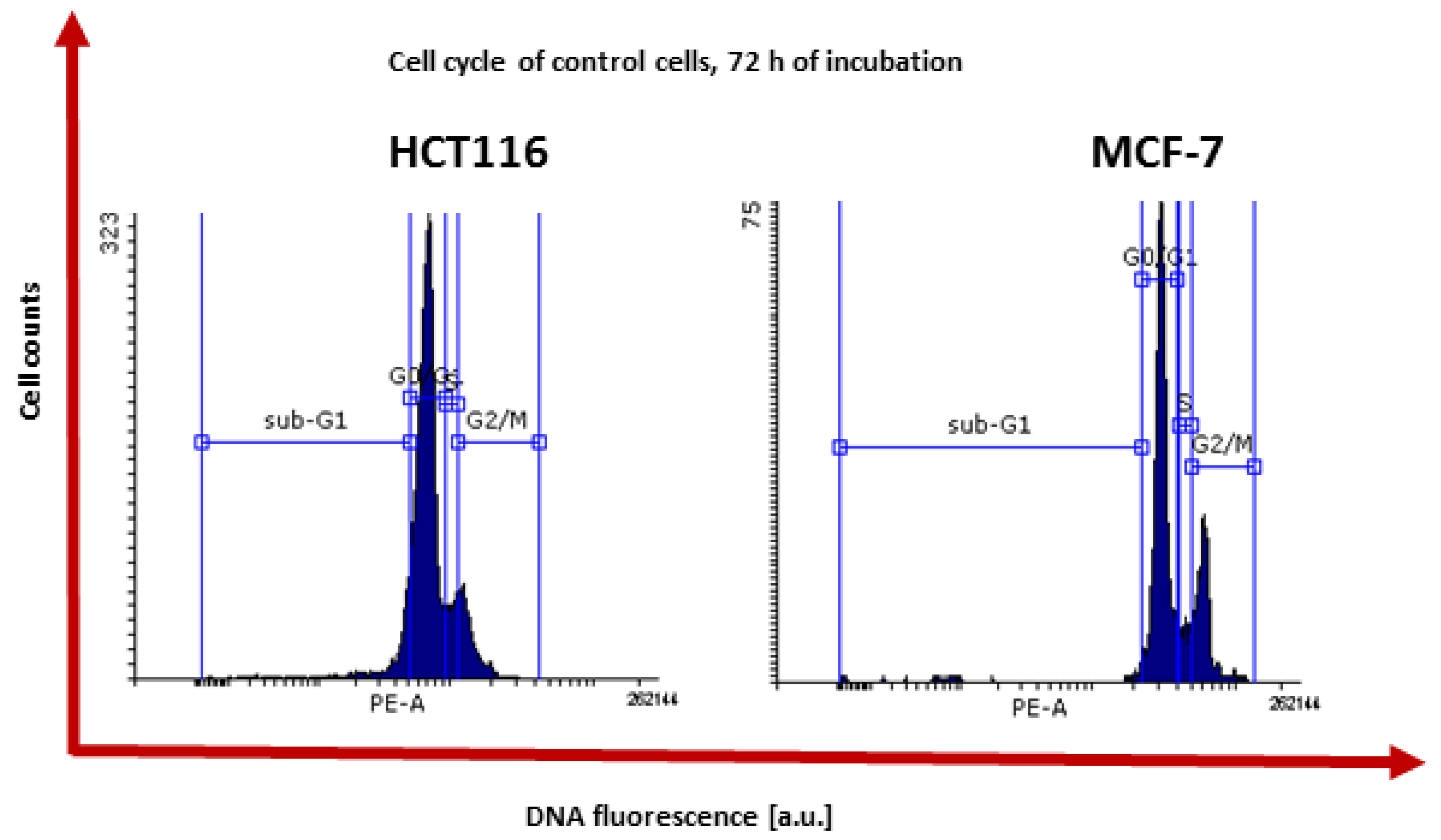
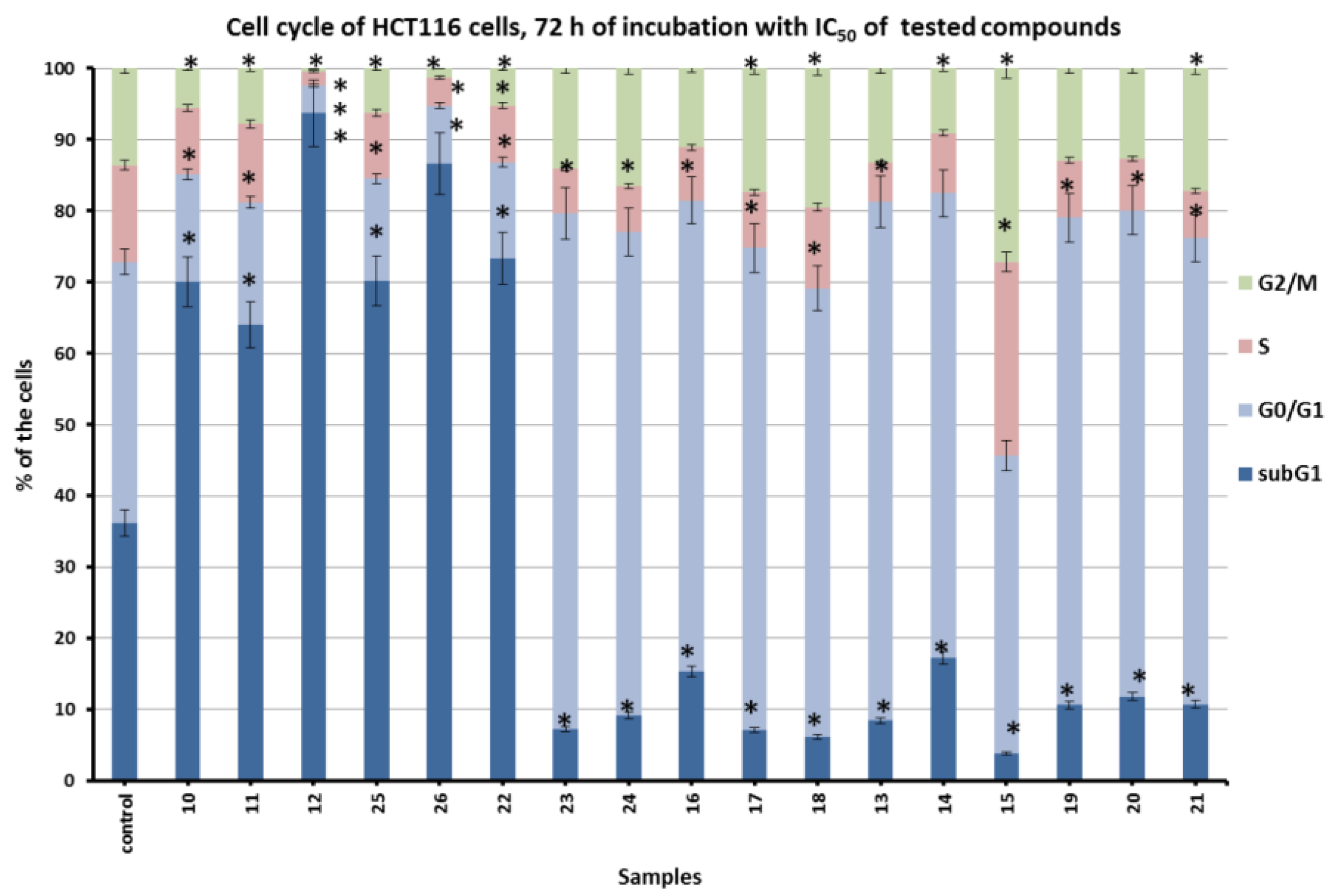
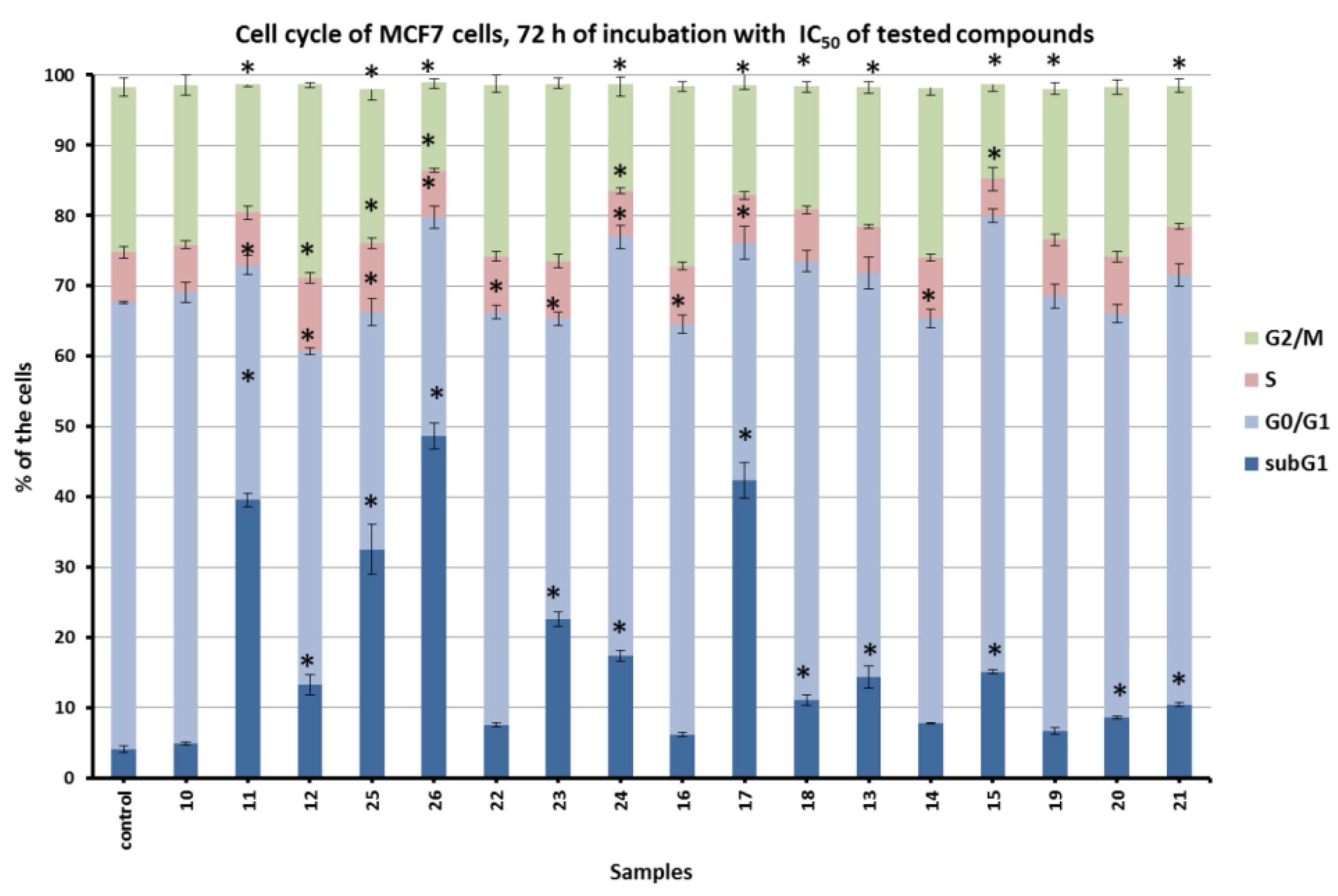
2.2.2. Inhibition of Cell Cycle and Pro-Apoptotic Action
3. Conclusions
4. Experimental Section
4.1. General Information
4.2. Chemistry
4.2.1. Synthesis of Sugar Azides
- 1-(3-Azidopropyloxy)-4-O-acetyl-2,3,6-trideoxy-L-erythro-hex-2-enopyranoside 1
- 1-(3-Azidopropyloxy)-2,3,4-tri-O-acetyl-β-L-rhamnopyranoside 2
- General procedure A
- 1-Azido-2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 3
- 1-Azido-2,3,4-tri-O-acetyl-β-L-fucopyranoside 4
- 1-Azido-2,3,4-tri-O-acetyl-L-rhamnopyranoside 5
- 1-Azido-4-O-acetyl-2,3,6-trideoxy-L-erythro-hex-2-enopyranoside 6
4.2.2. Synthesis of 3,4-Dichloro-Furan-2(5H)-One and 2H-Pyrrol-2-One Derivatives
- 3,4-Dichloro-5-(prop-2-yn-1-yloxy)-furan-2(5H)-one 7
- 3,4-Dichloro-1,5-dihydro-5-hydroxy-1-(prop-2-yn-1-yl)-2H-pyrrol-2-one 8
- 3,4-Dichloro-1,5-dihydro-1-(prop-2-yn-1-yl)-5-(triisopropylsilyloxy)-2H-pyrrol-2-one 9
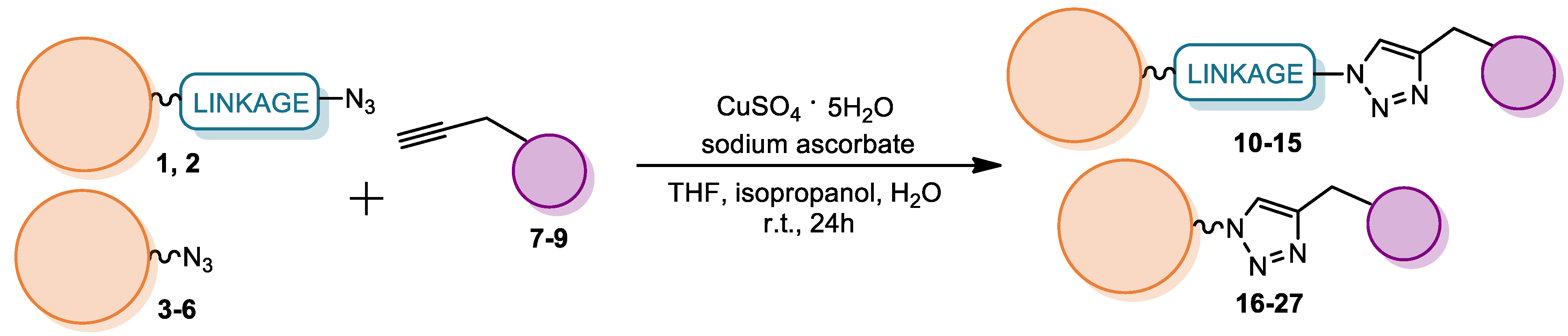
4.2.3. Synthesis of Glycoconjugates 10–26
- General procedure B
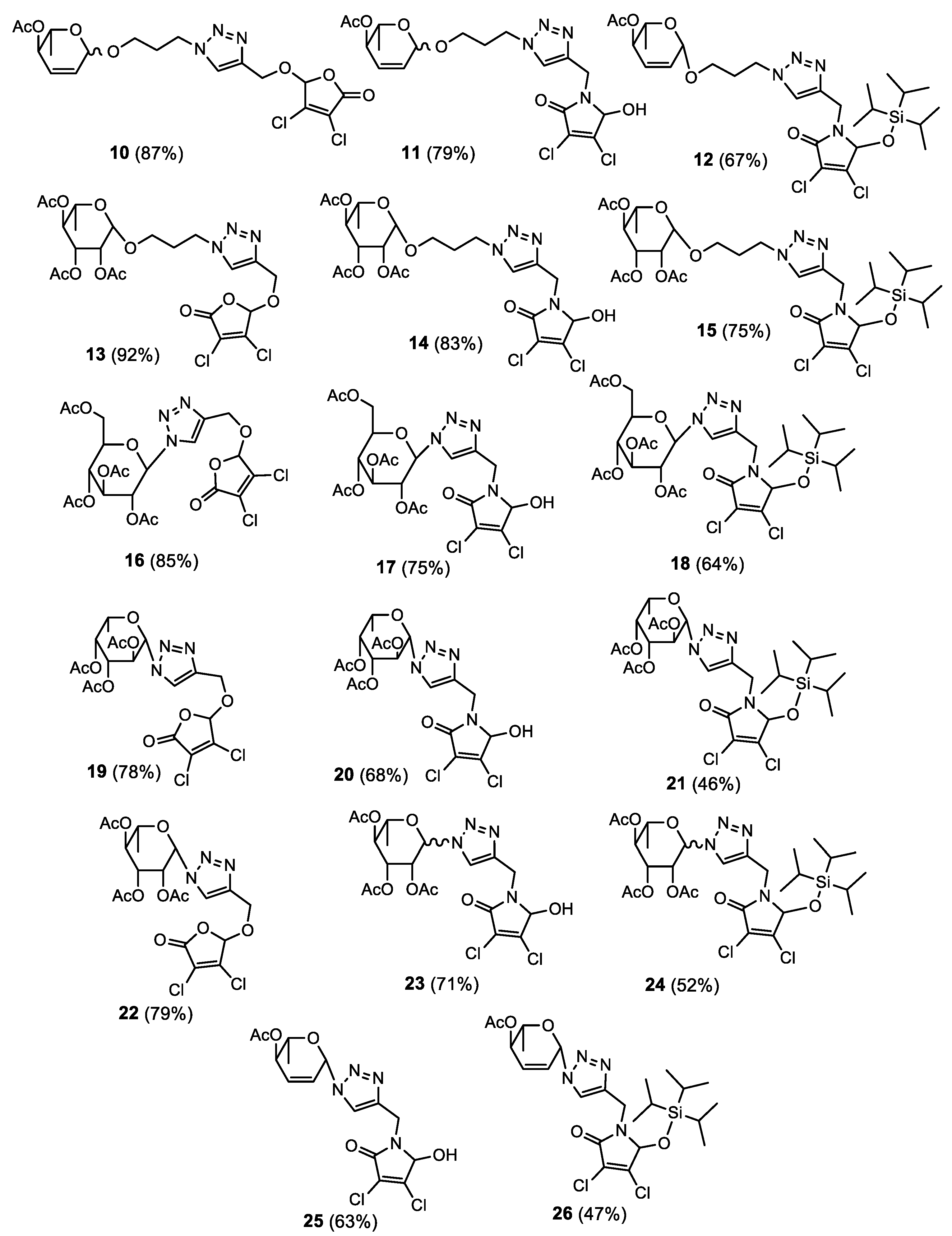
- 3,4-Dichloro-5-((1-(3-(1-O-propyl)-4-O-acetyl-2,3,6-trideoxy-L-erythro-hex-2-enopyranose)-1H-1,2,3-triazol-4-yl)methoxy)-furan-2(5H)-one 10
- 3,4-Dichloro-N-((1-(3-(1-O-propyl)-4-O-acetyl-2,3,6-trideoxy-L-erythro-hex-2-enopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-2H-pyrrol-2-one 11
- 3,4-Dichloro-N-((1-(3-(1-O-propyl)-4-O-acetyl-2,3,6-trideoxy-β-L-erythro-hex-2-enopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-5-isopropylsilyloxy-2H-pyrrol-2-one 12
- 3,4-Dichloro-5-((1-(3-(1-O-propyl)-2,3,4-tri-O-acetyl-β-L-rhamnopyranose)-1H-1,2,3-triazol-4-yl)methoxy)-furan-2(5H)-one 13
- 3,4-Dichloro-N-((1-(3-(1-O-propyl)-2,3,4-tri-O-acetyl-β-L-rhamnopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-2H-pyrrol-2-one 14
- 3,4-Dichloro-N-((1-(3-(1-O-propyl)-2,3,4-tri-O-acetyl-β-L-rhamnopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-5-triisopropylsilyloxy-2H-pyrrol-2-one 15
- 3,4-Dichloro-5-((1-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranose)-1H-1,2,3-triazol-4-yl)methoxy)-furan-2(5H)-one 16
- 3,4-Dichloro-N-((1-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-2H-pyrrol-2-one 17
- 3,4-Dichloro-N-((1-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-5-triisopropylsilyloxy-2H-pyrrol-2-one 18
- 3,4-Dichloro-5-((1-(2,3,4-tri-O-acetyl-β-L-fucopyranose)-1H-1,2,3-triazol-4-yl)methoxy)-furan-2(5H)-one 19
- 3,4-Dichloro-N-((1-(2,3,4-tri-O-acetyl-β-L-fucopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-2H-pyrrol-2-one 20
- 3,4-Dichloro-N-((1-(2,3,4-tri-O-acetyl-β-L-fucopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-5-triisopropylsilyloxy-2H-pyrrol-2-one 21
- 3,4-Dichloro-5-((1-(2,3,4-tri-O-acetyl-β-L-rhamnopyranose)-1H-1,2,3-triazol-4-yl) methoxy)-furan-2(5H)-one 22
- 3,4-Dichloro-N-((1-(2,3,4-tri-O-acetyl-L-rhamnopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-2H-pyrrol-2-one 23
- 3,4-Dichloro-N-((1-(2,3,4-tri-O-acetyl-L-rhamnopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-5-triisopropylsilyloxy-2H-pyrrol-2-one 24
- 3,4-Dichloro-N-((1-(4-O-acetyl-2,3,6-trideoxy-β-L-erythro-hex-2-enopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-2H-pyrrol-2-one 25
- 3,4-Dichloro-N-((1-(4-O-acetyl-2,3,6-trideoxy-β-L-erythro-hex-2-enopyranose)-1H-1,2,3-triazol-4-yl)-methyl)-5-triisopropylsilyloxy-2H-pyrrol-2-one 26
4.3. Biological Evaluation
4.3.1. Cells
4.3.2. MTT Cytotoxicity Assay
4.3.3. Cell Cycle and Cytometry Analysis of Apoptosis
4.3.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franco, C.M.; Borde, U.P.; Vijayakumar, E.K.; Chatterjee, S.; Blumbach, J.; Ganguli, B.N. Butalactin, a new butanolide antibiotic. Taxonomy, fermentation, isolation, and biological activity. J. Antibiot. 1991, 44, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Steenackers, H.P.; Levin, J.; Janssens, J.C.; De Weerdt, A.; Balzarini, J.; Vanderleyden, J.; De Vos, J.D.; De Keersmaecker, S.C. Structure–activity relationship of brominated 3-alkyl-5-methylene-2(5H)-furanones and alkylmaleic anhydrides as inhibitors of Salmonella biofilm formation and quorum sensing regulated bioluminescence in Vibrio harveyi. Bioorg. Med. Chem. 2010, 18, 5224–5233. [Google Scholar] [CrossRef]
- Gondela, E.; Walczak, K.Z. Synthesis and preliminary bioactivity assays of 3,4-dichloro-5-(ω-hydroxyalkylamino)-2(5H)-furanones. Eur. J. Med. Chem. 2010, 45, 3993–3997. [Google Scholar] [CrossRef]
- Lattmann, E.; Sattayasai, N.; Schwalbe, C.S.; Niamsanit, S.; Billington, D.C.; Lattmann, P.; Langley, C.A.; Singh, H.; Dunn, S. Novel anti-bacterials against MRSA: Synthesis of focussed combinatorial libraries of tri-substituted 2(5H)-furanones. Curr. Drug Discov. Technol. 2006, 3, 125–134. [Google Scholar] [CrossRef]
- Garcia-Pastor, P.; Randazzo, A.; Gomez-Paloma, L.; Alcaraz, M.J.; Paya, M. Effects of Petrosaspongiolide M, a Novel Phospholipase A2 Inhibitor, on Acute and Chronic Inflammation. J. Pharmacol. Exp. Therapeut. 1999, 289, 166–172. [Google Scholar]
- Calderón-Montaño, J.M.; Burgos-Morón, E.; Orta, M.L.; Pastor, N.; Austin, C.A.; Mateos, S.; Lopez-Lazaro, M. Alpha, beta-unsaturated lactones 2-furanone and 2-pyrone induce cellular DNA damage, formation of topoisomerase I- and II-DNA complexes and cancer cell death. Toxicol. Lett. 2013, 222, 64–71. [Google Scholar] [CrossRef]
- Shi, C.; Wang, J.; Chen, H.; Shi, D. Regioselective Synthesis and in Vitro Anticancer Activity of 4-Aza-podophyllotoxin Derivatives Catalyzed by l-Proline. J. Comb. Chem. 2010, 12, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Lattmann, E.; Ayuko, W.O.; Kinchington, D.; Langley, C.A.; Singh, H.; Karimi, L.; Tisdale, M.J. Synthesis and evaluation of 5-arylated 2(5H)-furanones and 2-arylated pyridazin-3(2H)-ones as anti-cancer agents. J. Pharm. Pharmacol. 2003, 55, 1259–1265. [Google Scholar] [CrossRef]
- Lattmann, E.; Kinchington, D.; Dunn, S.; Singh, H.; Ayuko, W.O.; Tisdale, M.J. Cytotoxicity of 3,4-dihalogenated 2(5H)-furanones. J. Pharm. Pharmacol. 2004, 56, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Luo, S.-H.; Mei, W.-J.; Cao, L.; Wu, H.-Q.; Wang, Z.-Y. Synthesis and biological evaluation of 4-biphenylamino-5-halo-2(5H)-furanones as potential anticancer agents. Eur. J. Med. Chem. 2017, 139, 84–94. [Google Scholar] [CrossRef]
- Pour, M.; Spulák, M.; Balsánek, V.; Kunes, J.; Buchta, V.; Waisser, K. 3-Phenyl-5-methyl-2H,5H-furan-2-ones: Tuning antifungal activity by varying substituents on the phenyl ring. Bioorg. Med. Chem. Lett. 2000, 10, 1893–1895. [Google Scholar] [CrossRef]
- Zapf, S.; Anke, T.; Sterner, O. Incrustoporin, a New Antibiotic from Incrustoporia carneola (Bres.) Ryv. (Basidiomycetes). Acta Chem. Scand. 1995, 49, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Hashem, A.I.; Youssef, A.S.A.; Kandeel, K.A.; Abou-Elmagd, W.S.I. Conversion of some 2(3H)-furanones bearing a pyrazolyl group into other heterocyclic systems with a study of their antiviral activity. Eur. J. Med. Chem. 2007, 42, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Eren, G.; Ünlü, S.; Nuñez, M.-T.; Labeaga, L.; Ledo, F.; Entrena, A.; Banoğlu, E.; Costantino, G.; Şahin, M.F. Synthesis, biological evaluation, and docking studies of novel heterocyclic diaryl compounds as selective COX-2 inhibitors. Bioorg. Med. Chem. 2010, 18, 6367–6376. [Google Scholar] [CrossRef]
- Weber, V.; Coudert, P.; Rubat, C.; Duroux, E.; Vallée-Goyet, D.; Gardette, D.; Bria, M.; Albuisson, E.; Leal, F.; Gramain, J.-C.; et al. Novel 4,5-Diaryl-3-hydroxy-2(5H)-furanones as Antioxidants and Anti-Inflammatory Agents. Bioorg. Med. Chem. 2002, 10, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Meli, R.; Bucci, M.; Cirino, G. Dual inhibitors of cyclooxygenase and 5-lipoxygenase. A new avenue in anti-inflammatory therapy? Biochem. Pharmacol. 2001, 62, 1433–1438. [Google Scholar] [CrossRef]
- Uddin, J.; Crews, B.C.; Ghebreselasie, K.; Tantawy, M.N.; Marnett, L. [123I]-Celecoxib Analogues as SPECT Tracers of Cyclooxygenase-2 in Inflammation. J. Med. Chem. Lett. 2011, 5, 1254–1258. [Google Scholar] [CrossRef]
- Bendich, A.; Machlin, L.J.; Scandurra, O.; Burton, W.G.; Wayner, D.D.M. The antioxidant role of vitamin C. Free Radic. Biol. Med. 1986, 2, 419–444. [Google Scholar] [CrossRef]
- Kitel, R.; Byczek-Wyrostek, A.; Hopko, K.; Kasprzycka, A.; Walczak, K. Effect of Selected Silyl Groups on the Anticancer Activity of 3,4-Dibromo-5-Hydroxy-Furan-2(5H)-one Derivatives. Pharmaceuticals 2021, 14, 1079. [Google Scholar] [CrossRef] [PubMed]
- Byczek-Wyrostek, A.; Kitel, R.; Rumak, K.; Skonieczna, M.; Kasprzycka, A.; Walczak, K. Simple 2(5H)-furanone derivatives with selective cytotoxicity towards non-small cell lung cancer cell line A549—Synthesis, structure-activity relationship, and biological evaluation. Eur. J. Med. Chem. 2018, 150, 687–697. [Google Scholar] [CrossRef]
- Biswas, K.; Gholap, R.; Srinivas, P.; Kanyal, S.; Das Sarma, K. β-Substituted γ-butyrolactams from mucochloric acid: Synthesis of (±)-baclofen and other γ-aminobutyric acids and useful building blocks. RSC Adv. 2014, 4, 2538–2545. [Google Scholar] [CrossRef]
- Dornan, M.; Krishnan, R.; Macklin, A.; Selman, M.; El Sayes, N.; Son, H.H.; Davis, C.; Chen, A.; Keillor, K.; Le, P.J.; et al. First-in-class small molecule potentiators of cancer virotherapy. Sci. Rep. 2016, 6, 26786–26796. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, E.; Bassi, G.; Ruffini, A.; Panseri, S.; Montesi, M.; Velasco-Torrijos, T.; Montagner, D. Click Pt(IV)-Carbohydrates Pro-Drugs for Treatment of Osteosarcoma. Front. Chem. 2021, 9, 795997–796009. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Lu, Q.; Xing, D.; Zhang, R. Exploring Carbohydrates for Therapeutics: A Review on Future Directions. Front. Pharmacol. 2021, 12, 756724. [Google Scholar] [CrossRef]
- Ritter, T.K.; Wong, C.-H. Carbohydrate-Based Antibiotics: A New Approach to Tackling the Problem of Resistance. Angew. Chem. Int. Ed. 2001, 40, 3508–3533. [Google Scholar] [CrossRef]
- Scheen, A.J. Clinical efficacy of acarbose in diabetes mellitus: A critical review of controlled trials. Diabetes Metab. 1998, 24, 311–320. [Google Scholar] [PubMed]
- Bokor, É.; Kun, S.; Goyard, D.; Tóth, M.; Praly, J.-P.; Vidal, S.; Somsák, L. C-Glycopyranosyl Arenes and Hetarenes: Synthetic Methods and Bioactivity Focused on Antidiabetic Potential. Chem. Rev. 2017, 117, 1687–1764. [Google Scholar] [CrossRef] [PubMed]
- Magano, J. Synthetic approaches to the neuraminidase inhibitors zanamivir (Relenza) and oseltamivir phosphate (Tamiflu) for the treatment of influenza. Chem. Rev. 2009, 109, 4398–4438. [Google Scholar] [CrossRef]
- Michiels, B.; Van Puyenbroeck, K.; Verhoeven, V.; Vermeire, E.; Coenen, S. The value of neuraminidase inhibitors for the prevention and treatment of seasonal influenza: A systematic review of systematic reviews. PLoS ONE 2013, 8, e60348. [Google Scholar] [CrossRef]
- Pawar, S.A.; Jabgunde, A.M.; Govender, P.; Maguire, G.E.M.; Kruger, H.G.; Parboosing, R.; Soliman, M.E.S.; Sayed, Y.; Dhavale, D.D.; Govender, T. Synthesis and molecular modelling studies of novel carbapeptide analogs for inhibition of HIV-1 protease. Eur. J. Med. Chem. 2012, 53, 13–21. [Google Scholar] [CrossRef]
- Jensen, K.J.; Brask, J. Carbohydrates in peptide and protein design. Pept. Sci. 2005, 80, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Gondela, E.; Walczak, K.Z. Convenient Synthesis of 3,4-Dichloro-5-hydroxy-2(5H)-Furanone Glycoconjugates. Molecules 2011, 16, 1011–1020. [Google Scholar] [CrossRef]
- Mao, L.; Sun, G.; Zhao, J.; Xu, G.; Yuan, M.; Li, Y.-M. Design, synthesis, and antitumor activity of icotinib derivatives. Bioorg. Chem. 2020, 105, 104421–104432. [Google Scholar] [CrossRef] [PubMed]
- Rezki, N.; Almehmadi, M.A.; Ihmaid, S.; Shehata, A.M.; Omar, A.M.; Ahmed, H.E.A.; Aouad, M.R. Novel scaffold hopping of potent benzothiazole and isatin analogues linked to 1,2,3-triazole fragment that mimic quinazoline epidermal growth factor receptor inhibitors: Synthesis, antitumor and mechanistic analyses. Bioorg. Chem. 2020, 103, 104133–104147. [Google Scholar] [CrossRef]
- Ihmaid, S.K.; Alraqa, S.Y.; Aouad, M.R.; Aljuhani, A.; Elbadawy, H.M.; Salama, S.A.; Rezki, N.; Ahmed, H.E.A. Design of molecular hybrids of phthalimide-triazole agents with potent selective MCF-7/HepG2 cytotoxicity: Synthesis, EGFR inhibitory effect, and metabolic stability. Bioorg. Chem. 2021, 111, 104835–104848. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, K.; Li, X.; Lu, G.; Xue, W.; Qian, X.; Mohamed, O.K.; Meng, F. Design, synthesis, and in vitro and in vivo anti-angiogenesis study of a novel vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor based on 1,2,3-triazole scaffold. Eur. J. Med. Chem. 2021, 211, 113083–113098. [Google Scholar] [CrossRef]
- Li, S.; Li, X.; Zhang, T.; Zhu, J.; Xue, W.; Qian, X.; Me, F. Design, synthesis, and biological evaluation of erythrina derivatives bearing a 1,2,3-triazole moiety as PARP-1 inhibitors. Bioorg. Chem. 2020, 96, 103575–103588. [Google Scholar] [CrossRef]
- Tanaka, H.; Yamaguchi, S.; Yoshizawa, A.; Takagi, M.; Shin-ya, K.; Takahashi, T. Combinatorial Synthesis of Deoxyhexasaccharides Related to the Landomycin a Sugar Moiety, Based on an Orthogonal Deprotection Strategy. Chem. Asian J. 2010, 5, 1407–1424. [Google Scholar] [CrossRef]
- Wu, B.; Wei, N.; Thon, V.; Wei, M.; Yu, Z.; Xu, Y.; Chen, X.; Liu, J.; Wang, P.G.; Li, T. Facile chemoenzymatic synthesis of biotinylated heparosan hexasaccharide. Org. Biomol. Chem. 2015, 13, 5098–5101. [Google Scholar] [CrossRef]
- Percec, V.; Leowanawat, P.; Sun, H.-J.; Kulikov, O.; Nusbaum, C.D.; Tran, T.M.; Bertin, A.; Wilson, D.A.; Peterca, M.; Zhang, S.; et al. Modular Synthesis of Amphiphilic Janus Glycodendrimers and Their Self-Assembly into Glycodendrimersomes and Other Complex Architectures with Bioactivity to Biomedically Relevant Lectins. J. Am. Chem. Soc. 2013, 135, 9055–9077. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.V.S.; Rao, C.V.; Chand, P.K.; Prasad, A.R. Iodine-Catalyzed Stereoselective Synthesis of Allylglycosides, Glycosyl Cyanides and Glycosyl azides. Synlett 2001, 2001, 1638–1640. [Google Scholar] [CrossRef]
- Tsutsui, N.; Tanabe, G.; Gotoh, G.; Morita, N.; Nomura, N.; Kita, A.; Sugiura, R.; Muraoka, O. Structure-activity relationship studies on acremomannolipin A, the potent calcium signal modulator with a novel glycolipid structure 2: Role of the alditol side chain stereochemistry. Bioorg. Med. Chem. 2014, 22, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Luo, T.; Zou, D.; Dong, H. Using DMF as Both a Catalyst and Cosolvent for the Regioselective Silylation of Polyols and Diols. Eur. J. Org. Chem. 2019, 2019, 6383–6395. [Google Scholar] [CrossRef]
- Batty, P.; Gerlich, D.W. Mitotic Chromosome Mechanics: How Cells Segregate Their Genome. Trends Cell Biol. 2019, 29, 717–726. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| R | Conditions | Product | Yield [%] | α/β |
| -OCH2CH2CH2N3 | HO(CH2)3N3, FeCl3, DCM, 1 h—r.t. | 1 | 87% | 1:6 |
| -OCH2CH2CH2N3 | HO(CH2)3N3, BF3OEt2, DCM, 2 h—0 °C, 8 h—r.t. | 2 | 82% | 0:100 |
| -N3 | TMSN3, SnCl4, DCM, 24 h—r.t. | 3 | 97% | 0:100 |
| -N3 | 4 | 98% | 0:100 | |
| -N3 | 5 | 97% | 1:8 | |
| -N3 | TMSN3, I2, DCM, 8 h—0 °C-r.t. | 6 | 78% | 1:1.5 |
| Compound | IC50(μM) | |
|---|---|---|
| HCT116 | MCF-7 | |
| 10 | No effect * | 29.9 ± 0.1 |
| 11 | 14.6 ± 3.7 | No effect * |
| 12 | 9.1 ± 2.4 | 10.8 ± 0.7 |
| 13 | 166.4 ± 0.7 | 212.1 ± 1.0 |
| 14 | 39.2 ± 17.8 | 11.9 ± 0.02 |
| 15 | 31.4 ± 6.4 | No effect * |
| 16 | 19.2 ± 3.3 | 19.7 ± 8.8 |
| 17 | No effect * | 258.9 ± 2.2 |
| 18 | 13.0 ± 4.3 | 11.1 ± 1.9 |
| 19 | 31.4 ± 6.4 | No effect * |
| 20 | 70.4 ± 10.8 | No effect * |
| 21 | 14.4 ± 4.3 | 8.9 ± 0.05 |
| 22 | 14.3 ± 1.3 | 35.6 ± 0.03 |
| 23 | No effect * | No effect * |
| 24 | 15.0 ± 2.2 | 12.5 ± 2.7 |
| 25 | 14.0 ± 0.2 | 19.0 ± 0.9 |
| 26 | 9.2 ± 0.6 | 17.0 ± 1.0 |
| MCA a | 8.3 ± 1.1 | 32.3 ± 4.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żurawska, K.; Burdalska, D.; Skonieczna, M.; Byczek-Wyrostek, A.; Dawicka, A.; Kasprzycka, A.; Walczak, K. Glycoconjugates of Mucochloric Acid—Synthesis and Biological Activity. Pharmaceuticals 2023, 16, 525. https://doi.org/10.3390/ph16040525
Żurawska K, Burdalska D, Skonieczna M, Byczek-Wyrostek A, Dawicka A, Kasprzycka A, Walczak K. Glycoconjugates of Mucochloric Acid—Synthesis and Biological Activity. Pharmaceuticals. 2023; 16(4):525. https://doi.org/10.3390/ph16040525
Chicago/Turabian StyleŻurawska, Katarzyna, Daria Burdalska, Magdalena Skonieczna, Anna Byczek-Wyrostek, Anahit Dawicka, Anna Kasprzycka, and Krzysztof Walczak. 2023. "Glycoconjugates of Mucochloric Acid—Synthesis and Biological Activity" Pharmaceuticals 16, no. 4: 525. https://doi.org/10.3390/ph16040525
APA StyleŻurawska, K., Burdalska, D., Skonieczna, M., Byczek-Wyrostek, A., Dawicka, A., Kasprzycka, A., & Walczak, K. (2023). Glycoconjugates of Mucochloric Acid—Synthesis and Biological Activity. Pharmaceuticals, 16(4), 525. https://doi.org/10.3390/ph16040525