
Natural Product-Based Screening for Lead Compounds Targeting SARS CoV-2 Mpro
Abstract
1. Introduction
2. Results
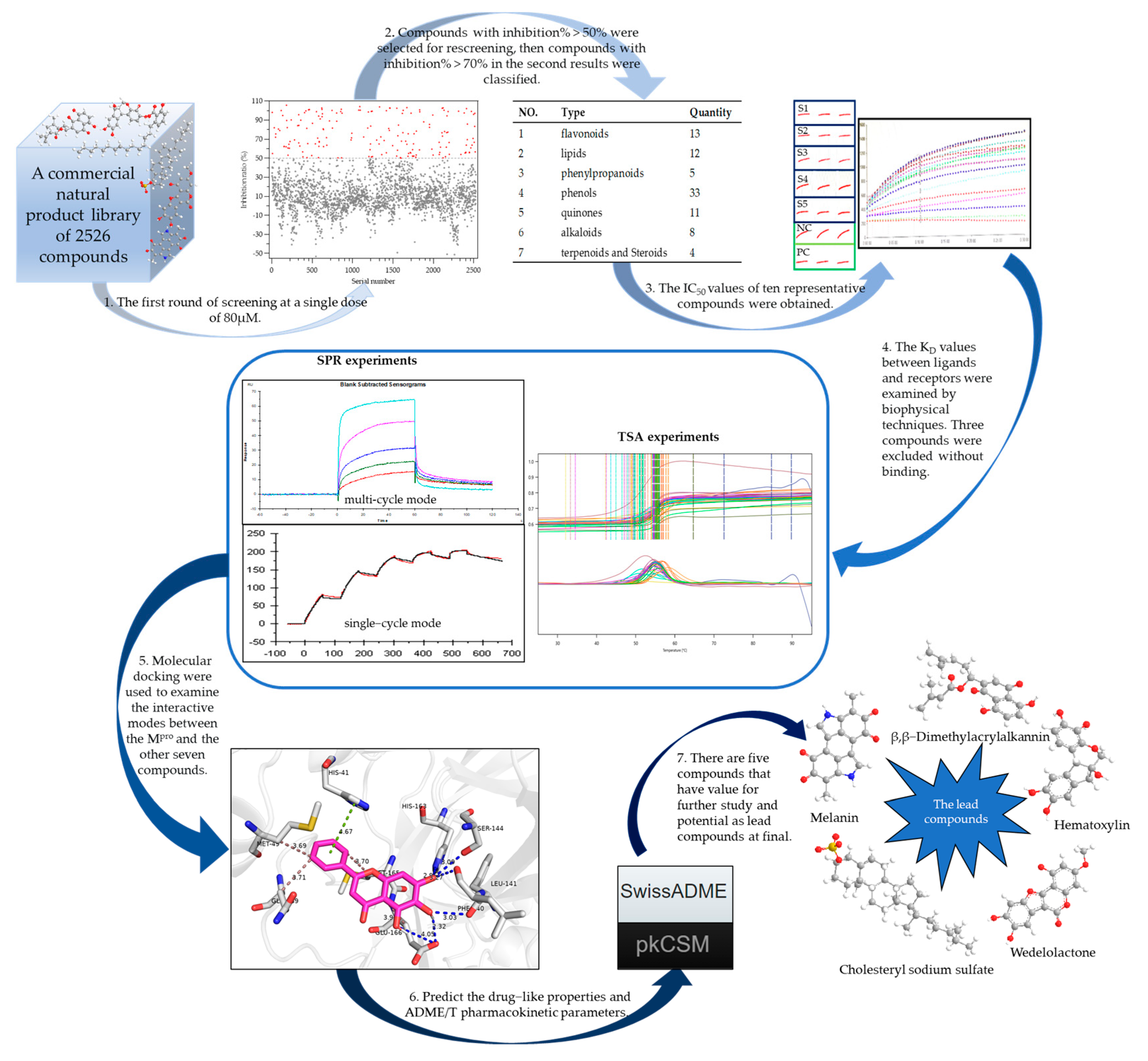
2.1. Building the High-Throughput Screening Model
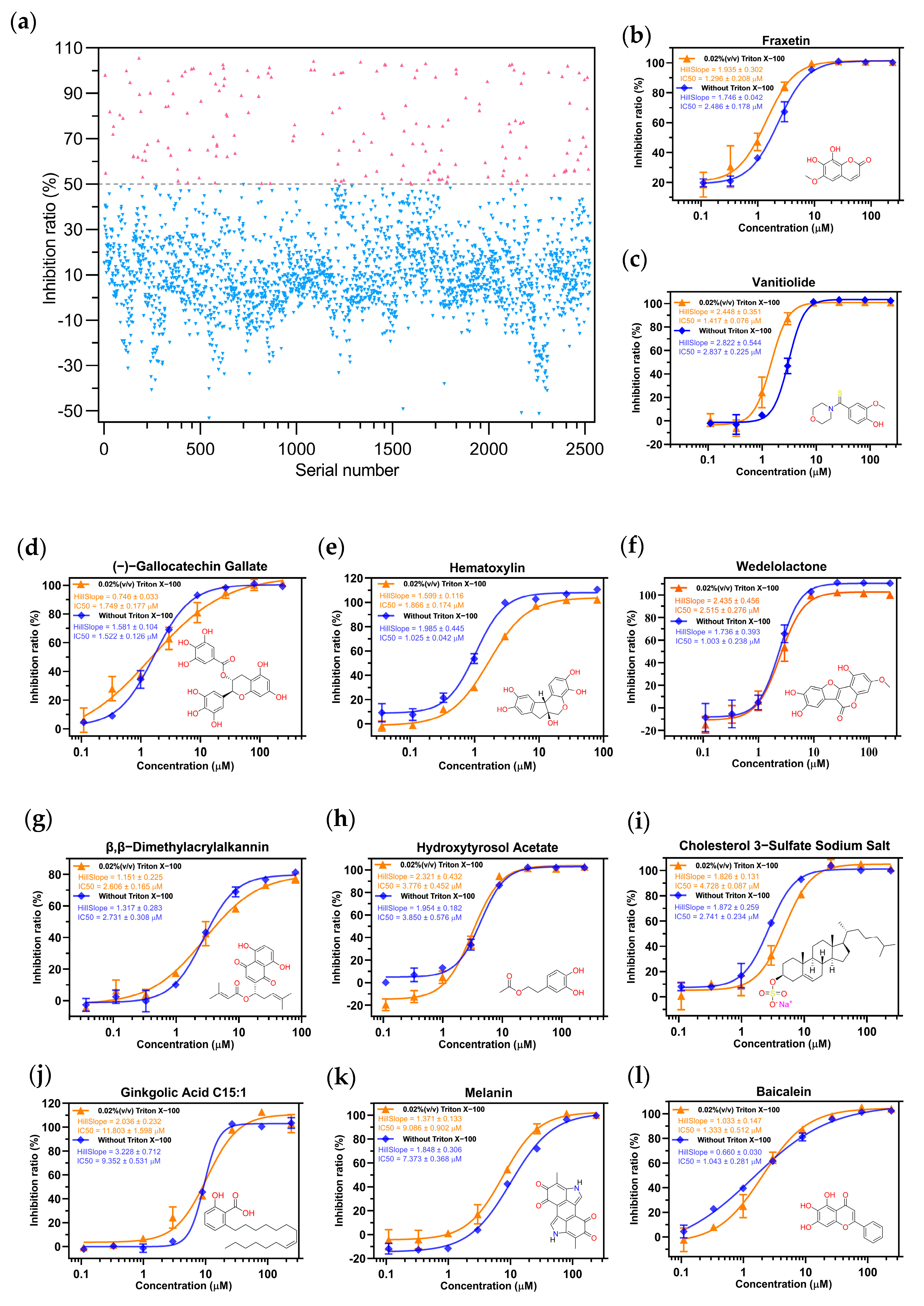
2.2. Screening the Prospective Natural Compounds
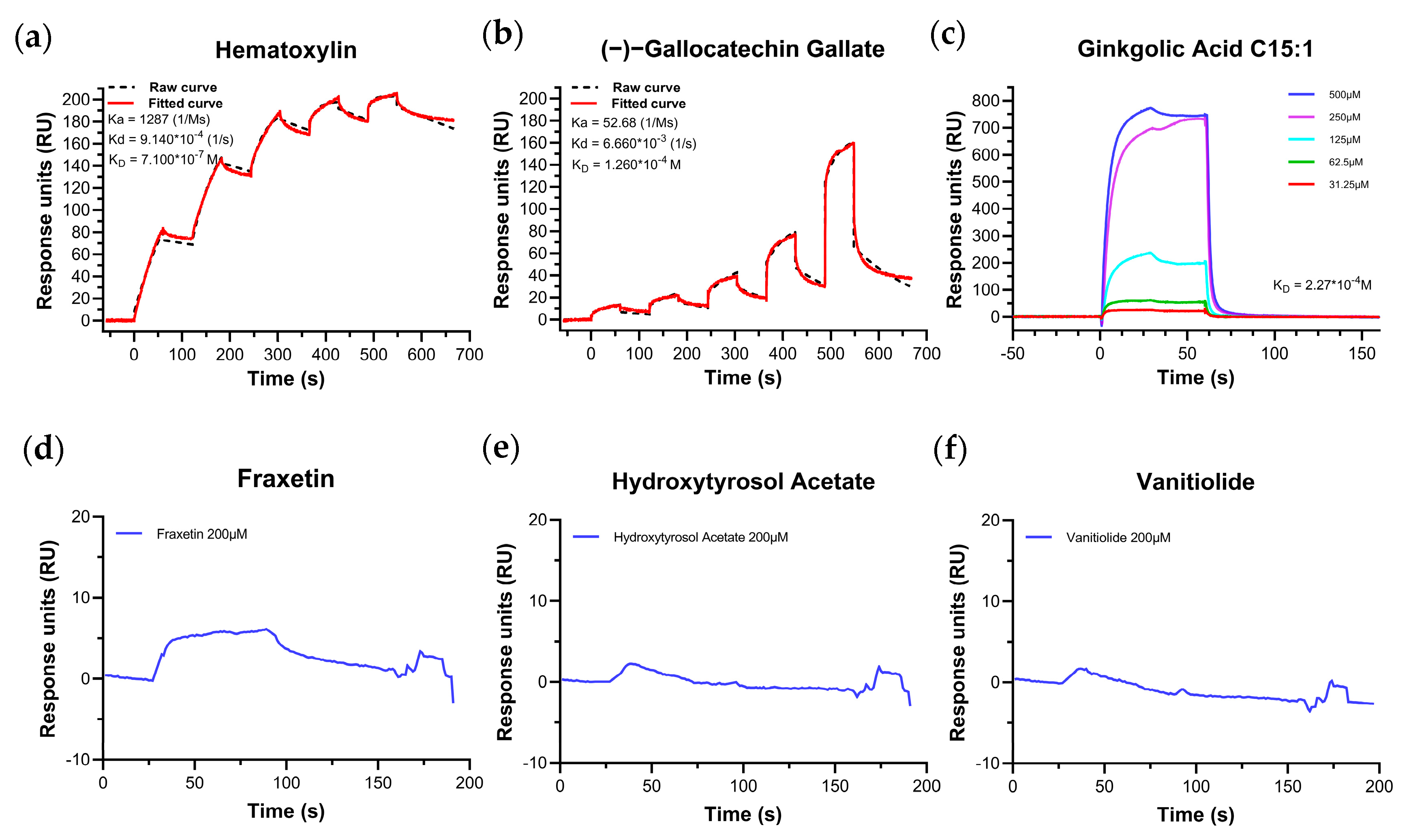
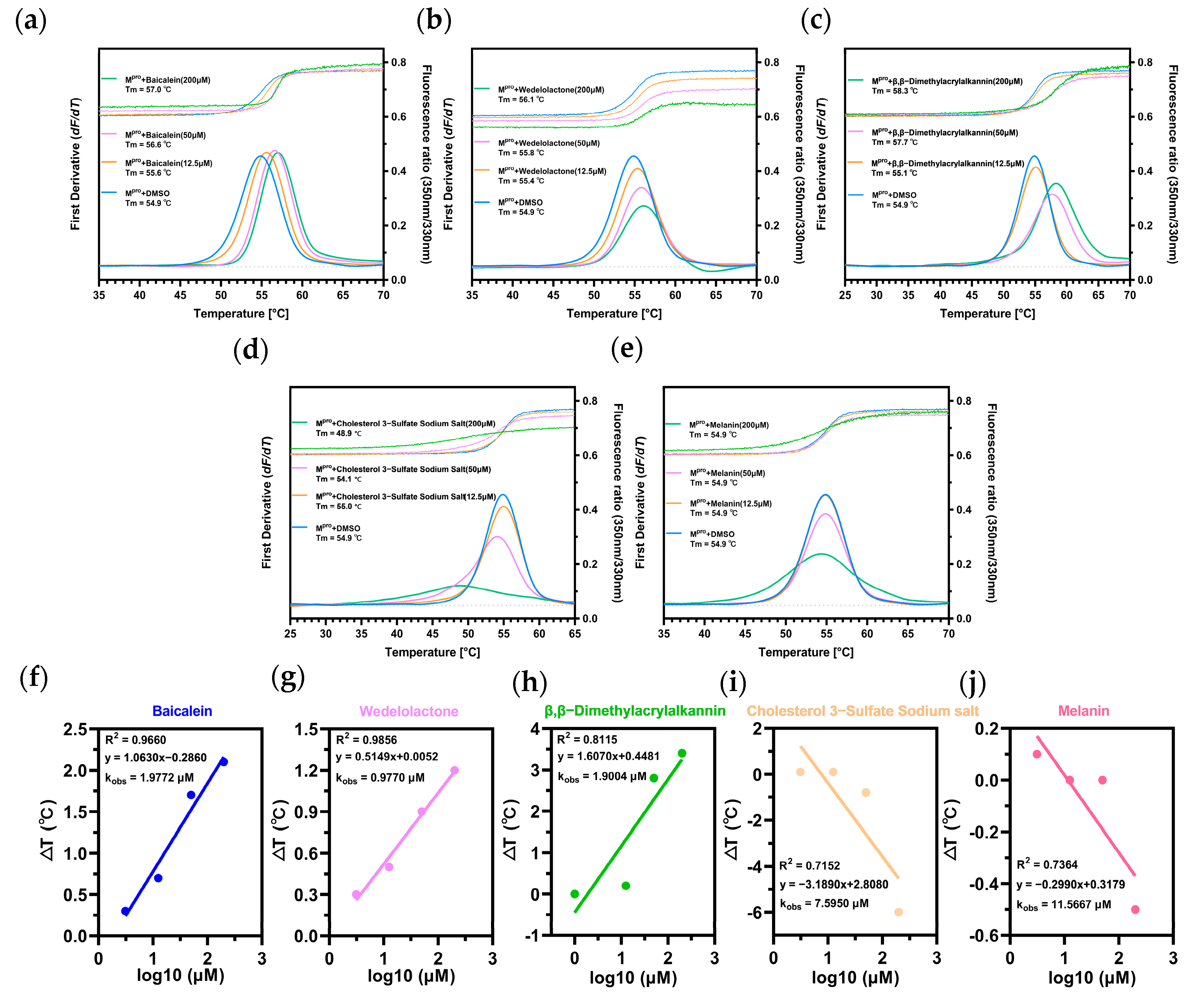
2.3. Biomolecular Interaction Experiments to Detect Binding
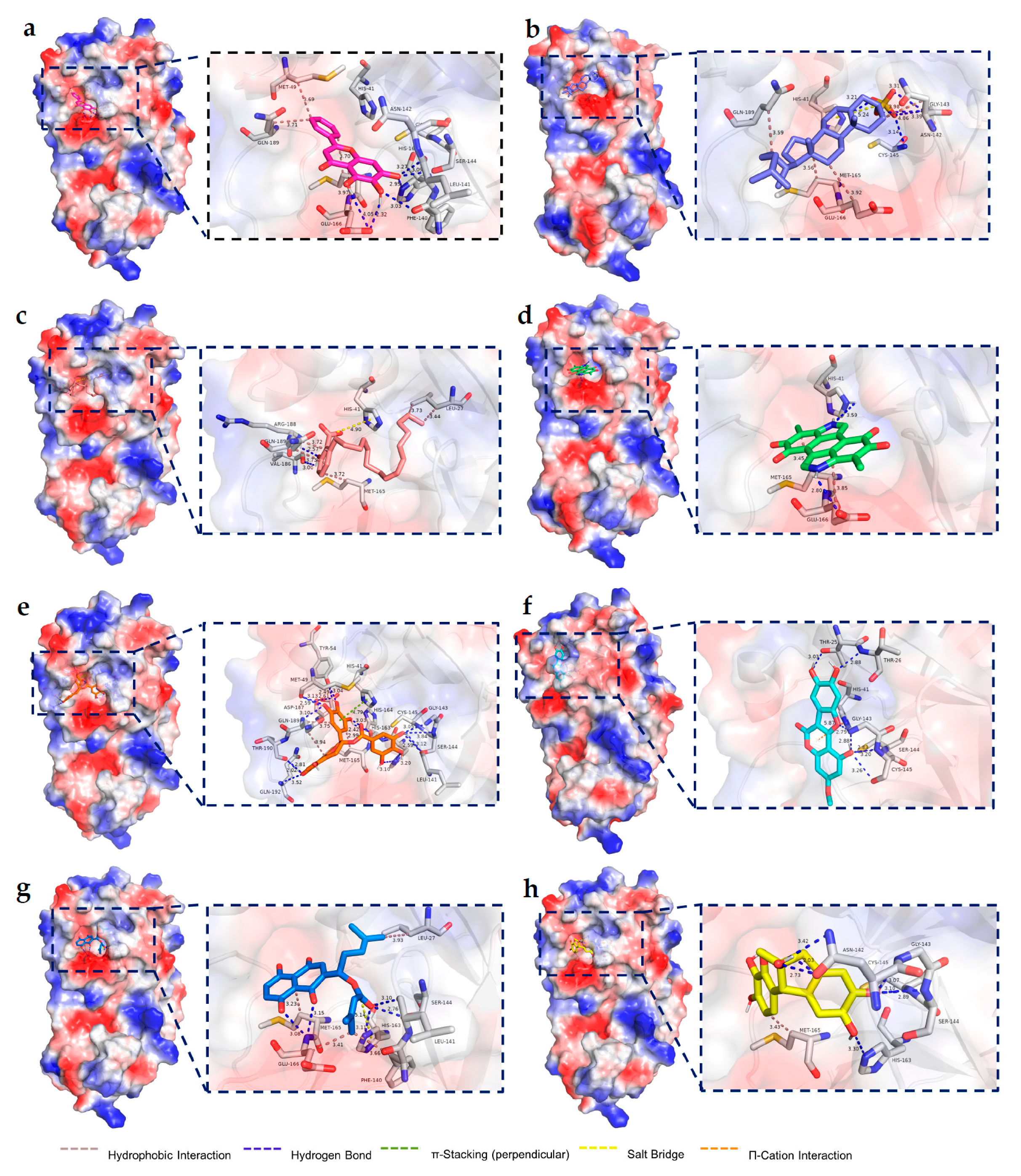
2.4. Molecular Docking Results
2.5. Assessing the Druggability
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Building the High-Throughput Screening Model
Protein Purification
Protein–Ligand Binding of Baicalein to Mpro Supported by ITC
Selection of FRET Systems
4.2.2. High-Throughput Screening
4.2.3. Biomolecular Interaction Experiments
SPR Experiments
Thermal Shift Assay
4.2.4. Molecular Docking
4.2.5. Assessing the Druggability
4.2.6. Ingredients-Herbal Counterpart
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating and Research Team. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324, Erratum in: PLoS Biol. 2005, 3, e428. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Pfizer Inc. Pfizer Shares Top-Line Results from Phase 2/3 EPIC-PEP Study of PAXLOVID™ for Post-Exposure Prophylactic Use. 29 April 2022. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-shares-top-line-results-phase-23-epic-pep-study#.YvzL5-Ynw5E.link (accessed on 6 May 2022).
- Wang, L.; Berger, N.A.; Davis, P.B.; Kaelber, D.C.; Volkow, N.D.; Xu, R. COVID-19 rebound after Paxlovid and Molnupiravir during January–June 2022. medRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Cui, J.-J.; Ouyang, Q.-Y.; Zhan, Y.; Guo, C.-X.; Yin, J.-Y. Remdesivir and COVID-19. Lancet 2020, 396, 953–954. [Google Scholar] [CrossRef]
- Grundeis, F.; Ansems, K.; Dahms, K.; Thieme, V.; Metzendorf, M.-I.; Skoetz, N.; Benstoem, C.; Mikolajewska, A.; Griesel, M.; Fichtner, F.; et al. Remdesivir for the treatment of COVID-19. Cochrane Database Syst. Rev. 2023, 1, CD014962. [Google Scholar] [CrossRef]
- Masyeni, S.; Iqhrammullah, M.; Frediansyah, A.; Nainu, F.; Tallei, T.; Bin Emran, T.; Ophinni, Y.; Dhama, K.; Harapan, H. Molnupiravir: A lethal mutagenic drug against rapidly mutating severe acute respiratory syndrome coronavirus 2—A narrative review. J. Med. Virol. 2022, 94, 3006–3016. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhuang, Z.; Zheng, J.; Li, K.; Wong, R.L.-Y.; Liu, D.; Huang, J.; He, J.; Zhu, A.; Zhao, J.; et al. Generation of a Broadly Useful Model for COVID-19 Pathogenesis, Vaccination, and Treatment. Cell 2020, 182, 734–743.e5. [Google Scholar] [CrossRef]
- Zhao, C.; Li, L.; Yang, W.; Lv, W.; Wang, J.; Guo, J.; Dong, Y.; Shi, N.; Lu, C.; Li, Z.; et al. Chinese Medicine Formula Huashibaidu Granule Early Treatment for Mild COVID-19 Patients: An Unblinded, Cluster-Randomized Clinical Trial. Front. Med. 2021, 8, 696976. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Tian, Y.; Ma, Y.; Liu, B.; Ruan, L.; Lu, C.; Huang, L.; National Traditional Chinese Medicine Medical Team. The effect of Huashibaidu formula on the blood oxygen saturation status of severe COVID-19: A retrospective cohort study. Phytomedicine 2022, 95, 153868. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, W.; Liu, Y.; Lu, C.; Ruan, L.; Zhao, C.; Huo, R.; Shen, X.; Miao, Q.; Lv, W.; et al. Combination of Hua Shi Bai Du granule (Q-14) and standard care in the treatment of patients with coronavirus disease 2019 (COVID-19): A single-center, open-label, randomized controlled trial. Phytomedicine 2021, 91, 153671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, C.; Li, H.; Qi, W.; Ruan, L.; Bian, Y.; Shi, H.; Song, H.; Tu, S.; Zhang, Y.; et al. Efficacy and safety assessment of severe COVID-19 patients with Chinese medicine: A retrospective case series study at early stage of the COVID-19 epidemic in Wuhan, China. J. Ethnopharmacol. 2021, 277, 113888. [Google Scholar] [CrossRef]
- Lyu, M.; Fan, G.; Xiao, G.; Wang, T.; Xu, D.; Gao, J.; Ge, S.; Li, Q.; Ma, Y.; Zhang, H.; et al. Traditional Chinese medicine in COVID-19. Acta Pharm. Sin. B 2021, 11, 3337–3363. [Google Scholar] [CrossRef]
- Shao, F.; Wilson, I.W.; Qiu, D. The Research Progress of Taxol in Taxus. Curr. Pharm. Biotechnol. 2021, 22, 360–366. [Google Scholar] [CrossRef]
- Tu, Y. Artemisinin-A Gift from Traditional Chinese Medicine to the World (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2016, 55, 10210–10226. [Google Scholar] [CrossRef]
- Hu, L.H. Re-examination of classical natural products. In Proceedings of the Seventh National Symposium on Natural Organic Chemistry, Chengdu, China, September 2008; Volume 38. [Google Scholar]
- Hasan, T.N.; Naqvi, S.S.; Rehman, M.U.; Ullah, R.; Ammad, M.; Arshad, N.; Ain, Q.U.; Perween, S.; Hussain, A. Ginger ring compounds as an inhibitor of spike binding protein of alpha, beta, gamma and delta variants of SARS CoV-2: An in-silico study. Narra J 2023, 3, e98. [Google Scholar] [CrossRef]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef] [PubMed]
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Liu, J.; Shang, W.J.; Xie, H.; Ke, C.Q.; Hu, H.C.; Gao, M.N.; et al. Anti-SARS CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef]
- Li, C.; Zhou, H.; Guo, L.; Xie, D.; He, H.; Zhang, H.; Liu, Y.; Peng, L.; Zheng, L.; Lu, W.; et al. Potential inhibitors for blocking the interaction of the coronavirus SARS CoV-2 spike protein and its host cell receptor ACE2. J. Transl. Med. 2022, 20, 314. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Li, Y.; Zhang, Y.; Liu, Z.; Mu, X.; Gu, C.; Liu, J.; Li, Y.; Li, G.; Chen, J. Ceftazidime is a potential drug to inhibit SARS CoV-2 infection in vitro by blocking spike protein-ACE2 interaction. Signal Transduct. Target. Ther. 2021, 6, 198, Erratum in: Signal Transduct. Target. Ther. 2021, 6, 230. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Verschueren, K.H.; Anand, K.; Shen, J.; Yang, M.; Xu, Y.; Rao, Z.; Bigalke, J.; Heisen, B.; Mesters, J.R.; et al. pH-dependent conformational flexibility of the SARS CoV main proteinase (M(pro)) dimer: Molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 2005, 354, 25–40. [Google Scholar] [CrossRef]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef]
- Aoyama, E.; Takigawa, M. Evaluation of Molecular Interaction between CCN2 Protein and Its Binding Partners by Surface Plasmon Resonance (SPR). Methods Mol. Biol. 2017, 1489, 169–176. [Google Scholar] [CrossRef]
- Magnusson, A.O.; Szekrenyi, A.; Joosten, H.; Finnigan, J.; Charnock, S.; Fessner, W. nanoDSF as screening tool for enzyme libraries and biotechnology development. FEBS J. 2019, 286, 184–204. [Google Scholar] [CrossRef]
- Lo, M.-C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Huynh, K.; Partch, C.L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 2015, 79, 28.9.1–28.9.14. [Google Scholar] [CrossRef]
- Sorenson, A.E.; Schaeffer, P.M. High-Throughput Differential Scanning Fluorimetry of GFP-Tagged Proteins. Methods Mol. Biol. 2020, 2089, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Lu, J.; Wang, Y.; Shen, Q.; Xiao, J.; Wang, F.; Du, Y.; Zheng, M.; Zhu, W.; Jiang, H.; et al. Prediction of the properties of several ADME/T. In Proceedings of the 28th Annual Meeting of the Chinese Chemical Society, Chengdu, China, 13 April 2012; Abstracts of the 14th session. Volume 122. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef]
- Li, J.; Zhou, X.; Zhang, Y.; Zhong, F.; Lin, C.; McCormick, P.J.; Jiang, F.; Luo, J.; Zhou, H.; Wang, Q.; et al. Crystal structure of SARS CoV-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci. Bull. 2021, 66, 661–663. [Google Scholar] [CrossRef]
- Xu, H.-Y.; Zhang, Y.-Q.; Liu, Z.-M.; Chen, T.; Lv, C.-Y.; Tang, S.; Zhang, X.-B.; Zhang, W.; Li, Z.-Y.; Zhou, R.-R.; et al. ETCM: An encyclopaedia of traditional Chinese medicine. Nucleic Acids Res. 2019, 47, D976–D982. [Google Scholar] [CrossRef]
- Neto, L.T.; Monteiro, M.L.G.; Galvan, D.; Conte-Junior, C.A. An Evaluation of the Potential of Essential Oils against SARS CoV-2 from In Silico Studies through the Systematic Review Using a Chemometric Approach. Pharmaceuticals 2021, 14, 1138. [Google Scholar] [CrossRef]
- Iqhrammullah, M.; Rizki, D.R.; Purnama, A.; Duta, T.F.; Harapan, H.; Idroes, R.; Ginting, B. Antiviral Molecular Targets of Essential Oils against SARS CoV-2: A Systematic Review. Sci. Pharm. 2023, 91, 15. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS CoV-2. main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Hematoxylin | Melanin | Wedelolactone | Ginkgolic Acid C15:1 | β,β–Dimethylacrylalkannin | Cholesteryl Sodium Sulfate | (−)–Gallocatechin Gallate |
|---|---|---|---|---|---|---|---|
| Molecular weight | 302.282 | 318.288 | 314.249 | 346.511 | 370.401 | 488.71 | 458.375 |
| LogP | 1.3205 | 1.32664 | 2.8178 | 6.5001 | 3.6375 | 3.8772 | 2.2332 |
| Rotatable bonds | 0 | 0 | 1 | 14 | 5 | 7 | 3 |
| Acceptors | 6 | 4 | 7 | 2 | 6 | 4 | 11 |
| Donors | 5 | 2 | 3 | 2 | 2 | 0 | 8 |
| Lipinski | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 1 violation | Yes; 0 violation | Yes; 0 violation | No; 2 violations |
| Water solubility (log mol/L) | −3.424 | −3.317 | −3.393 | −5.421 | −3.583 | −4.189 | −2.933 |
| Intestinal absorption (human) (% Absorbed) | 84.974 | 99.755 | 95.067 | 88.537 | 63.844 | 100 | 46.991 |
| Fraction bound (%) (human) | 91.4 | 87.4 | 97.5 | 92.5 | 79.6 | 93.8 | 76.1 |
| BBB permeability (log BB) | −1.135 | −0.078 | −1.262 | −0.403 | −0.076 | −0.437 | −2.058 |
| CYP2D6 substrate | No | No | No | No | No | No | No |
| CYP3A4 substrate | No | Yes | No | Yes | No | Yes | No |
| CYP1A2 inhibitor | No | Yes | Yes | No | No | No | Yes |
| CYP2C19 inhibitor | No | No | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | Yes | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | Yes | No | No | No | No |
| Total clearance (log mL/min/kg) | 0.046 | 0.521 | 0.677 | 1.59 | 0.402 | 0.685 | 0.431 |
| Max. tolerated dose (human)(log mg/kg/day) | 0.403 | 0.485 | 0.751 | 0.066 | −0.246 | −0.75 | 0.605 |
| hERG I inhibitor | No | No | No | No | No | No | No |
| Oral rat acute toxicity (ld50) (mol/kg) | 2.07 | 2.457 | 2.363 | 2.111 | 1.819 | 2.359 | 2.764 |
| Oral rat Chronic toxicity (LOAEL) (log mg/kg_bw/day) | 3.032 | 1.963 | 1.377 | 2.643 | 1.954 | 1.141 | 3.844 |
| Hepatotoxicity | No | No | No | No | No | No | No |
| Skin sensitization | No | No | No | No | No | No | No |
| Group | Protein (μL) | Sample (μL) | Substrate (μL) | Final Volume (μL) |
|---|---|---|---|---|
| Sample group | 40 | 40 sample | 20 | 100 |
| Positive control | 40 | 40 baicalein | 20 | 100 |
| Negative control | 40 | 40 buffer | 20 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Zhou, X.; Fu, L.; Xu, H. Natural Product-Based Screening for Lead Compounds Targeting SARS CoV-2 Mpro. Pharmaceuticals 2023, 16, 767. https://doi.org/10.3390/ph16050767
Chen J, Zhou X, Fu L, Xu H. Natural Product-Based Screening for Lead Compounds Targeting SARS CoV-2 Mpro. Pharmaceuticals. 2023; 16(5):767. https://doi.org/10.3390/ph16050767
Chicago/Turabian StyleChen, Jie, Xiang Zhou, Lifeng Fu, and Haiyu Xu. 2023. "Natural Product-Based Screening for Lead Compounds Targeting SARS CoV-2 Mpro" Pharmaceuticals 16, no. 5: 767. https://doi.org/10.3390/ph16050767
APA StyleChen, J., Zhou, X., Fu, L., & Xu, H. (2023). Natural Product-Based Screening for Lead Compounds Targeting SARS CoV-2 Mpro. Pharmaceuticals, 16(5), 767. https://doi.org/10.3390/ph16050767