1. Introduction
Flavonoids, compounds that are secondary metabolites from plants, have a nucleus which consists of A, B and C rings. A series of modification reactions, such as hydroxylation, glycosylation, prenylation, and methylation, can enhance multiple physiological functions corresponding to both their structural diversity and tissue specificities. The
O-methylation of aglycone flavonoids, such as Quercetin (Q), results in the reduction of the molecular activity of a hydroxyl fraction and the consequent increase in lipophilicity, which modifies its intracellular compartmentalization. Furthermore,
O-methylation provides a branch point in the biosynthesis of several metabolic pathways, including production of modified flavonoids with increased antimicrobial properties [
1]. Considered a post-modification product, these derivatives are formed through the methyl group fixation with oxygen at the flavonoid hydroxyl moiety. Due to the diverse hydroxyl groups in the flavonoid core, flavonoids’ methylation positions are diverse and provide multiple health benefits, such as increased bioavailability compared to flavonoid precursors [
2]. Therefore, these methylated flavonoids are potential candidates for use as anti-inflammatories by decreasing the enzymatic activity of enzymes such as cyclooxygenases (COX), pro-inflammatory interleukins, reactive oxygen species (ROS) and nitrogen (RNS) production [
2,
3,
4]. Previous analyzes using ChEMBL and SwissTargetPrediction tools reveals that both Rhamnetin (Rhm; 7-
O-Methylquercetin); 3-
O-Methylquercetin (3MQ; 3-
O-Methylquercetin) are compounds with an antioxidant capacity, besides to exhibits anti-inflammatory potential by strongly decreasing the cyclooxygenase (COX) and Lipoxygenase (LOX) activity. Rhamnazin (Rhz; 7,3’-Di-
O-methylquercetin) exhibits two methylations, which seem to considerably increase the ability to sequester free radicals in cells. Methylated derivatives of Q are also found in some plant species, such as
Coriandrum sativum, Achyrocline satureioides, and
Rhamnus petiolaris with Rhm, 3MQ, and Rhz compounds, respectively, which have already shown different responses against BthTX-II activities [
5].
Snake venoms consist of a complex mixture of biologically active molecules, and the phospholipase A2 (PLA2) group is one of the most studied toxins. There are two main groups of PLA2 (E.C. 3.1.1.4) in snake venoms: phospholipase A2-like (PLA2-like), such as the Lys49-PLA2 and the classic phospholipase A2, Asp49-PLA2. These proteins belong to the secreted PLA2 (sPLA2), a subgroup found in diverse secretions, body fluids and venom of bees, scorpions, and snakes [
6]. Therefore, these macromolecules hydrolyze the sn-2 position of glycerophospholipids, leading to a release of fatty acids, such as arachidonic acid (AA) and lysophospholipids, triggering the inflammatory process [
7,
8]. Bothropstoxin II (BthTX-II) is an Asp49-PLA2 from
Bothrops jararacussu (Bj), which belongs to the Viperidae family. This toxin is considered a PLA2-like due to some characteristics, such as the Ca
2+ binding loop distortion, leading to changes in the C-terminal region. In this way, this protein shows a low phospholipase A2 activity besides to exhibit myotoxic, edematogenic, and hemolytic effects [
9,
10]. Bothropstoxin I (BthTX-I) is a Lys49-PLA2, which reveals high myotoxic activity with a lack of enzymatic activity. The myotoxic mechanism of these enzymes has already been demonstrated and includes mainly some amino acids from the C-terminal region, which interacts with membrane [
8].
Crotalus durissus terrificus (Cdt) belongs to the same family and has an essential heterodimeric complex named Crotoxin in its venom, which is a potent β-neurotoxin with a phospholipase A2 activity. This toxin consists of two subunits: a basic pla2 with a weak neurotoxicity, named crotoxin B (CB) or Cdt PLA2, associated with a small acidic, nontoxic and nonenzymatic protein named crotapotin or crotoxin A (CA) [
11]. CB presents four isoforms, CBa2, CBb, CBc, and CBd, performing 16 different CA-CB complexes. To date, there are three crystal structures from these isomers: one of them includes CA2 and CBb, the other with CBd (tetramer), and the structure used in this study consists of isoforms CBa2 and CBc (tetramer) [
12].
Commercial inhibitors of PLA2, such as p-bromophenacyl bromide (BPB) and Varespladib (Var), have already shown their potential against PLA2 from diverse snake venoms. The classical inhibitor BPB has been used since 1970 to inhibit the catalytic PLA2 once it binds specifically with the His48 residue. However, this compound has already been shown to inhibit myotoxic activity of PrTX-I, a Lys49-PLA2 from
Bothrops pirajai, through a covalent binding to His48, leading to a Ca
2+-binding loop distortion and then, a C-terminus rearrangement [
13]. Var (LY315920) is a synthetic molecule which was clinically tested to block the inflammatory cascade initiated by secreted PLA2. There are several studies that reveal the efficacy of this compound in the treatment of phospholipase A2-rich snake venoms [
14]. In snake venoms, this compound has already been shown to inhibit the cytotoxic e myotoxic effect of MjTX-II from
Bothrops moojeni through the physical blockage of its allosteric activation [
15]. In addition to this, a synergic effect of this compound with the antivenom was observed to decrease neuromuscular blockage induced by crotamine, highlighting the broad-spectrum effect of this drug [
16].
Due to PLA2 role in the inflammatory process, the abundance of this toxin in the Cdt and Bj venoms, and the fact that these proteins show similar structures with sPLA2 from mammals [
12], it is essential to find new inhibitors of this enzyme. Hence, in this study, we performed some steps to better understand how these compounds could interact with these three phospholipases A2. First, we aim to elucidate the main protein’s regions to anchor with an inhibitor using CavityPlus. Afterwards, using the phosphonate transition-state analogue from 1POB crystal structure, named L-1-0-octyl-2-heptylphos-phonyl-sn-glycero-3-phosphoethanolamine (Analogue) [
17], we focus on the elucidation of the main residues that interact with each toxin. Moreover, two commercial inhibitors were used for comparison purposes to identify the main residues involved in the anchoring with all toxins. Finally, we intend to compare the compounds’ interactions with phospholipase A2 from
Bothrops jararacussu and
Crotalus durissus terrificus with the analogue and the inhibitors. Thus, we analyze if compounds anchor equally with all toxins, and if not, we try to elucidate main differences and the reasons for that.
3. Discussion
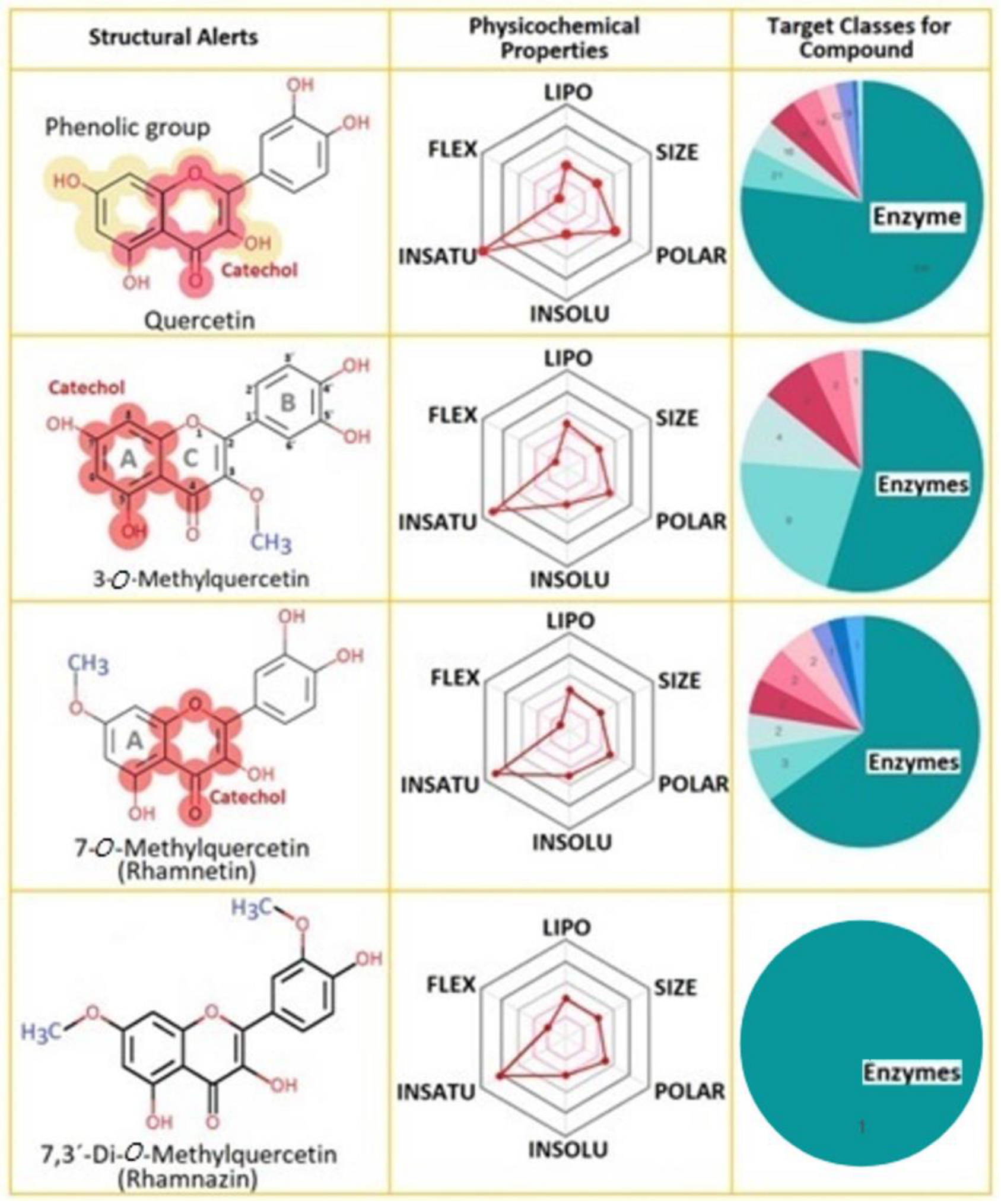
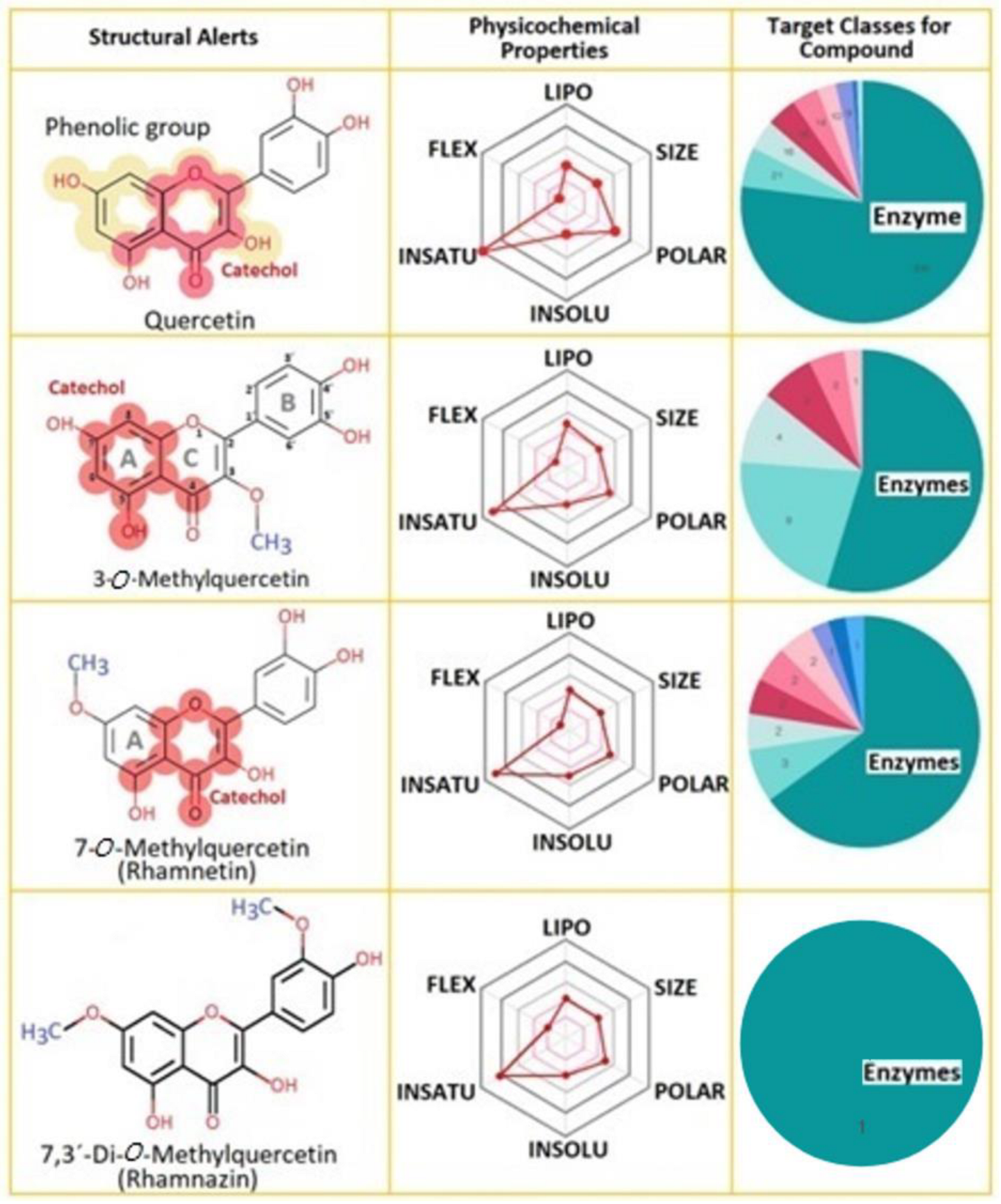
To date, diverse in silico tools help to evaluate molecular structures, mainly to select compounds which exhibit great possibility to become an effective drug [
13]. These tools also have an essential role in identifying the interaction of target compounds [
20]. The TPSA is an important descriptor to understand the specific regions of the protein-compound’s interactions, and Rhz shows the smallest value. Rhm and 3MQ show a similar value, and Q exhibits the highest polar area, which exceeds the range (20–130 Å
2) [
13]. Therefore, lipophilicity could be another descriptor to support the results obtained in the molecular docking analysis. Diverse natural compounds, such as flavonoids, exhibit an inhibitory potential against sPLA2 activity that seems to be dependent on the 5-hydroxyl group, besides the double bond and the double-bonded oxygen in the oxane ring and the hydroxyl groups at the 3′ and 4′ position [
21]. Q is widely found as a secondary metabolite in fruits, vegetables, and flowers. Its structure consists of three rings, consisting of a basic nucleus of a phenyl benzo (γ) pyrone and the side groups usually are hydroxyl, glycosyl, or methoxyl [
22].
In recent studies, the search to provide an alternative or complementary treatment to antivenom therapy using synthetic and natural compounds has been the subject of investigation. Var is a synthetic compound known for its inhibitory potential against human-secreted groups IIA PLA2. Due to their high structural homology with PLA2 from snake venoms, studies were made to verify its inhibitory potential [
10]. Furthermore, this compound has already been shown to inhibit myotoxins revealing great contacts with Gly30, Lys49 and His48 of MjTX-I and MjTX-II from
Bothrops moojeni [
15]. Additionally, Var also shows to anchor with other myotoxins, such as PrTX-I from
Bothrops pirajai and BthTX-I from
Bothrops jararacussu [
10]. Herein, results with inhibitor Var reveal that the compound shows higher affinity by BthTX-I from
B. jararacussu, with great contacts with His48, Lys49, and Gly30, residues also observed in the analogue interactions and in other phospholipases from some
Bothrops sp venoms [
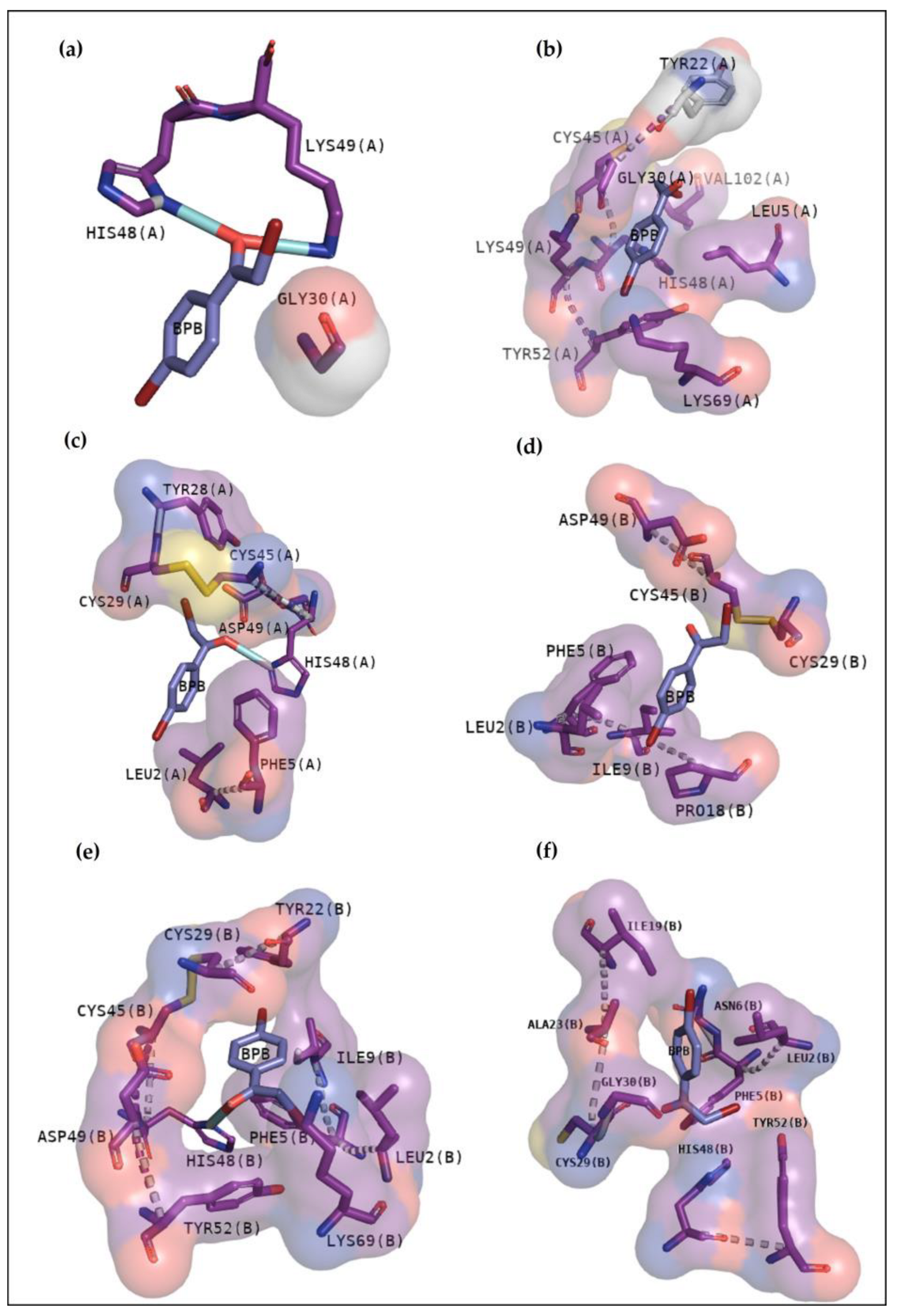
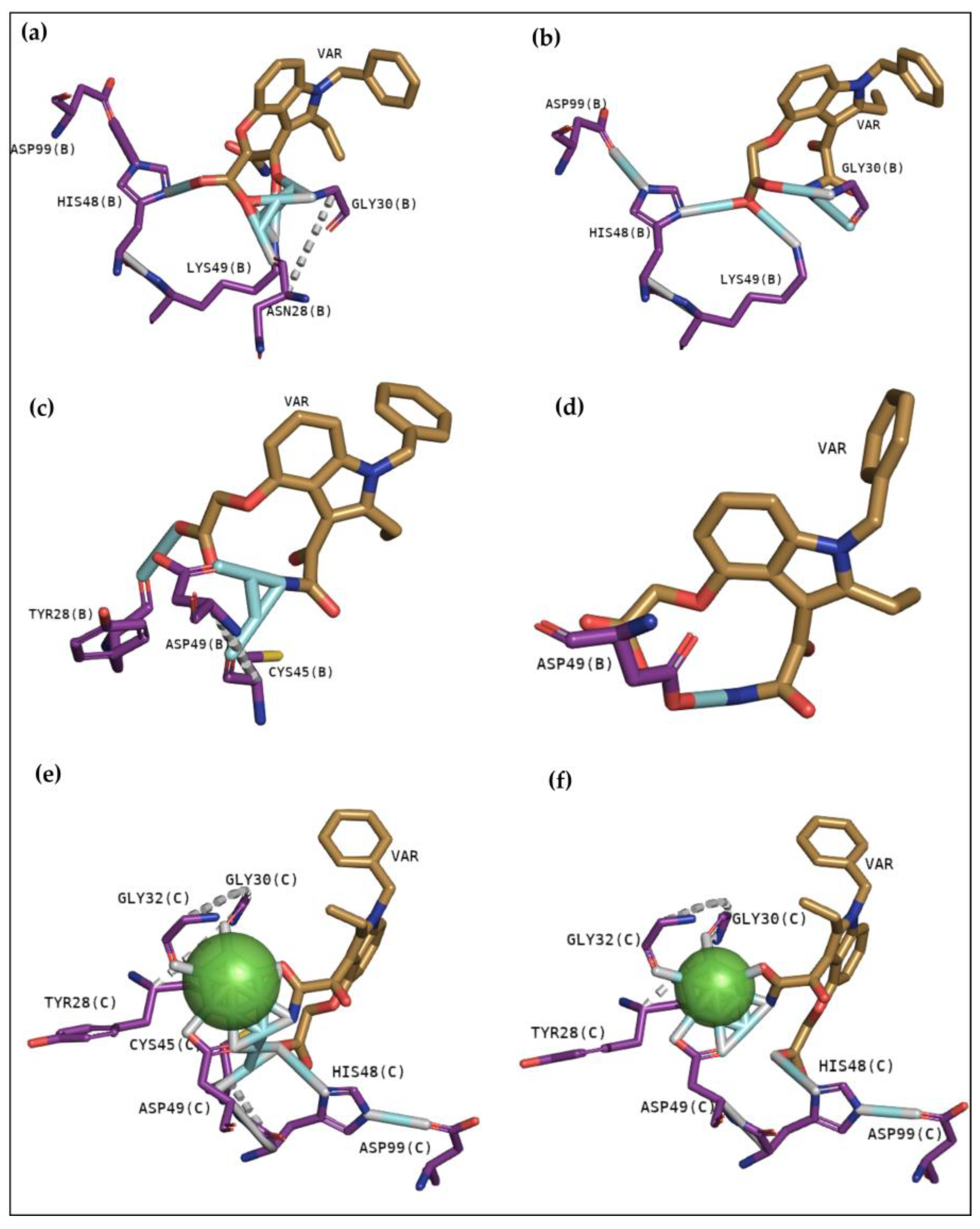
15]. Although BPB interacted with essential amino acids to anchor with the analogue (
Figure 3), it exhibits lower values of affinity when compared with Var. This inhibitor is known to decrease the enzymatic activity of phospholipase A2, binding covalently to His48 of PrTX-I [
13,
19]. This interaction leads to a distortion of the Ca
2+-binding loop plus a C-terminus rearrangement, decreasing the myotoxic activity of this protein. In this study, BPB also exhibits interactions with His48, Lys49, Leu5, Gly30, and Lys69 of BthTX-I and with the main residues of BthTX-II and CdtsPLA2 active site. In addition, earlier studies reveal that BPB also fits well in the hydrophobic channel with extensive hydrophobic interactions with the surrounding residues, especially Phe5, Cys45, and Gly30 from bovine pancreatic PLA
2, besides reducing edema induced by PrTX-I in rat and rabbit [
22,
23]. Moreover, it has already demonstrated the His48 chemical modification of the acid phospholipase A2 from
B. jararacussu using BPB [
24].
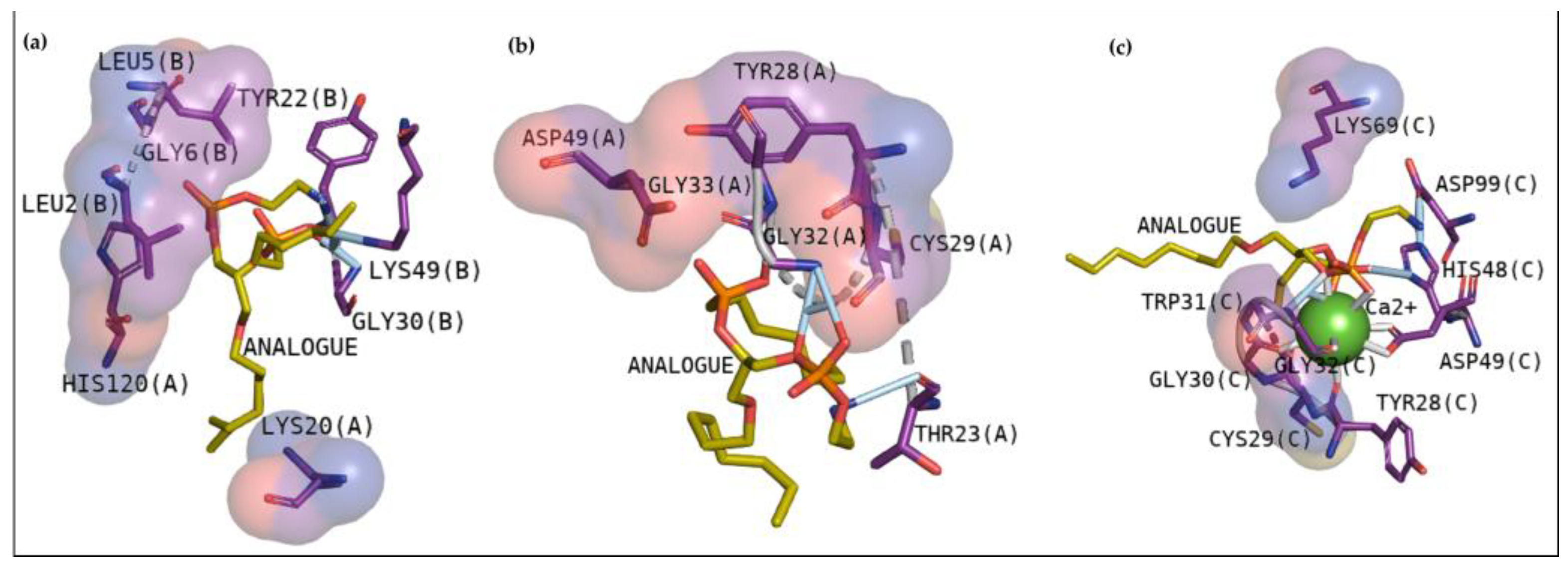
Cavity analysis has been used to identify potential binding sites on the protein surface besides ranking them based on ligandability and druggability scores [
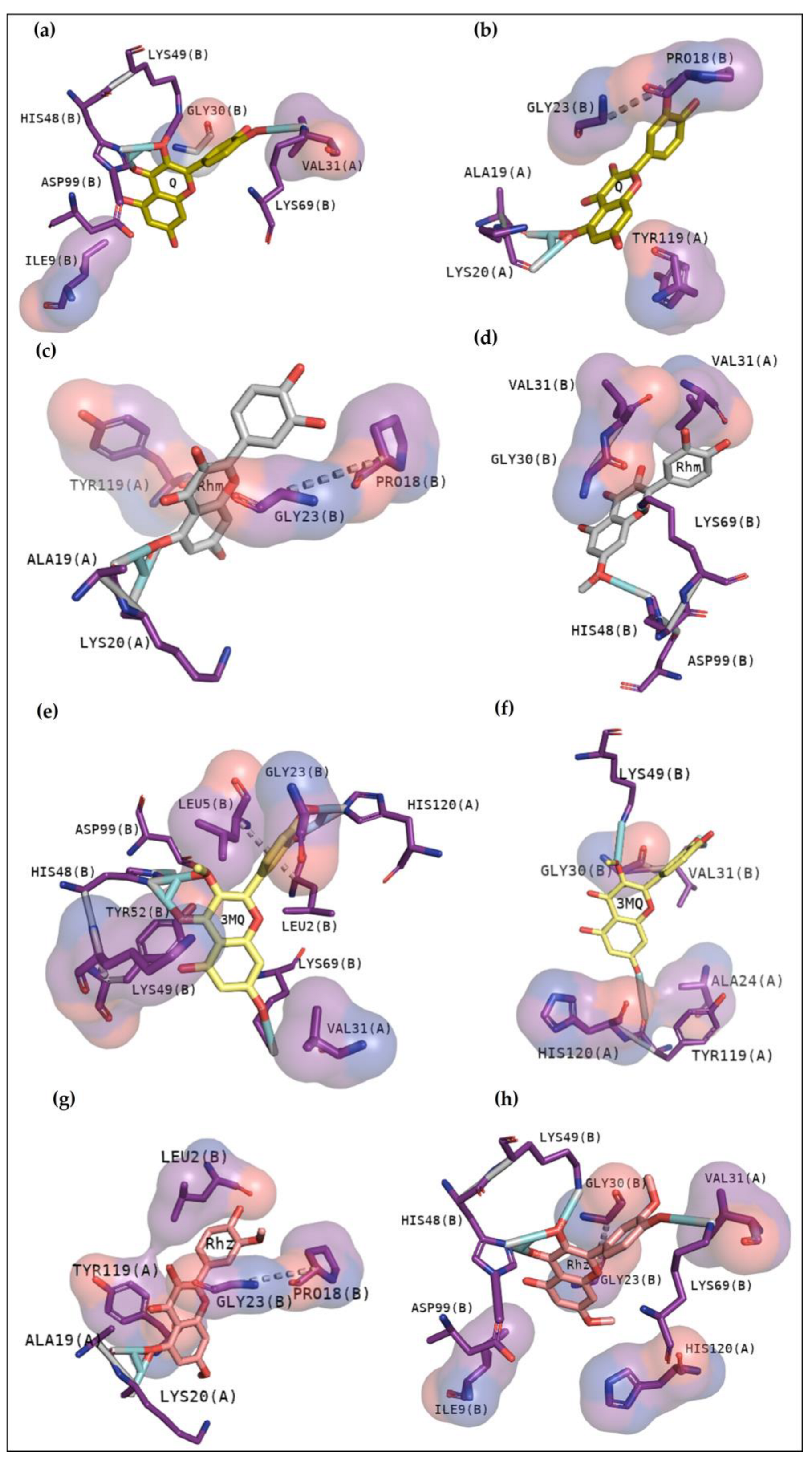
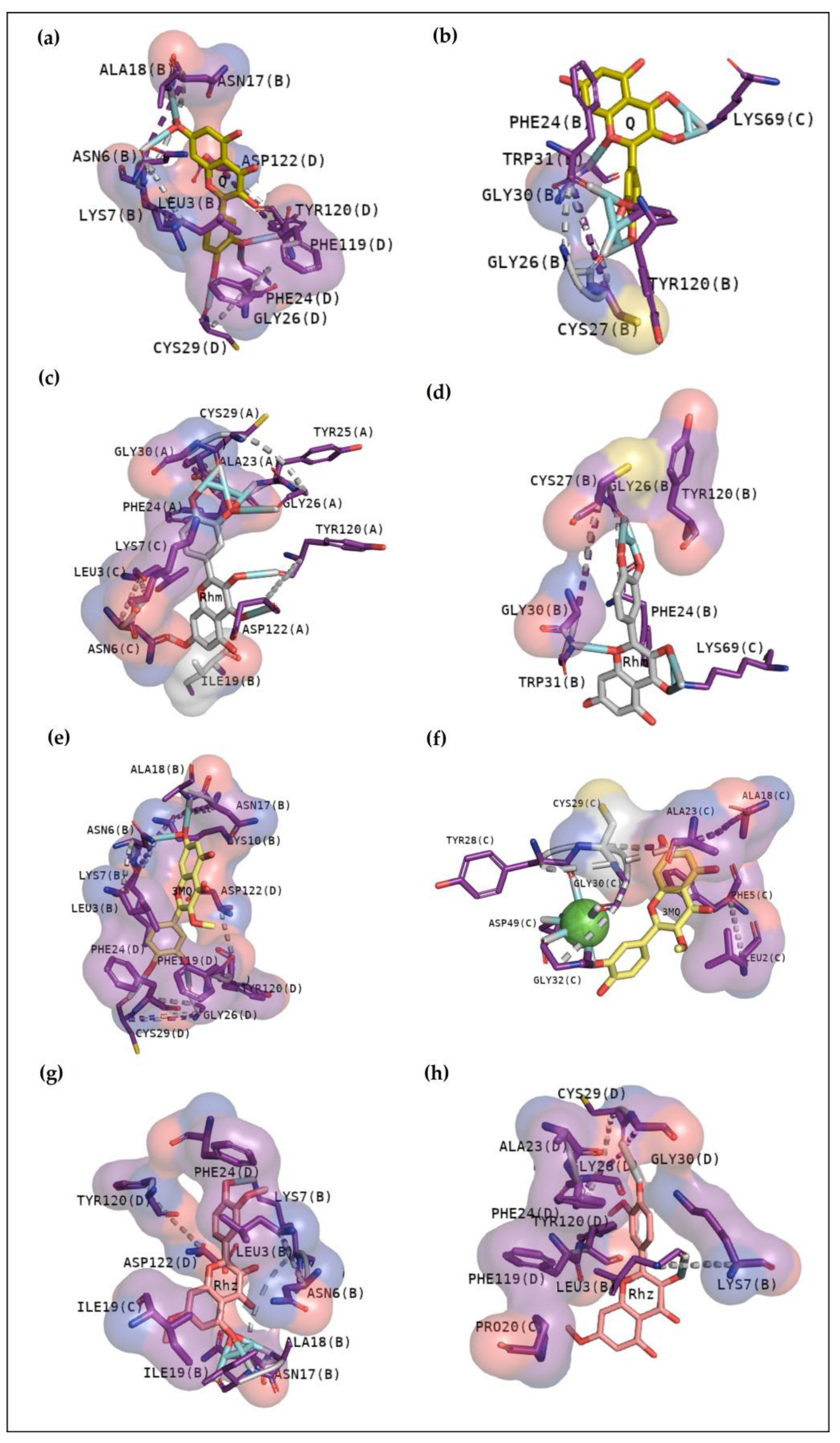
25]. In this study, this web server helped to confirm and guide the best sites of the protein to focus on molecular docking. To better understand the compounds’ potential to interact with these toxins, it is essential to emphasize the residues involved in the anchoring between these three toxins with the phosphonate transition-state analogue. In
Figure 2a, BthTX-I revealed that Lys20, Gly30, Lys49, Tyr22, His120, and some N-terminal residues are involved in anchoring the compound-toxin. BthTX-II exhibited contacts with Tyr28, Gly32,33, and Asp49, and CdtsPLA2 shows great interactions with Gly30, 32, His48, and Asp49. Similarly, Q shows great contact with some of these residues of BthTX-I, such as Lys20, Lys49, Gly30, and His48. Quercetin has already been shown to bind with the dimmer interface and active site of MTX-II, a Lys-49 PLA2 from the
Bothrops brazili venom [
26]. Furthermore, in this study, Q fits well in the active site of BthTX-II, and these data are supported by a previous study in which the compound inhibited the protein enzymatic activity [
5]. Q with CdtsPLA2 exhibited similar interactions with BPB, such as Lys69, Gly30, and some residues of the N-terminal region. In addition, Q anchors in the interface between monomers B and D in both analyses, with a great value of affinity, higher than that observed with BthTX-II. This toxin in the solution presents dimeric or tetrameric oligomers [
27]. Hence, it may be necessary to use a higher concentration of inhibitor compared to that was used in BthTX-II, once the protein is primarily in a monomeric form in its relaxed state with a fatty acid in its hydrophobic channel [
28].
Rhm reveals a different region with high affinity with BthTX-I, the interface between chain A and B, with hydrogen bonds with Lys20 and Gly30, residues that match the phospholipid analogue interaction. Hydrophobic contacts can also be compared since the Tyr119 residue next to His120 presented in
Figure 2b and Gly30 are included in Rhm with BthTX-II. Rhm has already revealed an antimyotoxic activity against BthTX-II in previous work [
5] and could be a potential inhibitor of BthTX-I. The dimeric BthTX-II (tense-state) is necessary for myotoxic activity [
8], hence, compounds that change this conformation could decrease its activity. This compound also shows contacts with Gly30 and other important residues, such as Trp31, Tyr120, and the N-terminal region, similar to the amino acids involved in the analogue and myotoxin, BthTX-I. There is no data concerning the inhibitory potential of this compound with CdtsPLA2; however, it has already been shown to decrease the inflammatory cytokines levels and oxidative stress in the mice aortic tissue, besides to inhibit enzymatic, edematogenic, and myotoxic effect of BthTX-II [
5,
29].
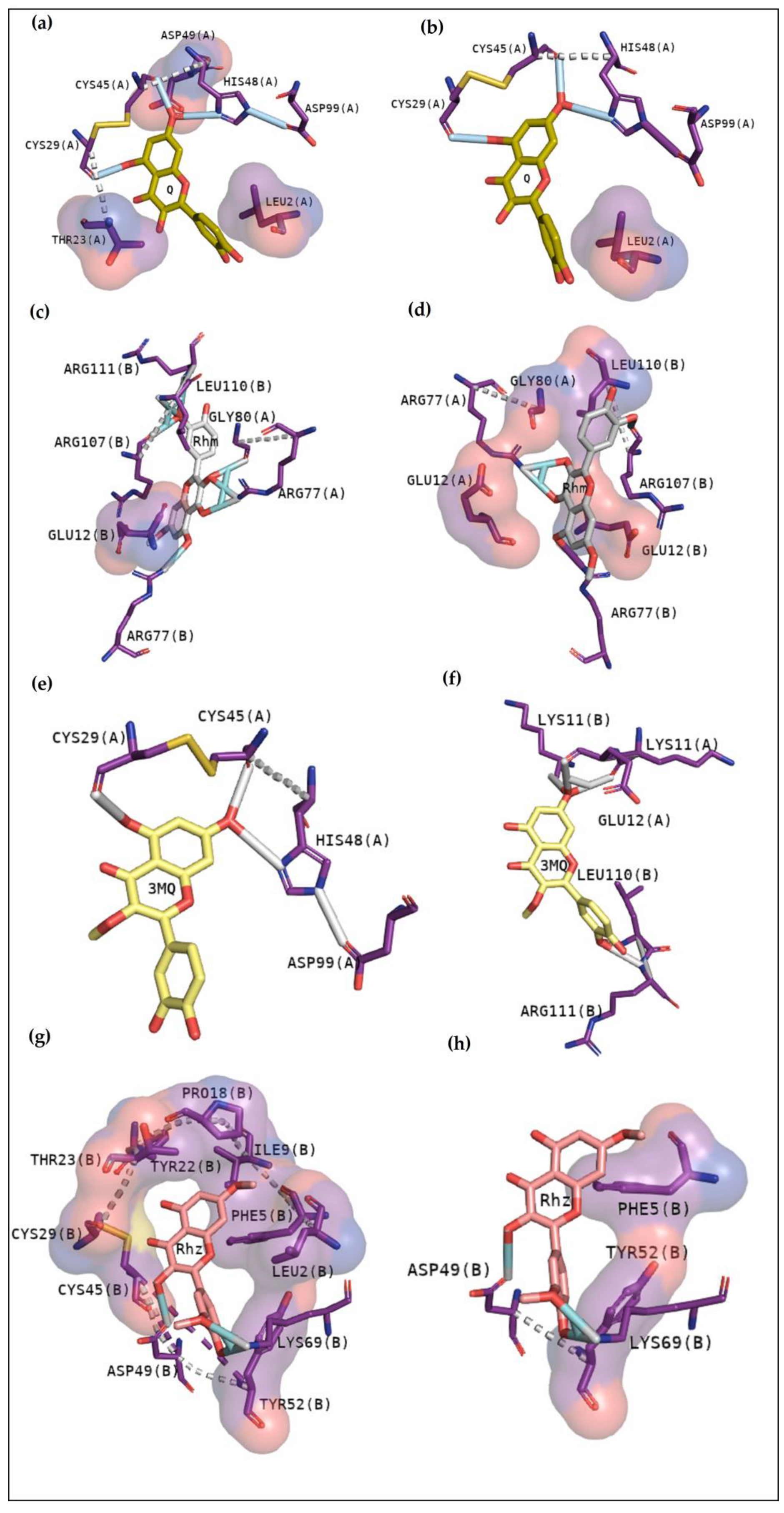
As observed in Rhm, Rhz shows similar interactions with CdtsPLA2. However, it is possible to notice more hydrophobic contact, and contacts with residues are not found in the active site. In addition, Rhz reveals an anchoring in the active site in BthTX-II; in fact, it has already been shown to inhibit the enzymatic activity of this protein [
5]. Rhz and Rhm anchor between chains A and B of the BthTX-I and exhibits some important residues in common that were involved in this contact, such as His120, Lys69, His48, Val31, Gly30, and Lys20.
Figure 3e,f exhibits that 3MQ also fits in the phospholipid channel, highlighting Lys49, N-terminal residues and His120 of BthTX-I. BthTX-II, besides its myotoxic activity, exhibits low enzymatic activity, and
Figure 2b, exhibits the phospholipid analogue in the catalytic site and 3MQ shows to bind in this region. However, it reveals a lower affinity value and fewer interactions. Therefore, this compound has already been shown to poorly inhibit BthTX-II catalytic activity [
5]. In a different manner, 3MQ exhibits great interactions in the active site of CdtsPLA2, with the same residues observed in the anchoring between the protein-analogue and protein-commercial inhibitors.
Considering that all these three toxins are from two different species of snake from the Viperidae family, and PLA2 is one of the most abundant components of both Cdt and Bj venoms, it is essential to investigate how distinct compounds can interact with different macromolecules [
30,
31]. These data indicate that Q, Rhm, 3MQ, and Rhz can anchor in different manners with each toxin. Studies concerning the structural interactions with in vitro analysis, besides the pharmacological assays, could help to better understand diverse mechanisms of inhibition of these compounds. Therefore, our results show that a great candidate to inhibit BthTX-I must interact with residues key-residues cited. BthTX-I docking analysis shows quercetin derivatives could potentially diminish the myotoxic activity by anchoring in the phospholipid hydrophobic channel or interacting with residues next to C-terminal region, which is an essential area to execute the biological effects of this toxin [
10]. Nevertheless, it is necessary to perform in vitro and in vivo assays with BthTX-I and CdtsPLA2 to correlate and confirm all the results.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}