
Network Biology-Inspired Machine Learning Features Predict Cancer Gene Targets and Reveal Target Coordinating Mechanisms

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
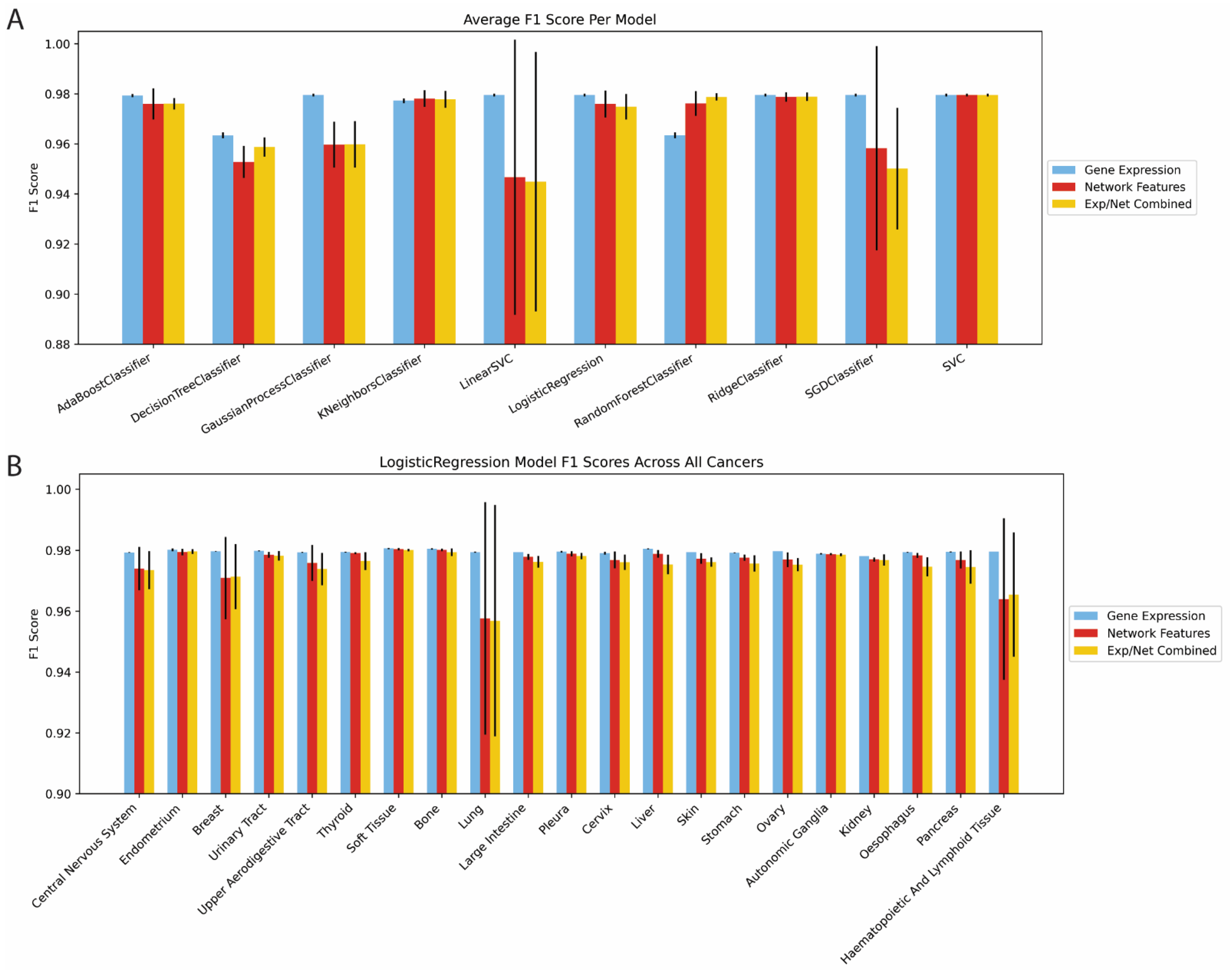
2.1. Network-Based Features Are Predictive of Binary Gene Dependency
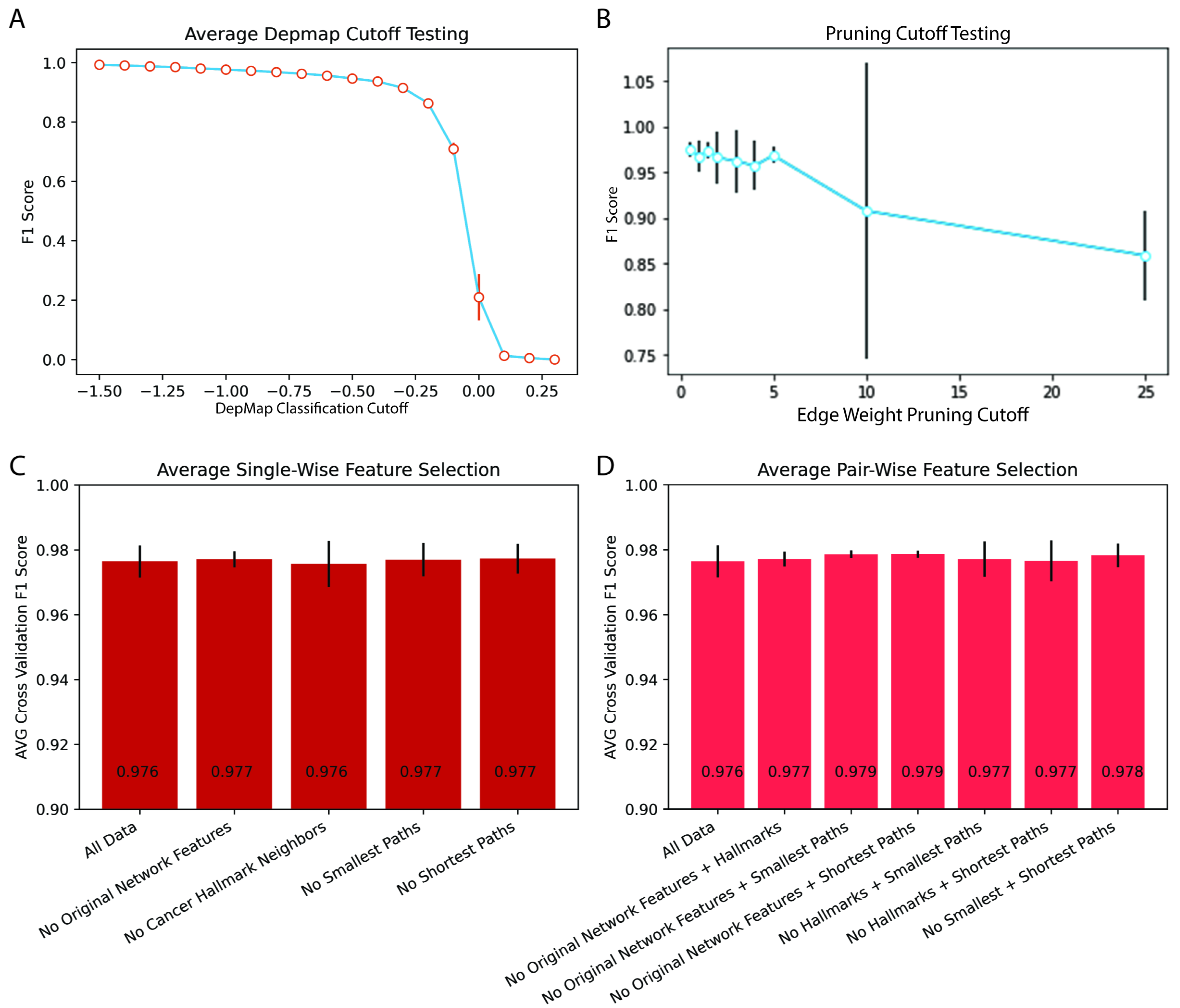
2.2. Network-Based Prediction Was Stable across a Range of Network Construction and Dependency Hyperparameters
2.3. Traditional Network and Biological Hybrid Features Encoded Overlapping Information Predictive of Gene Dependency
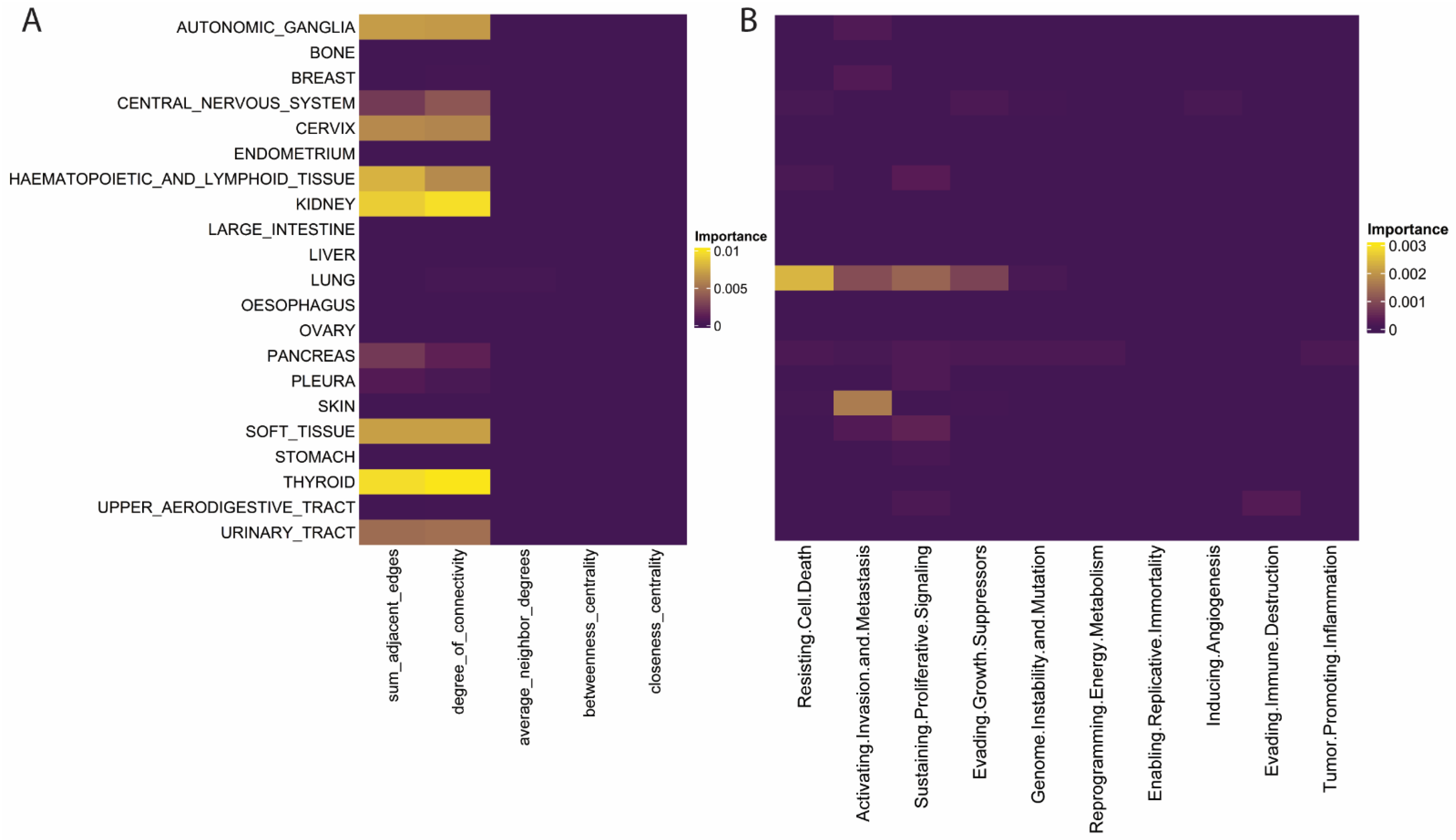
2.4. Specific Features within the Larger Feature Classes Demonstrate Histology Specific Importance
3. Discussion
4. Materials and Methods
4.1. Key Packages
4.2. Data Sources and Preprocessing
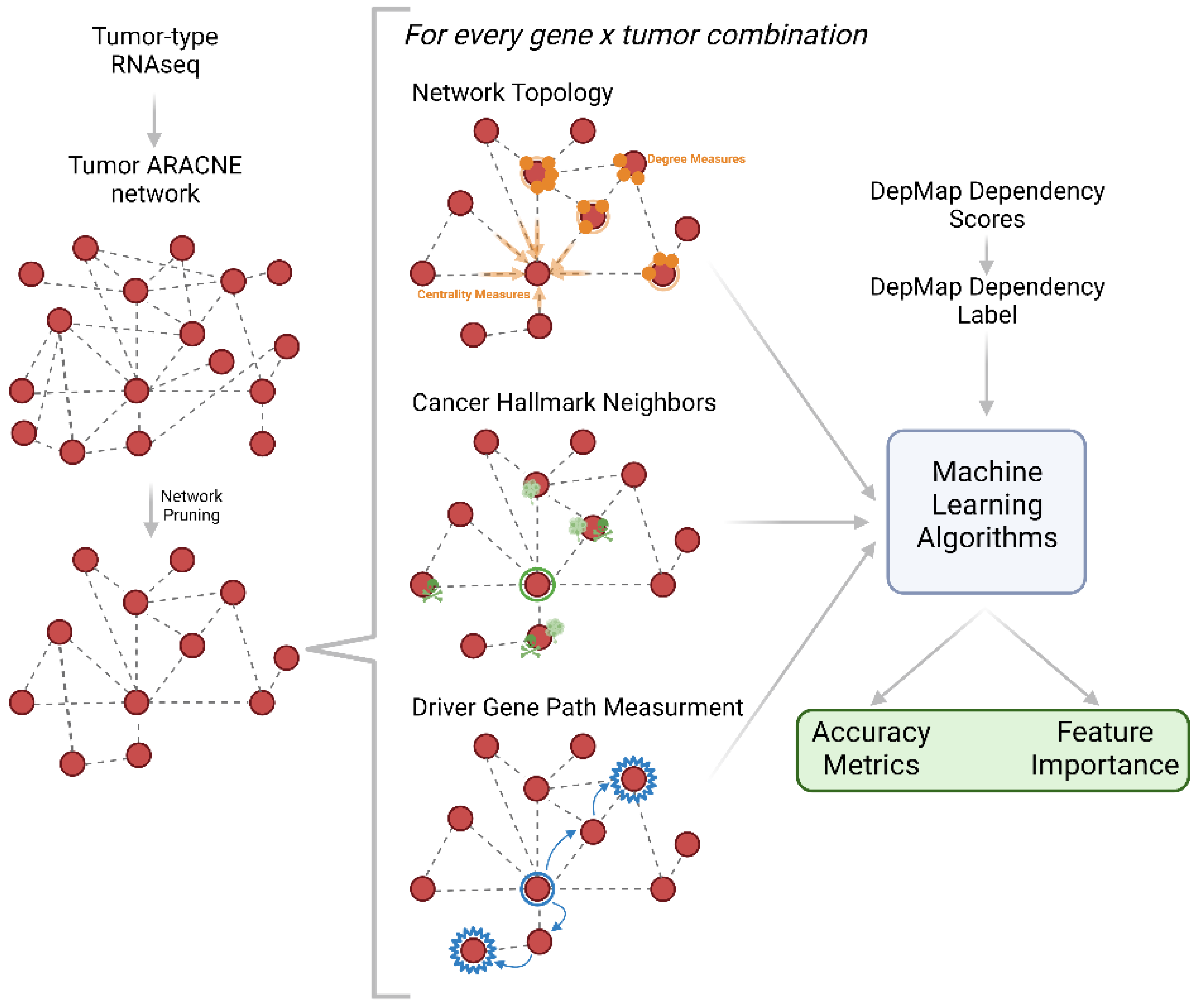
4.3. Cancer Type Network Construction
4.4. Training and Testing Data
4.5. Labeling Gene Dependency
4.6. Machine Learning Modeling and Feature Importance
4.7. Testing Hyperparameter Effects on Efficacy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Paolella, B.R.; Asfaw, A.; Rothberg, M.V.; Skipper, T.A.; Langan, C.; Mesa, G.; Gonzalez, A.; Surface, L.E.; Ito, K.; et al. Phosphate Dysregulation via the XPR1–KIDINS220 Protein Complex Is a Therapeutic Vulnerability in Ovarian Cancer. Nat. Cancer 2022, 3, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Krill-burger, J.M.; Dempster, J.M.; Borah, A.A.; Paolella, B.R.; David, E.; Golub, T.R.; Boehm, J.S.; Hahn, W.C.; Mcfarland, J.M.; Vazquez, F.; et al. Partial Gene Suppression Improves Identification of Cancer Vulnerabilities when CRISPR-Cas9 Knockout Is Pan-Lethal. bioRxiv, 2022; 02.482624. [Google Scholar] [CrossRef]
- Ito, T.; Young, M.J.; Li, R.; Jain, S.; Wernitznig, A.; Krill-Burger, J.M.; Lemke, C.T.; Monducci, D.; Rodriguez, D.J.; Chang, L.; et al. Paralog Knockout Profiling Identifies DUSP4 and DUSP6 as a Digenic Dependence in MAPK Pathway-Driven Cancers. Nat. Genet. 2021, 53, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, Z.; Zhang, Z.; Goss, L.; McFarland, J.; Nagaraja, A.; Xie, Y.; Gu, S.; Peng, K.; Zeng, Y.; et al. Pan-ERBB Kinase Inhibition Augments CDK4/6 Inhibitor Efficacy in Oesophageal Squamous Cell Carcinoma. Gut 2022, 71, 665–675. [Google Scholar] [CrossRef]
- Chiu, Y.C.; Zheng, S.; Wang, L.J.; Iskra, B.S.; Rao, M.K.; Houghton, P.J.; Huang, Y.; Chen, Y. Predicting and Characterizing a Cancer Dependency Map of Tumors with Deep Learning. Sci. Adv. 2021, 7, eabh1275. [Google Scholar] [CrossRef]
- Dempster, J.M.; Krill-Burger, J.M.; McFarland, J.M.; Warren, A.; Boehm, J.S.; Vazquez, F.; Hahn, W.C.; Golub, T.R.; Tsherniak, A. Gene Expression Has More Power for Predicting in Vitro Cancer Cell Vulnerabilities than Genomics. bioRxiv 2020, 21.959627. [Google Scholar] [CrossRef]
- Meng-Lin, K.; Ung, C.; Weiskittel, T.; Chen, A.; Zhang, C.; Correia, H.; Li, H. Machine Learning and Systems Biology Approaches to Characterizing Dosage Based Dependencies in Cancer Cells. J. Bioinform. Syst. Biol. 2021, 4, 13–32. [Google Scholar]
- Lin, C.H.; Lichtarge, O. Using Interpretable Deep Learning to Model Cancer Dependencies. Bioinformatics 2021, 37, 2675–2681. [Google Scholar] [CrossRef]
- Nabirotchkin, S.; Peluffo, A.E.; Rinaudo, P.; Yu, J.; Hajj, R.; Cohen, D. Next-Generation Drug Repurposing Using Human Genetics and Network Biology. Curr. Opin. Pharmacol. 2020, 51, 78–92. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting Potential Drug-Drug Interactions by Integrating Chemical, Biological, Phenotypic and Network Data. BMC Bioinform. 2017, 18, 18. [Google Scholar] [CrossRef]
- Guney, E.; Menche, J.; Vidal, M.; Barábasi, A.L. Network-Based in Silico Drug Efficacy Screening. Nat. Commun. 2016, 7, 10331. [Google Scholar] [CrossRef] [PubMed]
- Quan, P.; Wang, K.; Yan, S.; Wen, S.; Wei, C.; Zhang, X.; Cao, J.; Yao, L. Integrated Network Analysis Identifying Potential Novel Drug Candidates and Targets for Parkinson’s Disease. Sci. Rep. 2021, 11, 13154. [Google Scholar] [CrossRef]
- Kong, J.H.; Lee, H.; Kim, D.; Han, S.K.; Ha, D.; Shin, K.; Kim, S. Network-Based Machine Learning in Colorectal and Bladder Organoid Models Predicts Anti-Cancer Drug Efficacy in Patients. Nat. Commun. 2020, 11, 5485. [Google Scholar] [CrossRef] [PubMed]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Huo, D.; Xie, H.; Wu, L.; Zhang, J.; Liu, L.; Jin, Q.; Chen, X. CHG: A Systematically Integrated Database of Cancer Hallmark Genes. Front. Genet. 2020, 11, 29. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing Genetic Dysfunction across All Human Cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Dempster, J.M.; Pacini, C.; Pantel, S.; Behan, F.M.; Green, T.; Krill-Burger, J.; Beaver, C.M.; Younger, S.T.; Zhivich, V.; Najgebauer, H.; et al. Agreement between Two Large Pan-Cancer CRISPR-Cas9 Gene Dependency Data Sets. Nat. Commun. 2019, 10, 5817. [Google Scholar] [CrossRef]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A Cell Population Dynamics Model of CRISPR Experiments That Improves Inference of Gene Fitness Effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabási, A.L.; Oltvai, Z.N. Lethality and Centrality in Protein Networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Groves, S.M.; Ildefonso, G.V.; McAtee, C.O.; Ozawa, P.M.M.; Ireland, A.S.; Stauffer, P.E.; Wasdin, P.T.; Huang, X.; Qiao, Y.; Lim, J.S.; et al. Archetype Tasks Link Intratumoral Heterogeneity to Plasticity and Cancer Hallmarks in Small Cell Lung Cancer. Cell Syst. 2022, 13, 690–710.e17. [Google Scholar] [CrossRef] [PubMed]
- Fania, L.; Didona, D.; Di Pietro, F.R.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous Squamous Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef]
- Zheng, T.; Huang, J.; Xiang, X.; Li, S.; Yu, J.; Qu, K.; Xu, Z.; Han, P.; Dong, Z.; Liu, Y.; et al. Systematical Analysis Reveals a Strong Cancer Relevance of CREB1-Regulated Genes. Cancer Cell Int. 2021, 21, 530. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Rooper, L.M.; Dermawan, J.K.; Zhang, Y.; Suurmeijer, A.J.H.; Dickson, B.C.; Demicco, E.G.; Antonescu, C.R. Mesenchymal Chondrosarcoma of the Head and Neck with HEY1::NCOA2 Fusion: A Clinicopathologic and Molecular Study of 13 Cases with Emphasis on Diagnostic Pitfalls. Genes Chromosom. Cancer 2022, 61, 670–677. [Google Scholar] [CrossRef]
- Niedan, S.; Kauer, M.; Aryee, D.N.T.; Kofler, R.; Schwentner, R.; Meier, A.; Pötschger, U.; Kontny, U.; Kovar, H. Suppression of FOXO1 Is Responsible for a Growth Regulatory Repressive Transcriptional Sub-Signature of EWS-FLI1 in Ewing Sarcoma. Oncogene 2014, 33, 3927–3938. [Google Scholar] [CrossRef]
- Almeida, T.G.; Garcia, N.; Cunha, I.W.; Baiocchi, G.; Soares, F.A.; Maciel, G.A.; Baracat, E.C.; Carvalho, K.C. Lack of Expression of FOXO3 Correlates with Poor Prognosis on Uterine Sarcomas. In Proceedings of the 106th Annual Meeting of the American Association for Cancer Research, Philadelphia, PA, USA, 18–22 April 2015. [Google Scholar]
- Kauer, M.O.; Jug, G.; Niedan, S.; Walker, R.L.; Meltzer, P.S.; Kovar, H. A New Repressive Network Module in Ewing’s Sarcoma. In Proceedings of the 101st Annual Meeting of the American Association for Cancer Research, Washington, DC, USA, 17–21 April 2010. [Google Scholar]
- Singh, R.K.; Sivabalakrishnan, M. Feature Selection of Gene Expression Data for Cancer Classification: A Review. Procedia Comput. Sci. 2015, 50, 52–57. [Google Scholar] [CrossRef]
- Chuang, H.Y.; Lee, E.; Liu, Y.T.; Lee, D.; Ideker, T. Network-Based Classification of Breast Cancer Metastasis. Mol. Syst. Biol. 2007, 3, 140. [Google Scholar] [CrossRef]
- Galon, J.; Pagès, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer Classification Using the Immunoscore: A Worldwide Task Force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Z. Analysis of Topological Parameters of Complex Disease Genes Reveals the Importance of Location in a Biomolecular Network. Genes 2019, 10, 143. [Google Scholar] [CrossRef]
- Weiskittel, T.M.; Ung, C.Y.; Correia, C.; Zhang, C.; Li, H. De Novo Individualized Disease Modules Reveal the Synthetic Penetrance of Genes and Inform Personalized Treatment Regimens. Genome Res. 2022, 32, 124–134. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.R.; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. [Google Scholar]
- Onishi, I.; Yamamoto, K.; Kinowaki, Y.; Kitagawa, M.; Kurata, M. To Discover the Efficient and Novel Drug Targets in Human Cancers Using Crispr/Cas Screening and Databases. Int. J. Mol. Sci. 2021, 22, 12322. [Google Scholar] [CrossRef] [PubMed]
- Nibbe, R.K.; Markowitz, S.; Myeroff, L.; Ewing, R.; Chance, M.R. Discovery and Scoring of Protein Interaction Subnetworks Discriminative of Late Stage Human Colon Cancer. Mol. Cell. Proteom. 2009, 8, 827–845. [Google Scholar] [CrossRef]
- Zhang, W.; Ota, T.; Shridhar, V.; Chien, J.; Wu, B.; Kuang, R. Network-Based Survival Analysis Reveals Subnetwork Signatures for Predicting Outcomes of Ovarian Cancer Treatment. PLoS Comput. Biol. 2013, 9, e1002975. [Google Scholar] [CrossRef] [PubMed]
- Ashtiani, M.; Salehzadeh-Yazdi, A.; Razaghi-Moghadam, Z.; Hennig, H.; Wolkenhauer, O.; Mirzaie, M.; Jafari, M. A Systematic Survey of Centrality Measures for Protein-Protein Interaction Networks. BMC Syst. Biol. 2018, 12, 80. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiskittel, T.M.; Cao, A.; Meng-Lin, K.; Lehmann, Z.; Feng, B.; Correia, C.; Zhang, C.; Wisniewski, P.; Zhu, S.; Yong Ung, C.; et al. Network Biology-Inspired Machine Learning Features Predict Cancer Gene Targets and Reveal Target Coordinating Mechanisms. Pharmaceuticals 2023, 16, 752. https://doi.org/10.3390/ph16050752
Weiskittel TM, Cao A, Meng-Lin K, Lehmann Z, Feng B, Correia C, Zhang C, Wisniewski P, Zhu S, Yong Ung C, et al. Network Biology-Inspired Machine Learning Features Predict Cancer Gene Targets and Reveal Target Coordinating Mechanisms. Pharmaceuticals. 2023; 16(5):752. https://doi.org/10.3390/ph16050752
Chicago/Turabian StyleWeiskittel, Taylor M., Andrew Cao, Kevin Meng-Lin, Zachary Lehmann, Benjamin Feng, Cristina Correia, Cheng Zhang, Philip Wisniewski, Shizhen Zhu, Choong Yong Ung, and et al. 2023. "Network Biology-Inspired Machine Learning Features Predict Cancer Gene Targets and Reveal Target Coordinating Mechanisms" Pharmaceuticals 16, no. 5: 752. https://doi.org/10.3390/ph16050752
APA StyleWeiskittel, T. M., Cao, A., Meng-Lin, K., Lehmann, Z., Feng, B., Correia, C., Zhang, C., Wisniewski, P., Zhu, S., Yong Ung, C., & Li, H. (2023). Network Biology-Inspired Machine Learning Features Predict Cancer Gene Targets and Reveal Target Coordinating Mechanisms. Pharmaceuticals, 16(5), 752. https://doi.org/10.3390/ph16050752