Abstract
The epidermal growth factor receptor (EGFR) plays a critical role in the tumorigenesis of various forms of cancer. Targeting the mutant forms of EGFR has been identified as an attractive therapeutic approach and led to the approval of three generations of inhibitors. The quinazoline core has emerged as a favorable scaffold for the development of novel EGFR inhibitors due to increased affinity for the active site of EGFR kinase. Currently, there are five first-generation (gefitinib, erlotinib, lapatinib, vandetanib, and icotinib) and two second-generation (afatinib and dacomitinib) quinazoline-based EGFR inhibitors approved for the treatment of various types of cancers. The aim of this review is to outline the structural modulations favorable for the inhibitory activity toward both common mutant (del19 and L858R) and resistance-conferring mutant (T790M and C797S) EGFR forms, and provide an overview of the newly synthesized quinazoline derivatives as potentially competitive, covalent or allosteric inhibitors of EGFR.
1. Introduction
Cancer is one of the most studied public health problems in the modern world, yet it is still one of the leading causes of death among developed countries [1,2,3]. With multifactorial etiology counting both endogenous [4,5] and exogenous [6] factors as drivers for mutagenesis and carcinogenesis [7,8], tumoral cells often harbor hundreds of genomic alterations [9] that lead to uncontrolled proliferation, differentiation, and expansion [10,11]. “The hallmarks of cancer” has been defined by Hanahan and Weinberg as biological capabilities of the tumoral cell developed during the multistep process of tumor progression [12]. The distinctive features that lead to malignancy development are: sustained proliferative signaling, resistance to apoptosis, avoidance of growth suppressors, enabling replicative immortality, development of blood vessels through neoangiogenesis, invasion of the adjacent tissue and metastasis, reprogramming cellular metabolism, and avoidance of immune destruction [12,13,14].
Cytotoxic therapy has been for a long time the gold standard in the treatment of cancer [15]. The inconsistent therapeutic response and low safety profile led to the discovery of novel strategies to target cancer [16]. The development and widespread clinical trials of highly selective anticancer agents such as therapeutic antibodies [17], tyrosine kinase inhibitors (TKIs) [18], micro-RNA therapy [19], oncolytic viruses [20], or gene-editing therapies [21] have led to an expansion of the arsenal against various types of tumors.
Nitrogen-containing heterocycles are a valuable source of pharmacophores for the development of novel active molecules, and more than 75% of the Food and Drug Administration (FDA)-approved molecules contain in their structures N-heterocyclic moieties [22]. Small molecules bearing nitrogen-containing heterocycles are valuable scaffolds for the development of novel inhibitors of various cellular signaling biomolecules [22,23,24].
EGFR has been one of the most extensively studied tyrosine kinase receptors whose role in oncogenesis has been well established [25,26]. The quinazoline core has been one of the most used heterocycles in the development of novel EGFR TKIs and also as a building block for the development of small molecules with an increased selectivity [27,28] aspect that is further discussed in this review.
The main purpose of this review is to outline the general principles for the development of potent EGFR TKIs, exploring the impact of structural modulations in key positions of the quinazoline core on the anticancer effect. It is intended as a guide for the rational development of future generations of quinazoline-based EGFR TKIs that comprises both already established drug design principles and an overview of the experimental quinazoline series reported from 2017 to the present as EGFR TKIs. By performing a Scopus database search using the keywords “quinazoline AND EGFR AND synthesis” we identified an increased interest in this field in recent years (Figure S1—Supplementary Data) [29]. Due to the vast amount of knowledge in the field of quinazolines, we chose to focus on the main SAR principles, while the other important topics such as synthesis methods will be discussed in our future works. Therefore, we selected 42 experimental quinazoline series to be discussed in the following sections (that met the minimal inclusion criteria: in vitro or in silico evidence of an EGFR inhibition mechanism of action).
2. Epidermal Growth Factor Receptor (EGFR)
2.1. EGFR Implications in Cancer
EGFR (HER1) is a tyrosine kinase transmembrane receptor, a member of the ErbB family along with HER2, HER3, and HER4 [30], that plays a crucial role in cell cycle progression [31], differentiation [32], and cytoskeletal rearrangements [33]. Each member of the erythroblastosis oncogene B (ErbB) family consists of an extracellular ligand-binding domain (ectodomain), a hydrophobic transmembrane domain, and an intracellular fragment with a tyrosine kinase domain that possesses enzymatic activity [34]. Signal transduction is generated by ligand binding in the extracellular domain, followed by dimerization and trans-autophosphorylation of various tyrosine residues in the C-tail of the receiving receptor that act as docking sites for various transductor proteins [35,36].
EGFR is one of the most extensively studied receptors due to its role in oncogenesis [37]. The proto-oncogenic role of EGFR has been established in several types of cancers, such as non-small-cell lung cancer (NSCLC) [38], glioblastoma [39], squamous cell carcinoma of the head and neck [40], breast cancer [41], colorectal cancer [42], and pancreatic cancer [43]. Oncogenic transformation of EGFR in tumor cells can lead to angiogenesis [44], accelerated cell proliferation, inhibition of apoptosis [31], cell motility, and metastasis [45,46]. Hyperactivation of EGFR signaling mediates intracellular antiapoptotic and pro-survival signals through downstream targets such as phosphatidylinositol-3 kinase–Akt (PI3K-AKT), mitogen-activated kinase (ERK), and signal transducer and activator of transcription proteins 3 (STAT3), making the survival of these cells EGFR-dependent [47,48,49].
EGFR signaling alteration is often associated with gene amplification, in-frame deletions, and specific mutations. Mutations often occur in specific locations mainly around the kinase domain, ectodomain, and C-terminal tail [50]. Extensive studies of EGFR signaling in NSCLC have revealed that most mutations occur around the kinase domain in around 30% of adenocarcinoma cases and promote constitutive activation in a ligand-independent manner [37,46]. The development of common mutations (L858R) leads to increased affinity for ATP by up to 20-fold that also impacts the affinity for small molecule inhibitors [35,51]. This makes EGFR an attractive target in the development of novel therapeutic agents as potent anticancer drugs for the treatment of NSCLC [52]. In other types of cancer, such as glioblastoma, ectodomain mutations (EGFRvIII mutant found in 20% of the cases) can display ligand-independent signaling, low constitutive activity, and decreased down-regulation of endocytosis [39,53]. Increased gene amplification has been reported in several cases of colorectal cancer as an independent prognostic marker for anti-EGFR monoclonal antibodies [54].
2.2. EGFR Inhibition in Cancer
The EGFR inhibition of EGFR-dependent cancer cells leads to cell cycle arrest in the G1/S phase before DNA synthesis [55] by up-regulation of p27KIP1 activity [56,57] and up-regulation of pro-apoptotic molecules, such as Bcl-2-interacting mediator of cell death (BIM) that activates the intrinsic mitochondrial apoptotic pathway and cell death [58,59]. The blockage of EGFR signaling has been achieved by various strategies, among which two well-identified emerging categories of drugs have an established role in clinical practice: therapeutic antibodies, acting as competitive antagonists in the ectodomain [60], and TKIs, that disrupt the enzymatic activity of the kinase domain [18]. Other strategies such as immunotoxin conjugates [61], RNA-based therapeutics [62], vaccines [63], and protein degraders (PROTAC derivatives) [64] are still under clinical development. The following sections are mostly focused on the preclinical development and structure–activity relationship studies of EGFR TKIs in the last 6 years.
3. First Generation of EGFR TKIs
In-frame deletions in exon 19 (delE746_A750 or del19) and L858R insertion in exon 21, referred to as “common mutations”, are the oncogenic drivers for the development of more than 90% of the reported NSCLC cases found positive for a mutation of EGFR, reported with an increased incidence in Asian non-smoker females with a late-stage adenocarcinoma malignancy [65,66,67]. First-generation EGFR TKIs were specifically developed to inhibit the activation of common mutations bearing EGFR by replacing the adenosine triphosphate (ATP) in the catalytic site of the kinase in a competitive fashion and concentration-dependent manner [26].
Three first-generation inhibitors bearing a quinazoline core have been approved (Figure 1) for the treatment of NSCLC: gefitinib in 2003 (ZD1839, Iressa®—approved worldwide as first-line monotherapy for adults with locally advanced or metastatic NSCLC with del19 or L858R mutations detected by an FDA-approved test) [68,69,70]; erlotinib in 2004 (OSI-774, Tarceva®—approved for locally advanced or metastatic NSCLC with no improvement after four cycles of platinum-based chemotherapy and first-line therapy for locally advanced, complicated, or metastatic pancreatic cancer, combined with gemcitabine) [71,72,73]; and icotinib (approved in China for advanced or metastatic NSCLC) [74,75]. Phase III clinical trial IRESSA Pan-Asia Study (IPASS) demonstrated an increased period-free survival (PFS) for gefitinib compared to standard carboplatin/paclitaxel first-line therapy in an EGFR-mutation-positive group, higher objective response rate (ORR) (71.2% vs. 47.3%) [65,67], increased health-related quality of life index (HRQoL) [67,76], but no improvement in the overall survival (OR) (18.1 months vs. 18.3 months) [65,67]. Newer studies revealed that first-generation inhibitors are active on the L858R/C797S-harboring mutant following first-line treatment with osimertinib, a third-generation EGFR TKI, due to a non-covalent inhibition mechanism [77]. The main issue of the first-generation inhibitors is the development of secondary resistance-conferring mutations after a median period of 12 months by the replacement of Thr790 gatekeeper residue, which regulates access to the allosteric pocket, with a bulkier methionine [78,79,80]. Primary resistance was identified in patients harboring uncommon mutations (G719S, L861Q, S768I, and exon 20 insertions) [81,82,83,84]. In vitro studies suggest a 50-fold weaker affinity of gefitinib for EGFRG719S compared to EGFRL858R and a 14-fold higher affinity for ATP compared to EGFRwt which makes those mutations less sensitive to first-generation inhibitors [51].
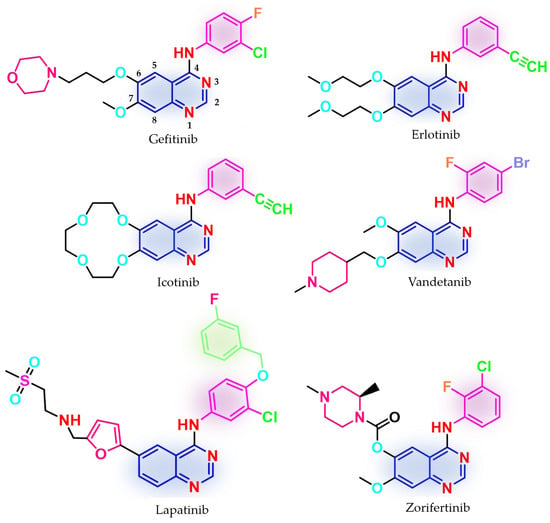
Figure 1.
Chemical structures of first-generation quinazoline-based EGFR inhibitors approved in therapy and under clinical trial: gefitinib, erlotinib, icotinib, lapatinib, vandetanib, and zorifertinib.
Other quinazoline-based TKIs, such as lapatinib (dual inhibitor of EGFR/HER2, approved in 2007 and used in association with capecitabine or letrozole for HER2-overexpressed advanced or metastatic breast cancer) [85,86] and vandetanib (pan-inhibitor of rearranged during transfection (RET), vascular epidermal growth factor receptor (VEGFR2 and VEGFR3), and EGFR, approved in 2011 for the treatment of unresectable, metastatic, or locally advanced medullary thyroid cancer) [87,88], whose therapeutic effects were proven to be associated with the inhibition of other targets [89], have emerged as models for the structural development of novel EGFR TKIs (Figure 1). Clinical trials are still underway for novel quinazoline derivatives such as zorifertinib, a first-generation EGFR TKI with increased lipophilicity and effectiveness in NSCLC with leptomeningeal metastasis (Figure 1) [90].
The molecular architecture of the ATP-binding pocket can be subdivided into specific regions according to the interactions between ATP and the key amino acids: hinge regions (adenine binding pocket) and solvent-accessible regions (sugar and phosphate binding region). Of particular interest for drug discovery are a series of hydrophobic pockets (I and II) and the back pocket (also called allosteric pocket) in the back of the gatekeeper residue that is in the proximity of the first mentioned regions [91,92,93]. Most of the approved quinazoline derivatives (gefitinib and erlotinib) follow a type I inhibition mechanism, binding to the active conformation of the receptor in a competitive manner with both ATP and Asp residue of the DFG-motif (Asp855-Phe656-Gly857) pointing inward to the binding site (αC-in conformation) [94], that narrows the efficiency spectrum of the EGFR mutants inhibited to activator mutations of the kinase domain [95]. Selected inhibitors (such as lapatinib) follow a type ½I inhibitory mechanism, binding mainly to the active conformation (αC-out and DFG-in) with access to the allosteric pocket and improved (but not clinically relevant) affinity for EGFR mutant forms stabilized in the inactive conformation [92,95].
The interactions of gefitinib in the active site of EGFR mutant L858R (Figure 2A,C) revealed that the quinazoline core is oriented in the back of the hinge region N-1 forming a hydrogen bond (H-bond) with the main amine chain of Met793. The 4-aniline moiety extends into the back of the ATP-binding cleft in the hydrophobic pocket forming a 45° angle with the quinazoline ring. The meta-chlorine and para-fluorine substituents increase the affinity through hydrophobic interactions. C-6 and C-7 extend into the solvent-accessible area forming an additional ionic bond through the tertiary amine of the morpholine moiety and Asp800. Erlotinib interacts with the active site of EGFR in a similar fashion, forming an H-bond in the hinge with Met769 at N-1 of the quinazoline ring and an extra H-bond through a water bridge with Gln767, Thr766, and Thr830 [96]. Lapatinib interacts differently with the residues in the active site (Figure 2B): while it maintains the H-bond with Met793, ionic bond with Asp800, and hydrophobic interactions at the aniline moiety, the presence of the (3-fluorophenyl)-methoxy moiety leads to novel interactions with the allosteric pocket and Thr790 (Figure 2D) compared to gefitinib.
The 4-anilino-quinazoline moiety has emerged as a privileged scaffold for developing novel first-generation EGFR TKIs due to a series of structural properties [27,97]. SAR studies of the already approved quinazoline-based EGFR TKI and novel synthesized compounds revealed that the formation of H-bonds between the N-1 quinazoline ring and a series of methionine residues (Met793 and Met769) and between N-3 and threonine residues through water bridges (Thr766 and Thr830) leads to a tighter binding conformation of the inhibitor to the active site of the kinase and also to increased potency [28,96,98].
The further substitution of the quinazoline ring in position 3 led to decreased activity through the steric hindrance of the H-bonds mentioned above. The presence of the aniline moiety [97] or another lipophilic moiety [27,99,100] in position 4 of the quinazoline core is mandatory for the affinity toward the ATP-binding pocket due to the extension in the hydrophobic pocket I and the generation of novel interactions unavailable to the ATP molecule [93,97]. The main focus in the development of novel first-generation EGFR TKIs is the further substitution of the aniline moiety and the replacement of the substituents in positions 6 and 7 [28], which will be discussed next.


Figure 2.
Visualization of gefitinib (A) and lapatinib (B) binding to the human EGFR kinase domain co-crystallized in the complexes 1XKK [101] and 2ITZ [51], from the Protein Data Bank [102] was performed using Chimera 1.10.2 (University of California, Oakland, CA, USA) [103]; regions of the active site and adjacent regions of the kinase domain occupied by gefitinib (C) and lapatinib (D).
Figure 2.
Visualization of gefitinib (A) and lapatinib (B) binding to the human EGFR kinase domain co-crystallized in the complexes 1XKK [101] and 2ITZ [51], from the Protein Data Bank [102] was performed using Chimera 1.10.2 (University of California, Oakland, CA, USA) [103]; regions of the active site and adjacent regions of the kinase domain occupied by gefitinib (C) and lapatinib (D).


4. Novel First-Generation Quinazoline EGFR TKIs (2017–Present)
4.1. 6,7-Dimorpholinoalkoxy-4-anilino-quinazolines
Chen et al. synthesized a series of 6,7-disubstituted 4-anilino-quinazoline derivatives with an (E)-propen-1-yl moiety as potent EGFR inhibitors. The compounds were evaluated for antiproliferative activity on four cell lines (A431, A549, NCI-H1975, and SW480). The newly synthesized compound 1 (Figure 3) showed significantly higher antiproliferative activity on all the cells than the reference drug gefitinib, and higher inhibition against EGFRwt kinase than the reference drug lapatinib (20.72 nM vs. 27.06 nM) (Table S1—Supplementary Data). Insertion of morpholine alkoxy groups in positions 6 and 7 led to increased antiproliferative activity, with a 7.5-fold higher activity for the compounds with three-carbon linkers compared to shorter two-carbon chains. The insertion of (E)-propen-1-yl group in the para-position of the aniline moiety led to a different binding mode of compound 1 in the active site of EGFR and the formation of a supplementary H-bond with Thr854 [104].
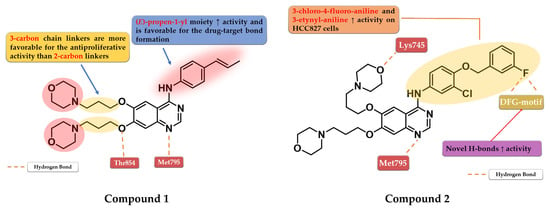
Figure 3.
Structure–activity relationship and molecular docking of compound 1 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported by Chen et al. [104]; structure–activity relationship and molecular docking of compound 2 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported by Zhang et al. [105].
Based on these results, Zhang et al. synthesized a series of novel 6,7-dimorpholinoalkoxy quinazoline derivatives with moderate to high antiproliferative activity. The compounds bearing a three-carbon chain linker between the quinazoline and morpholine cores were found more potent against the A431 cell line than the ones bearing a carbon chain of two atoms and the reference drugs, gefitinib and lapatinib. A longer chain allows the formation of an H-bond between the oxygen of the morpholine and the Lys745 residue of the kinase domain, leading to higher affinity toward the target. The most active compound 2 (Figure 3) is a 3-chloro-4-(3-fluorobenzyloxy) aniline derivative, with higher antiproliferative activity than those of the reference drugs on all the five cell lines and elevated inhibitory activity toward a large panel of mutated kinases (Table S1). The presence of the two morpholine alkoxy substituents in positions 6 and 7 of the quinazoline core determines a shift in the binding mode compared to lapatinib, leading to novel H-bond interactions between the fluorine residue and Asp855/Phe856 of the DFG-motif [105].
4.2. 6-Aryl-semicarbazone-4-anilino-quinazoline Derivatives
A novel series of quinazoline derivatives has been synthesized by Tu et al. by inserting an arylidene-semicarbazone moiety in the C-6 position of the lead compound afatinib. Remarkable results have been obtained with the 4-hydroxy substitution of the phenyl ring, the most active compound 3 (Figure 4) showing higher antiproliferative activity (HepG2—0.07 nM, MCF—0.91 nM, and PC-3—0.91 nM) than the reference drug afatinib (HepG2—1.40 nM, MCF—2.63 nM, and PC-3—2.62 nM) on three out of four cell lines in in vitro studies (Table S1). The replacement of the 4-hydroxy group with hydrophobic substituents (-F, -Cl, and -Br) or the further ortho- and meta-substitution with hydrophobic or hydrophilic (-OH and -OCH3) groups on the 4-hydroxy-phenyl moiety led to decreased activity [106]. The low potency of compound 3 toward the mutant form EGFRL858R/T790M (IC50 > 1000 nM) and its low stability led to further structural optimization focusing on the phenolic moiety. The best results were achieved by the replacement of the benzene ring with nitrogen-containing heterocycles to generate novel H-bonds with the target. Compound 4 (Figure 4) bearing a 2-(pyrrolidin-1-bestpyrimidine side chain had the most promising results, with a similar antiproliferative and EGFR L858R/T790M inhibitory activity (IC50 = 8.4 nM) to that of the reference drug afatinib (IC50 = 3.8 nM) (Table S1) [107].
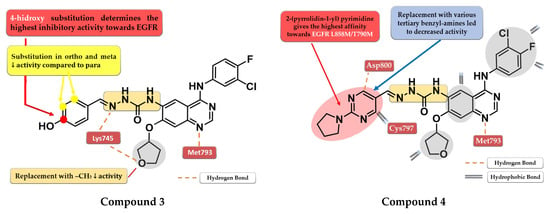
Figure 4.
Structure–activity relationship and molecular docking of compound 3 in the human EGFRT790M kinase domain (PDB ID 4G5J) [108], as reported by Tu et al. [106]; structure–activity relationship and molecular docking of compound 4 in the human EGFRT790M kinase domain (PDB ID 4G5J) [108], as reported by Wang et al. [107].
4.3. Disubstituted-Urea/Thiourea-Linked 4-Amino-quinazoline Derivatives
Zhang et al. synthesized a novel series of 4-anilino-quinazoline derivatives as dual inhibitors of EGFR and VEGFR2 by replacing the small hydrophobic substituents on the aniline with phenyl urea residues. The substitution of the terminal benzene ring in the para and meta positions with small hydrophobic substituents (-Cl and -CH3) led to elevated inhibitory activity toward both EGFR and VEGFR2, while the ortho-substitution led to less potent compounds. Further modification of the C-7 side chain residue concluded that the insertion of a 4-methyl-piperazine-containing residue gives the highest inhibitory activity toward EGFR due to the capability of generating an ionic bond with the carboxyl residues in the target’s structure. The replacement of the piperazine with morpholine or piperidine led to less potent EGFR inhibitors. The presence of the chloro-substituent in the meta position of the aniline residue further increased the inhibitory activity toward both EGFR and VEGFR2. The most active compound 5 (Figure 5) was an 11-fold more potent EGFR inhibitor (IC50 = 1 nM) compared to the reference drug vandetanib (IC50 = 11 nM) and had higher antiproliferative activity on all the tested cells (Table S1) [109].
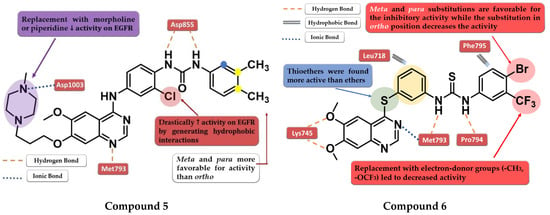
Figure 5.
Structure–activity relationship and molecular docking of compound 5 in the human EGFR kinase domain (PDB ID 2ITY) [51], as reported by Zhang et al. [109]; structure–activity relationship and molecular docking of compound 6 in the human EGFR kinase domain (PDB ID 2ITY) [51], as reported by Sun et al. [110].
A novel series of diaryl-thiourea-linked quinazoline derivatives have been synthesized as dual EGFR/VEGFR2 inhibitors by Sun et al. The presence of two electron-withdrawing groups (EWGs) on the terminal benzene ring led to elevated inhibitory activity toward both EGFR and VEGFR2 and higher antiproliferative activity on all three tested cell lines (HCT-116, MCF-7, and B16). The substitution with electron-donating groups (EDGs) (-CH3 and -OCF3) led to decreased inhibitory activity against EGFR/VEGFR2 kinases and antiproliferative activity. The most active compound 6 (Figure 5) displayed remarkable inhibitory activity against EGFR (IC50 = 10 nM) and VEGFR2 (IC50 = 80 nM) compared to the reference drug sorafenib (20 nM and 80 nM), and better antiproliferative activity on all the tested cell lines (Table S1). In vivo tests revealed a two-fold increase in the inhibition of tumor growth in the xenograft model of B16 melanoma for compound 6 (64.04%) compared to sorafenib (31.25%). Docking studies of compound 6 in the active site of EGFR revealed a tight binding due to the multiple H-bonds formed by the thiourea moiety and the molecular tail, the low-electron-density areas of the molecule being engaged in hydrophobic interactions [110].
Hamad et al. synthesized a series of novel dual inhibitors of EGFR kinase and nuclear factor kappa B (NF-κB) activities. The insertion of various hydrophilic substituents (sulfonamides, hydroxy groups) on the 4-anilino moiety or the total replacement with amino-pyridine led to decreased inhibitory activity for both EGFR and NF-κB. The substitution with small and lipophilic (-Br, -Cl, and -methyl) groups particularly in the meta position increased the inhibitory activity. The thiourea linker is mandatory for the inhibitory activity toward NF-κB. The replacement with a urea linker led to the decreased inhibitory activity of NF-κB but conserved affinity for EGFR kinase. The substitution of the lateral benzene ring at the C-6 position of the quinazoline core with small and lipophilic groups increased inhibition potency for both targets while the insertion of hydrophilic groups led to decreased activity. Total replacement of the lateral benzene ring with substituted thiadiazole or thiazole rings led to increased affinity toward NF-κB and less affinity for EGFR (>100 nM). In vitro data of the antiproliferative activity of prototype compounds “6c” and “6h” (structures not provided) on A549 cells (1.6 µM and 1.0 µM) suggested that the dual inhibitory mechanism might provide an advantage in EGFR-dependent cell lines. SAR studies led to the synthesis of compound 7 (Figure 6) with remarkable inhibitory activity toward NF-κB (IC50 = 0.3 µM) and EGFR kinase in a nanomolar range (IC50 = 60.1 nM). Compound 7 displayed elevated antiproliferative activity on MDA-MB-231 cells (0.9 µM), higher than that of the reference drug gefitinib (14.2 µM) (Table S1), and inhibited the in vivo development of a tumor in a xenograft model with fewer side effects [111].
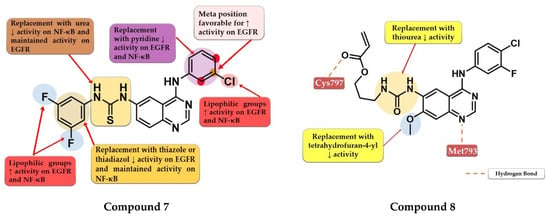
Figure 6.
Structure–activity relationship of compound 7 as reported by Hamad et al. [111]; structure–activity relationship and molecular docking of compound 8 in the human EGFRT790M kinase domain (PDB ID 4G5J) [108], as reported by Gan et al. [112].
Gan et al. synthesized a series of novel 4-arylamino-quinazoline derivatives as potential EGFRT790M/L858R inhibitors. Preliminary studies revealed that urea linkers are more favorable for inhibitory activity compared to thiourea-containing derivatives. The most active compound 8 (Figure 6) displayed comparable inhibitory activity toward EGFRwt (IC50 = 0.8 nM) and EGFRT790M/L858R (IC50 = 2.7 nM) with that of the reference drug afatinib (0.6 nM and 3.5 nM) and broad-spectrum cytotoxic activity (H1975, A549, HeLa, and MCF-7 cells) (Table S1). Compound 8 acts in a concentration-dependent manner similar to first-generation inhibitors by forming only H-bonds with Cys797 through the ester group at the C-6 position and not covalent adducts [112].
4.4. 6-Substituted-amide-4-amino-quinazoline Derivatives
A series of 6-benzamide quinazoline derivatives that reversibly inhibit the EGFR activity were synthesized by Hou et al. The introduction of a fluor-substituent in the C-2 position of the benzene ring is vital for inhibitory activity. Further substitution at the C-5 position of the benzamide moiety with a nitro group (-NO2) led to a two-fold increase in inhibitory activity on EGFRwt kinase. Replacement of the amide linker with a methyl-amino linker between phenyl and quinazoline led to an almost 50-fold decrease in inhibitory activity. Bulkier substituents in position C-7 of the quinazoline core are favorable for inhibitory activity. The replacement of morpholine with piperazine or dimethylamine led to conserved activity. The most promising compound 9 (Figure 7) displayed high antiproliferative activity against various cells harboring a mutation of the EGFR, minimal toxicity on HEK293 cells and EGFRwt bearing cells, high selectivity toward EGFR in kinase inhibition profiling (Table S1), and favorable pharmacokinetics [113].
To overcome the T790M mutation, Zhang et al. synthesized a series of novel cinnamamide-quinazoline derivatives. The highest affinity toward EGFRT790M was achieved through the substitution of the lateral phenyl moiety with 3-methoxy and 4-acetoxy groups by generating a higher selectivity profile and favorable pharmacokinetics. The further substitution of the lateral benzene ring, 3-acetoxy/4-methoxy substitution profile, and the hydrolysis of the ester group led to a decrease or total loss of antiproliferative activity on all three tested cell lines (A431, A549, and H1975). The most active compound 10 (Figure 7) displayed comparable antiproliferative activity on H1975 cells (1.22 µM) with reference drug osimertinib (11.29 µM) but a lower selectivity index (2.72 vs. 4.6) (Table S1) [114].

Figure 7.
Structure–activity relationship of compound 9 as reported by Hou et al. [113]; structure–activity relationship and molecular docking of compound 10 in the human EGFRT790M kinase domain (PDB ID 2JIU) [115], as reported by Zhang et al. [114].
Figure 7.
Structure–activity relationship of compound 9 as reported by Hou et al. [113]; structure–activity relationship and molecular docking of compound 10 in the human EGFRT790M kinase domain (PDB ID 2JIU) [115], as reported by Zhang et al. [114].

Tang et al. developed a novel series of reversible/irreversible pan-HER inhibitors bearing an N-(3-bromo-1H-indol-5-yl)-quinazolin-4-amine scaffold. Preliminary SAR studies revealed that the insertion of 1H-indol-5-amine in position 4 of the quinazoline core led to sub-nanomolar IC50 values. The further substitution of the indole moiety with various substituents concluded that the in positions 1 and 2 led to decreased in vitro activity while the insertion of a small lipophilic bromine in position 3 gave the best results. These data determined the synthesis of the most active compound 11 (Figure 8) as a pan-HER high-strength reversible inhibitor displaying comparable inhibitory activity toward EGFRwt (IC50 = 0.38 nM) and EGFRT90M/L858R (IC50 = 2.2 nM) with the reference drug afatinib (IC50 = 0.67 nM and IC50 = 3.7 nM). A multi-kinase assay revealed that compound 11 is also a potent HER2 and HER4 inhibitor. The compound displayed increased cytotoxic activity on high-expression EGFR cells (A431, H1975, and HCC82) and HER2 (SK-BR-3 and BT-474) similar to the reference drug afatinib (Table S1) [116].
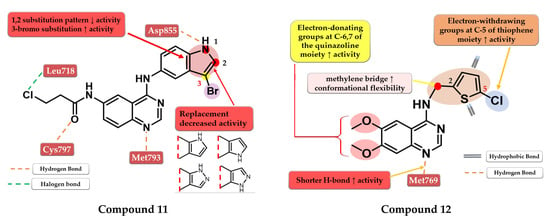
Figure 8.
Structure–activity relationship and molecular docking of compound 11 in the human EGFRT790M kinase domain (PDB ID 4G5J) [108], as reported by Tang et al. [116]; structure–activity relationship and molecular docking of compound 12 in the human EGFRT790M kinase domain (PDB ID 4HJO) [96], as reported by Zou et al. [117].
4.5. 6.7-Dimethoxy-4-amino-quinazoline Derivatives
Zou et al. synthesized a series of novel quinazoline derivatives by inserting a thiophene-2-ylmethanamine at the C-4 position of the quinazoline core to increase the conformational flexibility. The synthesized molecules displayed good antiproliferative activity on A431 cells, the most active compound 12 (Figure 8) having comparable results (IC50 = 3.4 µM) with the reference drug erlotinib (IC50 = 3 µM) (Table S1). The insertion of electron-donating groups at the 6 and 7 positions of the quinazoline core increased the activity of the compounds. The 6,7-dimethoxy substitution was more favorable for the inhibition of EGFR due to the slight deviation of the quinazoline core in the active site of EGFR generated by bulkier groups (2-methoxy-ethyl-oxy) leading to shorter H-bonds with Met769 and the N-1 of the quinazoline core. The further substitution of the C-5 position of the thiophene moiety with small lipophilic substituents (-Cl and -Br) led to a significant increase in the antiproliferative activity [117].
4.6. 6-Heteroaryl-4-amino-quinazoline Derivatives
A novel series of 4-anilino-quinazoline derivatives bearing a 5-substituted furan-2-yl moiety at the C-6 position of the quinazoline core was synthesized by Zhang et al. The most active compound 13 (Figure 9) displayed a two-fold higher antiproliferative activity on A549 cells (7.35 µM) compared to the reference drugs gefitinib (21.17 µM) and lapatinib (14.09 µM), and significantly higher antiproliferative activity on H1975 cells (3.01 µM, 9.08 µM, and 8.05 µM) (EGFRL858R/T790M) (Table S1). The acetyl-derivative of compound 13 displayed similar antiproliferative activity to that of compound 13 on SW480 and A549 cells but was found to be less potent on the other two cell lines. Compound 13 was found to be a highly potent EGFRwt inhibitor in vitro (IC50 = 5.06 nM) with a significantly higher activity compared to lapatinib (IC50 = 27.06 nM) and similar activity to gefitinib (IC50 = 3.22 nM) (Table S1). The docking studies of compound 13 in the active site of EGFR revealed a binding mode similar to that of lapatinib, with the five-fold difference in activity being correlated with shorter and stronger H-bonds generated [118].
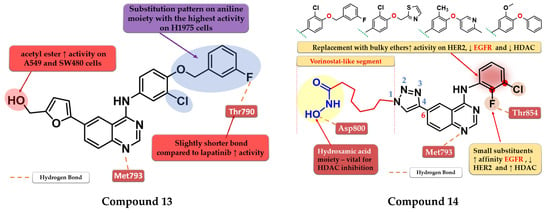
Figure 9.
Structure–activity relationship and molecular docking of compound 13 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported by Zhang et al. [118]; structure–activity relationship and molecular docking of compound 14 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported by Ding et al. [119].
By merging the 4-anilino-quinazoline structural motif and a hydroxamic acid moiety, Ding et al. synthesized a series of novel 6-(1,2,3-triazol-4-yl)-4-amino-quinazoline derivatives with multitarget inhibitory activity, including EGFR, HER2, histone deacetylases 1 (HDAC1), and 6 (HDAC6). Smaller and lipophilic substituents (-F and -Cl) on the aniline moiety led to enhanced affinity toward EGFR, moderate activity toward HER2, and higher potency toward both HDAC1/HDAC6 compared to the reference drug vorinostat. The substitution with various bulky ethers (3-fluorobenzyloxy, thiazol-2-ylmethoxy, 6-methylpyridinyl-3-oxy, and 3-methoxy-4-phenoxy) of the aniline moiety increased activity on HER2, decreased inhibitory activity for EGFR by more than 10-fold and drastically reduced the activity on HDAC1/HDAC6 due to the steric hindrance on the outside surface of the HDAC1. The insertion of the vorinostat-like segment at the C-6 of the quinazoline core led to a higher affinity toward EGFR compared to HER2. The most active compound 14 (Figure 9) displayed higher antiproliferative activity on A549 cells (0.63 µM) and lower antiproliferative activity on BT-474 cells (3.88 µM) compared to the reference drugs lapatinib (1.74 µM and 0.1 µM) and vorinostat (2.57 µM and 2.67 µM) (Table S1). The compound displayed a similar antiproliferative activity pattern on A431 cells (EGFR overexpressed) and SK-BR-3 (HER2 overexpressed) while showing lower activity on H1975 cells compared to lapatinib, which could be correlated with the lower affinity toward HER2 and therefore blocking the heterodimerization of EGFR-HER2 to a lesser extent (Table S1) [119].
4.7. Nitro-Substituted-Azole Linked 4-Amino-quinazoline Derivatives
By inserting the 3-nitro-1,2,4-triazole motif at the C-7 position of the quinazoline core Wei et al. synthesized a series of novel 4-anilino-quinazoline derivatives as dual EGFR/VEGFR2 inhibitors active in hypoxic conditions. A longer chain linker between the quinazoline core and the triazole moiety was found to be favorable for the inhibitory activity toward both EGFR and VEGFR2. Most of the synthesized compounds displayed increased inhibitory activity toward EGFR (IC50 = 0.37 to 12.93 nM). A 2,4-substitution on the aniline moiety with heavy and bulky halogen atoms increased the activity on VEGFR2 while a 3,4-substitution increased activity on EGFR even more. Compound 15 (Figure 10) displayed a 3.5-fold higher inhibitory activity on EGFR (5.9 nM) and similar inhibitory activity on VEGFR2 (36.78 nM) compared to the reference drug vandetanib (19.76 nM and 33.26 nM). It also displayed significantly higher antiproliferative activity on both hypoxic and hypoxic conditions associated with irradiation on the tested cell lines (A549 and H446) compared to vandetanib (Table S1) and remarkable inhibition on A549 tumor growth in the in vivo model [120].
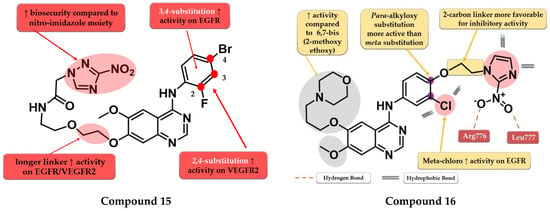
Figure 10.
Structure–activity relationship and molecular docking of compound 15 in the active site of EGFR as reported by Wei et al. [120]; structure–activity relationship and molecular docking of compound 16 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported by Cheng et al. [121].
Cheng et al. synthesized a series of lapatinib analogs by replacing the benzyloxy moiety with nitro-imidazole residues to obtain compounds with increased activity in hypoxic conditions. Compound 16 (Figure 10) displayed higher antiproliferative activity on the tested cells in both normoxia (1.59 µM and 2.46 µM) and hypoxia (1.09 µM and 1.35 µM) conditions compared to the reference drug lapatinib (11.3 µM and 13.26 µM; 6.81 and 8.85 µM) (Table S1). The meta-chlorine substitution on the aniline moiety was beneficial for the activity due to the hydrophobic interactions. The insertion of 2-nitro-imidazole-1H-alkyloxyl in the para position of the aniline moiety led to significantly higher activity on EGFR compared to meta substitution, generating a binding mode in the active site of EGFR similar to lapatinib. Compounds bearing a three-carbon alkyloxy linker were less potent than the ones bearing a two-carbon bridge due to an unfavorable binding mode within the active site of EGFR leading to a more “out” binding position compared to lapatinib [121].
Elkamhawy et al. developed a series of 4-anilino-quinazoline derivatives as dual EGFR/HER2 that incorporate the nitro-imidazole moiety active in hypoxic conditions. SAR studies revealed that the optimal length of the carbon chain linker for the highest dual inhibitory activity is four. The most active compound 17 (Figure 11) was synthesized by inserting the hydrophilic moiety of neratinib (structure not provided) in position 4 of the quinazoline core and displayed an increased inhibitory activity toward EGFR kinase (1.8 nM) compared to the reference drugs (lapatinib–10 nM and staurosporine–88.1 nM) and significantly less affinity toward HER2 kinase (Table S1). Replacement of the (3-chloro-4-(pyridin-2-ylmethoxy)-aniline moiety with quinoline led to conserved EGFR activity while the substitution with benzonitrile led to activity reduction on both kinases [122].
4.8. Fused 2,3-Dihydro-[1,4] Dioxino-[2,3-f] Quinazoline Derivatives
Quin et al. synthesized a series of novel 2,3-dihydro-[1,4]-dioxino-[2,3-f]-quinazoline derivatives as EGFR inhibitors. The cyclization at C-5 and C-6 of the quinazoline core led to smaller and more compact molecules, able to tolerate variations in the structure of the kinase domain due to secondary or tertiary mutations. The substitution of the aniline moiety with small and hydrophobic substituents (decreased activity in the order F < Br < Cl) increased the activity compared to smaller EDGs (25). The most active compound 18 (Figure 11) displayed antiproliferative activity (A549–9.95 µM and NCI-H15-11.66 µM) comparable to the reference drug erlotinib (7.26 µM and 6.88 µM), and inhibitory activity toward EGFRwt kinase (10.29 nM) similar to reference drugs gefitinib and erlotinib (10.41 nM and 11.65 nM) (Table S1) [123].

Figure 11.
Structure–activity relationship and molecular docking of compound 17 in the human EGFR kinase domain (PDB ID 1M17) [124], as reported by Elkamhawy et al. [122]; structure–activity relationship and molecular docking of compound 18 in the human EGFR kinase domain (PDB ID 1M17) [124], as reported by Quin et al. [123].
Figure 11.
Structure–activity relationship and molecular docking of compound 17 in the human EGFR kinase domain (PDB ID 1M17) [124], as reported by Elkamhawy et al. [122]; structure–activity relationship and molecular docking of compound 18 in the human EGFR kinase domain (PDB ID 1M17) [124], as reported by Quin et al. [123].

4.9. (2-Bromo-phenyl)-4-amino-quinazoline Derivatives
Zheng et al. evaluated the addition of various N-Boc amino acid moieties in position 6 of the 4-anilino-quinazoline scaffold on antiproliferative activity. The 3-bromo-aniline compound 19 (Figure 12) bearing an N-Boc glycine residue in position 6 showed moderate antiproliferative activity on HepG2 cells (IC50 = 8.3 µM) and a potent inhibitory activity against EGFR (IC50 = 3.2 nM) with a 2000-fold higher selectivity over various kinases related to liver cancer (Table S1). Molecular docking studies of compound 19 revealed a novel H-bond interaction between the nitrogen atom of the side chain in position 6 and Leu718. The steric hindrance of this bond by replacing N-Boc glycine with various bulkier N-Boc amino acids led to decreased activity [125].
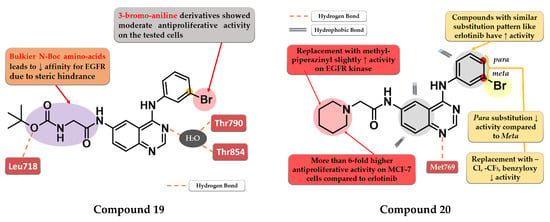
Figure 12.
Structure–activity relationship and molecular docking of compound 19 in the human EGFR kinase domain (PDB ID 2ITY) [51], as reported by Zheng et al. [125]; structure–activity relationship and molecular docking of compound 20 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported by Ismail et al. [126].
Ismail et al. synthesized a series of 4-anilino-quinazoline derivatives by attaching 2-substituted acetamido moieties at the C-6 of the quinazoline core. The most active compound 20 (Figure 12) displayed potent antiproliferative activity on both tested cell lines (HepG2 12 µM and MCF-7 3 µM) compared to the reference drug erlotinib (25 µM and 20 µM) (Table S1). Meta-bromoaniline derivatives displayed higher antiproliferative activity compared to para-substituted derivatives, especially when associated with piperidinyl moiety on the acetamide spacer at the C-6 position. The further replacement of -Br with -Cl, -CF3, or benzyloxy group led to decreased activity. A possible explanation is that the substitution pattern on the aniline moiety could be correlated with the results of the docking studies which indicated that the most active compounds presented a binding mode similar to erlotinib [126].
4.10. 2-Aryl-4-Substituted Quinazoline Derivatives
Ahmed et al. synthesized a novel series of 6-bromo-2-(pyridin-3-yl)-4-substituted quinazolines as potential dual EGFR-HER2 inhibitors. The 4-bromo-phenylethylidene-hydrazinyl compound 21 (Figure 13) displayed the highest inhibitory activity toward EGFRwt (46.1 nM) comparable with that of lapatinib (53.1 nM). The para-substitution of the phenyl moiety with various EWGs (-Cl, -Br, -F, and -SO2CH3) led to higher inhibitory activity compared to electron-donating groups (-CH3 and -OCH3). In vitro testing revealed an increased cytotoxic activity toward MDA-MB-231 cells compared to the reference drug lapatinib (Table S1) [127].
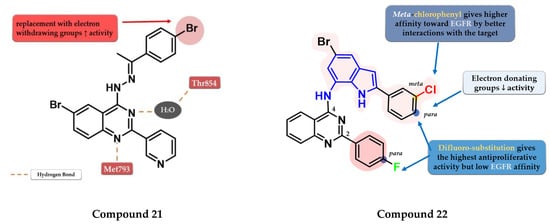
Figure 13.
Structure–activity relationship and molecular docking of compound 21 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported by Ahmed et al. [127]; structure–activity relationship of compound 22, as reported by Mphahlele et al. [128].
Mphahlele et al. synthesized a series of indole-amino-quinazolines with various substituents in positions 2 and 4 of the quinazoline core and evaluated their antiproliferative activity on 5 different cell lines (A549, Caco-2, C3A, MCF-7, and HeLa). The presence of small lipophilic substituents in para (-F) on both side benzene rings in positions 2 and 4 of the quinazoline core led to increased antiproliferative activity on all the tested cells, while the substitution with para-chlorophenyl on the indole moiety and unsubstituted benzene at position 2 led to potent selective toxicity on A549 and Caco-2 cells (structures not provided). The replacement of the halogen hydrophobic substituents with EWGs (-OCH3) led to a major decrease in the activity on all the cells. The highest affinity for EGFR was identified for compound 22 (IC50 = 40.7 nM) (Figure 13) which showed similar EGFR inhibitory activity compared to gefitinib (IC50 = 38.9 nM) and moderate antiproliferative activity (Table S1) [128].
4.11. Sulfonamide Linked Quinazoline Derivatives
A series of dual EGFR-PI3Kα inhibiting 4-amino-quinazoline derivatives were synthesized by Ding et al. by merging the methoxy-pyridil-3-yl-sulfonamide moiety (found in PI3Kα-specific inhibitor omipalisib—structure not provided) and 4-arylamino-quinazoline motif (Figure 1). Methyl-sulfonamide derivatives were more active than propyl or phenyl-substituted sulfonamide derivatives due to the steric hindrance and lack of new H-bond interactions in the sulfonamide group. The replacement of aniline residue at the C-4 position with 3-amino-pyridine led to conserved activity. The most active compound 23 (Figure 14) displayed similar inhibitory activity on the EGFR kinase to that of the reference drug gefitinib (IC50 = 2.4 nM) and less potent affinity toward PI3Kα (IC50 = 317 nM) compared to the reference drug dactolisib (IC50 = 16.4 nM) as well as moderate cytotoxic activity (Table S1) [129].
Zhang et al. synthesized a series of novel EGFR/carbonic anhydrase IX (CAIX) dual inhibitors by merging the sulfamoyl-aryl moiety found in various CAIX inhibitors with the 4-anilino-quinazoline scaffold. The meta substitution of the aniline ring with the trifluoromethyl group gave the highest antiproliferative activity on all cells (A549, A431, and H1975), while the insertion of a second substituent in para or mono-para substitution led to decreased activity. A flexible four-carbon linker between the sulfamoyl-aryl moiety and quinazoline core was found to increase the antiproliferative and inhibitory activities toward both EGFRwt and EGFRT790M kinase. The most active compound 24 (Figure 14) displayed higher antiproliferative activity on EGFRwt positive cell lines (A549 6.54 µM and A431 4.04 µM) than the reference drug gefitinib (15.59 µM and 8.37 µM) and slightly less potent antiproliferative activity on EGFRT790M-positive cells (1.94 µM) compared to osimertinib (0.98 µM) (Table S1). Compound 24 is a highly specific inhibitor of EGFR (IC50 = 9.2 nM) with a comparable affinity to that of the reference drug osimertinib (IC50 = 8.1 nM) and lower inhibitory activity toward EGFRwt (IC50 = 27 nM) compared to gefitinib (IC50 = 17.1 nM). The selectivity of compound 24 toward the mutant form of EGFR could be correlated with the multiple H-bonds generated by the sulfamoyl moiety in the solvent-accessible region of the mutant form of the kinase compared to just one H-bond in the EGFRwt kinase [130].

Figure 14.
Structure–activity relationship and molecular docking of compound 23 in the human EGFR kinase domain (PDB ID 4WKQ) [131], as reported By Ding et al. [129]; structure–activity relationship and molecular docking of compound 24 in the human EGFRT790M kinase domain (PDB ID 2JIU) [115], as reported by Zang et al. [130].
Figure 14.
Structure–activity relationship and molecular docking of compound 23 in the human EGFR kinase domain (PDB ID 4WKQ) [131], as reported By Ding et al. [129]; structure–activity relationship and molecular docking of compound 24 in the human EGFRT790M kinase domain (PDB ID 2JIU) [115], as reported by Zang et al. [130].

4.12. (4-Cyano-phenyl)-4-amino-quinazoline Derivatives
A series of 4-anilino-quinazoline derivatives were synthesized by Chang et al. by modifying the substituent in the C-7 position of the quinazoline core. Highly rigid substituents (methylbenzyl-amino, hydroxyethyl-benzylamino, and 2-hydroxyethyl-piperidinyl) and amino alcohol groups (4-hydroxypiperidinyl and 4-hydroxyethylpiperidinyl) linked in the C-7 position of the quinazoline core led to weak antiproliferative activity. Remarkable results were obtained with the 4-piperidinyl-piperidinyl, 4-phenylpiperidinyl, 4-ethyl-piperazinyl, and pyrrolyl residues. The most active compound 25 (Figure 15) has a broad anticancer effect, higher antiproliferative activity, and inhibitory activity toward EGFR similar to the reference drug gefitinib (3.62 nM) (Table S1). The binding mode of compound 25 into the kinase domain of EGFR showed multiple interactions of the C-7 side chain, including hydrophobic and π–π interactions with Phe771 and Tyr777 residues [132].
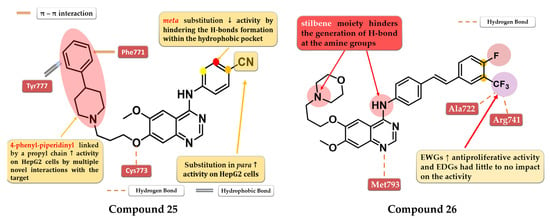
Figure 15.
Structure–activity relationship and molecular docking of compound 25 in the human EGFR kinase domain (PDB ID 1M17) [124], as reported by Chang et al. [132]; structure–activity relationship and molecular docking of compound 26 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported by Wang et al. [133].
4.13. 6,7-Dialkoxy-4-stilbenyl-amino-quinazolines
A series of novel 4-stilbenyl-amino-quinazolines derivatives were synthesized by Wang et al. The substitution of the classical halogen-substituted anilines with (E)-4-styrylanilines at position 4 of the quinazoline core can lead to enhanced stability and lipophilicity while the further substitution of the stilbene moiety with EWGs (-F, -Br, and -CF3) augmented the antiproliferative activity. All the tested compounds showed high antiproliferative activity (IC50 = 1.23–5.17 μM) compared to the reference drug gefitinib on all the tested cells (A431 12.93 µM, A549 13.75 µM, and BGC-823 > 10 µM). The most active was the trifluoromethyl styryl compound 26 (Figure 15, Table S1). The presence of -CF3 in position 3 or 4 of the side benzene ring enhanced the antiproliferative activity by generating novel acceptor H-bonds with arginine and glycine in the hydrophobic pocket of the active site of EGFR [133].
4.14. Macrocyclic Quinazoline Derivatives
The low electron density of the morpholine core in the structure of gefitinib (Figure 1) led Ju et al. to synthesize a novel series of quinazoline derivatives with a macrocyclic polyamine side chain at the C-6 position of the quinazoline core. The replacement of the morpholine moiety with 1,4,7,10-tetraazacyclododecane (cyclen) or 1,4,7-triazonane (tacn) led to a two-fold increase in the antiproliferative activity on A431 cells compared to the reference drug gefitinib, while the cyclen derivatives were found the most potent on A549 cells. Further structural optimizations led to the synthesis of compound 27 (Figure 16) by replacing the halogenated aniline moiety at C-4 with 3-chloro-4-((3-fluorobenzyl) oxy)-aniline. Compound 27 was found to effectively deplete intracellular ATP at 8 μM. It displayed a significant increase in the antiproliferative activity on both cell lines (A431 0.95 µM and A549 3.4 µM) compared to gefitinib (2.47 µM and 11.08 µM) and lapatinib (2.66 µM and 3.64 µM). Compound 27 is a potent inhibitor of both EGFR (1.4 nM) and HER2 (2.1 nM) with a nine-fold higher affinity for EGFRT790M (16.5 nM) compared to gefitinib (148.7 nM) (Table S1) [134].
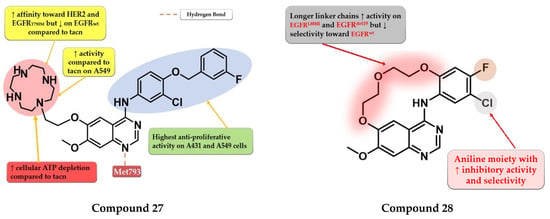
Figure 16.
Structure–activity relationship and molecular docking of compound 27 in the human EGFR kinase domain (PDB ID 1XKK) [101], as reported By Ju et al. [134]; structure–activity relationship compound 28, as reported by Amerheim et al. [135].
Amrhein et al. synthesized a series of novel macrocyclic quinazoline derivatives as potential EGFR inhibitors with increased specificity toward the common mutant forms of EGFR. Preliminary SAR studies revealed that the length of the macrocycle had an impact on the kinase inhibition profile: shorter linker chains led to increased EGFRdel19 activity (IC50 = 20–236 nM), lower activity on EGFRL858R (IC50 = 128–821 nM), and undetectable activity on EGFRwt, while longer linker chains led to an even greater increase in the inhibitory activity (EGFRdel19 and EGFRL858R) but with less selectivity toward EGFRwt. The most promising compound 28 (Figure 16) displayed an increased selectivity toward EGFRdel19 kinase (IC50 = 119.1 nM) with an undetectable affinity for EGFRwt (IC50 ≥ 10 µM) and an excellent selectivity profile kinome-wide. Cytotoxicity assays revealed that compound 28 had no activity on Ba/F3-EGFRwt (IC50 ≥ 10 µM) and elevated activity toward Ba/F3-EGFRL858R (IC50 = 385.6 nM), Ba/F3-EGFRL858R/C797 (IC50 = 749.6 nM), Ba/F3-EGFR del19 (IC50 = 147.9 nM), and Ba/F3-EGFR del19/C797S (IC50 = 197.5 nM) (Table S1) [135].
4.15. Acetyl Glucose-Modified 4-Anilino-quinazoline Derivatives
To enhance the radio-sensitizing effect while maintaining the inhibitory activity toward EGFR, Yamahana et al. synthesized a novel series of acetyl glucose-modified gefitinib derivatives. The compounds displayed antiproliferative activity (IC50 = 34.3–39.4 nM) similar to gefitinib (IC50 = 31.2 nM) and a significantly higher radio-sensitizing effect. A longer alkyloxy linker (2–4 carbons) between the quinazoline core and the acetyl glucose moiety allows the break of the O-glycosidic bond and the generation of an alkyl-7-ol derivative of gefitinib metabolite (which keeps the inhibitory activity toward EGFR). The most active compound 29 (Figure 17) displayed increased EGFR inhibitory activity and cytotoxicity (Table S1) [136].
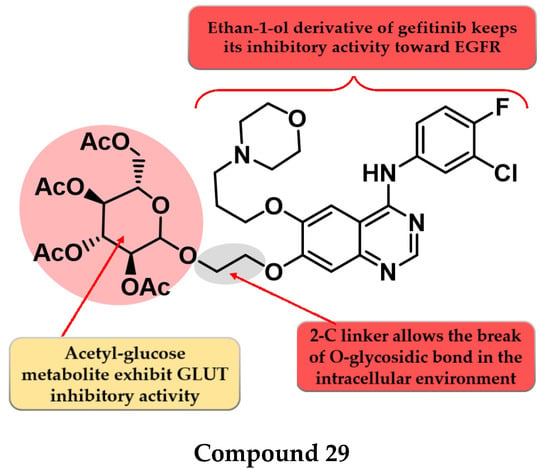
Figure 17.
Structure–activity relationship of compound 29 as reported by Yamahana et al. [136].
5. Second Generation of EGFR TKIs
The second-generation EGFR TKIs are defined as covalent inhibitors capable of binding to the active site of the EGFR tyrosine kinase and forming an irreversible bond with specific amino acids through an electrophilic warhead called “Michael acceptor” [137,138]. Their development was possible after the discovery through bioinformatics analysis of the Cys797 residue of the EGFR kinase domain in the solvent-accessible region that became the key nucleophilic target for the next generation of covalent inhibitors [138]. Targeting cysteine residues for the development of covalent inhibitors has been extensively studied due to the low prevalence in the proteome and overall better selectivity index [139]. The theoretical advantages of the second-generation inhibitors over the first-generation inhibitors include an increased affinity for the mutant target forms, a more complete blockage of EGFR signaling by inhibiting the heterodimerization partners such as HER2/HER4, and long-term binding that could lead to delay or suppression of the secondary gatekeeper mutation T790M development [137,140].
Covalent binding of the protein leads to enzymatic inactivation maintaining the activity depending on the re-synthesis of the protein [140]. The covalent bond formation follows a two-step interaction with the biological target as depicted in Figure 18.
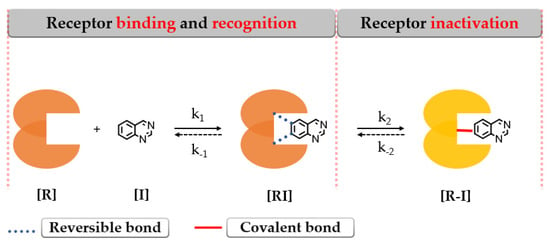
Figure 18.
The 2-step formation of a covalent bond between the target [R] and inhibitor [I] [141].
The first step addresses the formation of an intermediary complex [RI] (described by k1) like in the case of the first-generation inhibitors by binding to the active site of EGFR and forming weak reversible interactions in a manner that the electrophilic warhead is brought into the proximity of the targeted cysteine residue. The second step is represented by the spontaneous formation of a covalent bond between the electrophilic warhead and the nucleophilic moiety of the inhibitor [R–I] (described by k2) (Figure 18) [141,142,143].
Several attempts have been made to develop second-generation EGFR TKIs (Figure 19) [137]. Two covalent 4-anilino-quinazoline inhibitors have been approved for clinical use in the treatment of NSCLC: afatinib in 2013 (BIBW2992, Gilotrif®—approved worldwide as first-line monotherapy for adults with locally advanced or metastatic NSCLC with del19 or L858R mutations detected by an FDA-approved test) [144] and dacomitinib in 2018 (PF-002998044, Vizimpro®—the same therapeutic indications described for afatinib) [145]. Compared to the first-generation inhibitors, clinical data suggest the utility of second-generation inhibitors such as afatinib as first-line treatment for patients harboring uncommon EGFR mutations (G719S, S768I, and L861Q) [146,147]. Other uncommon mutations, such as exon 20 insertions, can be targeted with second-generation inhibitors. Dacomitinib was found particularly active in glycine insertion at position 770 [148] while a broad-spectrum inhibition of various exon 20 insertions was achieved by the combination of afatinib and cetuximab [149].
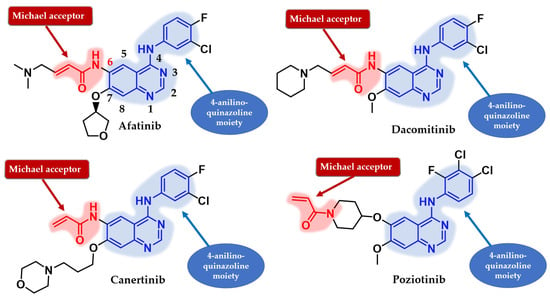
Figure 19.
Chemical structure of second-generation EGFR inhibitors quinazoline derivatives approved in therapy or under clinical trial: afatinib, dacomitinib, canertinib, and poziotinib.
The preliminary results of the LUX-Lung 7 clinical trial revealed an increased PFS (47% vs. 41%), higher ORR (70% vs. 54%), and time to treatment failure (TTF) in the afatinib treatment group compared to gefitinib for patients with adenocarcinoma and activating EGFR mutations with minimal OS differences (27.9 vs. 25 months) [150,151]. Better overall clinical usage of second-generation inhibitors compared to first-generation inhibitors is related to their increased potency [151,152] and pan-inhibitory effect over the entire ErbB family of kinases, the overexpression of receptors such as HER2 being associated with tumor resistance in NSCLC patients treated with first-generation inhibitors [153,154]. Even if second-generation inhibitors display a broad-spectrum inhibitory activity toward a large panel of ErbB family kinases, including EGFRT970M mutants [155], the clinical utility for NSCLC patients with developed resistance after treatment with first-generation inhibitors is limited due to increased inhibitory activity toward EGFRwt and significant gastrointestinal and dermatological side effects [156,157].
LUX-Lung 3 and LUX-Lung 6 evaluated the efficiency of the first-line treatment afatinib compared to pemetrexed + cisplatin and gemcitabine + cisplatin. The results indicated a significantly higher ORR in the afatinib group (56% vs. 23% and 67% vs. 23%) and median duration of response (11.1 vs. 5 months and 9.7 vs. 4.3 months), but no improvement for OS compared to chemotherapy in the EGFR-mutation-positive lung adenocarcinoma group [158,159,160].
The efficiency of a covalent inhibitor is quantitatively evaluated by the second order of rate constant that describes the protein covalent modification k2/k1 [161]. The ideal covalent inhibitor is characterized by increased k1 (nM range), which increases the residence time of the inhibitor near the nucleophilic moiety, and low to moderate reactivity and value of k2 (Figure 18) [141,162]. The increased intrinsic affinity of the inhibitor for the target is achieved for EGFR TKIs by following the general principles of drug design for the first-generation EGFR TKIs [28,91]. Most second-generation inhibitors have a 4-anilino-quinazoline base structure for increased affinity (Figure 19) and form similar bonds within the hinge region and the hydrophobic pockets of the active site [108,163,164]. The binding mode of afatinib in the active site of EGFRT790M is presented in Figure 20.
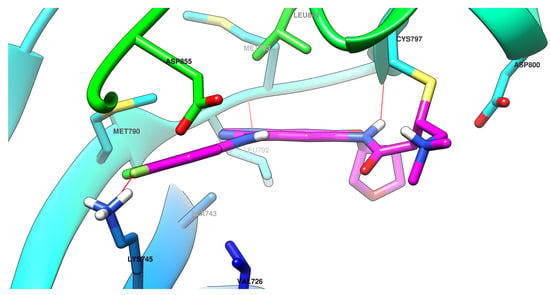
Figure 20.
Visualization of afatinib binding to the human EGFRT790M kinase domain co-crystallized in the complexes 4G5P [108] from Protein Data Bank [102] performed using Chimera 1.10.2 (University of California, USA) [103].
The position and the nature of the electrophilic moiety in the structure of the covalent inhibitor are detrimental to the development of novel clinical candidates [165,166]. Insertion of the electrophilic moiety in the C-6 position of the quinazoline core leads to proximity to the Cys797 residue in the temporary complex [R–I] and is favorable for the formation of the covalent bond [RI] (Figure 18) [141,166,167]. The reactivity of the electrophilic warhead is often a predictor for the off-target effect and side effects [168]. The acrylamide warhead has become extensively used in covalent inhibitor development due to its electrophilic “softness” that has electron conjugation by resonance over the carbonyl and alkenyl moieties [162,169,170,171]. Further modulation of the electronic density and reactivity can be achieved by inserting various substituents at the β-end of the alkenyl moiety that affects the distribution of the electronic cloud [142,171]. The most common modulation is the attachment of an alkaline group to the acrylamide moiety (afatinib and dacomitinib—Figure 19) that elevates the biological activity through intramolecular catalysis of the covalent bond formation [172].
6. Novel Second-Generation Quinazoline EGFR TKIs (2017–Present)
6.1. α-Chlorofluoro Acetamide Derivatives
Shindo et al. synthesized a series of novel quinazoline derivatives bearing an α-chloro-fluor-acetamide covalent binding group, that displayed significantly less reactivity and higher selectivity toward EGFR and mutant forms of EGFR. The presence of a D-proline linker is vital for inhibitory activity by generating the optimal distance between the electrophilic moiety and the reactive thiol group of Cys797. Preliminary SAR studies led to the synthesis of compound 30 (Figure 21) which displayed antiproliferative activity on H1975 cells similar to afatinib (0.19 µM), and less potency toward A431 (7.3 vs. 1.79 µM) and two EGFR-independent cells (HEK293 9.06 vs. 1.78 µM and SW620 10.4 vs. 2.98 µM) (Table S1). The compound displayed a four-fold higher selectivity and lower toxicity with similar excellent potency in growth inhibition in a mouse xenograft model (H1975 cells) compared to the reference drug afatinib. X-ray crystallography data revealed that compound 30 effectively inhibits EGFRL858R/T790M double mutant kinase activity irreversibly by binding the Cys797 residue of the active site with a higher selectivity compared to afatinib due to the shorter length between the electrophilic warhead and the reactive thiol group (3.83 Å vs. 4.29 Å) (Figure 21) [173].
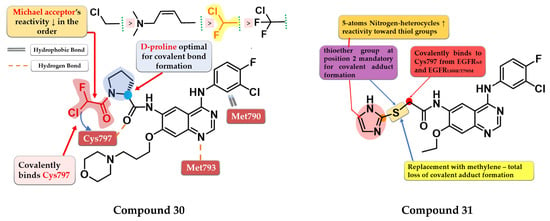
Figure 21.
Structure–activity relationship and molecular docking of compound 30 covalently bonded in the human EGFRL858R/T790M kinase domain (PDB ID 5Y25), as reported by Shindo et al. [173]; structure–activity relationship of compound 31, as reported by Castelli et al. [174].
6.2. 6-Heteroaryl-thioacetamide-4-anilino-quinazoline Derivatives
Castelli et al. synthesized a novel series of heteroaryl-thioacetamide derivatives as potent covalent inhibitors of EGFR. Preliminary studies revealed that the thioacetamide derivatives displayed a significantly lower reactivity (t1/2 > 1440 min) compared to chloroacetamide and acrylamide homologs and a higher selectivity toward the EGFR kinase domain. SAR studies of the synthesized compounds concluded that a pentacyclic nitrogen-bearing heterocycle (imidazole and tetrazole) with the thioether group in position 2 increases the reactivity and the chance of forming the covalent adduct. The covalent adduct formation of the most active compound 31 (Figure 21) was confirmed by both QM/MM simulations and EGFR inhibitory activity on A549 cells after 8 h of washout. Compound 31 displayed a sub-nanomolecular inhibition potency toward EGFRwt kinase (0.62 nM) comparable to the reference drug gefitinib (0.47 nM) and with less potency toward EGFRL858R/T790M after 5 h incubation (≈190 nM) compared to chloroacetamide homolog “8” (structure not provided) in the same conditions (≈4 nM). Compound 31 displayed significant antiproliferative activity on H1975 cells (1.4 µM) compared to the reference drug gefitinib (9.1 µM) (Table S1) [174].
6.3. 6-Acrylamide-4-amino-quinazoline Derivatives
OuYang et al. synthesized a series of novel quinazoline derivatives as potential inhibitors of EGFR kinase. The replacement of the aniline moiety with novel bicyclic amines led to conserved inhibitory activity toward EGFR. Compound 32 (Figure 22) displayed the highest antiproliferative activity on all the tested cells (A549 1.09 µM, MCF-7 1.34 µM, and PC-3 1.23 µM) comparable with that of the reference drug afatinib (0.71 µM, 0.93 µM, and 2.51 µM) (Table S1). The insertion of a tertiary amine at the C-6 position of the quinazoline side chain led to increased activity compared to unsubstituted enamide derivatives due to favorable pharmacokinetics. Compound 32 was identified as a potent inhibitor of EGFRwt kinase (IC50 = 5 nM) comparable to the reference drug afatinib (IC50 = 5 nM) while exhibiting moderate inhibitory activity toward EGFRT790M kinase (IC50 = 26 nM) [175].
Pawara et al. synthesized a series of novel N-(4-substituted-quinazolin-6-yl)-acrylamide derivatives as irreversible EGFR inhibitors by inserting various amino groups containing heterocyclic and homocyclic moieties at the C-4 position of the quinazoline core. All the compounds displayed a significantly higher antiproliferative activity on NCI-H1975 (0.171–0.250 μM) compared to gefitinib (11.71 μM) and similar activity to the reference drug WZ4002 (0.202 μM). Compound 33 (Figure 22) displayed the highest antiproliferative activity on NCI-H1975 cells and a 3.25-fold higher inhibitory activity toward EGFRL858R/T790M compared to EGFRwt, with a selectivity pattern like that of WZ4002 (3.15 vs. 2.87) (Table S1). The high selectivity of compound 33 could be correlated with the flexible hydrazine moiety which allows the generation of novel interactions of the pyridine moiety with the DFG motif of the EGFRL858R/T790M [176].

Figure 22.
Structure–activity relationship and molecular docking of compound 32 covalently bonded in the active site of EGFRT790M, as reported by OuYang et al. [175]; structure–activity relationship and molecular docking of compound 33 covalently bonded in the human EGFRL858R/T790M kinase domain (PDB ID 3IKA) [177], as reported by Pawara et al. [176].
Figure 22.
Structure–activity relationship and molecular docking of compound 32 covalently bonded in the active site of EGFRT790M, as reported by OuYang et al. [175]; structure–activity relationship and molecular docking of compound 33 covalently bonded in the human EGFRL858R/T790M kinase domain (PDB ID 3IKA) [177], as reported by Pawara et al. [176].

Zhao et al. synthesized a series of novel quinazoline derivatives as multitarget ErbB/HDAC inhibitors. Smaller lipophilic substituents on the aniline moiety increased the affinity toward EGFRT790M/L858R compared to bulky ethers. The insertion of a propyl linker between the triazole core and hydroxamic acid moiety led to increased inhibitory activity on EGFRT790M/L858R and HDAC. The most active compound 34 (Figure 23) displayed a higher affinity toward EGFRT790M/L858R (IC50 = 1.5 nM) compared to the reference drug afatinib (IC50 = 3 nM) and similar affinity for EGFRwt and EGFRL858R (0.35 nM and 1.1 nM). Compound 34 displayed a high selectivity for NCI-H1975 cells with minimal antiproliferative activity on A549, NCI-H838, SK-BR-3, and A431 cells (Table S1). The increased antiproliferative activity on EGFRT790M-bearing cells is correlated with the dual mechanism of action of both direct inhibition of EGFRT790M-phosphorylation and inhibition of EGFRT790M and Akt intracellular synthesis (hyperacetylation of HSP90 and reduction in its chaperone activity by inhibition of HDACs) [178].
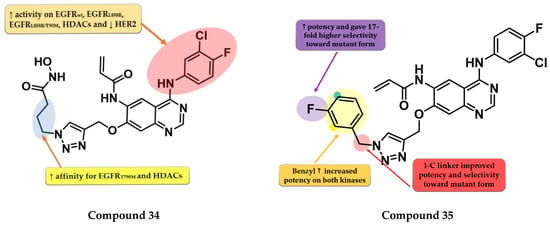
Figure 23.
Structure–activity relationship of compound 34, as reported by Zhao et al. [178]; structure–activity relationship of compound 35, as reported by Song et al. [179].
Song et al. synthesized a series of novel quinazoline-triazole hybrids with a higher affinity toward the mutant form EGFRT790M/L858R. The substitution in position 1 of the triazole ring with small and hydrophilic substituents (4-hydroxypropyl and morpholino-propyl) showed increased activity but a lower specificity ratio toward EGFRT790M/L858R. The replacement of those groups with a benzene ring linked by a one-carbon spacer led to the highest inhibitory activity on all the kinases and the best selectivity ratio. The substitution of the benzyl moiety with small hydrophobic substituents (-F) led to the highest selectivity toward the mutant form. Further modifications led to the conclusion that the meta-fluor-substituted compound 35 (Figure 23) had the highest selectivity ratio out of all the synthesized compounds, with a 17-fold higher affinity for EGFRT790M/L858R than EGFRwt. The triazole–quinazoline hybrids displayed lower antiproliferative activity (0.5 µM) than afatinib (0.131 µM) on H1975, but significantly higher compared to gefitinib (>10 µM) (Table S1) [179].
6.4. Dihydro-6H-[1,4]Oxazino [3,2-g]Quinazoline Acrylamide Derivatives
To overcome the developed tumor resistance for the first- and second-generation EGFR TKIs, Sun et al. synthesized a novel series of tricyclic oxazine fused quinazoline derivatives as dual HER2 and EGFR inhibitors with an irreversible binding mode. The oxazine derivatives were found to be more potent inhibitors of both kinases compared to oxazepine derivatives due to a lack of cell permeability. Dimethyl-amino substitution of the acrylamide moiety led to a flexible side chain and potent dual EGFR/HER2 inhibitors, but low stability due to rapid degradation through hydrolysis and therefore low antiproliferative activity in vivo. Highly potent dual inhibitors were obtained by replacing the dimethyl-amino moiety with bulkier heterocycle amines that led to steric hindrance and more stable compounds. The synthesized compounds displayed higher antiproliferative activity on NCI-H1975 cells harboring EGFRL858R/T790M (IC50 = 1.22–4.35 μM) compared to the reference drugs lapatinib, gefitinib, and erlotinib (7.37 µM, >10 µM, and 5.51 µM) (Table S1). The most active compound 36 (Figure 24) displayed remarkable antiproliferative activity on cells harboring both overexpression of EGFRwt or EGFRL858R/T790M and HER2 compared to the reference drug lapatinib and a three-fold more potent inhibition of EGFR in the enzyme assay (7 nM vs. 22 nM) [180].
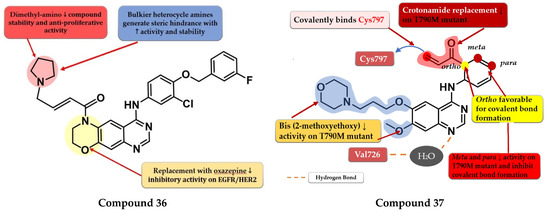
Figure 24.
Structure–activity relationship of compound 36, as reported by Sun et al. [180]; structure–activity relationship and molecular docking of compound 37 covalently bonded in the human EGFRT790M kinase domain (PDB ID 4P5G), as reported by Liu et al. [181].
6.5. N-(3-(Quinazolin-4-yl-amino)phenyl)acrylamide Derivatives
Liu et al. synthesized a series of novel quinazoline derivatives as potent inhibitors of EGFRL858R/T90M by incorporating the acrylamide moiety in various positions of the aniline ring. Ortho-substitution of the aniline moiety with acrylamide led to the synthesis of the most promising compound 37 (Figure 24). The meta or para insertion of the acrylamide moiety or its replacement with a crotonamide group led to decreased activity and incapability to generate covalent bonds with Cys797. Compound 37 displayed significant inhibitory activity toward EGFRL858R/T90M (IC50 = 4.3 nM), low potency toward EGFRwt (IC50 = 104 nM), and a comparable selectivity index with rociletinib (24.4 vs. 25). Molecular docking of compound 37 revealed a shift in the binding position in the active site of EGFRL858R/T790M with the C-6 substituent occupying the hydrophobic pocket I, C-7 substituent trapped in the purine binding area, and the acrylamide-substituted aniline moiety extended in the hydrophilic area capable of generating a covalent bond with Cys797. Compound 37 displayed moderate antiproliferative activity on H1975, A431, and HCC827 (8.7 µM, 13.45 µM, and 0.65 µM) cells and it was less active than the reference drugs gefitinib and rociletinib (0.137 µM, 1.29 µM, and 0.031 µM) (Table S1) [181].
6.6. 2,5-Diazabicyclo [2.2.1]Heptane Linked 4-Anilino-quinazoline Acrylates
Jiao et al. synthesized a series of novel 4-anilino-quinazoline as dual covalent inhibitors of EGFR/HER2. The further replacement of the 3-chloro-4-fluoroaniline moiety with various aryl-amine derivatives (polyhalogenated anilines and aromatic heteroaryl amines) failed to improve the inhibitory activity toward EGFR and provided low affinity toward HER2. An increase in the inhibitory activity of HER2, while maintaining the high affinity toward EGFR, was obtained by totally removing the piperazine moiety (which led to low antiproliferative activity due to the low stability of the ester) and the limitation of the conformation of the piperazine by replacing it with 2,5-diazabicyclo-[2.2.1]heptane. The replacement of the Michael acceptor with a 2-fluoro-acrylamide or methyl-acrylamide moieties led to decreased activity on HER2 and conserved activity on EGFR. Further substitution with trifluoromethyl or cyan of the acrylamide moiety led to increased HER2 inhibitory activity but lower antiproliferative activity on all the tested cells due to the low half-life of the covalently bonded warhead. The most promising compound 38 (Figure 25) displayed similar inhibitory activity toward EGFR (IC50 = 0.3 nM) and two-fold lower activity on HER2 (IC50 = 6.07 nM) compared to the reference drug afatinib (0.27 nM and 3.88 nM) as well as promising antiproliferative activity (Table S1) [182].
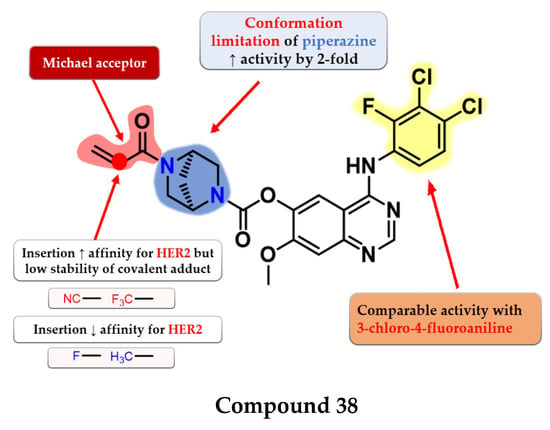
Figure 25.
Structure–activity relationship of compound 38, as reported by Jiao et al. [182].
7. Third Generation of EGFR TKIs
Gatekeeper mutation T790M leads to increased affinity for ATP (lower Michaelis Menten constant—Km) that mediates the resistance to quinazoline derivatives acting as first- and second-generation inhibitors of EGFR [115]. Superior affinity for the T790M form has been achieved by replacing the quinazoline nucleus with a more flexible core (pyrimidine) which led to the development of the third generation of EGFR TKIs [183,184,185,186]. Third-generation EGFR inhibitors are defined as covalent inhibitors with an increased binding affinity for EGFRT790M, significantly less affinity for EGFRwt, and maintained sub-nanomolecular IC50 for common activating mutations (L858R and del19) [187,188,189].
Osimertinib (Figure 26) (AZD9291, Tagrisso®) is the first third-generation EGFR TKI approved as first-line therapy for EGFR-positive activating mutations (L858R and del19), NSCLC, and metastatic EGFRT790M-positive NSCLC patients detected by an approved FDA test [190]. Its efficiency in patients with a T790M-acquired mutation was established in the FLAURA 3 clinical trial which compared osimertinib with platinum–pemetrexed therapy. Clinical data revealed an increased PFS for osimertinib (10.1 vs. 4.4 months) among all groups (including patients with CNS metastasis) and higher ORR (71% vs. 31%) [191]. Mobocertinib (TAK-788, Exkivity®) (Figure 26) is the latest third-generation EGFR inhibitor approved for the treatment of locally advanced or metastatic NSCLC with exon 20 insertion, following an accelerated approval by the FDA in 2021 [192,193,194,195].
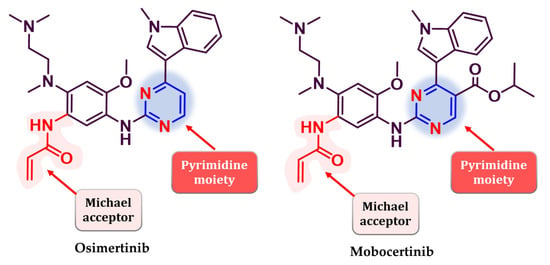
Figure 26.
Chemical structure of third-generation EGFR approved in therapy: osimertinib andmobocertinib.
Resistance to third-generation inhibitors occurs after 9–13 months of treatment and is mediated by the development of secondary mutations (L858R/C797S) when used as first-line treatment for EGFR-positive NSCLC with common mutations, and tertiary mutations (L858R/T790M/C797S) when used in patients that harbor the T790M mutant after treatment with first and second generation inhibitors by replacing the cysteine covalently bound by the Michael acceptor with a less reactive serine residue [77,196,197,198]. While the L858R/C797S mutant can be overcome by the switch to a first-generation inhibitor that does not rely on a covalent bind to Cys797, of particular interest is the triple L858R/T790M/C797S cis mutant (with the two resistance-conferring mutations on the same allele) which is refractory to EGFR TKI from first generation to third generation [199,200]. Given the definition of third-generation inhibitors, to the best of our knowledge, no quinazoline derivative has been reported as a third-generation inhibitor in the given time (2017–present) even though Section 5 and Section 6 of this work count several molecules active as inhibitors for EGFRT790M.
8. Fourth Generation of EGFR TKIs
C797S mutation was found to be responsible for 20% of reported cases of resistance to third-generation inhibitors [201,202]. Fourth-generation inhibitors were specifically developed to overcome tumor resistance conferred by triple mutant EGFRL858R/T790M/C797S by binding to other regions of the receptor that are not affected by mutations [203,204]. Cellular signaling is disrupted by the binding of inhibitors into the allosteric pocket (“allosteric inhibitors”) created by the displacement of the regulatory C-helix in the inactive conformation of EGFR kinase with increased affinity for mutant forms and minimal disruption of EGFRwt [205]. Most of the fourth-generation inhibitors displayed good in vitro activity toward EGFRL858R/T790M/C797S kinase but low in vivo effectiveness, leading to the combination with EGFR-targeted antibodies [202,205]. Most fourth-generation inhibitors display minimal to no effect on EGFRdel19/T790M/C797S kinase [205]. In a novel approach, inhibitors that bind both to the allosteric pocket and to the ligand-binding region have been developed (ATP-competitive) [206,207,208].
The discovery of the fourth-generation inhibitors such as EAI045 (Figure 27) led to a novel approach to EGFR inhibition [205]. Increased affinity for the allosteric pocket is conferred due to the “Y’ shape of the molecule that fits into the region and specifically binds to a Met790 residue of the mutant form [204].
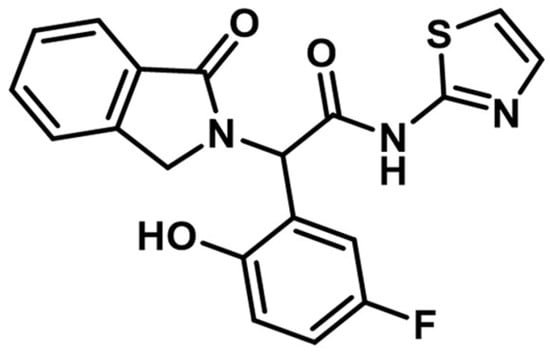
Figure 27.
Chemical structure of fourth-generation allosteric EGFR inhibitor EAI045.
9. Novel Fourth-Generation Quinazoline EGFR TKIs (2017–Present)
9.1. 2-Aryl-4-amino-quinazoline Derivatives
Park et al. synthesized a series of novel 2,4-disubstituted quinazoline derivatives as potent inhibitors of the triple EGFR d746−750/T790M/C797S mutant. In vitro studies revealed a more than 1000-fold selectivity ratio toward the triple mutant compared to EGFRwt kinase. The presence of 2-hydroxy-phenyl moiety at the C-2 position of the quinazoline core is vital for the inhibitory activity due to multiple H-bonds generated with the ATP-binding pocket. The further substitution of the 2-hydroxy-phenyl moiety in position 5 of the ring (hydroxyl, nitrile, acetyl, methoxy, or methyl-ester) led to a further increase in the inhibitory activity toward the triple mutant, with a conserved selectivity ratio. The terminal groups at the C-5 position of the phenyl moiety occupy the peripheral binding pocket, and increase the inhibitory activity by affecting the functions of the Gly-loop and DFG motif of the kinase domain. Docking simulations of the most active compound 39 (Figure 28) revealed that the presence of an alkaline 6-member heteroaryl moiety and a secondary amine linker is crucial for inhibitory activity by generating strong H-bond interactions with Met793. Compound 39 displayed the highest selectivity ratio toward the triple mutant (IC50 = 17.9 nM) compared to EGFRwt (IC50 ≥ 50 µM) [208].
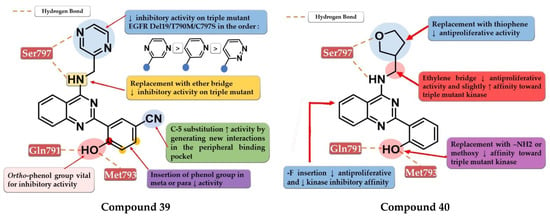
Figure 28.
Structure–activity relationship and molecular docking of compound 39 in the EGFRdel19/T790M/C797S kinase domain, constructed by homology using EGFRL858R/T790M (PDB ID 3W2R) [209], as reported by Park et al. [208]; structure–activity relationship and molecular docking of compound 40 in the human EGFRT790M/C797S kinase domain (PDB ID 5XGN) [210], as reported by Zhou et al. [207].
Further structural optimizations on compound 39 [208] were realized by Zhou et al. by maintaining the 2,4-substitution pattern on the quinazoline core. The compounds displayed higher antiproliferative activity on A549 cells than on NCI-H460 cells, less potency toward H1975, with more than 10-fold less activity overall compared to the reference drug AZD9291 on all three tested cell lines. The replacement of the phenol group with amino or methoxy groups led to decreased antiproliferative activity and inhibitory activity toward EGFRL858R/T790M. The substitution of the C-6 positions of the quinazoline core with fluorine led to decreased activity. Further modulation of the C-4 substituent of the quinazoline core led to the identification of compound 40 (Figure 28), synthesized by the replacement of the lateral pyridine ring of the lead compound with a tetrahydrofuran-3-yl moiety. The replacement of the methyl-amino bridge with longer linkers led to decreased activity. Compound 40 had a 10-fold higher selectivity toward EGFRL8585R/T790M (compared to EGFRwt—740 nM and >105 nM) than the reference drug afatinib (10 nM and 6 nM). Compound 40 displayed the highest antiproliferative activity on all the tested cells (A549, NCI-H460, and H1975) and 91% of EGFRDel19/T790M/C797S kinase at 10 µM [207].
9.2. 6-Methoxy-7-[(1-methylpiperidin-4-yl) methoxy]quinazolin-4-aniline Derivatives
Dou et al. developed a series of 4-anilino-quinazoline derivatives as potential fourth-generation EGFRC797S kinase inhibitors. The structural optimization by inserting various groups in the para-position of the 4-aniline moiety in the structure of the reference drug vandetanib (Figure 1), a moderate inhibitor of EGFRL858R/T790M/C797S kinase (IC50 = 369 nM), led to the synthesis of a series of novel potent compounds capable of spanning both the ATP-binding pocket and the allosteric site. The best results were obtained by inserting a highly lipophilic group linked by a flexible linker (ether or thioether bridge) capable of generating a suitable binding posture and torsion angle for a strong interaction with the allosteric site. The most active compound 41 (Figure 29) displayed an increased inhibitory activity toward EGFRL858R/T790M/C797S kinase (IC50 = 0.128 µM) with good selectivity and cytotoxic activity on BaF3-EGFRL858R/T790M/C797S (0.75 µM) and BaF3-EGFR19del/T790M/C797S (0.09 µM) cells, comparable with brigatinib (0.56 µM and 0.17 µM) (Table S1). The replacement of the lateral phenyl group with other groups (pyridine and cyclohexyl) led to a drastically decreased in vitro activity, while the further substitution with various small lipophilic substituents (-F, -Cl, and -Br) and EDGs (-CH3) led to increased kinase inhibitory activity but less potent cytotoxic activity due to poor pharmacokinetics [211].
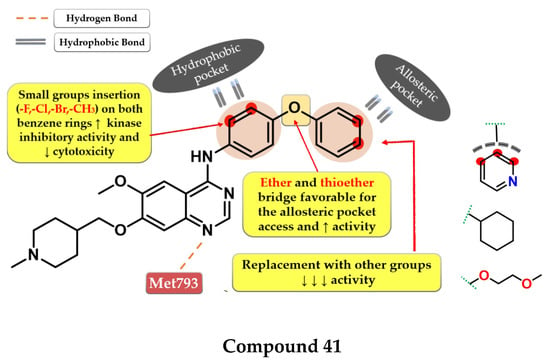
Figure 29.
Structure–activity relationship and molecular docking of compound 41 in the molecular model of EGFRdel19/T790M/C797SATP-binding domain, build using Maestro (PDB ID 1XKK used as a template) [101], as reported by Dou et al. [211].
Li et al. synthesized a series of novel reversible EGFRL858R/T790M/C797S quinazoline derivatives by merging the structural design of vandetanib (Figure 1) and EAI045 (Figure 26). Increased inhibitory activity was achieved by inserting the oxoisoindolin-2-phenylacetamide residue in the para-position of the 4-amino-phenyl moiety by generating a “Y-shaped” conformation capable of occupying the allosteric pocket. Moving the substituent on the aniline moiety in meta-position led to a four-fold decrease in the inhibitory activity, while the replacement with other more hydrophilic amide-linked groups led to a total loss of activity. The 2,5-difluoro substitution of the lateral phenyl ring led to increased binding affinity and the synthesis of compound 42 (Figure 30), the most active of the series. Compound 42 displayed a significant inhibitory activity toward EGFRL858R/T790M/C797S (IC50 = 2.2 nM) and an increased cytotoxic effect on BaF3-EGFRL858R/T790M/C797S (0.64 µM), comparable to the reference drug brigatinib (0.42 µM) and higher than that of afatinib (3.93 µM) (Table S1) [206].
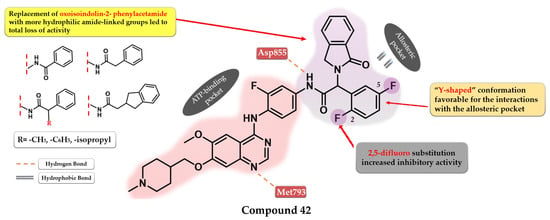
Figure 30.
Structure–activity relationship and molecular docking of compound 42 in the human EGFRdel19/T790M/C797S kinase domain (PDB code 5D41), as reported by Li et al. [206].
10. Conclusions
The identification of the quinazoline core as a valuable scaffold for EGFR inhibition led to the development of several TKIs with tremendous clinical utility in various types of cancer (especially NSCLC). The identification of the key structural features of an ideal EGFR TKI (increased potency toward mutant forms of EGFR, minimal affinity for EGFRwt, and low off-target effect) has been a hot topic for many research groups for some time. This work provides a collection of general drug design rules for the development of novel quinazoline derivatives as potential EGFR inhibitors based on new quinazoline derivatives synthesized in the last 6 years.
The substitution pattern on the quinazoline core plays a crucial role in the development of novel potent EGFR TKIs. Along with the already-established structure design principles (aniline moiety in position 4, electron-donating groups in positions 6 and 7, Michael acceptor inserted from the C-6 substituents) for the development of EGFR TKIs, this work outlines novel approaches to increasing the inhibition potential even further while maintaining a high selectivity index. Newly synthesized first-generation quinazoline inhibitors have lower IC50 for common mutation forms of EGFR (del19 and L858R) and increased affinity for secondary mutations (T790M) compared to the inhibitors already approved in therapy. This was achieved by structural modulations such as the replacement of various substituents in order to generate novel bonds with the target, and the development of multi-target inhibitors with favorable pharmacokinetics. A better understanding of the covalent bond formation for the second-generation inhibitors led to the discovery of novel Michael acceptors (α-chlorofluoro-acetamides, thioacetamides, and novel substitution patterns on the already-established acrylamide moiety), used for the development of novel EGFR quinazoline TKIs with improved balance between reactivity and affinity for the target.
A breakthrough is the discovery of the fourth-generation inhibitors, bearing a quinazoline core, as allosteric inhibitors of the triple mutant L858R/T790M/C797S highly resistant to the first three generations of inhibitors. This was achieved by the further substitution of the aniline moiety with various bulky substituents capable of occupying the allosteric pocket, or a non-canonical 2,4-substitution profile capable of generating a new binding mode into the active site of the kinase.
For our research group, this work will provide a solid base for the development of a series of original articles focused on the synthesis of novel quinazoline derivatives as potential EGFR TKIs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph16040534/s1. Figure S1. Bibliometric graphic of the reported series of quinazoline derivatives as EGFR TKIs found in the Scopus database (blue) and selected series for the discussion of SAR (red) in the period 2017–present; Table S1: In vitro efficiency of novel quinazoline derivatives (2017–present) as potential EGFR inhibitors.
Author Contributions
Conceptualization, A.Ș. and I.I.; writing—original draft, A.Ș., I.I. and O.O.; writing—review and editing, I.I., G.M., I.O. and D.E.; supervision, O.O. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania, grant numbers PCD 881/45/12.01.2022 and 772/36/11.01.2023.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Lowe, S.W. The Microcosmos of Cancer. Nature 2012, 482, 347–355. [Google Scholar] [CrossRef]
- Devita, V.T.; Rosenberg, S.A. Two Hundred Years of Cancer Research. N. Engl. J. Med. 2012, 366, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Emamalipour, M.; Seidi, K.; Zununi Vahed, S.; Jahanban-Esfahlan, A.; Jaymand, M.; Majdi, H.; Amoozgar, Z.; Chitkushev, L.T.; Javaheri, T.; Jahanban-Esfahlan, R.; et al. Horizontal Gene Transfer: From Evolutionary Flexibility to Disease Progression. Front. Cell Dev. Biol. 2020, 8, 245. [Google Scholar] [CrossRef]
- Rahman, N. Realizing the Promise of Cancer Predisposition Genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Irigaray, P.; Newby, J.A.; Clapp, R.; Hardell, L.; Howard, V.; Montagnier, L.; Epstein, S.; Belpomme, D. Lifestyle-Related Factors and Environmental Agents Causing Cancer: An Overview. Biomed. Pharmacother. 2007, 61, 640–658. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Gold, L.S. DNA Lesions, Inducible DNA Repair, and Cell Division: Three Key Factors in Mutagenesis and Carcinogenesis. Environ. Health Perspect. 1993, 101, 35–44. [Google Scholar] [CrossRef]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Russo, J.; Mailo, D.; Hu, Y.F.; Balogh, G.; Sheriff, F.; Russo, I.H.; Ingle, J.; Brodie, A.; Santen, R.; Colditz, G.; et al. Breast Differentiation and Its Implication in Cancer Prevention. Clin. Cancer Res. 2005, 11, 931s–936s. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, Cell Cycle and Apoptosis in Cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The Future of Cancer Treatment: Immunomodulation, CARs and Combination Immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. A Downside of Chemotherapy. Nat. Rev. Cancer 2012, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging New Therapeutic Antibody Derivatives for Cancer Treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA Silencing for Cancer Therapy Targeted to the Tumour Microenvironment. Nature 2014, 518, 107–110. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic Virus Therapy: A New Era of Cancer Treatment at Dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Troike, K.; Lathia, J.D. Optimising Gene Editing for Cancer Therapy. Nat. Cell Biol. 2020, 22, 259–261. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, H.A.M. A Comprehensive Review of Recent Advances in the Biological Activities of Quinazolines. Chem. Biol. Drug Des. 2022, 100, 639–655. [Google Scholar] [CrossRef]
- Haider, K.; Shafeeque, M.; Yahya, S.; Yar, M.S. A Comprehensive Review on Pyrazoline Based Heterocyclic Hybrids as Potent Anticancer Agents. Eur. J. Med. Chem. Rep. 2022, 5, 100042. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal Growth Factor Receptor Mutations in Lung Cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER Family of Protein-Tyrosine Kinases and Cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Jin, H.; Dan, H.G.; Rao, G.W. Research Progress in Quinazoline Derivatives as Multi-Target Tyrosine Kinase Inhibitors. Heterocycl. Commun. 2018, 24, 1–10. [Google Scholar] [CrossRef]
- Bhatia, P.; Sharma, V.; Alam, O.; Manaithiya, A.; Alam, P.; Kahksha; Alam, M.T.; Imran, M. Novel Quinazoline-Based EGFR Kinase Inhibitors: A Review Focussing on SAR and Molecular Docking Studies (2015–2019). Eur. J. Med. Chem. 2020, 204, 112640. [Google Scholar] [CrossRef]
- Reports Regarding the New Experimental Quinazoline Derivatives Series as EGFR Tirosin-Kinase Inhibitors. Available online: https://www.scopus.com (accessed on 24 March 2023).
- Tebbutt, N.; Pedersen, M.W.; Johns, T.G. Targeting the ERBB Family in Cancer: Couples Therapy. Nat. Rev. Cancer 2013, 13, 663–673. [Google Scholar] [CrossRef]
- Pennock, S.; Wang, Z. Stimulation of Cell Proliferation by Endosomal Epidermal Growth Factor Receptor as Revealed through Two Distinct Phases of Signaling. Mol. Cell. Biol. 2003, 23, 5803–5815. [Google Scholar] [CrossRef]
- Barberán, S.; Cebrià, F. The Role of the EGFR Signaling Pathway in Stem Cell Differentiation during Planarian Regeneration and Homeostasis. Semin. Cell Dev. Biol. 2019, 87, 45–57. [Google Scholar] [CrossRef]
- Chiasson-MacKenzie, C.; McClatchey, A.I. EGFR-Induced Cytoskeletal Changes Drive Complex Cell Behaviors: The Tip of the Iceberg. Sci. Signal. 2018, 11, eaas9473. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A Structural Perspective on the Regulation of the Epidermal Growth Factor Receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.W.; Cho, H.S.; Eigenbrot, C.; Ferguson, K.M.; Garrett, T.P.J.; Leahy, D.J.; Lemmon, M.A.; Sliwkowski, M.X.; Ward, C.W.; Yokoyama, S. An Open-and-Shut Case? Recent Insights into the Activation of EGF/ErbB Receptors. Mol. Cell 2003, 12, 541–552. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging Functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Chmielecki, J. Rational, Biologically Based Treatment of EGFR-Mutant Non-Small-Cell Lung Cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.Y.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y.; et al. Epidermal Growth Factor Receptor Activation in Glioblastoma through Novel Missense Mutations in the Extracellular Domain. PLoS Med. 2006, 3, e485. [Google Scholar] [CrossRef]
- Rogers, S.J.; Harrington, K.J.; Rhys-Evans, P.; O-Charoenrat, P.; Eccles, S.A. Biological Significance of C-ErbB Family Oncogenes in Head and Neck Cancer. Cancer Metastasis Rev. 2005, 24, 47–69. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Hung, M.C. EGFR Signaling Pathway in Breast Cancers: From Traditional Signal Transduction to Direct Nuclear Translocalization. Breast Cancer Res. Treat. 2006, 95, 211–218. [Google Scholar] [CrossRef]
- Spano, J.P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.F.; et al. Impact of EGFR Expression on Colorectal Cancer Patient Prognosis and Survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal Growth Factor Receptor in Pancreatic Cancer. Cancers 2011, 3, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Schmidt, M.H.H. EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. Int. J. Mol. Sci. 2017, 18, 1295. [Google Scholar] [CrossRef]
- Kedrin, D.; van Rheenen, J.; Hernandez, L.; Condeelis, J.; Segall, J.E. Cell Motility and Cytoskeletal Regulation in Invasion and Metastasis. J. Mammary Gland Biol. Neoplasia 2007, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.Y.; Na, I.I.; Kim, C.H.; Park, S.; Baek, H.; Yang, S.H. EGFR Mutation and Brain Metastasis in Pulmonary Adenocarcinomas. J. Thorac. Oncol. 2014, 9, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Sarris, E.G.; Saif, M.W.; Syrigos, K.N. The Biological Role of PI3K Pathway in Lung Cancer. Pharmaceuticals 2012, 5, 1236–1264. [Google Scholar] [CrossRef]
- Codony-Servat, C.; Codony-Servat, J.; Karachaliou, N.; Molina, M.A.; Chaib, I.; Ramirez, J.L.; Gil, M.d.l.L.; Solca, F.; Bivona, T.G.; Rosell, R. Activation of Signal Transducer and Activator of Transcription 3 (STAT3) Signaling in EGFR Mutant Non-Small-Cell Lung Cancer (NSCLC). Oncotarget 2017, 8, 47316. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Troiani, T.; Napolitano, S.; Della Corte, C.M.; Martini, G.; Martinelli, E.; Morgillo, F.; Ciardiello, F. Therapeutic Value of EGFR Inhibition in CRC and NSCLC: 15 Years of Clinical Evidence. ESMO Open 2016, 1, e000088. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The Epidermal Growth Factor Receptor Variant III (EGFRvIII): Where Wild Things Are Altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Martinelli, E.; Ciardiello, D.; Martini, G.; Troiani, T.; Cardone, C.; Vitiello, P.P.; Normanno, N.; Rachiglio, A.M.; Maiello, E.; Latiano, T.; et al. Implementing Anti-Epidermal Growth Factor Receptor (EGFR) Therapy in Metastatic Colorectal Cancer: Challenges and Future Perspectives. Ann. Oncol. 2020, 31, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.F.; Liu, Z.C.; Xie, B.F.; Li, Z.M.; Feng, G.K.; Yang, D.; Zeng, Y.X. EGFR Tyrosine Kinase Inhibitor AG1478 Inhibits Cell Proliferation and Arrests Cell Cycle in Nasopharyngeal Carcinoma Cells. Cancer Lett. 2001, 169, 27–32. [Google Scholar] [CrossRef]
- Gonzales, A.J.; Fry, D.W. G1 Cell Cycle Arrest Due to the Inhibition of ErbB Family Receptor Tyrosine Kinases Does Not Require the Retinoblastoma Protein. Exp. Cell Res. 2005, 303, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Jeong, E.H.; Lee, T.G.; Kim, S.Y.; Kim, H.R.; Kim, C.H. Gefitinib Induces Cytoplasmic Translocation of the CDK Inhibitor P27 and Its Binding to a Cleaved Intermediate of Caspase 8 in Non-Small Cell Lung Cancer Cells. Cell. Oncol. 2014, 37, 377–386. [Google Scholar] [CrossRef]
- Gong, Y.; Somwar, R.; Politi, K.; Balak, M.; Chmielecki, J.; Jiang, X.; Pao, W. Induction of BIM Is Essential for Apoptosis Triggered by EGFR Kinase Inhibitors in Mutant EGFR-Dependent Lung Adenocarcinomas. PLoS Med. 2007, 4, e294. [Google Scholar] [CrossRef]
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM Mediates EGFR Tyrosine Kinase Inhibitor-Induced Apoptosis in Lung Cancers with Oncogenic EGFR Mutations. PLoS Med. 2007, 4, e315. [Google Scholar] [CrossRef]
- Santos, E.d.S.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.d.B.V.; Júnior, I.J.d.S.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR Targeting for Cancer Therapy: Pharmacology and Immunoconjugates with Drugs and Nanoparticles. Int. J. Pharm. 2021, 592, 120082. [Google Scholar] [CrossRef]
- Wu, S.; Deng, C.; Zhang, C.; Xiong, J.; Gu, X.; Wang, Z.; Tu, J.; Xie, J. Preparation of a Novel EGFR Specific Immunotoxin and Its Efficacy of Anti-Colorectal Cancer In Vitro and In Vivo. Clin. Transl. Oncol. 2021, 23, 1549–1560. [Google Scholar] [CrossRef]
- Bauman, J.E.; Duvvuri, U.; Thomas, S.; Gooding, W.E.; Clump, D.A.; Karlovits, B.; Wehbe, A.; Miller, F.R.; Kim, S.; Sen, M.; et al. Phase 1 Study of EGFR-Antisense DNA, Cetuximab, and Radiotherapy in Head and Neck Cancer with Preclinical Correlatives. Cancer 2018, 124, 3881–3889. [Google Scholar] [CrossRef] [PubMed]
- Malkki, H. Glioblastoma Vaccine Therapy Disappointment in Phase III Trial. Nat. Rev. Neurol. 2016, 12, 190. [Google Scholar] [CrossRef]
- Du, Y.; Chen, Y.; Wang, Y.; Chen, J.; Lu, X.; Zhang, L.; Li, Y.; Wang, Z.; Ye, G.; Zhang, G. HJM-561, a Potent, Selective, and Orally Bioavailable EGFR PROTAC That Overcomes Osimertinib-Resistant EGFR Triple Mutations. Mol. Cancer Ther. 2022, 21, 1060–1066. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Li, A.R.; Chitale, D.; Riely, G.J.; Pao, W.; Miller, V.A.; Zakowski, M.F.; Rusch, V.; Kris, M.G.; Ladanyi, M. Clinical Testing Experience and Relationship to EGFR Gene Copy Number and Immunohistochemical Expression. J. Mol. Diagn. 2008, 10, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Chu, D.T.; Han, B.; Liu, X.; Zhang, L.; Zhou, C.; Liao, M.; Mok, T.; Jiang, H.; Duffield, E.; et al. Phase III, Randomized, Open-Label, First-Line Study in Asia of Gefitinib versus Carboplatin/Paclitaxel in Clinically Selected Patients with Advanced Non-Small-Cell Lung Cancer: Evaluation of Patients Recruited from Mainland China. Asia. Pac. J. Clin. Oncol. 2012, 8, 232–243. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA) Label—IRESSA® (Gefitinib) 250mg Tablets for Oral Use. Available online: www.fda.gov/medwatch (accessed on 15 February 2023).
- Patil, V.M.; Noronha, V.; Joshi, A.; Choughule, A.B.; Bhattacharjee, A.; Kumar, R.; Goud, S.; More, S.; Ramaswamy, A.; Karpe, A.; et al. Phase III Study of Gefitinib or Pemetrexed with Carboplatin in EGFR-Mutated Advanced Lung Adenocarcinoma. ESMO Open 2017, 2, eooo25. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.H.; Lee, K.H.; Lu, S.; et al. Afatinib versus Gefitinib as First-Line Treatment of Patients with EGFR Mutation-Positive Non-Small-Cell Lung Cancer (LUX-Lung 7): A Phase 2B, Open-Label, Randomised Controlled Trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA) Label—TARCEVA® (Erlotinib) 25 Mg, 100 Mg and 150 Mg Tablets. Available online: www.fda.gov/medwatch (accessed on 15 February 2023).
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus Gemcitabine Compared with Gemcitabine Alone in Patients with Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Pereira, J.; Ciuleanu, T.E.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Bezjak, A.; Tu, D.; Santabárbara, P.; Seymour, L. A Randomized Placebo-Controlled Trial of Erlotinib in Patients with Advanced Non-Small Cell Lung Cancer (NSCLC) Following Failure of 1st Line or 2nd Line Chemotherapy. A National Cancer Institute of Canada Clinical Trials Group (NCIC CTG) Trial. J. Clin. Oncol. 2004, 22, 7022. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, L.; Han, B.; Li, W.; Yu, P.; Liu, Y.; Ding, C.; Song, X.; Yong, M.Z.; Ren, X.; et al. First-Line Icotinib versus Cisplatine/Pemetrexed plus Pemetrexed Maintenance Therapy in Lung Adenocarcinoma Patients with Sensitizing EGFR Mutation (CONVINCE). J. Clin. Oncol. 2016, 34, 9041. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Liu, X.; Zhou, C.; Zhang, S.; Wang, D.; Li, Q.; Qin, S.; Hu, C.; Zhang, Y.; et al. Icotinib versus Gefitinib in Previously Treated Advanced Non-Small-Cell Lung Cancer (ICOGEN): A Randomised, Double-Blind Phase 3 Non-Inferiority Trial. Lancet Oncol. 2013, 14, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Thongprasert, S.; Duffield, E.; Saijo, N.; Wu, Y.L.; Yang, J.C.H.; Chu, D.T.; Liao, M.; Chen, Y.M.; Kuo, H.P.; Negoro, S.; et al. Health-Related Quality-of-Life in a Randomized Phase III First-Line Study of Gefitinib Versus Carboplatin/Paclitaxel in Clinically Selected Patients from Asia with Advanced NSCLC (IPASS). J. Thorac. Oncol. 2011, 6, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, D.; To, C.; Shpilsky, J.E.; VanderLaan, P.A.; Kobayashi, S.S.; Mushajiang, M.; Lau, C.J.; Paweletz, C.P.; Oxnard, G.R.; Jänne, P.A.; et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14, 1995–2002. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, 0225–0235. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Arcila, M.E.; Sima, C.S.; Riely, G.J.; Chmielecki, J.; Kris, M.G.; Pao, W.; Ladanyi, M.; Miller, V.A. Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Lung Cancer: Distinct Natural History of Patients with Tumors Harboring the T790M Mutation. Clin. Cancer Res. 2011, 17, 1616–1622. [Google Scholar] [CrossRef]
- Ogino, A.; Kitao, H.; Hirano, S.; Uchida, A.; Ishiai, M.; Kozuki, T.; Takigawa, N.; Takata, M.; Kiura, K.; Tanimoto, M. Emergence of Epidermal Growth Factor Receptor T790M Mutation during Chronic Exposure to Gefitinib in a Non–Small Cell Lung Cancer Cell Line. Cancer Res. 2007, 67, 7807–7814. [Google Scholar] [CrossRef]
- Chiu, C.H.; Yang, C.T.; Shih, J.Y.; Huang, M.S.; Su, W.C.; Lai, R.S.; Wang, C.C.; Hsiao, S.H.; Lin, Y.C.; Ho, C.L.; et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment Response in Advanced Lung Adenocarcinomas with G719X/L861Q/S768I Mutations. J. Thorac. Oncol. 2015, 10, 793–799. [Google Scholar] [CrossRef]
- Kancha, R.K.; Von Bubnoff, N.; Peschel, C.; Duyster, J. Functional Analysis of Epidermal Growth Factor Receptor (EGFR) Mutations and Potential Implications for EGFR Targeted Therapy. Clin. Cancer Res. 2009, 15, 460–467. [Google Scholar] [CrossRef]
- Lei, L.; Wang, W.; Zhu, Y.; Li, J.; Fang, Y.; Wang, H.; Zhuang, W.; Zhang, Y.; Wang, L.; Fang, M.; et al. Potential Mechanism of Primary Resistance to Icotinib in Patients with Advanced Non–Small Cell Lung Cancer Harboring Uncommon Mutant Epidermal Growth Factor Receptor: A Multi-Center Study. Cancer Sci. 2020, 111, 679–686. [Google Scholar] [CrossRef]
- Vyse, S.; Huang, P.H. Targeting EGFR Exon 20 Insertion Mutations in Non-Small Cell Lung Cancer. Signal Transduct. Target. Ther. 2019, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, H.P.; Im, S.A.; Kang, S.; Hur, H.S.; Yoon, Y.K.; Oh, D.Y.; Kim, J.H.; Lee, D.S.; Kim, T.Y.; et al. The Growth Inhibitory Effect of Lapatinib, a Dual Inhibitor of EGFR and HER2 Tyrosine Kinase, in Gastric Cancer Cell Lines. Cancer Lett. 2008, 272, 296–306. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA) Label—TYKERB® (Lapatinib) 250 Mg Tablets. Available online: www.fda.gov/medwatch (accessed on 15 February 2023).
- Morabito, A.; Piccirillo, M.C.; Falasconi, F.; De Feo, G.; Del Giudice, A.; Bryce, J.; Di Maio, M.; De Maio, E.; Normanno, N.; Perrone, F. Vandetanib (ZD6474), a Dual Inhibitor of Vascular Endothelial Growth Factor Receptor (VEGFR) and Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinases: Current Status and Future Directions. Oncologist 2009, 14, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA) Label—CAPRELSA® (Vandetanib) 100 Mg and 300 Mg Tablets for Oral Use. Available online: www.fda.gov/medwatch (accessed on 15 February 2023).
- Zhang, D.; Pal, A.; Bornmann, W.G.; Yamasaki, F.; Esteva, F.J.; Hortobagyi, G.N.; Bartholomeusz, C.; Ueno, N.T. Activity of Lapatinib Is Independent of EGFR Expression Level in HER2-Overexpressing Breast Cancer Cells. Mol. Cancer Ther. 2008, 7, 1846–1850. [Google Scholar] [CrossRef]
- Ahn, M.J.; Kim, D.W.; Cho, B.C.; Kim, S.W.; Lee, J.S.; Ahn, J.S.; Kim, T.M.; Lin, C.C.; Kim, H.R.; John, T.; et al. Activity and Safety of AZD3759 in EGFR-Mutant Non-Small-Cell Lung Cancer with CNS Metastases (BLOOM): A Phase 1, Open-Label, Dose-Escalation and Dose-Expansion Study. Lancet Respir. Med. 2017, 5, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Milik, S.N.; Lasheen, D.S.; Serya, R.A.T.; Abouzid, K.A.M. How to Train Your Inhibitor: Design Strategies to Overcome Resistance to Epidermal Growth Factor Receptor Inhibitors. Eur. J. Med. Chem. 2017, 142, 131–151. [Google Scholar] [CrossRef]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J. Med. Chem. 2010, 53, 2681–2694. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.-C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural Analysis of the Mechanism of Inhibition and Allosteric Activation of the Kinase Domain of HER2 Protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef]
- Tong, M.; Seeliger, M.A. Targeting Conformational Plasticity of Protein Kinases. ACS Chem. Biol. 2015, 10, 190–200. [Google Scholar] [CrossRef]
- Vivanco, I.; Ian Robins, H.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N.; et al. Differential Sensitivity of Glioma- versus Lung Cancer-Specific EGFR Mutations to EGFR Kinase Inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib Binds Both Inactive and Active Conformations of the EGFR Tyrosine Kinase Domain. Biochem. J. 2012, 448, 423. [Google Scholar] [CrossRef] [PubMed]
- Ward, W.H.J.; Cook, P.N.; Slater, A.M.; Davies, D.H.; Holdgate, G.A.; Green, L.R. Epidermal Growth Factor Receptor Tyrosine Kinase: Investigation of Catalytic Mechanism, Structure-Based Searching and Discovery of a Potent Inhibitor. Biochem. Pharmacol. 1994, 48, 659–666. [Google Scholar] [CrossRef]
- Verma, N.; Rai, A.K.; Kaushik, V.; Brünnert, D.; Chahar, K.R.; Pandey, J.; Goyal, P. Identification of Gefitinib Off-Targets Using a Structure-Based Systems Biology Approach; Their Validation with Reverse Docking and Retrospective Data Mining. Sci. Rep. 2016, 6, 33949. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, M.; Qu, Y.; Tang, W.; Zheng, Y.; Lian, J.; Ji, M.; Xu, L. Design and Synthesis of Novel Gefitinib Analogues with Improved Anti-Tumor Activity. Bioorg. Med. Chem. 2010, 18, 3812–3822. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Sun, M.; Wu, X.; Chen, J.; Wang, P.; Zong, X.; Ji, M. Design and Synthesis of Novel 4-Benzothiazole Amino Quinazolines Dasatinib Derivatives as Potential Anti-Tumor Agents. Eur. J. Med. Chem. 2013, 63, 702–712. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib). Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.; Liu, J.; Wang, W.; Li, X.; Zhao, L.; Wang, W.; Li, B. Novel 4-Arylaminoquinazoline Derivatives with (E)-Propen-1-Yl Moiety as Potent EGFR Inhibitors with Enhanced Antiproliferative Activities against Tumor Cells. Eur. J. Med. Chem. 2017, 138, 689–697. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, L.; Xu, H.; Li, X.; Zhao, L.; Wang, W.; Li, B.; Zhang, X. 6,7-Dimorpholinoalkoxy Quinazoline Derivatives as Potent EGFR Inhibitors with Enhanced Antiproliferative Activities against Tumor Cells. Eur. J. Med. Chem. 2018, 147, 77–89. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, C.; Xu, S.; Lan, Z.; Li, W.; Han, J.; Zhou, Y.; Zheng, P.; Zhu, W. Design, Synthesis, and Docking Studies of Quinazoline Analogues Bearing Aryl Semicarbazone Scaffolds as Potent EGFR Inhibitors. Bioorg. Med. Chem. 2017, 25, 3148–3157. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, S.; Peng, L.; Zhang, B.; Zhang, H.; Hu, Y.; Zheng, P.; Zhu, W. Design, Synthesis and Biological Evaluation of Novel 4-Anlinoquinazoline Derivatives as EGFR Inhibitors with the Potential to Inhibit the Gefitinib-Resistant Nonsmall Cell Lung Cancers. J. Enzyme Inhib. Med. Chem. 2019, 34, 204–218. [Google Scholar] [CrossRef]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Gong, F.H.; Ye, J.Q.; Zhang, C.; Yue, X.H.; Li, C.G.; Xu, Y.G.; Sun, L.P. Design and Discovery of 4-Anilinoquinazoline-Urea Derivatives as Dual TK Inhibitors of EGFR and VEGFR-2. Eur. J. Med. Chem. 2017, 125, 245–254. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, J.; Wang, N.; Kong, X.; Fu, F.; Wang, H.; Yao, J. Design and Discovery of Quinazoline- and Thiourea-Containing Sorafenib Analogs as EGFR and VEGFR-2 Dual TK Inhibitors. Molecules 2017, 23, 24. [Google Scholar] [CrossRef]
- Hamed, M.M.; Darwish, S.S.; Herrmann, J.; Abadi, A.H.; Engel, M. First Bispecific Inhibitors of the Epidermal Growth Factor Receptor Kinase and the NF-ΚB Activity as Novel Anticancer Agents. J. Med. Chem. 2017, 60, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Wang, C.; Pan, Q.; Li, Y.; Guo, Y.; Fan, D.; Peng, Y.; Rao, Z.; Xu, S.; Zheng, P.; et al. Discovery of Novel 4-Arylamino-Quinazoline Derivatives as EGFRL858R/T790M Inhibitors with the Potential to Inhibit the Non-Small Cell Lung Cancers. Bioorg. Chem. 2022, 127, 106011. [Google Scholar] [CrossRef]
- Hou, W.; Ren, Y.; Zhang, Z.; Sun, H.; Ma, Y.; Yan, B. Novel Quinazoline Derivatives Bearing Various 6-Benzamide Moieties as Highly Selective and Potent EGFR Inhibitors. Bioorg. Med. Chem. 2018, 26, 1740–1750. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, Z.; Liu, Q.; Xia, S.; Liu, Z.; Liao, Z.; Gou, S. Design, Synthesis and Biological Evaluation of Cinnamamide-Quinazoline Derivatives as Potential EGFR Inhibitors to Reverse T790M Mutation. Bioorg. Chem. 2021, 117, 105432. [Google Scholar] [CrossRef]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070. [Google Scholar] [CrossRef]
- Tang, Q.; Peng, T.; Hu, J.; Zhang, T.; Chen, P.; Chen, D.; Wang, Y.; Chen, L.; Tong, L.; Chen, Y.; et al. Discovery of N-(3-Bromo-1H-Indol-5-Yl)-Quinazolin-4-Amine as an Effective Molecular Skeleton to Develop Reversible/Irreversible Pan-HER Inhibitors. Eur. J. Med. Chem. 2022, 233, 114262. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Jin, B.; Liu, Y.; Chen, H.; Zhang, Z.; Zhang, C.; Zhao, Z.; Zheng, L. Synthesis and Biological Evaluation of Some Novel Thiophene-Bearing Quinazoline Derivatives as EGFR Inhibitors. Lett. Drug Des. Discov. 2018, 16, 102–110. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Liu, J.; Chen, L.; Zhao, L.; Li, B.; Wang, W. Synthesis and in Vitro Biological Evaluation of Novel Quinazoline Derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 1584–1587. [Google Scholar] [CrossRef]
- Ding, C.; Chen, S.; Zhang, C.; Hu, G.; Zhang, W.; Li, L.; Chen, Y.Z.; Tan, C.; Jiang, Y. Synthesis and Investigation of Novel 6-(1,2,3-Triazol-4-Yl)-4-Aminoquinazolin Derivatives Possessing Hydroxamic Acid Moiety for Cancer Therapy. Bioorg. Med. Chem. 2017, 25, 27–37. [Google Scholar] [CrossRef]
- Wei, H.; Duan, Y.; Gou, W.; Cui, J.; Ning, H.; Li, D.; Qin, Y.; Liu, Q.; Li, Y. Design, Synthesis and Biological Evaluation of Novel 4-Anilinoquinazoline Derivatives as Hypoxia-Selective EGFR and VEGFR-2 Dual Inhibitors. Eur. J. Med. Chem. 2019, 181, 111564. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, S.; Yang, Z.; Tian, X.; Hu, Y. Design, Synthesis, and Biological Study of 4-[(2-Nitroimidazole-1H-Alkyloxyl)Aniline]-Quinazolines as EGFR Inhibitors Exerting Cytotoxicities Both under Normoxia and Hypoxia. Drug Des. Devel. Ther. 2019, 13, 3079–3089. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Son, S.; Lee, H.Y.; El-Maghrabey, M.H.; El Hamd, M.A.; Alshammari, S.O.; Abdelhameed, A.A.; Alshammari, Q.A.; Abdeen, A.; Ibrahim, S.F.; et al. Design, Synthesis, Biological Evaluation, and Molecular Dynamics Studies of Novel Lapatinib Derivatives. Pharmaceuticals 2022, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Yang, L.; Liu, P.; Yang, L.; Chen, L.; Hu, L.; Jiang, M. Design, Synthesis and Biological Evaluation of 2,3-Dihydro-[1,4]Dioxino[2,3-f]Quinazoline Derivatives as EGFR Inhibitors. Bioorg. Chem. 2021, 110, 104755. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Zheng, Y.G.; Su, J.; Gao, C.Y.; Jiang, P.; An, L.; Xue, Y.S.; Gao, J.; Liu, Y. Design, Synthesis, and Biological Evaluation of Novel 4-Anilinoquinazoline Derivatives Bearing Amino Acid Moiety as Potential EGFR Kinase Inhibitors. Eur. J. Med. Chem. 2017, 130, 393–405. [Google Scholar] [CrossRef]
- Ismail, R.S.M.; Abou-Seri, S.M.; Eldehna, W.M.; Ismail, N.S.M.; Elgazwi, S.M.; Ghabbour, H.A.; Ahmed, M.S.; Halaweish, F.T.; Abou El Ella, D.A. Novel Series of 6-(2-Substitutedacetamido)-4-Anilinoquinazolines as EGFR-ERK Signal Transduction Inhibitors in MCF-7 Breast Cancer Cells. Eur. J. Med. Chem. 2018, 155, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Farouk, A.K.B.A.W.; Abdelrasheed Allam, H.; Rashwan, E.; George, R.F.; Abbas, S.E.S. Design and Synthesis of Some New 6-Bromo-2-(Pyridin-3-Yl)-4-Substituted Quinazolines as Multi Tyrosine Kinase Inhibitors. Bioorg. Chem. 2022, 128, 10710. [Google Scholar] [CrossRef] [PubMed]
- Mphahlele, M.J.; Mmonwa, M.M.; Aro, A.; McGaw, L.J.; Choong, Y.S. Synthesis, Biological Evaluation and Molecular Docking of Novel Indole-Aminoquinazoline Hybrids for Anticancer Properties. Int. J. Mol. Sci. 2018, 19, 2232. [Google Scholar] [CrossRef]
- Ding, H.W.; Deng, C.L.; Li, D.D.; Liu, D.D.; Chai, S.M.; Wang, W.; Zhang, Y.; Chen, K.; Li, X.; Wang, J.; et al. Design, Synthesis and Biological Evaluation of Novel 4-Aminoquinazolines as Dual Target Inhibitors of EGFR-PI3Kα. Eur. J. Med. Chem. 2018, 146, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, Z.; Xia, S.; Liu, Q.; Gou, S. Design, Synthesis and Biological Evaluation of Sulfamoylphenyl-Quinazoline Derivatives as Potential EGFR/CAIX Dual Inhibitors. Eur. J. Med. Chem. 2021, 216, 113300. [Google Scholar] [CrossRef] [PubMed]
- Yosaatmadja, Y.; Squire, C.J. 1.85 Angstrom Structure of EGFR Kinase Domain with Gefitinib. Available online: https://www.rcsb.org/structure/4WKQ (accessed on 22 March 2023).
- Chang, J.; Ren, H.; Zhao, M.; Chong, Y.; Zhao, W.; He, Y.; Zhao, Y.; Zhang, H.; Qi, C. Development of a Series of Novel 4-Anlinoquinazoline Derivatives Possessing Quinazoline Skeleton: Design, Synthesis, EGFR Kinase Inhibitory Efficacy, and Evaluation of Anticancer Activities in Vitro. Eur. J. Med. Chem. 2017, 138, 669–688. [Google Scholar] [CrossRef]
- Wang, L.; Li, P.; Li, B.; Wang, Y.; Li, J.; Song, L. Design, Synthesis, and Antitumor Activity of Novel Quinazoline Derivatives. Molecules 2017, 22, 1624. [Google Scholar] [CrossRef]
- Ju, Y.; Wu, J.; Yuan, X.; Zhao, L.; Zhang, G.; Li, C.; Qiao, R. Design and Evaluation of Potent EGFR Inhibitors through the Incorporation of Macrocyclic Polyamine Moieties into the 4-Anilinoquinazoline Scaffold. J. Med. Chem. 2018, 61, 11372–11383. [Google Scholar] [CrossRef]
- Amrhein, J.A.; Beyett, T.S.; Feng, W.W.; Krämer, A.; Weckesser, J.; Schaeffner, I.K.; Rana, J.K.; Jänne, P.A.; Eck, M.J.; Knapp, S.; et al. Macrocyclization of Quinazoline-Based EGFR Inhibitors Leads to Exclusive Mutant Selectivity for EGFR L858R and Del19. J. Med. Chem. 2022, 65, 15679–15697. [Google Scholar] [CrossRef]
- Yamahana, H.; Kunieda, Y.; Tominaga, M.; Yamada, H.; Uto, Y. Development of a Novel Acetyl Glucose-Modified Gefitinib Derivative to Enhance the Radiosensitizing Effect. Bioorg. Med. Chem. 2021, 29, 115889. [Google Scholar] [CrossRef]
- Hossam, M.; Lasheen, D.S.; Abouzid, K.A.M. Covalent EGFR Inhibitors: Binding Mechanisms, Synthetic Approaches, and Clinical Profiles. Arch. Pharm. 2016, 349, 573–593. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Dobrusin, E.M.; Fry, D.W.; Haske, T.; Whitty, A.; McNamara, D.J. Structure-Based Design of a Potent, Selective, and Irreversible Inhibitor of the Catalytic Domain of the ErbB Receptor Subfamily of Protein Tyrosine Kinases. J. Med. Chem. 1997, 40, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, Q.; Bliven, S.; Xie, L.; Bourne, P.E. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem. 2017, 60, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.I. Second-Generation Irreversible Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs): A Better Mousetrap? A Review of the Clinical Evidence. Crit. Rev. Oncol. Hematol. 2012, 83, 407–421. [Google Scholar] [CrossRef]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Lonsdale, R.; Burgess, J.; Colclough, N.; Davies, N.L.; Lenz, E.M.; Orton, A.L.; Ward, R.A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57, 3124–3137. [Google Scholar] [CrossRef]
- Tuley, A.; Fast, W. The Taxonomy of Covalent Inhibitors. Biochemistry 2018, 57, 3326–3337. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA) Label—GILOTRIFTM (Afatinib): 40 Mg, 30 Mg, and 20 Mg Tablets, for Oral Use. Available online: www.fda.gov/medwatch (accessed on 18 February 2023).
- Food and Drug Administration (FDA) Label—VIZIMPRO® (Dacomitinib) 15 Mg, 30 Mg, and 45 Mg Tablets, for Oral Use. Available online: www.fda.gov/medwatch (accessed on 18 February 2023).
- Shen, Y.C.; Tseng, G.C.; Tu, C.Y.; Chen, W.C.; Liao, W.C.; Chen, W.C.; Li, C.H.; Chen, H.J.; Hsia, T.C. Comparing the Effects of Afatinib with Gefitinib or Erlotinib in Patients with Advanced-Stage Lung Adenocarcinoma Harboring Non-Classical Epidermal Growth Factor Receptor Mutations. Lung Cancer 2017, 110, 56–62. [Google Scholar] [CrossRef]
- Hsu, P.C.; Lee, S.H.; Chiu, L.C.; Lee, C.S.; Wu, C.E.; Kuo, S.C.H.; Ju, J.S.; Huang, A.C.C.; Li, S.H.; Ko, H.W.; et al. Afatinib in Untreated Stage IIIB/IV Lung Adenocarcinoma with Major Uncommon Epidermal Growth Factor Receptor (EGFR) Mutations (G719X/L861Q/S768I): A Multicenter Observational Study in Taiwan. Target. Oncol. 2023, 18, 195–207. [Google Scholar] [CrossRef]
- Kosaka, T.; Tanizaki, J.; Paranal, R.M.; Endoh, H.; Lydon, C.; Capelletti, M.; Repellin, C.E.; Choi, J.; Ogino, A.; Calles, A.; et al. Response Heterogeneity of EGFR and HER2 Exon 20 Insertions to Covalent EGFR and HER2 Inhibitors. Cancer Res. 2017, 77, 2712–2721. [Google Scholar] [CrossRef]
- van Veggel, B.; de Langen, A.J.; Hashemi, S.M.S.; Monkhorst, K.; Heideman, D.A.M.; Thunnissen, E.; Smit, E.F. Afatinib and Cetuximab in Four Patients with EGFR Exon 20 Insertion-Positive Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Metro, G.; Crinò, L. The LUX-Lung Clinical Trial Program of Afatinib for Non-Small-Cell Lung Cancer. Expert Rev. Anticancer Ther. 2014, 11, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Sequist, L.V.; Tan, E.H.; Geater, S.L.; Orlov, S.; Zhang, L.; Lee, K.H.; Tsai, C.M.; Kato, T.; Barrios, C.H.; et al. Afatinib as First-Line Treatment of Older Patients with EGFR Mutation-Positive Non-Small-Cell Lung Cancer: Subgroup Analyses of the LUX-Lung 3, LUX-Lung 6, and LUX-Lung 7 Trials. Clin. Lung Cancer 2018, 19, e465–e479. [Google Scholar] [CrossRef] [PubMed]
- Tagliamento, M.; Genova, C.; Rijavec, E.; Rossi, G.; Biello, F.; Dal Bello, M.G.; Alama, A.; Coco, S.; Boccardo, S.; Grossi, F. Afatinib and Erlotinib in the Treatment of Squamous-Cell Lung Cancer. Expert Opin. Pharmacother. 2018, 19, 2055–2062. [Google Scholar] [CrossRef]
- Ohashi, K.; Ninomiya, K.; Yoshioka, H.; Bessho, A.; Shibayama, T.; Aoe, K.; Ishikawa, N.; Kozuki, T.; Kawai, H.; Kuyama, S.; et al. Impact of HER2 Expression on EGFR-TKI Treatment Outcomes in Lung Tumors Harboring EGFR Mutations: A HER2-CS Study Subset Analysis. Lung Cancer 2020, 150, 83–89. [Google Scholar] [CrossRef]
- Nagasaka, M.; Singh, V.; Baca, Y.; Sukari, A.; Kim, C.; Mamdani, H.; Spira, A.I.; Uprety, D.; Bepler, G.; Kim, E.S.; et al. The Effects of HER2 Alterations in EGFR Mutant Non-Small Cell Lung Cancer. Clin. Lung Cancer 2022, 23, 52–59. [Google Scholar] [CrossRef]
- Wind, S.; Schnell, D.; Ebner, T.; Freiwald, M.; Stopfer, P. Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. 2017, 56, 235–250. [Google Scholar] [CrossRef]
- Ding, P.N.; Lord, S.J.; Gebski, V.; Links, M.; Bray, V.; Gralla, R.J.; Yang, J.C.H.; Lee, C.K. Risk of Treatment-Related Toxicities from EGFR Tyrosine Kinase Inhibitors: A Meta-Analysis of Clinical Trials of Gefitinib, Erlotinib, and Afatinib in Advanced EGFR-Mutated Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 633–643. [Google Scholar] [CrossRef]
- Han, W.; Du, Y. Recent Development of the Second and Third Generation Irreversible Epidermal Growth Factor Receptor Inhibitors. Chem. Biodivers. 2017, 14, e1600393. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin plus Pemetrexed in Patients with Metastatic Lung Adenocarcinoma with EGFR Mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef]
- Yang, J.C.H.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus Cisplatin-Based Chemotherapy for EGFR Mutation-Positive Lung Adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of Overall Survival Data from Two Randomised, Phase 3 Trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus Cisplatin plus Gemcitabine for First-Line Treatment of Asian Patients with Advanced Non-Small-Cell Lung Cancer Harbouring EGFR Mutations (LUX-Lung 6): An Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Miyahisa, I.; Sameshima, T.; Hixon, M.S. Rapid Determination of the Specificity Constant of Irreversible Inhibitors (Kinact/KI) by Means of an Endpoint Competition Assay. Angew. Chem. 2015, 127, 14305–14308. [Google Scholar] [CrossRef]
- Schwartz, P.A.; Kuzmic, P.; Solowiej, J.; Bergqvist, S.; Bolanos, B.; Almaden, C.; Nagata, A.; Ryan, K.; Feng, J.; Dalvie, D.; et al. Covalent EGFR Inhibitor Analysis Reveals Importance of Reversible Interactions to Potency and Mechanisms of Drug Resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the Aberrant Activity of Mutant EGFR Kinase Domain and Drug Recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef]
- Lategahn, J.; Tumbrink, H.L.; Schultz-Fademrecht, C.; Heimsoeth, A.; Werr, L.; Niggenaber, J.; Keul, M.; Parmaksiz, F.; Baumann, M.; Menninger, S.; et al. Insight into Targeting Exon20 Insertion Mutations of the Epidermal Growth Factor Receptor with Wild Type-Sparing Inhibitors. J. Med. Chem. 2022, 65, 6643–6655. [Google Scholar] [CrossRef]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent Inhibitors Design and Discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef]
- Wissner, A.; Fraser, H.L.; Ingalls, C.L.; Dushin, R.G.; Floyd, M.B.; Cheung, K.; Nittoli, T.; Ravi, M.R.; Tan, X.; Loganzo, F. Dual Irreversible Kinase Inhibitors: Quinazoline-Based Inhibitors Incorporating Two Independent Reactive Centers with Each Targeting Different Cysteine Residues in the Kinase Domains of EGFR and VEGFR-2. Bioorg. Med. Chem. 2007, 15, 3635–3648. [Google Scholar] [CrossRef]
- Blair, J.A.; Rauh, D.; Kung, C.; Yun, C.H.; Fan, Q.W.; Rode, H.; Zhang, C.; Eck, M.J.; Weiss, W.A.; Shokat, K.M. Structure-Guided Development of Affinity Probes for Tyrosine Kinases Using Chemical Genetics. Nat. Chem. Biol. 2007, 3, 229–238. [Google Scholar] [CrossRef]
- Martin, J.S.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising Covalent Warhead Reactivity. Bioorg. Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef]
- Flanagan, M.E.; Abramite, J.A.; Anderson, D.P.; Aulabaugh, A.; Dahal, U.P.; Gilbert, A.M.; Li, C.; Montgomery, J.; Oppenheimer, S.R.; Ryder, T.; et al. Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem. 2014, 57, 10072–10079. [Google Scholar] [CrossRef] [PubMed]
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A Road Map for Prioritizing Warheads for Cysteine Targeting Covalent Inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Abdeldayem, A.; Raouf, Y.S.; Constantinescu, S.N.; Moriggl, R.; Gunning, P.T. Advances in Covalent Kinase Inhibitors. Chem. Soc. Rev. 2020, 49, 2617–2687. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.R.; Mamuya, N.; Johnson, B.D.; Reich, M.F.; Gruber, B.C.; Ye, F.; Nilakantan, R.; Shen, R.; Discafani, C.; DeBlanc, R.; et al. 6-Substituted-4-(3-Bromophenylamino) Quinazolines as Putative Irreversible Inhibitors of the Epidermal Growth Factor Receptor (EGFR) and Human Epidermal Growth Factor Receptor (HER-2) Tyrosine Kinases with Enhanced Antitumor Activity. J. Med. Chem. 2001, 44, 2719–2734. [Google Scholar] [CrossRef] [PubMed]
- Shindo, N.; Fuchida, H.; Sato, M.; Watari, K.; Shibata, T.; Kuwata, K.; Miura, C.; Okamoto, K.; Hatsuyama, Y.; Tokunaga, K.; et al. Selective and Reversible Modification of Kinase Cysteines with Chlorofluoroacetamides. Nat. Chem. Biol. 2019, 15, 250–258. [Google Scholar] [CrossRef]
- Castelli, R.; Bozza, N.; Cavazzoni, A.; Bonelli, M.; Vacondio, F.; Ferlenghi, F.; Callegari, D.; Silva, C.; Rivara, S.; Lodola, A.; et al. Balancing Reactivity and Antitumor Activity: Heteroarylthioacetamide Derivatives as Potent and Time-Dependent Inhibitors of EGFR. Eur. J. Med. Chem. 2019, 162, 507–524. [Google Scholar] [CrossRef]
- OuYang, Y.; Zou, W.; Peng, L.; Yang, Z.; Tang, Q.; Chen, M.; Jia, S.; Zhang, H.; Lan, Z.; Zheng, P.; et al. Design, Synthesis, Antiproliferative Activity and Docking Studies of Quinazoline Derivatives Bearing 2,3-Dihydro-Indole or 1,2,3,4-Tetrahydroquinoline as Potential EGFR Inhibitors. Eur. J. Med. Chem. 2018, 154, 29–43. [Google Scholar] [CrossRef]
- Pawara, R.; Ahmad, I.; Nayak, D.; Wagh, S.; Wadkar, A.; Ansari, A.; Belamkar, S.; Surana, S.; Nath Kundu, C.; Patil, C.; et al. Novel, Selective Acrylamide Linked Quinazolines for the Treatment of Double Mutant EGFR-L858R/T790M Non-Small-Cell Lung Cancer (NSCLC). Bioorg. Chem. 2021, 115, 105234. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel Mutant-Selective EGFR Kinase Inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Zhao, L.; Fan, T.; Shi, Z.; Ding, C.; Zhang, C.; Yuan, Z.; Sun, Q.; Tan, C.; Chu, B.; Jiang, Y. Design, Synthesis and Evaluation of Novel ErbB/HDAC Multitargeted Inhibitors with Selectivity in EGFRT790M Mutant Cell Lines. Eur. J. Med. Chem. 2021, 213, 113173. [Google Scholar] [CrossRef]
- Song, J.; Jang, S.; Lee, J.W.; Jung, D.; Lee, S.; Min, K.H. Click Chemistry for Improvement in Selectivity of Quinazoline-Based Kinase Inhibitors for Mutant Epidermal Growth Factor Receptors. Bioorg. Med. Chem. Lett. 2019, 29, 477–480. [Google Scholar] [CrossRef]
- Sun, M.; Jia, J.; Sun, H.; Wang, F. Design and Synthesis of a Novel Class EGFR/HER2 Dual Inhibitors Containing Tricyclic Oxazine Fused Quinazolines Scaffold. Bioorg. Med. Chem. Lett. 2020, 30, 127060. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, L.; Feng, M.; Yi, Y.; Zhang, W.; Liu, W.; Li, L.; Liu, Z.; Li, Y.; Ma, X. New Acrylamide-Substituted Quinazoline Derivatives with Enhanced Potency for the Treatment of EGFR T790M-Mutant Non-Small-Cell Lung Cancers. Bioorg. Chem. 2018, 77, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Zhang, Q.; Zhang, Y.; Shao, J.; Ding, L.; Tang, C.; Feng, B. Synthesis and Biological Evaluation of New Series of Quinazoline Derivatives as EGFR/HER2 Dual-Target Inhibitors. Bioorg. Med. Chem. Lett. 2022, 67, 128703. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Kumarakulasinghe, N.B.; Huang, Y.Q.; Ang, Y.L.E.; Choo, J.R.E.; Goh, B.C.; Soo, R.A. Third Generation EGFR TKIs: Current Data and Future Directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Pawara, R.; Ansari, A.; Surana, S. Recent Updates on Third Generation EGFR Inhibitors and Emergence of Fourth Generation EGFR Inhibitors to Combat C797S Resistance. Eur. J. Med. Chem. 2017, 142, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Murtuza, A.; Bulbul, A.; Shen, J.P.; Keshavarzian, P.; Woodward, B.D.; Lopez-Diaz, F.J.; Lippman, S.M.; Husain, H. Novel Third-Generation EGFR Tyrosine Kinase Inhibitors and Strategies to Overcome Therapeutic Resistance in Lung Cancer. Cancer Res. 2019, 79, 689–698. [Google Scholar] [CrossRef]
- Remon, J.; Steuer, C.E.; Ramalingam, S.S.; Felip, E. Osimertinib and Other Third-Generation EGFR TKI in EGFR-Mutant NSCLC Patients. Ann. Oncol. 2018, 29, i20–i27. [Google Scholar] [CrossRef]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Ward, R.A.; Anderton, M.J.; Ashton, S.; Bethel, P.A.; Box, M.; Butterworth, S.; Colclough, N.; Chorley, C.G.; Chuaqui, C.; Cross, D.A.E.; et al. Structure- and Reactivity-Based Development of Covalent Inhibitors of the Activating and Gatekeeper Mutant Forms of the Epidermal Growth Factor Receptor (EGFR). J. Med. Chem. 2013, 56, 7025–7048. [Google Scholar] [CrossRef]
- Yver, A. Osimertinib (AZD9291)-a Sciencedriven, Collaborative Approach to Rapid Drug Design and Development. Ann. Oncol. 2016, 27, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA) Label—TAGRISSO® (Osimertinib) 80 Mg and 40 Mg Tablets, for Oral Use. Available online: www.fda.gov/medwatch (accessed on 21 February 2023).
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR -Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Zhou, C.; Kim, T.M.; Kim, S.-W.; Yang, J.C.-H.; Riely, G.J.; Mekhail, T.; Nguyen, D.; García Campelo, M.R.; Felip, E.; et al. Mobocertinib (TAK-788) in EGFR Exon 20 Insertion (Ex20ins)+ Metastatic NSCLC (MNSCLC): Additional Results from Platinum-Pretreated Patients (Pts) and EXCLAIM Cohort of Phase 1/2 Study. J. Clin. Oncol. 2021, 39, 9014. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Vincent, S.; Baker, T.E.; Gould, A.E.; Li, S.; Wardwell, S.D.; Nadworny, S.; Ning, Y.; Zhang, S.; Huang, W.S.; et al. Mobocertinib (Tak-788): A Targeted Inhibitor of Egfr Exon 20 Insertion Mutants in Non–Small Cell Lung Cancer. Cancer Discov. 2021, 11, 1672–1687. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA) Label—EXKIVITY® (Mobocertinib) 40 Mg Capsules, for Oral Use. Available online: www.fda.gov/medwatch (accessed on 7 March 2023).
- Zhang, S.S.; Zhu, V.W. Spotlight on Mobocertinib (TAK-788) in NSCLC with EGFR Exon 20 Insertion Mutations. Lung Cancer Targets Ther. 2021, 12, 61–65. [Google Scholar] [CrossRef]
- Starrett, J.H.; Guernet, A.A.; Cuomo, M.E.; Poels, K.E.; van Alderwerelt van Rosenburgh, I.K.; Nagelberg, A.; Farnsworth, D.; Price, K.S.; Khan, H.; Ashtekar, K.D.; et al. Drug Sensitivity and Allele Specificity of First-Line Osimertinib Resistance EGFR Mutations. Cancer Res. 2020, 80, 2017–2030. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non-Small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [PubMed]
- Chic, N.; Mayo-de-las-Casas, C.; Reguart, N. Successful Treatment with Gefitinib in Advanced Non–Small Cell Lung Cancer after Acquired Resistance to Osimertinib. J. Thorac. Oncol. 2017, 12, e78–e80. [Google Scholar] [CrossRef]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef]
- Sullivan, I.; Planchard, D. Osimertinib in the Treatment of Patients with Epidermal Growth Factor Receptor T790M Mutation-Positive Metastatic Non-Small Cell Lung Cancer: Clinical Trial Evidence and Experience. Ther. Adv. Respir. Dis. 2016, 10, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Scalvini, L.; Castelli, R.; La Monica, S.; Tiseo, M.; Alfieri, R. Fighting Tertiary Mutations in EGFR-Driven Lung-Cancers: Current Advances and Future Perspectives in Medicinal Chemistry. Biochem. Pharmacol. 2021, 190, 114643. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xu, B.; Wang, J.; Gao, Y.; He, X.; Xie, T.; Ye, X.Y. Recent Advances of Novel Fourth Generation EGFR Inhibitors in Overcoming C797S Mutation of Lung Cancer Therapy. Eur. J. Med. Chem. 2023, 245, 114900. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Yao, M.Y.; Zhu, S.J.; Chen, J.Y.; Yun, C.H. Crystal Structure of EGFR T790M/C797S/V948R in Complex with EAI045. Biochem. Biophys. Res. Commun. 2018, 502, 332–337. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, T.; Li, S.; Tong, L.; Li, J.; Su, Z.; Feng, F.; Sun, D.; Tong, Y.; Wang, X.; et al. Discovery of Potent and Noncovalent Reversible EGFR Kinase Inhibitors of EGFRL858R/T790M/C797S. ACS Med. Chem. Lett. 2019, 10, 869–873. [Google Scholar] [CrossRef]
- Zhou, Z.; He, J.; Yang, F.; Pan, Q.; Yang, Z.; Zheng, P.; Xu, S.; Zhu, W. Design, Synthesis and Evaluation of Anti-Proliferative Activity of 2-Aryl-4-Aminoquinazoline Derivatives as EGFR Inhibitors. Bioorg. Chem. 2021, 112, 104848. [Google Scholar] [CrossRef]
- Park, H.; Jung, H.Y.; Mah, S.; Hong, S. Discovery of EGF Receptor Inhibitors That Are Selective for the D746-750/T790M/C797S Mutant through Structure-Based de Novo Design. Angew. Chem. Int. Ed. Engl. 2017, 56, 7634–7638. [Google Scholar] [CrossRef]
- Sogabe, S.; Kawakita, Y.; Igaki, S.; Iwata, H.; Miki, H.; Cary, D.R.; Takagi, T.; Takagi, S.; Ohta, Y.; Ishikawa, T. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]Pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Med. Chem. Lett. 2012, 4, 201–205. [Google Scholar] [CrossRef]
- Kong, L.L.; Ma, R.; Yao, M.Y.; Yan, X.E.; Zhu, S.J.; Zhao, P.; Yun, C.H. Structural Pharmacological Studies on EGFR T790M/C797S. Biochem. Biophys. Res. Commun. 2017, 488, 266–272. [Google Scholar] [CrossRef]
- Dou, D.; Wang, J.; Qiao, Y.; Wumaier, G.; Sha, W.; Li, W.; Mei, W.; Yang, T.; Zhang, C.; He, H.; et al. Discovery and Optimization of 4-Anilinoquinazoline Derivatives Spanning ATP Binding Site and Allosteric Site as Effective EGFR-C797S Inhibitors. Eur. J. Med. Chem. 2022, 244, 114856. [Google Scholar] [CrossRef] [PubMed]

























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).