
Dapagliflozin Ameliorates Cognitive Impairment in Aluminum-Chloride-Induced Alzheimer’s Disease via Modulation of AMPK/mTOR, Oxidative Stress and Glucose Metabolism
 , and
, and
Abstract

1. Introduction
2. Results
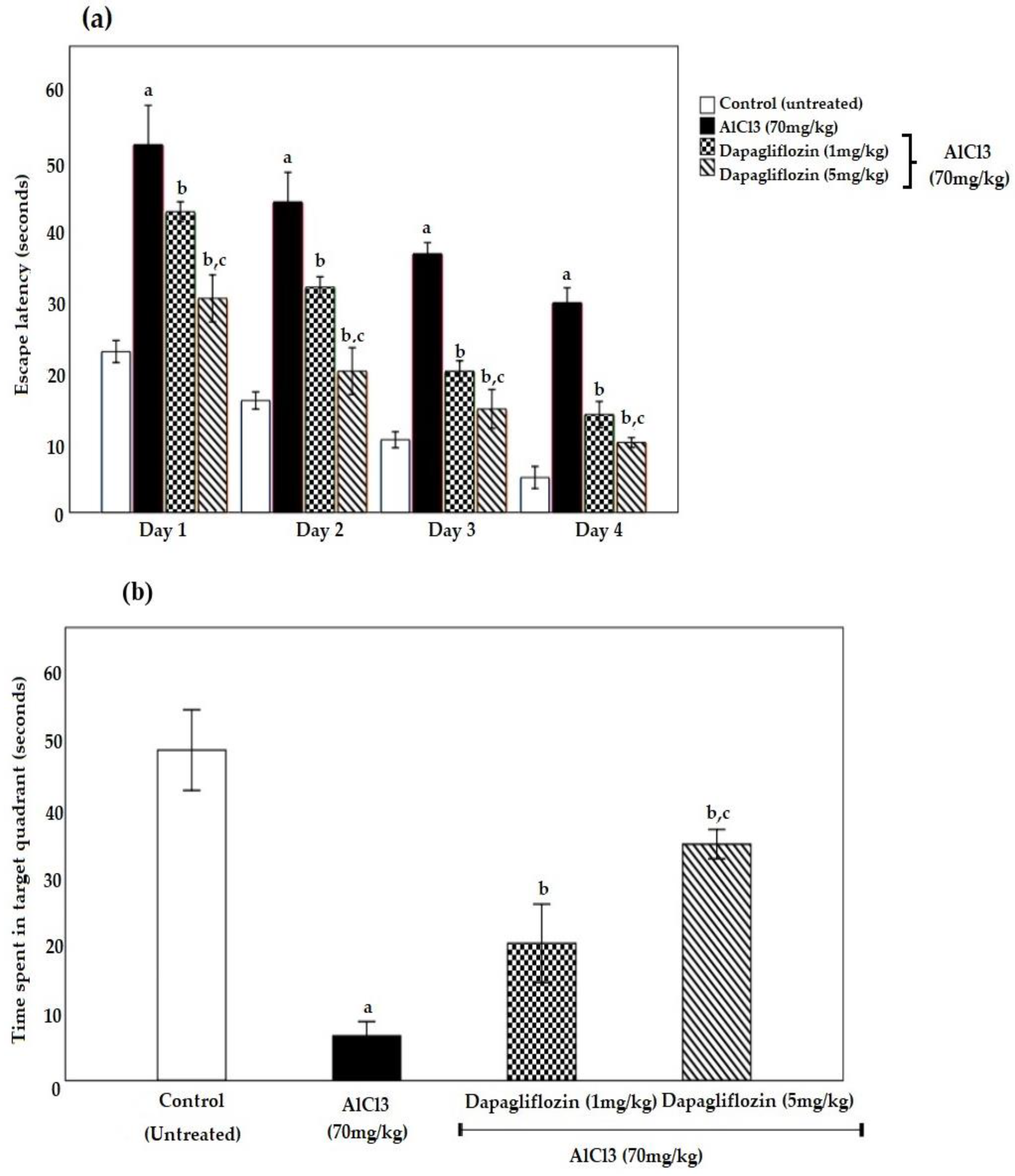
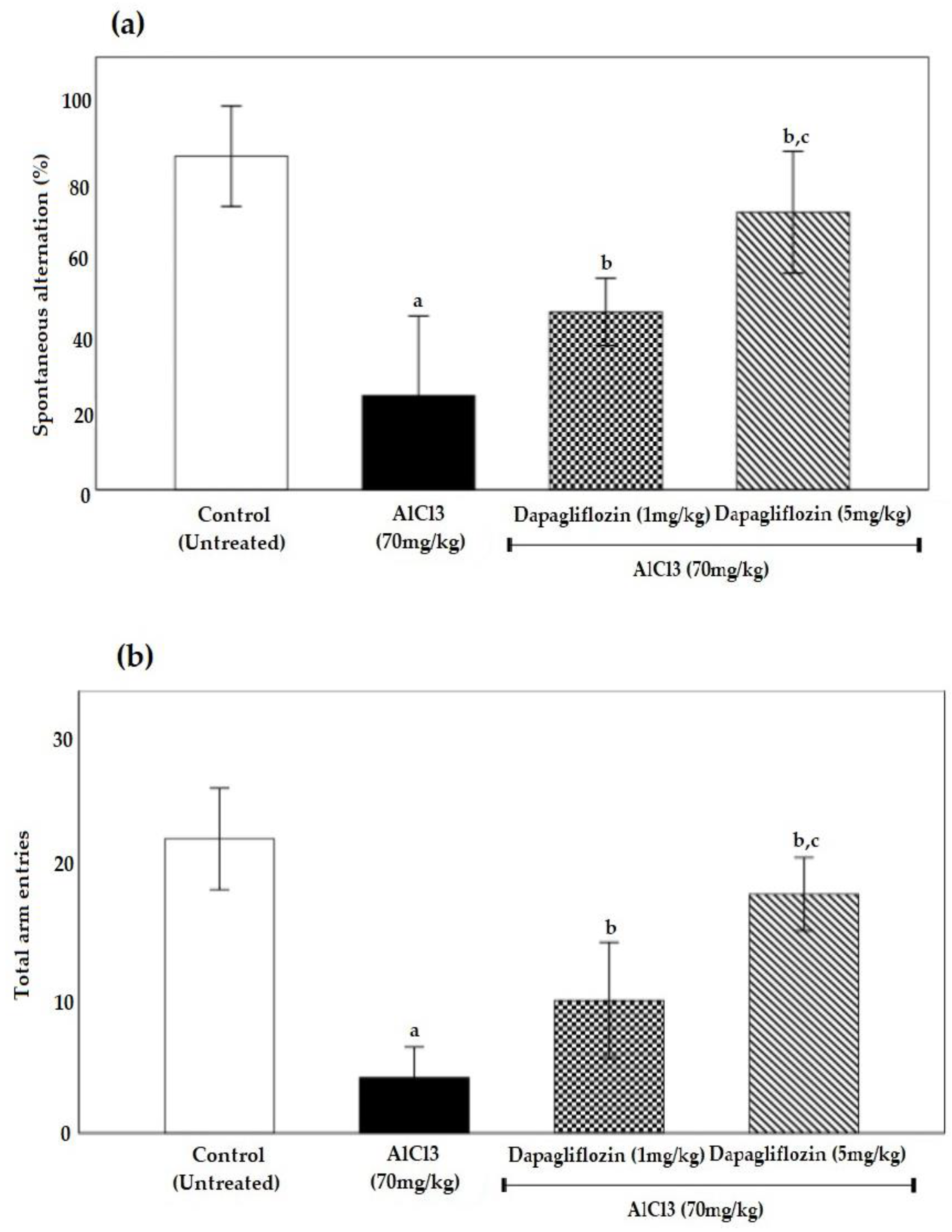
2.1. Dapagliflozin Improved Acquisition Impairment and Depressive Behavior
2.1.1. Dapagliflozin Effect on Morris Water Maze
2.1.2. Dapagliflozin Effect on Y-Maze Spontaneous Alternation
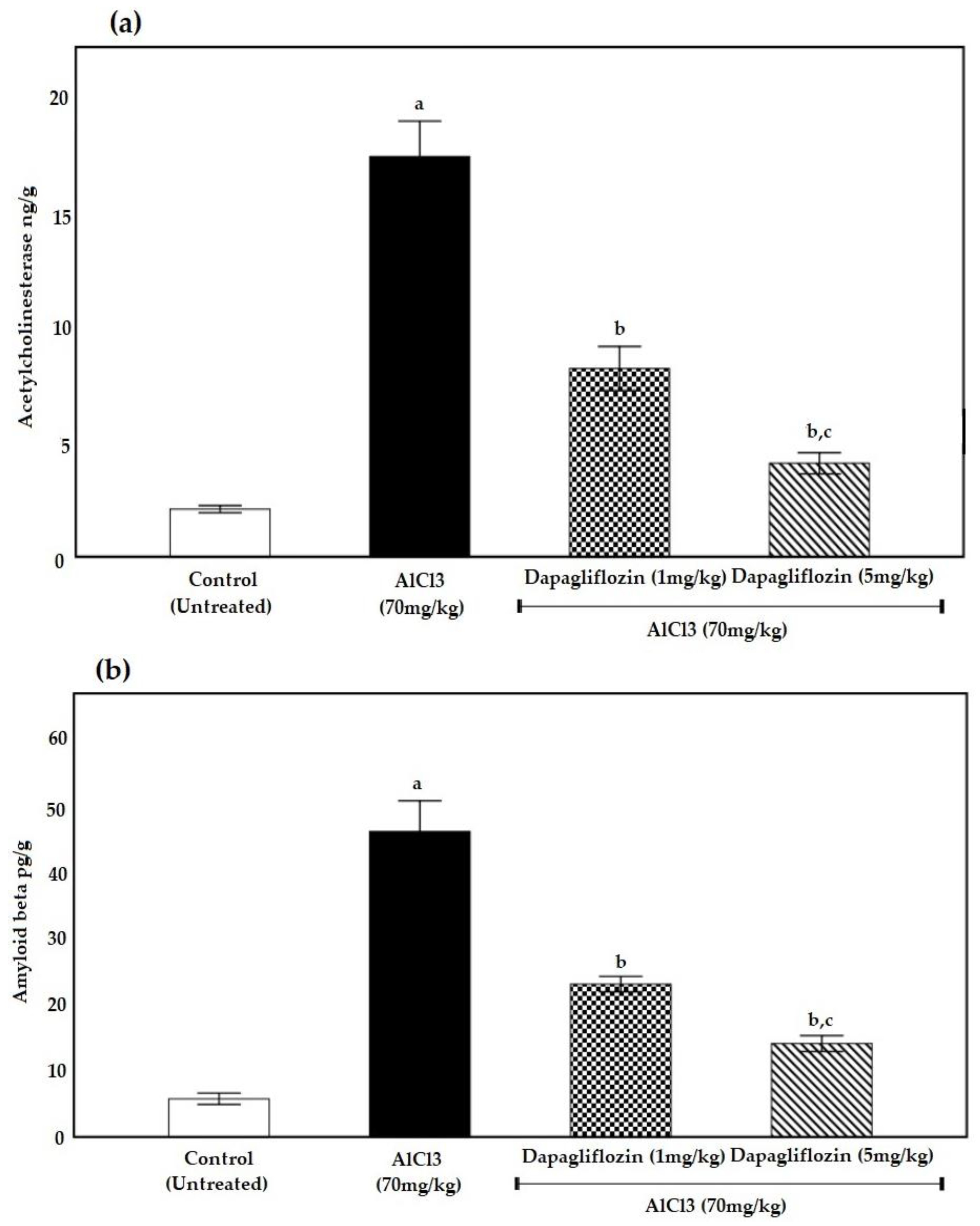
2.2. Biochemical Parameters
2.2.1. Dapagliflozin Therapy Reduces Acetylcholinesterase
2.2.2. Dapagliflozin Suppresses Brain Amyloid β Protein Deposition
2.2.3. Dapagliflozin Inhibits Oxidative Stress Markers
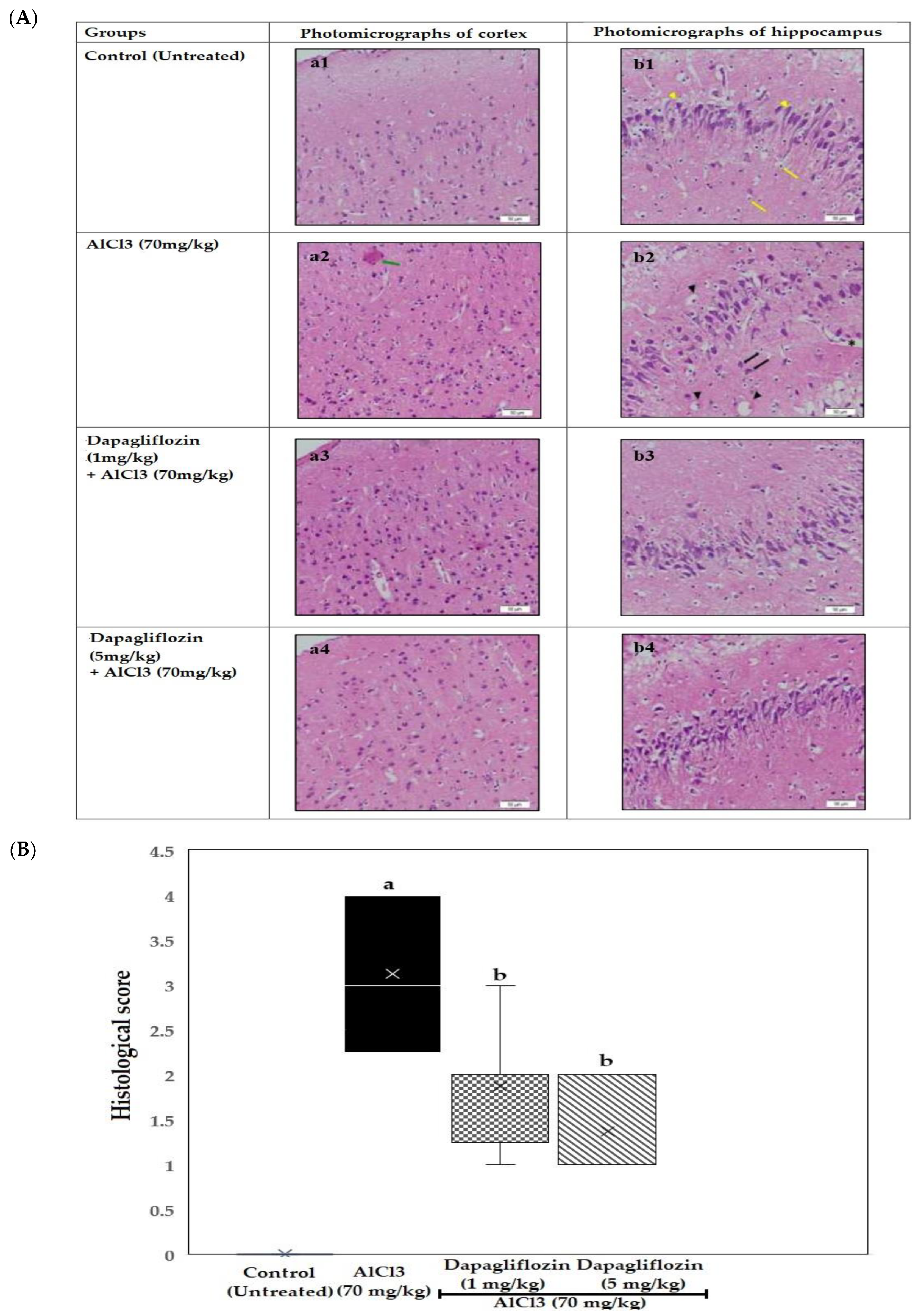
2.3. Histopathological Examination of the Brain
2.3.1. Dapagliflozin Effect on Cerebral Cortex
2.3.2. Dapagliflozin Effect on Hippocampus
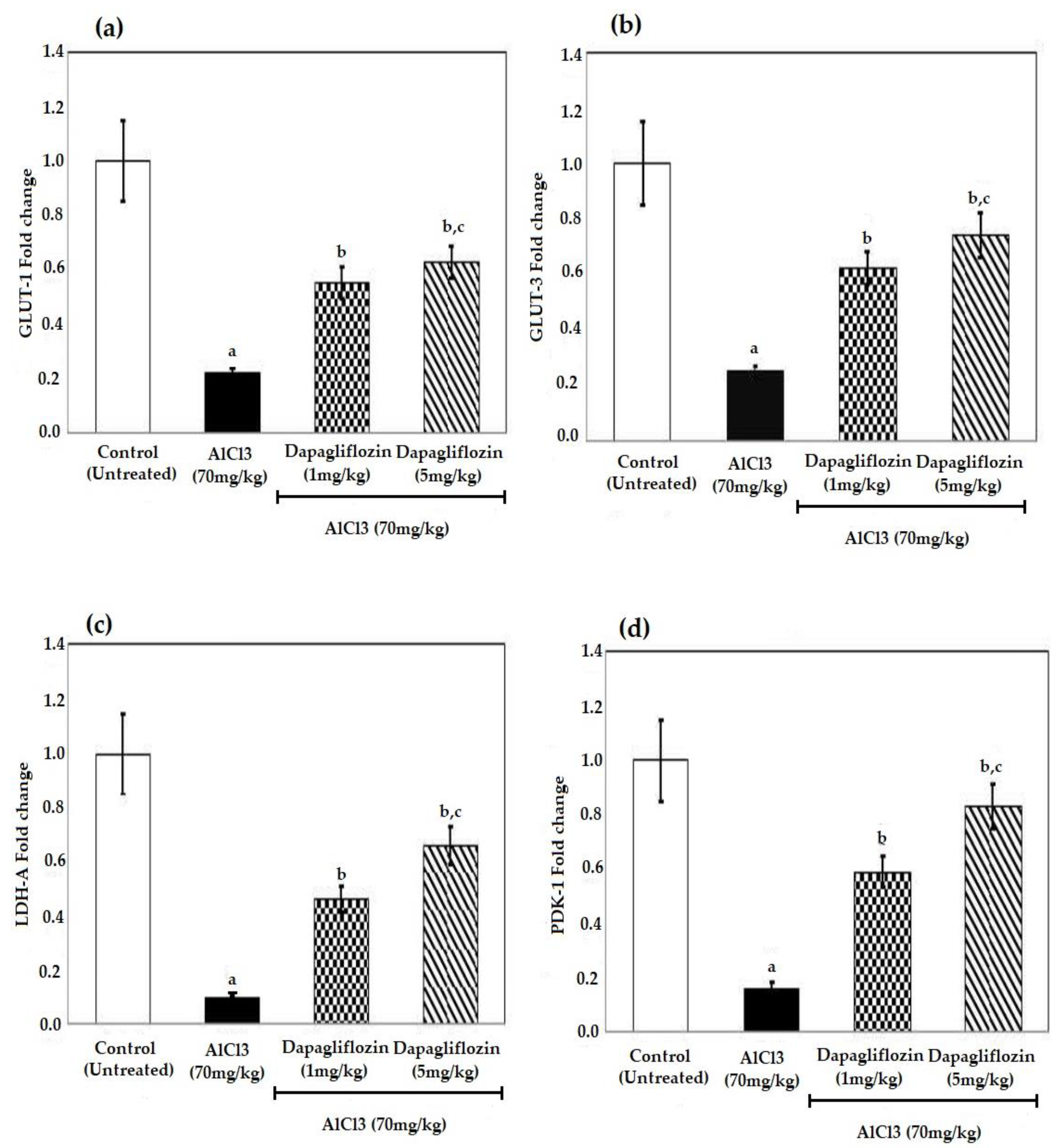
2.4. Dapagliflozin Alleviates Brain Glucose Levels
2.5. Dapagliflozin Increases the Levels of Glucose Transporters and Glycolytic Enzymes
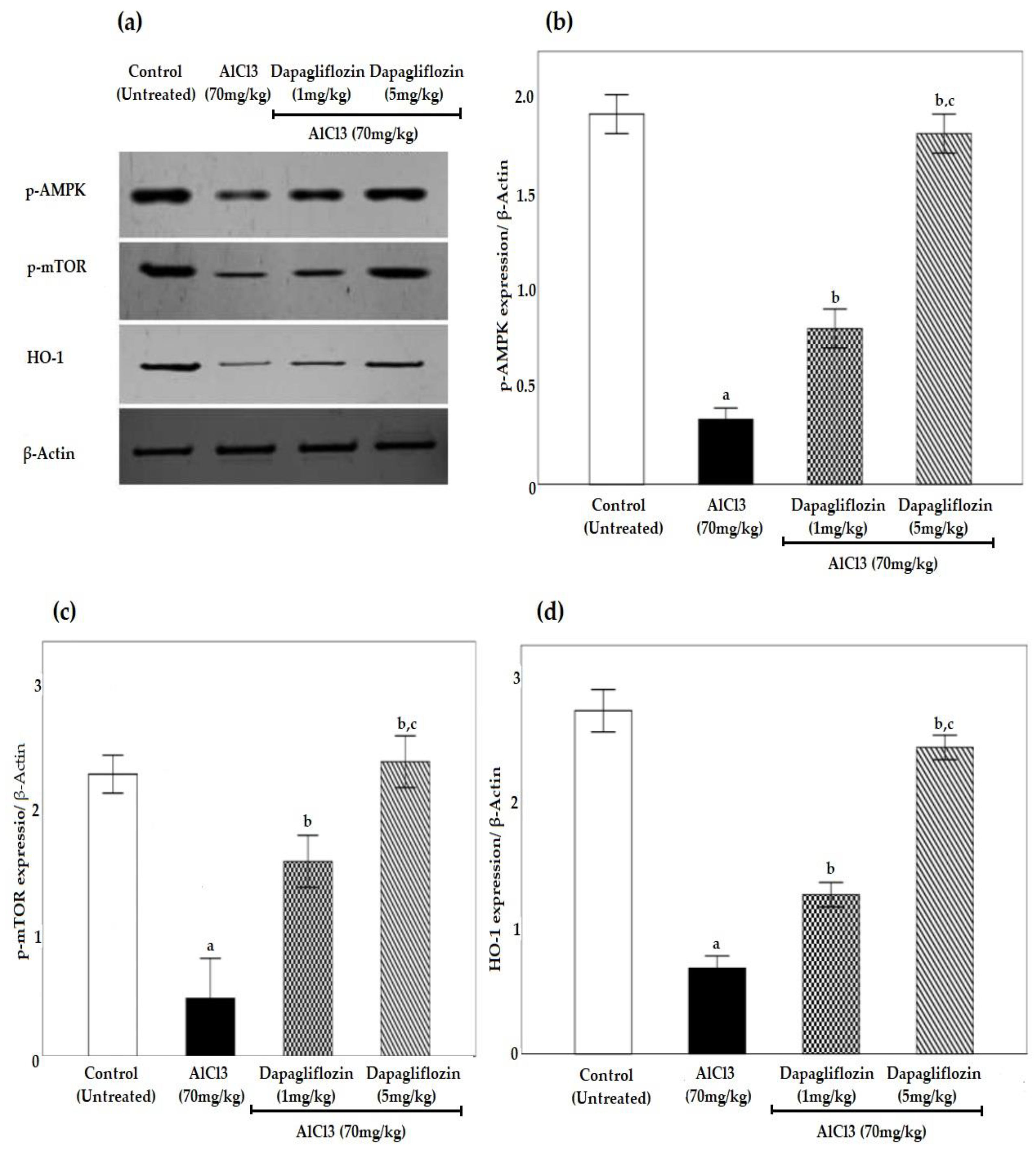
2.6. Dapagliflozin Upregulates Phosphorylated 5’ AMP-Activated Protein Kinase, Phosphorylated Mammalian Target of Rapamycin and Heme Oxygenase-1 Signaling Pathways
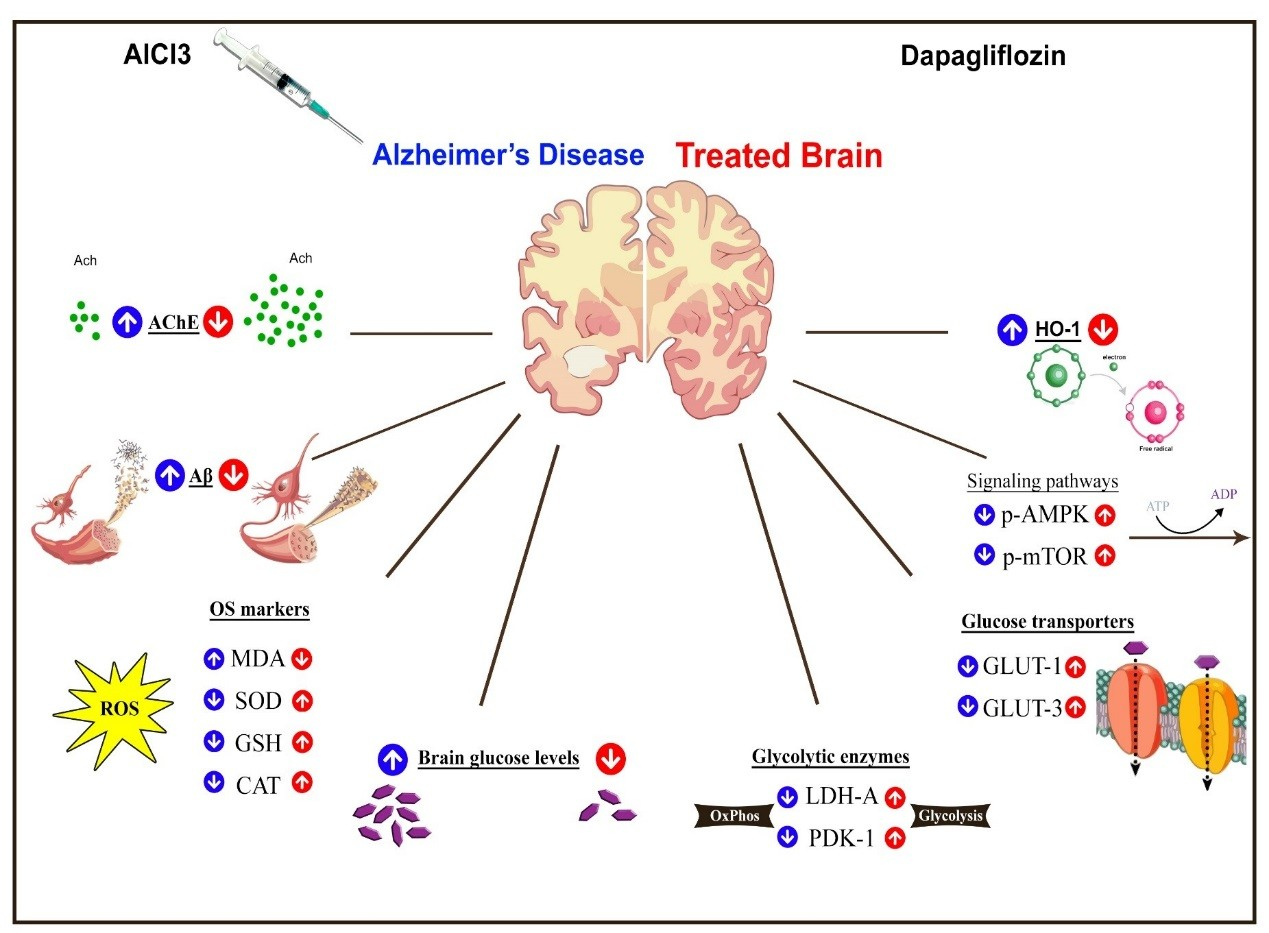
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Drugs and Chemicals
4.3. Induction of AD
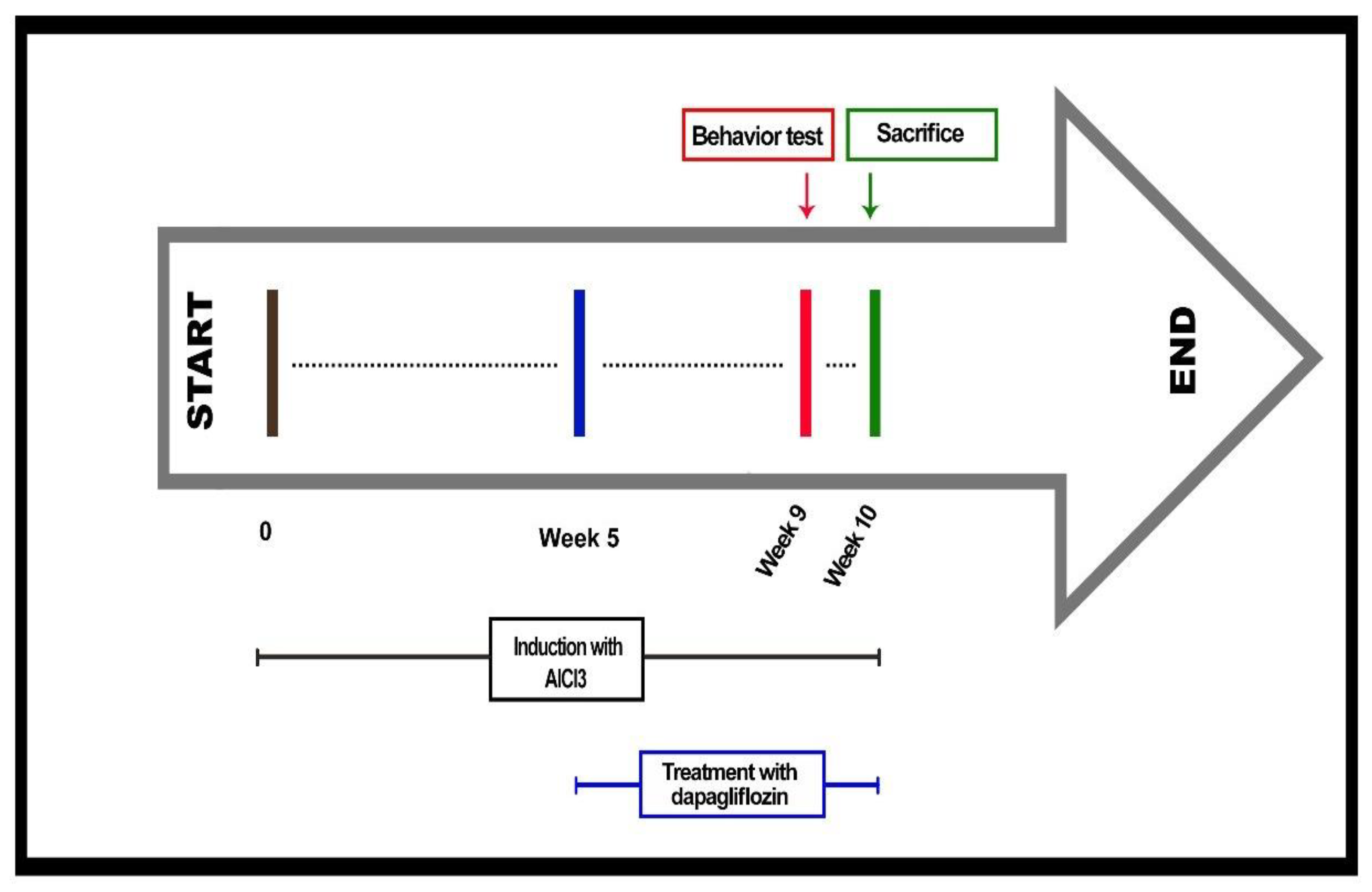
4.4. Experimental Design
4.5. Sample Preparation
4.6. Behavioral Experiment
4.6.1. Morris Water Maze Task
4.6.2. Y-Maze Spontaneous Alternation
4.7. Biochemical Parameters
Determination of AChE and Aβ Level
4.8. Assessment of Brain OS Markers
4.9. Histopathological Investigation of the Brain
4.10. Brain Glucose Levels
4.11. Quantitative Polymerase Chain Reaction Assay
4.12. Western Blot
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prajapati, A.; Mehan, S.; Khan, Z. The Role of Smo-Shh/Gli Signaling Activation in the Prevention of Neurological and Ageing Disorders. Biogerontology 2023. [Google Scholar] [CrossRef] [PubMed]
- Mahaman, Y.A.R.; Huang, F.; Salissou, M.T.M.; Yacouba, M.B.M.; Wang, J.-Z.; Liu, R.; Zhang, B.; Li, H.-L.; Zhu, F.; Wang, X. Ferulic Acid Improves Synaptic Plasticity and Cognitive Impairments by Alleviating the PP2B/DARPP-32/PP1 Axis-Mediated STEP Increase and Aβ Burden in Alzheimer’s Disease. Neurotherapeutics 2023. [Google Scholar] [CrossRef] [PubMed]
- Daly, T. Learning from the Amyloid Hypothesis: Adaptability, Brevity, and Clarity of Ideas. Neurol. Sci. 2023, 44, 2177–2178. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; Lazarov, O.; Fitzsimons, C.P.; Tanzi, R.; Lucassen, P.J.; Choi, S.H. Adult Hippocampal Neurogenesis in Alzheimer’s Disease: A Roadmap to Clinical Relevance. Cell. Stem. Cell. 2023, 30, 120–136. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.; Chafee, M.V.; Lee, E.; Morishita, H. Psychosis Spectrum Illnesses as Disorders of Prefrontal Critical Period Plasticity. Neuropsychopharmacology 2023, 48, 168–185. [Google Scholar] [CrossRef]
- Ferreira-Vieira, H.T.M.; Guimaraes, I.R.; Silva, F.M.; Ribeiro, F. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- O’Hara, R.; Derouesné, C.; Fountoulakis, K.N.; Yesavage, J.A. Therapeutic Approaches to Age-Associated Neurocognitive Disorders. Dialogues. Clin. Neurosci. 2001, 3, 191–213. [Google Scholar] [CrossRef]
- Small, S.A.; Duff, K. Linking Aβ and Tau in Late-Onset Alzheimer’s Disease: A Dual Pathway Hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef]
- Cervellati, C.; Romani, A.; Seripa, D.; Cremonini, E.; Bosi, C.; Magon, S.; Passaro, A.; Bergamini, C.M.; Pilotto, A.; Zuliani, G. Oxidative Balance, Homocysteine, and Uric Acid Levels in Older Patients with Late Onset Alzheimer’s Disease or Vascular Dementia. J. Neurol. Sci. 2014, 337, 156–161. [Google Scholar] [CrossRef]
- Ramasamy, A.; Anandakumar, K.; Kathiresan, K. In-Vitro Antioxidant Potential and Acetylcholinesterase Inhibitory Effect of Ficus Benghalensis Aerial Root Extract. Afr. Health. Sci. 2022, 22, 291–299. [Google Scholar] [CrossRef]
- Håkansson, L.M. Modulation of Acetylcholine Release in Brain: Studies of Mechanisms in Experimental Animals and Alzheimer’s Disease; Acta Universitatis Upsaliensis Comprehensive Summaries of Uppsala Dissertations from the Faculty of Pharmacy; Uppsala University: Uppsala, Sweden, 1990. [Google Scholar]
- Mimuro, M.; Yoshida, M.; Kuzuhara, S.; Kokubo, Y. Amyotrophic Lateral Sclerosis and Parkinsonism-Dementia Complex of the Hohara Focus of the Kii Peninsula: A Multiple Proteinopathy? Kii ALS/PDC: A Multiple Proteinopathy. Neuropathology 2018, 38, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato-Negishi, M. Link between Aluminum and the Pathogenesis of Alzheimer’s Disease: The Integration of the Aluminum and Amyloid Cascade Hypotheses. Int. J. Alzheimer’s Dis. 2011, 2011, 276393. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Barton, A.J.L.; Najlerahim, A.; McDonald, B.; Pearson, R.C.A. Increased Muscarinic Receptor Messenger RNA in Alzheimer’s Disease Temporal Cortex Demonstrated by in Situ Hybridization Histochemistry. Mol. Brain Res. 1991, 9, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kiryu, H. Identification of Oxidative Stress-Related Genes Differentially Expressed in Alzheimer’s Disease and Construction of a Hub Gene-Based Diagnostic Model. Sci. Rep. 2023, 13, 6817. [Google Scholar] [CrossRef]
- Demetrius, L.A.; Magistretti, P.J.; Pellerin, L. Alzheimer’s Disease: The Amyloid Hypothesis and the Inverse Warburg Effect. Front. Physiol. 2014, 5, 522. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Shiou, Y.-L.; Jhuo, S.-J.; Chang, C.-Y.; Liu, P.-L.; Jhuang, W.-J.; Dai, Z.-K.; Chen, W.-Y.; Chen, Y.-F.; Lee, A.-S. The Sodium–Glucose Co-Transporter 2 Inhibitor Empagliflozin Attenuates Cardiac Fibrosis and Improves Ventricular Hemodynamics in Hypertensive Heart Failure Rats. Cardiovasc. Diabetol. 2019, 18, 45. [Google Scholar] [CrossRef]
- Lee, D.S.U.; Lee, H. Adherence and Persistence Rates of Major Antidiabetic Medications: A Review. Diabetol. Metab. Syndr. 2022, 14, 12. [Google Scholar] [CrossRef]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic Control with Empagliflozin, a Novel Selective SGLT2 Inhibitor, Ameliorates Cardiovascular Injury and Cognitive Dysfunction in Obese and Type 2 Diabetic Mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, H.; Liu, X.; Guo, X. Hypoglycemic Medicines in the Treatment of Alzheimer’s Disease: Pathophysiological Links between AD and Glucose Metabolism. Front. Pharmacol. 2023, 14, 1138499. [Google Scholar] [CrossRef]
- Ibrahim, W.W.; Kamel, A.S.; Wahid, A.; Abdelkader, N.F. Dapagliflozin as an Autophagic Enhancer via LKB1/AMPK/SIRT1 Pathway in Ovariectomized/D-Galactose Alzheimer’s Rat Model. Inflammopharmacology 2022, 30, 2505–2520. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Garcez, M.L.; Falchetti, A.C.B.; Mina, F.; Budni, J. Alzheimer´s Disease Associated with Psychiatric Comorbidities. An. Acad. Bras. Ciênc. 2015, 87 (Suppl. 2), 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Krewski, D.; Yokel, R.A.; Nieboer, E.; Borchelt, D.; Cohen, J.; Harry, J.; Kacew, S.; Lindsay, J.; Mahfouz, A.M.; Rondeau, V. Human Health Risk Assessment for Aluminium, Aluminium Oxide, and Aluminium Hydroxide. J. Toxicol. Environ. Health B Crit. Rev. 2007, 10 (Suppl. 1), 1–269. [Google Scholar] [CrossRef] [PubMed]
- LeBel, C.P.; Bondy, S.C. Oxygen Radicals: Common Mediators of Neurotoxicity. Neurotoxicology Teratol. 1991, 13, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Julka, D.; Vasishta, R.K.; Gill, K.D. Distribution of Aluminum in Different Brain Regions and Body Organs of Rat. Biol. Trace Elem. Res. 1996, 52, 181–192. [Google Scholar] [CrossRef]
- Nguyen, L.T.T.; Le, X.T.; Pham, H.N.T.; Van Nguyen, T.; Nguyen, P.T.; Van Thi Pham, A.; Nguyen, T.B.T.; Matsumoto, K. Therapeutic Effects of a Standardized-Flavonoid Diospyros Kaki L.f. Leaf Extract on Transient Focal Cerebral Ischemia-Induced Brain Injury in Mice. J. Nat. Med. 2023. [Google Scholar] [CrossRef]
- Gulya, K.; Rakonczay, Z.; Kása, P. Cholinotoxic Effects of Aluminum in Rat Brain. J. Neurochem. 1990, 54, 1020–1026. [Google Scholar] [CrossRef]
- Talesa, V.N. Acetylcholinesterase in Alzheimer’s Disease. Mech. Ageing Dev. 2001, 122, 1961–1969. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kaufer, D.I.; Hendrickson, R.; Ivanco, L.S.; Lopresti, B.; Davis, J.G.; Constantine, G.; Mathis, C.A.; Moore, R.Y.; DeKosky, S.T. Cognitive Correlates of Alterations in Acetylcholinesterase in Alzheimer’s Disease. Neurosci. Lett. 2005, 380, 127–132. [Google Scholar] [CrossRef]
- ELBini-Dhouib, I.; Doghri, R.; Ellefi, A.; Degrach, I.; Srairi-Abid, N.; Gati, A. Curcumin Attenuated Neurotoxicity in Sporadic Animal Model of Alzheimer’s Disease. Molecules 2021, 26, 3011. [Google Scholar] [CrossRef]
- Elsaid, F.G.; Shati, A.A.; Hafez, E.E. The Protective Role of Coffea Arabica L. and Crocus Sativus L. Against the Neurotoxicity Induced by Chronic Administration of Aluminium Chloride. J. Pharmacol. Toxicol. 2011, 6, 647–663. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, F.; Ni, Y.; Kokot, S. Effects of Aluminum on Amyloid-Beta Aggregation in the Context of Alzheimer’s Disease. Arab. J. Chem. 2019, 12, 2897–2904. [Google Scholar] [CrossRef]
- Prakash, A.; Kumar, A. Mitoprotective Effect of Centella Asiatica against Aluminum-Induced Neurotoxicity in Rats: Possible Relevance to Its Anti-Oxidant and Anti-Apoptosis Mechanism. Neurol. Sci. 2013, 34, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Sreekumaran, E.; Ramakrishna, T.; Madhav, T.R.; Anandh, D.; Prabhu, B.M.; Sulekha, S.; Bindu, P.N.; Raju, T.R. Loss of Dendritic Connectivity in CA1, CA2, and CA3 Neurons in Hippocampus in Rat under Aluminum Toxicity: Antidotal Effect of Pyridoxine. Brain Res. Bull. 2003, 59, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Sethi, P.; Jyoti, A.; Singh, R.; Hussain, E.; Sharma, D. Aluminium-Induced Electrophysiological, Biochemical and Cognitive Modifications in the Hippocampus of Aging Rats. NeuroToxicology 2008, 29, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Castellani, R.J.; Zhu, X.; Moreira, P.I.; Perry, G.; Smith, M.A. Involvement of Oxidative Stress in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2006, 65, 631–641. [Google Scholar] [CrossRef]
- Pocernich, C.B.; Butterfield, D.A. Elevation of Glutathione as a Therapeutic Strategy in Alzheimer Disease. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2012, 1822, 625–630. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative Stress in Alzheimer’s Disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Zaman, K.; Zaman, W.; Siddique, H. Hematological and Enzymatic Results of Aluminum Intoxication in Rats. Comp. Biochem. Physiol. Part C Comp. Pharmacol. 1993, 105, 73–76. [Google Scholar] [CrossRef]
- Shah, K.; DeSilva, S.; Abbruscato, T. The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease. IJMS 2012, 13, 12629–12655. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Decreased Glucose Transporters Correlate to Abnormal Hyperphosphorylation of Tau in Alzheimer Disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Kodam, P.; Sai Swaroop, R.; Pradhan, S.S.; Sivaramakrishnan, V.; Vadrevu, R. Integrated Multi-Omics Analysis of Alzheimer’s Disease Shows Molecular Signatures Associated with Disease Progression and Potential Therapeutic Targets. Sci. Rep. 2023, 13, 3695. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Pang, Z.; Geddes, J.W.; Uchida, K.; Mattson, M.P. Amyloid β-Peptide Impairs Glucose Transport in Hippocampal and Cortical Neurons: Involvement of Membrane Lipid Peroxidation. J. Neurosci. 1997, 17, 1046–1054. [Google Scholar] [CrossRef]
- Gong, C.-X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Impaired Brain Glucose Metabolism Leads to Alzheimer Neurofibrillary Degeneration through a Decrease in Tau O-GlcNAcylation. J. Alzheimers Dis. 2006, 9, 1–12. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for Brain Glucose Dysregulation in Alzheimer’s Disease. Alzheimers Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef]
- Cummings, J.; Ortiz, A.; Castellino, J.; Kinney, J. Diabetes: Risk Factor and Translational Therapeutic Implications for Alzheimer’s Disease. Eur. J. Neurosci. 2022, 56, 5727–5757. [Google Scholar] [CrossRef]
- Chen, H.; Tran, D.; Yang, H.-C.; Nylander, S.; Birnbaum, Y.; Ye, Y. Dapagliflozin and Ticagrelor Have Additive Effects on the Attenuation of the Activation of the NLRP3 Inflammasome and the Progression of Diabetic Cardiomyopathy: An AMPK–MTOR Interplay. Cardiovasc. Drugs Ther. 2020, 34, 443–461. [Google Scholar] [CrossRef]
- Cannon, J.R.; Greenamyre, J.T. The Role of Environmental Exposures in Neurodegeneration and Neurodegenerative Diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef]
- Babylon, L.; Schmitt, F.; Franke, Y.; Hubert, T.; Eckert, G.P. Effects of Combining Biofactors on Bioenergetic Parameters, Aβ Levels and Survival in Alzheimer Model Organisms. Int. J. Mol. Sci. 2022, 23, 8670. [Google Scholar] [CrossRef]
- Newington, J.T.; Pitts, A.; Chien, A.; Arseneault, R.; Schubert, D.; Cumming, R.C. Amyloid Beta Resistance in Nerve Cell Lines Is Mediated by the Warburg Effect. PLoS ONE 2011, 6, e19191. [Google Scholar] [CrossRef] [PubMed]
- Newington, J.T.; Harris, R.A.; Cumming, R.C. Reevaluating Metabolism in Alzheimer’s Disease from the Perspective of the Astrocyte-Neuron Lactate Shuttle Model. J. Neurodegener. Dis. 2013, 2013, 234572. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative Toxicity in Diabetes and Alzheimer’s Disease: Mechanisms behind ROS/ RNS Generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, Z.; Niu, Z.; Cheng, Y.; Wei, Z.; Cai, Y.; Ma, F.; Hu, L.; Zhu, J.; Zhang, W. A Neuroprotective Role of Ufmylation through Atg9 in the Aging Brain of Drosophila. Cell. Mol. Life Sci. 2023, 80, 129. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, Y.; Shi, L.; Zhong, D.; Li, Y.; Li, J.; Jin, R. AMPK: Potential Therapeutic Target for Alzheimer’s Disease. CPPS 2020, 21, 66–77. [Google Scholar] [CrossRef]
- Li, H.; Xue, X.; Li, L.; Li, Y.; Wang, Y.; Huang, T.; Wang, Y.; Meng, H.; Pan, B.; Niu, Q. Aluminum-Induced Synaptic Plasticity Impairment via PI3K-Akt-MTOR Signaling Pathway. Neurotox. Res. 2020, 37, 996–1008. [Google Scholar] [CrossRef]
- Liu, Q.; Qiu, J.; Liang, M.; Golinski, J.; van Leyen, K.; Jung, J.E.; You, Z.; Lo, E.H.; Degterev, A.; Whalen, M.J. Akt and MTOR Mediate Programmed Necrosis in Neurons. Cell. Death Dis. 2014, 5, e1084. [Google Scholar] [CrossRef]
- Gupta, P.; Strange, K.; Telange, R.; Guo, A.; Hatch, H.; Sobh, A.; Elie, J.; Carter, A.M.; Totenhagen, J.; Tan, C.; et al. Genetic Impairment of Succinate Metabolism Disrupts Bioenergetic Sensing in Adrenal Neuroendocrine Cancer. Cell. Rep. 2022, 40, 111218. [Google Scholar] [CrossRef]
- Do, T.D.; Economou, N.J.; Chamas, A.; Buratto, S.K.; Shea, J.-E.; Bowers, M.T. Interactions between Amyloid-β and Tau Fragments Promote Aberrant Aggregates: Implications for Amyloid Toxicity. J. Phys. Chem. B. 2014, 118, 11220–11230. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary Regulation of PI3K/AKT/GSK-3β Pathway in Alzheimer’s Disease. Alzheimers Res. Ther. 2014, 6, 35. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: A Key Regulator of Energy Balance in the Single Cell and the Whole Organism. Int. J. Obes. 2008, 32 (Suppl. 4), S7–S12. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Cai, Z.; Yan, L.-J.; Li, K.; Quazi, S.H.; Zhao, B. Roles of AMP-Activated Protein Kinase in Alzheimer’s Disease. Neuromol. Med. 2012, 14, 1–14. [Google Scholar] [CrossRef]
- Assefa, B.T.; Tafere, G.G.; Wondafrash, D.Z.; Gidey, M.T. The Bewildering Effect of AMPK Activators in Alzheimer’s Disease: Review of the Current Evidence. BioMed Res. Int. 2020, 2020, 9895121. [Google Scholar] [CrossRef]
- An, J.; Haile, W.B.; Wu, F.; Torre, E.; Yepes, M. Tissue-Type Plasminogen Activator Mediates Neuroglial Coupling in the Central Nervous System. Neuroscience 2014, 257, 41–48. [Google Scholar] [CrossRef]
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. IJMS 2020, 21, 2428. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK Is Abnormally Activated in Tangle- and Pre-Tangle-Bearing Neurons in Alzheimer’s Disease and Other Tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef]
- Noriega, G.O.; Balestrasse, K.B.; Batlle, A.; Tomaro, M.L. Heme Oxygenase Exerts a Protective Role against Oxidative Stress in Soybean Leaves. Biochem. Biophys. Res. Commun. 2004, 323, 1003–1008. [Google Scholar] [CrossRef]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and Aggregation Properties of Synthetic Alzheimer’s A4/Beta Amyloid Peptide Analogs. J. Biol. Chem. 1992, 267, 546–554. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, J.; Xuan, W.; Xie, Y. Up-Regulation of Heme Oxygenase-1 Contributes to the Amelioration of Aluminum-Induced Oxidative Stress in Medicago Sativa. J. Plant. Physiol. 2013, 170, 1328–1336. [Google Scholar] [CrossRef]
- Wiciński, M.; Wódkiewicz, E.; Górski, K.; Walczak, M.; Malinowski, B. Perspective of SGLT2 Inhibition in Treatment of Conditions Connected to Neuronal Loss: Focus on Alzheimer’s Disease and Ischemia-Related Brain Injury. Pharmaceuticals 2020, 13, 379. [Google Scholar] [CrossRef] [PubMed]
- Vukadinović, D.; Abdin, A.; Anker, S.D.; Rosano, G.M.C.; Mahfoud, F.; Packer, M.; Butler, J.; Böhm, M. Side Effects and Treatment Initiation Barriers of Sodium–Glucose Cotransporter 2 Inhibitors in Heart Failure: A Systematic Review and Meta-analysis. Eur. J. Heart Fail. 2022, 24, 1625–1632. [Google Scholar] [CrossRef]
- Sarnyai, Z.; Sibille, E.L.; Pavlides, C.; Fenster, R.J.; McEwen, B.S.; Toth, M. Impaired Hippocampal-Dependent Learning and Functional Abnormalities in the Hippocampus in Mice Lacking Serotonin(1A) Receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 14731–14736. [Google Scholar] [CrossRef] [PubMed]
- El-Sahar, A.E.; Rastanawi, A.A.; El-Yamany, M.F.; Saad, M.A. Dapagliflozin Improves Behavioral Dysfunction of Huntington’s Disease in Rats via Inhibiting Apoptosis-Related Glycolysis. Life Sci. 2020, 257, 118076. [Google Scholar] [CrossRef] [PubMed]
- Sa-nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-Inhibitor and DPP-4 Inhibitor Improve Brain Function via Attenuating Mitochondrial Dysfunction, Insulin Resistance, Inflammation, and Apoptosis in HFD-Induced Obese Rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef]
- Shaikh, S.; Rizvi, S.M.; Suhail, T.; Shakil, S.; Abuzenadah, A.; Anis, R.; Naaz, D.; Dallol, A.; Haneef, M.; Ahmad, A.; et al. Predictionof Anti-Diabetic Drugs as Dual Inhibitors Against Acetylcholinesterase and Beta-Secretase: A Neuroinformatics Study. CNS Neurol. Disord.-Drug Targets 2016, 15, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Sim, A.Y.; Barua, S.; Kim, J.Y.; Lee, Y.; Lee, J.E. Role of DPP-4 and SGLT2 Inhibitors Connected to Alzheimer Disease in Type 2 Diabetes Mellitus. Front. Neurosci. 2021, 15, 708547. [Google Scholar] [CrossRef]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Chowdhury, F.I.; Nayan, S.I.; Rahman, M.M.; Khan, F.; Subhan, N.; et al. Canagliflozin Attenuates Isoprenaline-Induced Cardiac Oxidative Stress by Stimulating Multiple Antioxidant and Anti-Inflammatory Signaling Pathways. Sci. Rep. 2020, 10, 14459. [Google Scholar] [CrossRef]
- Duelli, R.; Kuschinsky, W. Brain Glucose Transporters: Relationship to Local Energy Demand. News Physiol. Sci. 2001, 16, 71–76. [Google Scholar] [CrossRef]
- Erdogan, M.A.; Yusuf, D.; Christy, J.; Solmaz, V.; Erdogan, A.; Taskiran, E.; Erbas, O. Highly Selective SGLT2 Inhibitor Dapagliflozin Reduces Seizure Activity in Pentylenetetrazol-Induced Murine Model of Epilepsy. BMC Neurol. 2018, 18, 81. [Google Scholar] [CrossRef]
- Mustroph, J.; Lücht, C.M.; Wagemann, O.; Sowa, T.; Hammer, K.P.; Sag, C.M.; Tarnowski, D.; Holzamer, A.; Pabel, S.; Beuthner, B.E.; et al. Empagliflozin Enhances Human and Murine Cardiomyocyte Glucose Uptake by Increased Expression of GLUT1. Diabetologia 2019, 62, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Safhi, M.M.; Anwer, T.; Khan, G.; Siddiqui, R.; Moni Sivakumar, S.; Alam, M.F. The Combination of Canagliflozin and Omega-3 Fatty Acid Ameliorates Insulin Resistance and Cardiac Biomarkers via Modulation of Inflammatory Cytokines in Type 2 Diabetic Rats. Korean J. Physiol. Pharmacol. 2018, 22, 493. [Google Scholar] [CrossRef] [PubMed]
- Kamel, A.S.; Wahid, A.; Abdelkader, N.F.; Ibrahim, W.W. Boosting Amygdaloid GABAergic and Neurotrophic Machinery via Dapagliflozin-Enhanced LKB1/AMPK Signaling in Anxious Demented Rats. Life Sci. 2022, 310, 121002. [Google Scholar] [CrossRef] [PubMed]
- Sayour, A.A.; Korkmaz-Icöz, S.; Loganathan, S.; Ruppert, M.; Sayour, V.N.; Oláh, A.; Benke, K.; Brune, M.; Benkő, R.; Horváth, E.M.; et al. Acute Canagliflozin Treatment Protects against in Vivo Myocardial Ischemia–Reperfusion Injury in Non-Diabetic Male Rats and Enhances Endothelium-Dependent Vasorelaxation. J. Transl. Med. 2019, 17, 127. [Google Scholar] [CrossRef]
- Dalle Pezze, P.; Ruf, S.; Sonntag, A.G.; Langelaar-Makkinje, M.; Hall, P.; Heberle, A.M.; Razquin Navas, P.; van Eunen, K.; Tölle, R.C.; Schwarz, J.J.; et al. A Systems Study Reveals Concurrent Activation of AMPK and MTOR by Amino Acids. Nat. Commun. 2016, 7, 13254. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme Oxygenase-1, Oxidation, Inflammation, and Atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef]
- Albus, U. Guide for the Care and Use of Laboratory Animals (8th Edn). Lab. Anim. 2012, 46, 267–268. [Google Scholar] [CrossRef]
- Ali, A.A.; Ahmed, H.I.; Abu, K. Modeling Stages Mimic Alzheimer’s Disease Induced by Different Doses of Aluminum in Rats: Focus on Progression of the Disease in Response to Time. J. Alzheimer’s Park. Dement. 2016, 1, 12. [Google Scholar]
- Yuan, H.-X.; Pang, Y.-F.; Wang, J.-L.; Chen, W.-C. Impacts of aluminum on sperm quality and sperm mitochondria in male rats. Natl. J. Androl. 2019, 25, 579–585. [Google Scholar]
- Devrukhakar, P.S.; Shankar, M.S. Degradation Pathway Proposal, Structure Elucidation, and In Silico Toxicity Prediction of Dapagliflozin Propane Diol Hydrolytic Degradation Products. Chromatographia 2020, 83, 1233–1245. [Google Scholar] [CrossRef]
- Ali, A.A.I.; Ahmed, H. The Potential Effect of Caffeine and Nicotine Co-Administration against Aluminum-Induced Alzheimer’s Disease in Rats. J. Alzheimers Dis. Park. 2016, 6, 236. [Google Scholar] [CrossRef]
- Saad, M.A.; Rastanawi, A.A.; El-Yamany, M.F. Alogliptin Abates Memory Injuries of Hepatic Encephalopathy Induced by Acute Paracetamol Intoxication via Switching-off Autophagy-Related Apoptosis. Life Sci. 2018, 215, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.Z.; Fujita, M. Purification of a Phi-Type Glutathione S -Transferase from Pumpkin Flowers, and Molecular Cloning of Its CDNA. Biosci. Biotechnol. Biochem. 2002, 66, 2068–2076. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | MDA ng/g | SOD u/g | GSH pg/g | CAT u/g |
|---|---|---|---|---|
| Control (untreated) | 3.1 ± 0.2 | 41.2 ± 3.4 | 52.7 ± 2.4 | 37.5 ± 2.3 |
| AlCl3 (70 mg/kg) | 20.6 ± 1.9 a | 8 ± 1 a | 8.6 ± 1.1 a | 7.1 ± 0.8 a |
| AlCl3 (70 mg/kg) + Dapagliflozin (1 mg/kg) | 10.2 ± 1 b | 15.9 ± 1.7 b | 21.7 ± 1.3 b | 14.7 ± 1.6 b |
| AlCl3 (70 mg/kg) + Dapagliflozin (5 mg/kg) | 5.6 ± 0.6 b,c | 27.1 ± 0.5 b,c | 39.3 ± 0.8 b,c | 24.9 ± 1.4 b,c |
| Groups | Brain Glucose Level mg/dL |
|---|---|
| Control (untreated) | 13.7 ± 0.1 |
| AlCl3 (70 mg/kg) | 23.54 ± 1 a |
| AlCl3 (70 mg/kg) + Dapagliflozin (1 mg/kg) | 18.6 ± 0.5 b |
| AlCl3 (70 mg/kg) + Dapagliflozin (5 mg/kg) | 15.5 ± 0.2 b,c |
| GenBank® Accession No. | Gene | Primers | Annealing Temperature |
|---|---|---|---|
| NM_138827.2 | GLUT-1 | Forward: 5′-TGGCCAAGGACACACGAATACTGA-3′ Reverse: 5′-TGGAAGAGACAGGAATGGGCGAAT-3′ | 58 °C |
| NM_017102.2 | GLUT-3 | Forward: 5′-TGGCTACAACACCGGAGTCATCAA-3′ Reverse: 5′-CTGCCAAAGCGGTTGACAAAGAGT-3′ | 58 °C |
| NM_017025.2 | LDH-A | Forward: 5′-GATCTCGCGCACGCTACT-3′ Reverse: 5′-CACAATCAGCTGGTCCTTGAG-3′ | 53 °C |
| NM_053826.2 | PDK-1 | Forward: 5′-TCCCCCGATTCAGGTTCAC-3′ Reverse: 5′-CCCGGTCACTCATCTTCACA-3′ | 54 °C |
| NM_017008.4 | GAPDH | Forward: 5′-ATGACTCTACCCACGGCAAG-3′ Reverse: 5′-GATCTCGCTCCTGGAAGATG-3′ | 52 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samman, W.A.; Selim, S.M.; El Fayoumi, H.M.; El-Sayed, N.M.; Mehanna, E.T.; Hazem, R.M. Dapagliflozin Ameliorates Cognitive Impairment in Aluminum-Chloride-Induced Alzheimer’s Disease via Modulation of AMPK/mTOR, Oxidative Stress and Glucose Metabolism. Pharmaceuticals 2023, 16, 753. https://doi.org/10.3390/ph16050753
Samman WA, Selim SM, El Fayoumi HM, El-Sayed NM, Mehanna ET, Hazem RM. Dapagliflozin Ameliorates Cognitive Impairment in Aluminum-Chloride-Induced Alzheimer’s Disease via Modulation of AMPK/mTOR, Oxidative Stress and Glucose Metabolism. Pharmaceuticals. 2023; 16(5):753. https://doi.org/10.3390/ph16050753
Chicago/Turabian StyleSamman, Waad A., Salma M. Selim, Hassan M. El Fayoumi, Norhan M. El-Sayed, Eman T. Mehanna, and Reem M. Hazem. 2023. "Dapagliflozin Ameliorates Cognitive Impairment in Aluminum-Chloride-Induced Alzheimer’s Disease via Modulation of AMPK/mTOR, Oxidative Stress and Glucose Metabolism" Pharmaceuticals 16, no. 5: 753. https://doi.org/10.3390/ph16050753
APA StyleSamman, W. A., Selim, S. M., El Fayoumi, H. M., El-Sayed, N. M., Mehanna, E. T., & Hazem, R. M. (2023). Dapagliflozin Ameliorates Cognitive Impairment in Aluminum-Chloride-Induced Alzheimer’s Disease via Modulation of AMPK/mTOR, Oxidative Stress and Glucose Metabolism. Pharmaceuticals, 16(5), 753. https://doi.org/10.3390/ph16050753