

Recent Advances in the Green Synthesis of Active N-Heterocycles and Their Biological Activities

Abstract
:1. Introduction
2. Greener Access to Bioactive N-Heterocyclic Compounds
2.1. Microwave-Assisted Synthesis
2.1.1. Pyrazole Derivatives
2.1.2. Tetrazole Derivatives
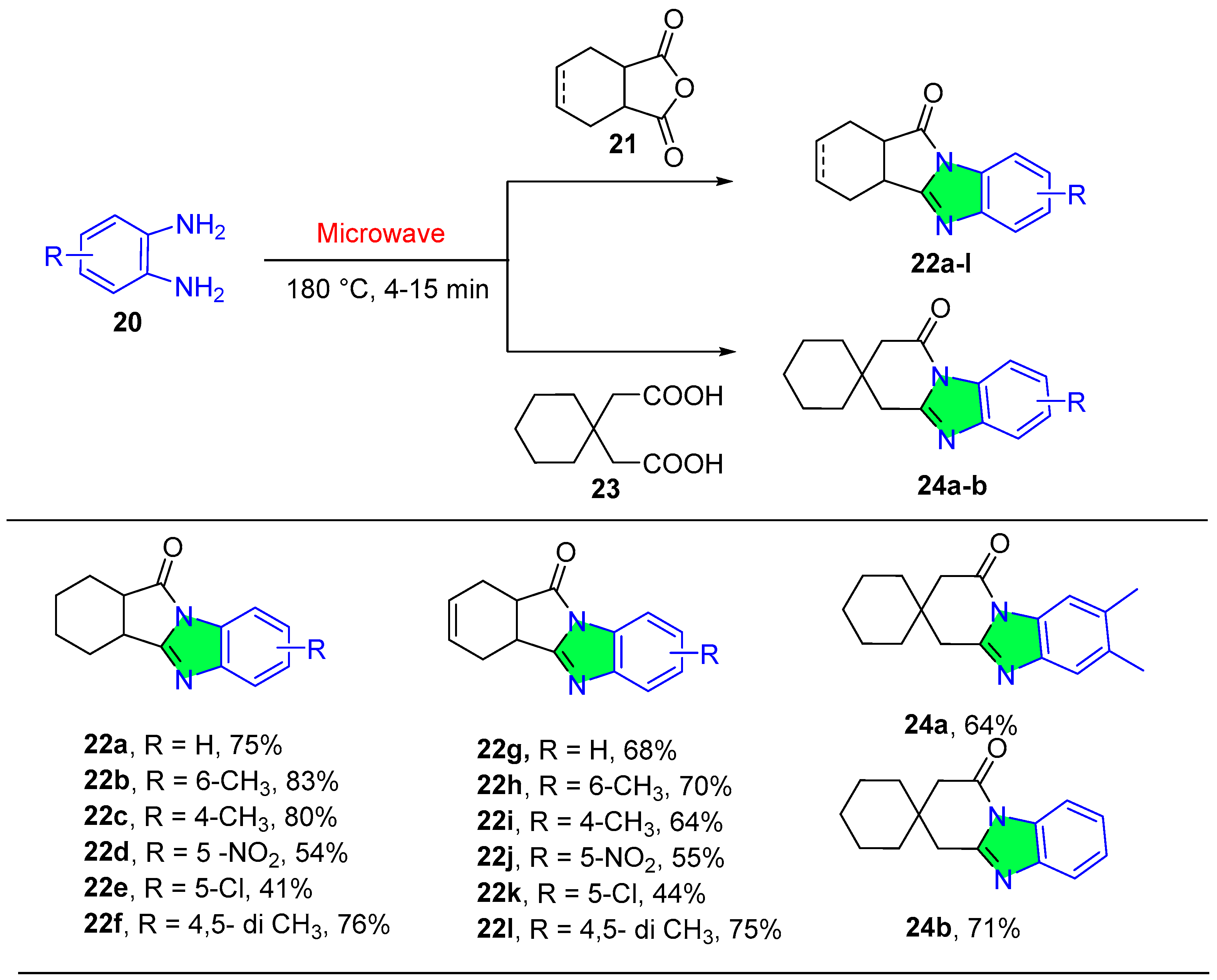
2.1.3. Benzimidazole Derivatives
2.1.4. Pyrimidine Derivatives
2.1.5. Quinoline Derivatives
2.1.6. Pyrido-Pyrimidine Derivatives
2.1.7. 1,2,3-Triazole-Conjugated Benzodiazepine
2.1.8. Tetrazole-Conjugated Benzodiazepines
2.2. Solvent-Free Synthesis
3. Heterogeneous Catalysis
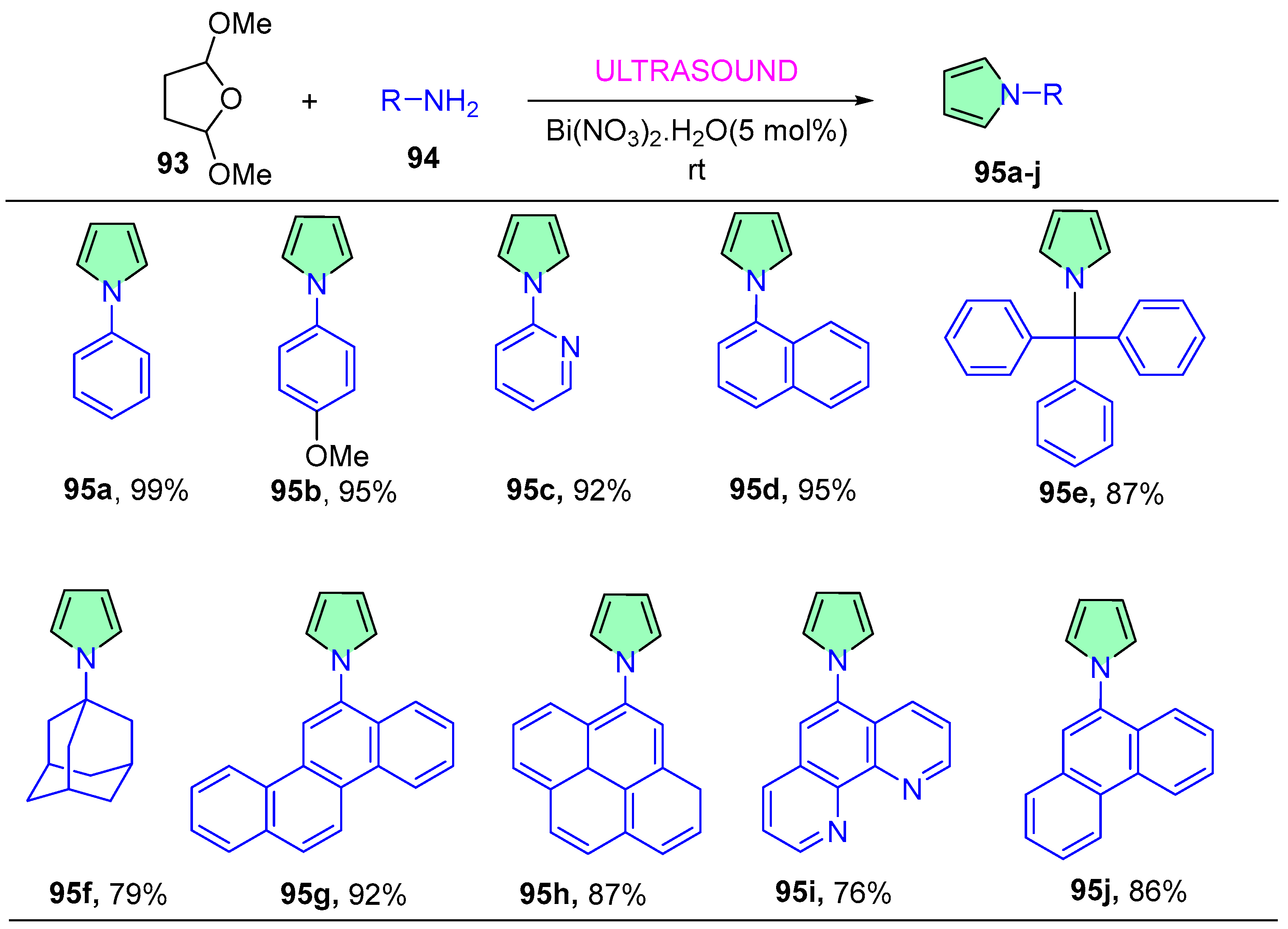
4. Ultrasound-Mediated Reactions
5. Biocatalyst-Mediated Reaction
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robb, M.J.; Moore, J.S. A Retro-Staudinger Cycloaddition: Mechanochemical Cycloelimination of a β-Lactam Mechanophore. J. Am. Chem. Soc. 2015, 137, 10946–10949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baiula, M.; Galletti, P.; Martelli, G.; Soldati, R.; Belvisi, L.; Civera, M.; Dattoli, S.D.; Spampinato, S.M.; Giacomini, D. New β-Lactam Derivatives Modulate Cell Adhesion and Signaling Mediated by RGD-Binding and Leukocyte Integrins. J. Med. Chem. 2016, 59, 9721–9742. [Google Scholar] [CrossRef] [PubMed]
- Mohamadzadeh, M.; Zarei, M.; Vessal, M. Synthesis, in Vitro Biological Evaluation and in Silico Molecular Docking Studies of Novel β-Lactam-Anthraquinone Hybrids. Bioorg. Chem. 2020, 95, 103515. [Google Scholar] [CrossRef]
- Sun, L.; Huang, T.; Dick, A.; Meuser, M.E.; Zalloum, W.A.; Chen, C.-H.; Ding, X.; Gao, P.; Cocklin, S.; Lee, K.-H.; et al. Design, Synthesis and Structure-Activity Relationships of 4-Phenyl-1H-1,2,3-Triazole Phenylalanine Derivatives as Novel HIV-1 Capsid Inhibitors with Promising Antiviral Activities. Eur. J. Med. Chem. 2020, 190, 112085. [Google Scholar] [CrossRef]
- Zhu, W.-J.; Cui, B.-W.; Wang, H.M.; Nan, J.-X.; Piao, H.-R.; Lian, L.-H.; Jin, C.H. Design, Synthesis, and Antifibrosis Evaluation of 4-(Benzo-[c][1,2,5]Thiadiazol-5-Yl)-3(5)-(6-Methyl- Pyridin-2-Yl)Pyrazole and 3(5)-(6-Methylpyridin-2-Yl)-4-(Thieno-[3,2,-c]Pyridin-2-Yl)Pyrazole Derivatives. Eur. J. Med. Chem. 2019, 180, 15–27. [Google Scholar] [CrossRef]
- Taha, M.; Sultan, S.; Imran, S.; Rahim, F.; Zaman, K.; Wadood, A.; Ur Rehman, A.; Uddin, N.; Mohammed Khan, K. Synthesis of Quinoline Derivatives as Diabetic II Inhibitors and Molecular Docking Studies. Bioorg. Med. Chem. 2019, 27, 4081–4088. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Singh, A.; Agarwal, A.; Chakraborty, A.; Bhardwaj, R.; Sutradhar, S.; Kumar Mittal, A.; Kumar Rajput, S.; Gupta, M.; Ray, D.; Mukherjee, M. Click Chemistry Tailored Benzimidazole Functionalized Triazole Block-Co-Polymer for Emergence of Exotic Chimaeric Nano-Crystalsomes. Eur. Polym. J. 2022, 178, 111503. [Google Scholar] [CrossRef]
- Kumar, V.; Saxena, A.; Patra, R.; Ray, D.; Li, H.; Saha, B. Synthesis of Fused Polycyclic β-Carboline Derivatives Using Ugi-4CR Followed by Cascade Cyclization. Mol. Divers. 2023, 27, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Naresh Yadav, R.; Krishna Banik, B. Vitamin C-Catalyzed Hantzsch Reaction under Microwave Condition: A Greener Access to 1,4-Dihydropyridines. Results Chem. 2022, 4, 100330. [Google Scholar] [CrossRef]
- Kumar, V.; Sachdeva, C.; Waidha, K.; Sharma, S.; Ray, D.; Kumar Kaushik, N.; Saha, B. In Vitro and In Silico Anti-plasmodial Evaluation of Newly Synthesized Β-Carboline Derivatives. ChemistrySelect 2021, 6, 5338–5342. [Google Scholar] [CrossRef]
- Waidha, K.; Saxena, A.; Kumar, P.; Sharma, S.; Ray, D.; Saha, B. Design and Identification of Novel Annomontine Analogues against SARS-CoV-2: An in-Silico Approach. Heliyon 2021, 7, e06657. [Google Scholar] [CrossRef]
- Kumar, V.; Banert, K.; Ray, D.; Saha, B. An Atom-Economical and Regioselective Metal-Free C-5 Chalcogenation of 8-Aminoquinolines under Mild Conditions. Org. Biomol. Chem. 2019, 17, 10245–10250. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.M.; Banik, B.K.; Kumar, B.V.V.R.; Panda, K.C.; Tiwari, A.; Tiwari, V.; Singh, S.; Kumar, M. Microwave Induced Green Synthesis: Sustainable Technology for Efficient Development of Bioactive Pyrimidine Scaffolds. Curr. Med. Chem. 2023, 30, 1029–1059. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Banik, B.K. Microwave-Induced Biocatalytic Reactions toward Medicinally Important Compounds. Phys. Sci. Rev. 2022, 7, 507–538. [Google Scholar] [CrossRef]
- Banik, B.K.; Sahoo, B.M.; Kumar, B.R.; Panda, K.C. Microwave Induced Green Chemistry Approach Towards the Synthesis of Heterocyclic Compounds via C-N Bond Forming Reactions. Curr. Microw. Chem. 2021, 8, 204–214. [Google Scholar] [CrossRef]
- Pantaine, L.R.E.; Milligan, J.A.; Matsui, J.K.; Kelly, C.B.; Molander, G.A. Photoredox Radical/Polar Crossover Enables Construction of Saturated Nitrogen Heterocycles. Org. Lett. 2019, 21, 2317–2321. [Google Scholar] [CrossRef]
- Cagir, A.; Jones, S.H.; Gao, R.; Eisenhauer, B.M.; Hecht, S.M.; Luotonin, A. A Naturally Occurring Human DNA Topoisomerase I Poison. J. Am. Chem. Soc. 2003, 125, 13628–13629. [Google Scholar] [CrossRef]
- Santos, A.S.; Raydan, D.; Cunha, J.C.; Viduedo, N.; Silva, A.M.S.; Marques, M.M.B. Advances in Green Catalysis for the Synthesis of Medicinally Relevant N-Heterocycles. Catalysts 2021, 11, 1108. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Banik, B.K. Microwave-Assisted Synthesis of Medicinally Privileged Heterocycles. In Green Synthetic Approaches for Biologically Relevant Heterocycles; Elsevier: Amsterdam, The Netherlands, 2021; pp. 49–110. [Google Scholar] [CrossRef]
- Ray, D. A Greener Synthetic Approach to Tetrazoles via Multicomponent Reactions. Curr. Organocatalysis 2023, 10. [Google Scholar] [CrossRef]
- Zárate-Zárate, D.; Aguilar, R.; Hernández-Benitez, R.I.; Labarrios, E.M.; Delgado, F.; Tamariz, J. Synthesis of α-Ketols by Functionalization of Captodative Alkenes and Divergent Preparation of Heterocycles and Natural Products. Tetrahedron 2015, 71, 6961–6978. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetto Tiz, D.; Bagnoli, L.; Rosati, O.; Marini, F.; Santi, C.; Sancineto, L. FDA-Approved Small Molecules in 2022: Clinical Uses and Their Synthesis. Pharmaceutics 2022, 14, 2538. [Google Scholar] [CrossRef] [PubMed]
- Kurkin, A.V.; Curreli, F.; Iusupov, I.R.; Spiridonov, E.A.; Ahmed, S.; Markov, P.O.; Manasova, E.V.; Altieri, A.; Debnath, A.K. Design, Synthesis, and Antiviral Activity of the Thiazole Positional Isomers of a Potent HIV-1 Entry Inhibitor NBD-14270. ChemMedChem 2022, 17, e202200344. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Ingarra, A.M.; Raimondi, M.V.; Spanò, V.; Piccionello, A.P.; De Franco, M.; Menilli, L.; Gandin, V.; Miolo, G.; Barraja, P.; et al. New Tricyclic Systems as Photosensitizers towards Triple Negative Breast Cancer Cells. Arch. Pharm. Res. 2022, 45, 806–821. [Google Scholar] [CrossRef]
- Oniciuc, L.; Amăriucăi-Mantu, D.; Diaconu, D.; Mangalagiu, V.; Danac, R.; Antoci, V.; Mangalagiu, I.I. Benzoquinoline Derivatives: An Attractive Approach to Newly Small Molecules with Anticancer Activity. Int. J. Mol. Sci. 2023, 24, 8124. [Google Scholar] [CrossRef]
- Becerra, D.; Abonia, R.; Castillo, J.-C. Recent Applications of the Multicomponent Synthesis for Bioactive Pyrazole Derivatives. Molecules 2022, 27, 4723. [Google Scholar] [CrossRef]
- Grillone, K.; Riillo, C.; Rocca, R.; Ascrizzi, S.; Spanò, V.; Scionti, F.; Polerà, N.; Maruca, A.; Barreca, M.; Juli, G.; et al. The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 10222. [Google Scholar] [CrossRef]
- Walsh, C.T. Nature Loves Nitrogen Heterocycles. Tetrahedron Lett. 2015, 56, 3075–3081. [Google Scholar] [CrossRef]
- Zhang, B.; Studer, A. Recent Advances in the Synthesis of Nitrogen Heterocycles via Radical Cascade Reactions Using Isonitriles as Radical Acceptors. Chem. Soc. Rev. 2015, 44, 3505–3521. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Zhang, Z.; Sharma, A.R.; Nakajima-Shimada, J.; Harunari, E.; Oku, N.; Trianto, A.; Igarashi, Y. Bulbiferamide, an Antitrypanosomal Hexapeptide Cyclized via an N -Acylindole Linkage from a Marine Obligate Microbulbifer. J. Nat. Prod. 2023, 86, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sheng, H.; Wang, Y.; Lai, Z.; Wang, Y.; Cui, S. Scaffold Hybrid of the Natural Product Tanshinone I with Piperidine for the Discovery of a Potent NLRP3 Inflammasome Inhibitor. J. Med. Chem. 2023, 66, 2946–2963. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-J.; Li, P.; Wu, B.-W.; Cui, X.-X.; Zhao, C.-B.; Zhang, S.-Y. Molecular Diversity of Trimethoxyphenyl-1,2,3-Triazole Hybrids as Novel Colchicine Site Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2019, 165, 309–322. [Google Scholar] [CrossRef]
- Ashour, H.F.; Abou-zeid, L.A.; El-Sayed, M.A.-A.; Selim, K.B. 1,2,3-Triazole-Chalcone Hybrids: Synthesis, in Vitro Cytotoxic Activity and Mechanistic Investigation of Apoptosis Induction in Multiple Myeloma RPMI-8226. Eur. J. Med. Chem. 2020, 189, 112062. [Google Scholar] [CrossRef]
- Asgari, M.S.; Mohammadi-Khanaposhtani, M.; Kiani, M.; Ranjbar, P.R.; Zabihi, E.; Pourbagher, R.; Rahimi, R.; Faramarzi, M.A.; Biglar, M.; Larijani, B.; et al. Biscoumarin-1,2,3-Triazole Hybrids as Novel Anti-Diabetic Agents: Design, Synthesis, in Vitro α-Glucosidase Inhibition, Kinetic, and Docking Studies. Bioorg. Chem. 2019, 92, 103206. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, M.; Mohammadi-Khanaposhtani, M.; Pourrabia, P.; Razzaghi, N.; Ghadimi, R.; Imanparast, S.; Faramarzi, M.A.; Bandarian, F.; Esfahani, E.N.; Safavi, M.; et al. Design and Synthesis of Novel Quinazolinone-1,2,3-Triazole Hybrids as New Anti-Diabetic Agents: In Vitro α-Glucosidase Inhibition, Kinetic, and Docking Study. Bioorg. Chem. 2019, 83, 161–169. [Google Scholar] [CrossRef]
- Kim, D.; Kang, M.-S.; Kim, J.S.; Jeong, J.-H. An Efficient Synthesis of Risperidonevia Stille Reaction: Antipsychotic, 5-HT2, and Dopamine-D2-Antagonist. Arch. Pharm. Res. 2005, 28, 1019–1022. [Google Scholar] [CrossRef]
- Campos, K.R.; Woo, J.C.S.; Lee, S.; Tillyer, R.D. A General Synthesis of Substituted Indoles from Cyclic Enol Ethers and Enol Lactones. Org. Lett. 2004, 6, 79–82. [Google Scholar] [CrossRef]
- Li, M.-M.; Chen, X.; Deng, Y.; Lu, J. Recent Advances of N-Heterocyclic Carbenes in the Applications of Constructing Carbo- and Heterocyclic Frameworks with Potential Biological Activity. RSC Adv. 2021, 11, 38060–38078. [Google Scholar] [CrossRef]
- Sreedevi, R.; Saranya, S.; Rohit, K.R.; Anilkumar, G. Recent Trends in Iron-Catalyzed Reactions towards the Synthesis of Nitrogen-Containing Heterocycles. Adv. Synth. Catal. 2019, 361, 2236–2249. [Google Scholar] [CrossRef]
- Li, H.; Guo, H.; Fang, Z.; Aida, T.M.; Smith, R.L. Cycloamination Strategies for Renewable N-Heterocycles. Green Chem. 2020, 22, 582–611. [Google Scholar] [CrossRef] [Green Version]
- Srinath, S.; Abinaya, R.; Prasanth, A.; Mariappan, M.; Sridhar, R.; Baskar, B. Reusable, Homogeneous Water Soluble Photoredox Catalyzed Oxidative Dehydrogenation of N-Heterocycles in a Biphasic System: Application to the Synthesis of Biologically Active Natural Products. Green Chem. 2020, 22, 2575–2587. [Google Scholar] [CrossRef]
- Gulati, S.; Singh, R.; Sangwan, S. A Review on Green Synthesis and Biological Activities of Medicinally Important Nitrogen and Oxygen Containing Heterocycles. Curr. Org. Chem. 2022, 26, 1848–1894. [Google Scholar] [CrossRef]
- Arya, N.; Jagdale, A.Y.; Patil, T.A.; Yeramwar, S.S.; Holikatti, S.S.; Dwivedi, J.; Shishoo, C.J.; Jain, K.S. The Chemistry and Biological Potential of Azetidin-2-Ones. Eur. J. Med. Chem. 2014, 74, 619–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Peng, X.-M.; Damu, G.L.V.; Geng, R.-X.; Zhou, C.-H. Comprehensive Review in Current Developments of Imidazole-Based Medicinal Chemistry. Med. Res. Rev. 2014, 34, 340–437. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman, S. Review: Biologically Active Pyrazole Derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Jain, S.; Chandra, V.; Kumar Jain, P.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive Review on Current Developments of Quinoline-Based Anticancer Agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.; Baxendale, I.R.; Ley, S.V.; Nikbin, N. An Overview of the Key Routes to the Best Selling 5-Membered Ring Heterocyclic Pharmaceuticals. Beilstein J. Org. Chem. 2011, 7, 442–495. [Google Scholar] [CrossRef]
- Gomha, S.M.; Edrees, M.M.; Faty, R.A.M.; Muhammad, Z.A.; Mabkhot, Y.N. Microwave-Assisted One Pot Three-Component Synthesis of Some Novel Pyrazole Scaffolds as Potent Anticancer Agents. Chem. Cent. J. 2017, 11, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddivari, C.K.R.; Devineni, S.R.; Venkateshwarulu, J.K.M.; Baki, V.B.; Chippada, A.R.; Wudayagiri, R.; Venkata, R.R.Y.; Chamarthi, N.R. ZnBr2-SiO2 Catalyzed Green Synthesis of Tetrazoles: Molecular Docking and Antioxidant Activity Studies. Eur. J. Chem. 2017, 8, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Bui, H.T.B.; Ha, Q.T.K.; Oh, W.K.; Vo, D.D.; Chau, Y.N.T.; Tu, C.T.K.; Pham, E.C.; Tran, P.T.; Tran, L.T.; Mai, H. Van Microwave Assisted Synthesis and Cytotoxic Activity Evaluations of New Benzimidazole Derivatives. Tetrahedron Lett. 2016, 57, 887–891. [Google Scholar] [CrossRef]
- Krim, J.; Grünewald, C.; Taourirte, M.; Engels, J.W. Efficient Microwave-Assisted Synthesis, Antibacterial Activity and High Fluorescence of 5 Benzimidazolyl-2′-Deoxyuridines. Bioorg. Med. Chem. 2012, 20, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Banerjee, S.; Roy, P.; Sondhi, S.M.; Sharma, A. Solvent-Free Synthesis and Anticancer Activity Evaluation of Benzimidazole and Perimidine Derivatives. Mol. Divers. 2018, 22, 113–127. [Google Scholar] [CrossRef]
- Aguado, L.; Canela, M.-D.; Thibaut, H.J.; Priego, E.-M.; Camarasa, M.-J.; Leyssen, P.; Neyts, J.; Pérez-Pérez, M.-J. Efficient Synthesis and Anti-Enteroviral Activity of 9-Arylpurines. Eur. J. Med. Chem. 2012, 49, 279–288. [Google Scholar] [CrossRef]
- Elumalai, K.; Ali, M.A.; Elumalai, M.; Eluri, K.; Srinivasan, S. Acetylcholinesterase Enzyme Inhibitor Activity of Some Novel Pyrazinamide Condensed 1,2,3,4-Tetrahydropyrimidines. Biotechnol. Rep. 2015, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Basiri, A.; Murugaiyah, V.; Osman, H.; Kumar, R.S.; Kia, Y.; Awang, K.B.; Ali, M.A. An Expedient, Ionic Liquid Mediated Multi-Component Synthesis of Novel Piperidone Grafted Cholinesterase Enzymes Inhibitors and Their Molecular Modeling Study. Eur. J. Med. Chem. 2013, 67, 221–229. [Google Scholar] [CrossRef]
- Basiri, A.; Murugaiyah, V.; Osman, H.; Kumar, R.S.; Kia, Y.; Ali, M.A. Microwave Assisted Synthesis, Cholinesterase Enzymes Inhibitory Activities and Molecular Docking Studies of New Pyridopyrimidine Derivatives. Bioorg. Med. Chem. 2013, 21, 3022–3031. [Google Scholar] [CrossRef]
- Hosamani, K.M.; Reddy, D.S.; Devarajegowda, H.C. Microwave-Assisted Synthesis of New Fluorinated Coumarin-Pyrimidine Hybrids as Potent Anticancer Agents, Their DNA Cleavage and X-ray Crystal Studies. RSC Adv. 2015, 5, 11261–11271. [Google Scholar] [CrossRef]
- Panda, K.C.; Kumar, B.V.V.R.; Sahoo, B.S.; Banik, B.K.; Tiwari, A. Microwave Irradiated Eco-friendly Synthesis of Pyridine Derivatives as Potential Antitubercular Agents. Asian J. Chem. 2022, 34, 907–911. [Google Scholar] [CrossRef]
- Liberto, N.A.; Simões, J.B.; de Paiva Silva, S.; da Silva, C.J.; Modolo, L.V.; de Fátima, Â.; Silva, L.M.; Derita, M.; Zacchino, S.; Zuñiga, O.M.P.; et al. Quinolines: Microwave-Assisted Synthesis and Their Antifungal, Anticancer and Radical Scavenger Properties. Bioorg. Med. Chem. 2017, 25, 1153–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, P.; Insuasty, B.; Ortiz, A.; Abonia, R.; Sortino, M.; Zacchino, S.A.; Quiroga, J. Solvent-Free Microwave-Assisted Synthesis of Novel Pyrazolo[4′,3′:5,6]Pyrido[2,3-d]Pyrimidines with Potential Antifungal Activity. Arab. J. Chem. 2016, 9, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Jaafar, Z.; Chniti, S.; Ben Sassi, A.; Dziri, H.; Marque, S.; Lecouvey, M.; Gharbi, R.; Msaddek, M. Design and Microwave-Assisted Synthesis of Dimers of 1,5-Benzodiazepine-1,2,3-Triazole Hybrids Bearing Alkyl/Aryl Spacers and Their Biological Assessment. J. Mol. Struct. 2019, 1195, 689–701. [Google Scholar] [CrossRef]
- Abdallah, W.; Znati, M.; Regazzetti, A.; Dargère, D.; Laprévote, O.; Ben Jannet, H.; Gharbi, R. Synthesis of S-Mono- and S,O-Bis-1,2,3-Triazole Linked 1,5-Benzodiazepine Conjugates and Evaluation of Their Cytotoxic, Anti-Tyrosinase, and Anti-Cholinesterase Activities. Phosphorus Sulfur. Silicon. Relat. Elem. 2017, 192, 835–844. [Google Scholar] [CrossRef]
- Bhoge, N.D.; Magare, B.K.; Mohite, P.B.; Jangale, M.S. Green Chemistry Approach for the Synthesis of Novel Tetrazole Derivatives and Evaluation of Antifungal Activity. Eur. Chem. Bull. 2019, 8, 265. [Google Scholar] [CrossRef] [Green Version]
- Tupare, S.D.; Pawar, R.P. Highly Efficient Synthesis and Antibacterial of 1,5-Benzodiazepines under Microwave Irradiation. Int. J. Appl. Chem. 2017, 13, 369–376. [Google Scholar]
- Sorra, K.; Chen, C.S.; Chang, C.F.; Pusuluri, S.; Mukkanti, K.; Wu, C.R.; Chuang, T.H. Synthesis, Anticonvulsant, Sedative and Anxiolytic Activities of Novel Annulated Pyrrolo[1,4]Benzodiazepines. Int. J. Mol. Sci. 2014, 15, 16500–16510. [Google Scholar] [CrossRef] [Green Version]
- Mariki, A.A.; Anaeigoudari, A.; Zahedifar, M.; Pouramiri, B.; Ayati, A.; Lotfi, S. Design, Green Synthesis, and Biological Evaluation of New Substituted Tetrahydropyrimidine Derivatives as Acetylcholinesterase Inhibitors. Polycycl. Aromat. Compd. 2022, 42, 5231–5241. [Google Scholar] [CrossRef]
- Elangovan, S.; Afanasenko, A.; Haupenthal, J.; Sun, Z.; Liu, Y.; Hirsch, A.K.H.; Barta, K. From Wood to Tetrahydro-2-Benzazepines in Three Waste-Free Steps: Modular Synthesis of Biologically Active Lignin-Derived Scaffolds. ACS Cent. Sci. 2019, 5, 1707–1716. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.S.; Kumar, P.M.; Kumar, K.A.; Sreenivasulu, M.; Jafar, A.A.; Rambabu, D.; Krishna, G.R.; Reddy, C.M.; Kapavarapu, R.; Shivakumar, K.; et al. A New Three-Component Reaction: Green Synthesis of Novel Isoindolo[2,1-a]Quinazoline Derivatives as Potent Inhibitors of TNF-α. Chem. Commun. 2011, 47, 5010–5012. [Google Scholar] [CrossRef]
- Kumar, G.; Mogha, N.K.; Kumar, M.; Subodh; Masram, D.T. NiO Nanocomposites/RGO as a Heterogeneous Catalyst for Imidazole Scaffolds with Applications in Inhibiting the DNA Binding Activity. Dalt. Trans. 2020, 49, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Parveen, M.; Ahmad, F.; Mohammed Malla, A.; Azaz, S. SiO2–H3BO3 Promoted Solvent-Free, Green and Sustainable Synthesis of Bioactive 1-Substituted-1H-Tetrazole Analogues. New J. Chem. 2015, 39, 2028–2041. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Mukherjee, S.; Granados, J.C.; Short, J.D.; Banik, B.K. Ultrasound-Assisted Bismuth Nitrate-Induced Green Synthesis of Novel Pyrrole Derivatives and Their Biological Evaluation as Anticancer Agents. Eur. J. Med. Chem. 2012, 50, 209–215. [Google Scholar] [CrossRef]
- Tiwari, S.V.; Seijas, J.A.; Vazquez-Tato, M.P.; Sarkate, A.P.; Lokwani, D.K.; Nikalje, A.P.G. Ultrasound Mediated One-Pot, Three Component Synthesis, Docking and ADME Prediction of Novel 5-Amino-2-(4-Chlorophenyl)-7-Substituted Phenyl-8,8a-Dihydro-7H-(1,3,4)Thiadiazolo(3,2-α) Pyrimidine-6-Carbonitrile Derivatives as Anticancer Agents. Molecules 2016, 21, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawandi, S.J.; Desai, V.G.; Joshi, S.; Shingade, S.; Pissurlenkar, R.R. Assessment of Elementary Derivatives of 1,5-Benzodiazepine as Anticancer Agents with Synergy Potential. Bioorg. Chem. 2021, 117, 105331. [Google Scholar] [CrossRef] [PubMed]































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) ± SEM | |
|---|---|---|
| AChE | BuChE | |
| 31a | 5.35 ± 0.01 | 7.21 ± 0.01 |
| 31b | 2.54 ± 0.01 | 5.93 ± 0.01 |
| 31c | 1.21 ± 0.01 | 4.96 ± 0.01 |
| 31d | 0.86 ± 0.01 | 4.84 ± 0.01 |
| 31e | 0.94 ± 0.01 | 4.75 ± 0.01 |
| 31f | 5.26 ± 0.01 | 6.75 ± 0.01 |
| 31g | 1.82 ± 0.01 | 5.38 ± 0.01 |
| 31h | 1.05 ± 0.01 | 4.31 ± 0.01 |
| 31i | 0.75 ± 0.01 | 3.93 ± 0.01 |
| 31j | 0.88 ± 0.01 | 4.13 ± 0.01 |
| 31k | 0.19 ± 0.01 | 3.92 ± 0.01 |
| 31l | 0.11 ± 0.01 | 3.46 ± 0.01 |
| Donepezil HCl | 0.13 ± 0.01 | 3.58 ± 0.01 |
| Compound | X | Ar | Yield% | IC50 (μΜ) | |
|---|---|---|---|---|---|
| AChE | BChE | ||||
| 40a | O | C6H5 | 94 | 68.73 | 2.86 |
| 40b | O | 2-CH3C6H4 | 91 | 40.42 | 5.22 |
| 40c | O | 2-ClC6H4 | 90 | 24.14 | 2.51 |
| 40d | O | 2-FC6H4 | 89 | 18.59 | 3.31 |
| 40e | O | 2-OCH3C6H4 | 93 | 40.23 | 10.70 |
| 40f | O | 3-NO2C6H4 | 91 | 28.27 | 10.34 |
| 40g | O | 4-BrC6H4 | 88 | 15.86 | 6.22 |
| 40h | O | 4-CH3C6H4 | 90 | 46.52 | 7.09 |
| 40i | O | 4-ClC6H4 | 92 | 36.84 | 16.73 |
| 40j | O | 4-FC6H4 | 94 | 44.23 | 18.90 |
| 40k | O | 2,4-Cl2C6H4 | 91 | 29.86 | 23.51 |
| 40l | O | 1-napthyl | 92 | 13.16 | 5.39 |
| 40m | S | C6H5 | 94 | 19.27 | 3.78 |
| 40n | S | 2-CH3C6H4 | 85 | 39.72 | 3.07 |
| 40o | S | 2-ClC6H4 | 95 | 32.72 | 2.91 |
| 40p | S | 2-FC6H4 | 89 | 40.43 | 6.50 |
| 40q | S | 2-OCH3C6H4 | 94 | 0.80 | 1.18 |
| 40r | S | 3-NO2C6H4 | 88 | 34.31 | 8.09 |
| 40s | S | 4-BrC6H4 | 93 | 11.88 | 1.65 |
| 40t | S | 4-CH3C6H4 | 95 | 37.47 | 6.27 |
| 40u | S | 4-ClC6H4 | 92 | 2.25 | 6.26 |
| 40v | S | 4-FC6H4 | 90 | 37.22 | 28.82 |
| 40w | S | 2,4-Cl2C6H4 | 91 | 26.25 | 49.2 |
| 40x | S | 1-napthyl | 92 | 1.37 | 5.58 |
| Galantamine | - | - | - | 2.09 | 19.34 |
| Compound | Cytotoxicity (IC50) in μM | |
|---|---|---|
| A-549 | MDA-MB-231 | |
| 43a | 16.73 ± 1.42 | 4.16 ± 0.37 |
| 43b | 16.11 ± 1.21 | 2.23 ± 0.19 |
| 43c | 4.32 ± 0.53 | 24.43 ± 2.56 |
| 43d | 2.15 ± 0.12 | 16.53 ± 1.61 |
| 43e | 24.31 ± 2.38 | 16.42 ± 1.42 |
| 43f | 22.41 ± 2.51 | 8.42 ± 0.73 |
| 43g | 8.43 ± 0.64 | 26.79 ± 2.79 |
| 43h | 4.64 ± 0.59 | 8.31 ± 0.83 |
| 43i | 25.63 ± 2.58 | 4.62 ± 0.59 |
| 43j | 21.72 ± 2.11 | 16.16 ± 1.31 |
| 43k | 8.73 ± 0.84 | 28.57 ± 2.43 |
| 43l | 8.56 ± 0.76 | 16.74 ± 1.82 |
| Cis-platin | 1.89 ± 0.09 | 3.5 ± 0.21 |
| Compound | Reduction in RLU (%) | |||
|---|---|---|---|---|
| M. tuberculosis H37Rv | Clinical Isolate: S, H, R, and E Resistant M. tuberculosis | |||
| 50 μg mL−1 | 100 μg mL−1 | 50 μg mL−1 | 100 μg mL−1 | |
| 47a | 41.62 | 47.48 | 43.62 | 47.76 |
| 47b | 44.46 | 48.64 | 44.37 | 49.83 |
| 47c | 44.85 | 51.68 | 38.76 | 47.24 |
| 47d | 54.76 | 58.46 | 43.35 | 54.85 |
| 47e | 61.45 | 67.84 | 40.78 | 47.43 |
| 47f | 47.65 | 53.76 | 48.87 | 52.66 |
| 47g | 52.67 | 57.86 | 54.87 | 58.48 |
| 47h | 62.47 | 66.82 | 51.62 | 56.64 |
| 47i | 50.36 | 62.73 | 38.84 | 42.77 |
| 47j | 56.84 | 61.68 | 56.64 | 61.46 |
| Isoniazid | 81.57 | 84.58 | ||
| Compound | R | Pyridine Substitution in 48 | Yield% | Conc. (μg/mL) | % of Inhibition | Conc. (μg/mL) | % of Inhibition | ||
|---|---|---|---|---|---|---|---|---|---|
| C. neoformans | C. albicans | C. neoformans | C. albicans | ||||||
| 55a | Cl | Pyridin-4-yl | 61 | 250 | 91.7 ± 2.8 | 78.3 ± 0.3 | 125 | 57.3 ± 0.7 | 31.0 ± 1.9 |
| 55b | Cl | Pyridin-3-yl | 57 | 250 | 17.5 ± 2.7 | 1.85 ± 0.1 | 125 | 16.9 ± 1.8 | 0 |
| 55c | Cl | Pyridin-2-yl | 42 | 250 | 81.6 ± 1.9 | 8.12 ± 0.7 | 125 | 148.6 ± 0.38 | 4.3 ± 0.4 |
| 55d | OMe | Pyridin-4-yl | 60 | 250 | 29.9 ± 1.9 | 7.3 ± 1.2 | 125 | 19.7 ± 0.6 | 3.0 ± 0.6 |
| 55e | OMe | Pyridin-3-yl | 61 | 250 | 72.5 ± 0.9 | 62.2 ± 2.3 | 125 | 20.7 ± 0.4 | 27.12 ± 1.1 |
| 55f | OMe | Pyridin-2-yl | 42 | 250 | 79.7 ± 1.8 | 63.8 ± 2.0 | 125 | 51.3 ± 1.5 | 23.7 ± 1.8 |
| 55g | Me | Pyridin-4-yl | 62 | 250 | 57.3 ± 1.1 | 76.9 ± 1.3 | 125 | 33.09 ± 0.3 | 25.4 ± 1.5 |
| 55h | Me | Pyridin-3-yl | 56 | 250 | 24.7 ± 1.4 | 58.5 ± 1.3 | 125 | 7.9 ± 2.1 | 31.2 ± 1.8 |
| 55i | Me | Pyridin-2-yl | 48 | 250 | 64.5 ± 2.9 | 11.0 ± 0.6 | 125 | 44.6 ± 1.6 | 3.2 ± 0.9 |
| 55j | 3,4-OCH2O | Pyridin-4-yl | 59 | 250 | 12.9 ± 1.1 | 22.1 ± 1.5 | 125 | 10.5 ± 0.9 | 11.3 ± 0.9 |
| 55k | 3,4-OCH2O | Pyridin-3-yl | 50 | 250 | 64.5 ± 1.5 | 50.2 ± 1.7 | 125 | 62.5 ± 1.0 | 34.9 ± 1.7 |
| Amphotericin B | - | - | - | 250 | 100 | 100 | 125 | 100 | 100 |
| Compounds | IC50 (μM) | ||
|---|---|---|---|
| MCF-7 | HeLa | A549 | |
| 60a | 42.0 ± 2.0 | 47.0 ± 3.0 | >100 |
| 60b | 55.0 ± 1.0 | >100 | >100 |
| 60c | 70.0 ± 2.0 | >100 | >100 |
| 60d | 59.0 ± 2.0 | >100 | >100 |
| 60e | 45.0 ± 1.0 | >100 | >100 |
| 60f | 18.0 ± 1.0 | 25.0 ± 2.0 | 38.0 ± 3.0 |
| 60g | 29.0 ± 2.0 | 50.0 ± 2.0 | 32.0 ± 2.0 |
| 60h | 18.0 ± 2.0 | 13.0 ± 1.0 | >100 |
| 60i | >100 | >100 | >100 |
| 60j | 15.0 ± 1.0 | 40.0 ± 2.0 | 58.0 ± 2.0 |
| 63a | 33.0 ± 2.0 | 62.0 ± 2.0 | >100 |
| 63b | 40.0 ± 2.0 | >100 | >100 |
| 63c | 51.0 ± 3.0 | >100 | >100 |
| 63d | 42.0 ± 2.0 | >100 | >100 |
| 63e | 30.0 ± 2.0 | >100 | >100 |
| 63f | 16.0 ± 2.0 | 35.0 ± 2.0 | 21.0 ± 2.0 |
| 63g | 23.0 ± 1.0 | 57.0 ± 3.0 | 17.0 ± 1.0 |
| 63h | 15.0 ± 1.0 | 22.0 ± 1.0 | >100 |
| 63j | 11.0 ± 1.0 | 52.0 ± 3.0 | 36.0 ± 2.0 |
| Doxorubicin | 0.38 ± 0.03 | 0.36 ± 0.03 | - |
| Ellipticine | - | - | 0.31 ± 0.04 |
| Compound | IC50 (μM) | |
|---|---|---|
| Tyrosinase | Cholinesterase | |
| 60a | 146.0 ± 1.0 | |
| 60b | 101.0 ± 2.0 | |
| 60f | 10.0 ± 0.2 | 29.0 ± 1.0 |
| 60g | 49.0 ± 0.5 | |
| 60j | 19.0 ± 0.3 | |
| 63a | 181.0 ± 1.0 | |
| 63e | 152.0 ± 1.7 | |
| 63f | 85.0 ± 1.0 | 105.0 ± 1.0 |
| 63g | 103.0 ± 0.9 | 152.0 ± 2.0 |
| 63j | 95.0 ± 1.0 | |
| Hydroquinone | 27.0 ± 0.2 | − |
| Galantamine | - | 0.38 × 10−3 ± 0.002 × 10−3 |
| Compound | Dose (mg/kg) | Picrotoxin | Strychnine | ||
|---|---|---|---|---|---|
| Latency (s) | Duration (s) | Latency (s) | Duration (s) | ||
| Vehicle | - | 294.4 ± 26.6 | 182.9 ± 14.3 | 304.6 ± 12.4 | 184.7 ± 14.9 |
| 74 | 1 | 297.5 ± 3.2 | 181.1 ± 36.3 | 357.5 ± 30.5 | 325.5 ± 4.6 *** |
| 75 | 1 | 312.8 ± 11.2 | 395.8 ± 38.9 *** | 304.6 ± 21.7 | 171.6 ± 6.1 |
| 76 | 1 | 313.0 ± 18.7 | 296.9 ± 35.6 * | 313.0 ± 18.7 | 185.6 ± 2.0 |
| Diazepam | 1 | 440.5 ± 20.1 ** | 481.1 ± 18.7 *** | 367.1 ± 31.0 * | 459.7 ± 24.0 *** |
| Compound | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| HepG2 | Hepa1–6 | Caco-2 | HT-29 | HeLa | NIG3T3 | |
| 95f | 38.6 ± 11.5 | 19.9 ± 6.1 | >50 | 11.9 ± 1.0 | 12.9 ± 5.9 | 24.0 ± 18.5 |
| 95g | >50 | 0.7 ± 0.8 | >50 | >50 | >50 | >50 |
| 95h | >50 | 10.7 ± 0.4 | >50 | 24.3 ± 0.7 | 17.7 ± 9.9 | ND |
| 95i | 3.0 ± 1.6 | 3.4 ± 0.4 | ND | 4.2 ± 0.5 | 27.9 ± 20.7 | 1.9 ± 1.5 |
| 95j | 13.4 ± 4.8 | 3.9 ± 0.3 | >50 | >50 | >50 | 2.1 ± 1.3 |
| Cis platin | 7.0 | 4.0 | 10.8 | 16.8 | 11.7 | 8.5 |
| Compound | GI50 μM | |||
|---|---|---|---|---|
| MCF-7 | K-562 | HeLa | PC-3 | |
| 99a | 38.9 | 58.3 | 38.7 | 34.7 |
| 99b | 88.5 | 47.9 | 56.2 | 38.9 |
| 99c | 80.6 | >100 | 58.1 | 30.2 |
| 99d | 38.9 | 54.2 | 43.8 | 26.7 |
| 99e | 43.8 | 57.1 | 54.3 | 37.9 |
| 99f | 55.0 | 60.1 | 55.7 | 38.4 |
| 99g | 38.3 | 58.1 | 48.6 | 25.4 |
| 99h | 34.8 | 54.3 | 47.9 | 25.3 |
| 99i | 32.7 | 55.3 | 34.3 | 28.9 |
| 99j | 82.5 | >100 | 60.9 | 55.3 |
| 5-FU | 32.18 | 47.03 | 43.71 | 12.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majee, S.; Shilpa; Sarav, M.; Banik, B.K.; Ray, D. Recent Advances in the Green Synthesis of Active N-Heterocycles and Their Biological Activities. Pharmaceuticals 2023, 16, 873. https://doi.org/10.3390/ph16060873
Majee S, Shilpa, Sarav M, Banik BK, Ray D. Recent Advances in the Green Synthesis of Active N-Heterocycles and Their Biological Activities. Pharmaceuticals. 2023; 16(6):873. https://doi.org/10.3390/ph16060873
Chicago/Turabian StyleMajee, Suman, Shilpa, Mansi Sarav, Bimal Krishna Banik, and Devalina Ray. 2023. "Recent Advances in the Green Synthesis of Active N-Heterocycles and Their Biological Activities" Pharmaceuticals 16, no. 6: 873. https://doi.org/10.3390/ph16060873