

Computer-Aided Identification of Kinase-Targeted Small Molecules for Cancer: A Review on AKT Protein


Abstract
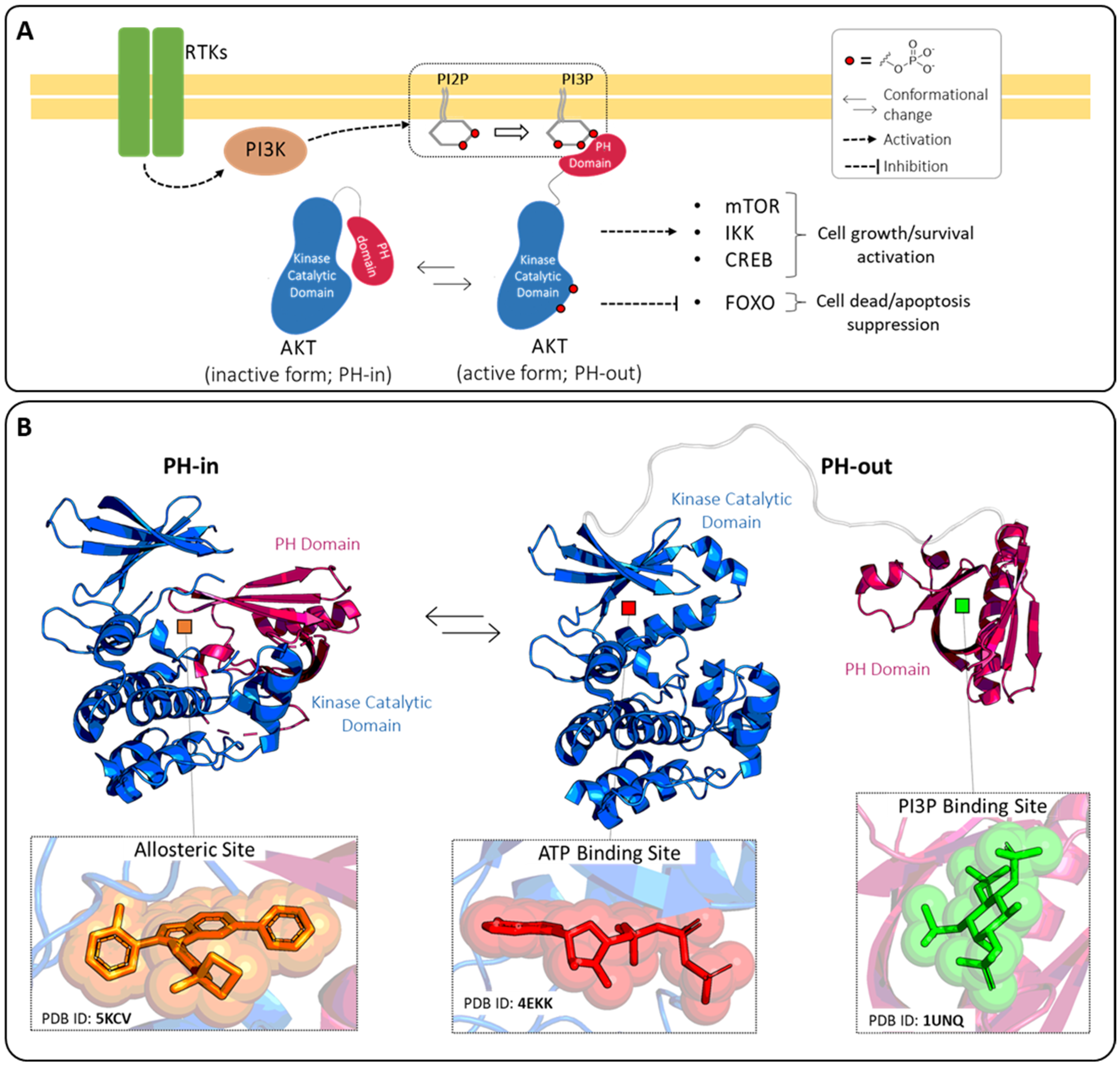
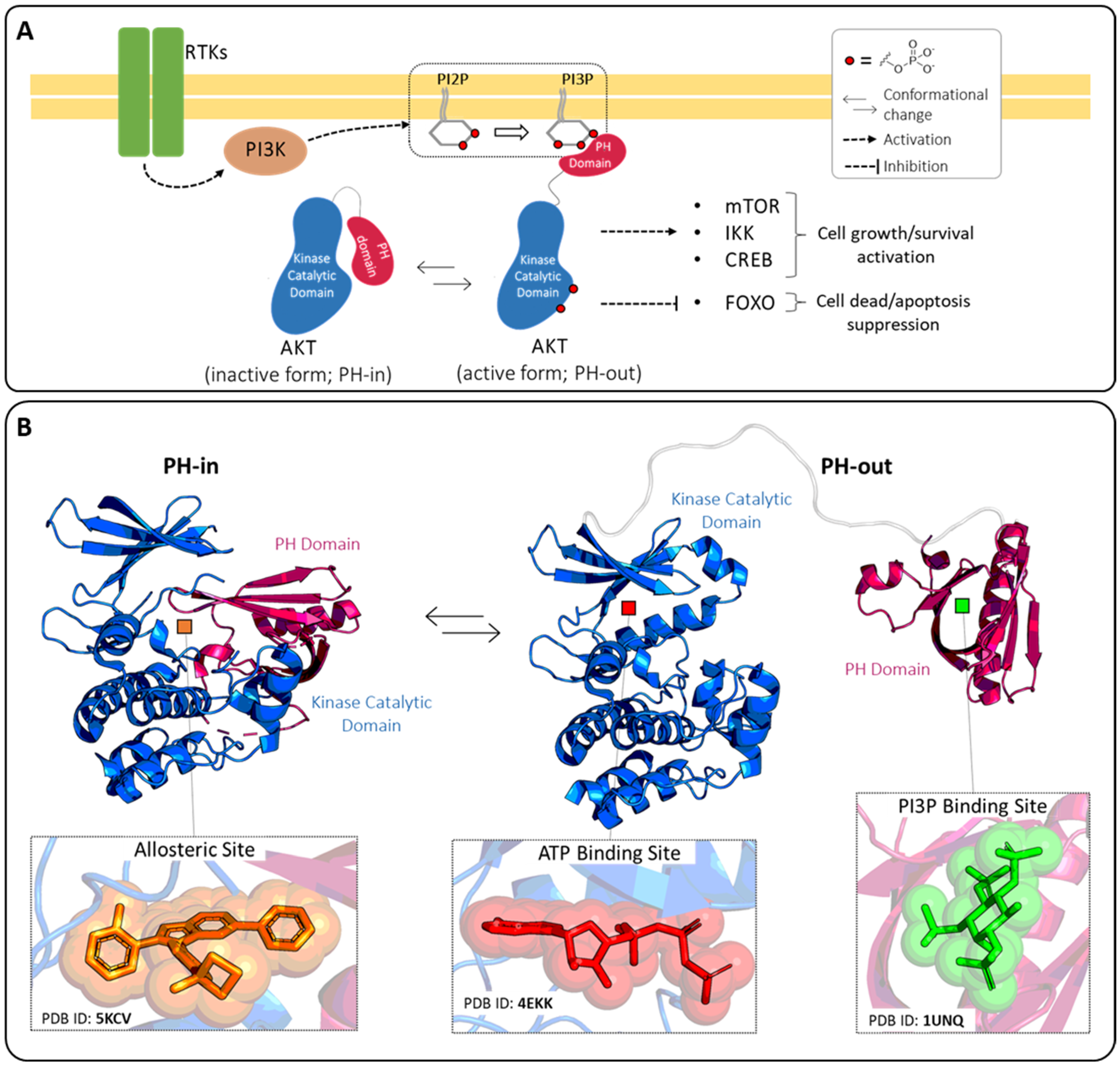
:1. Introduction
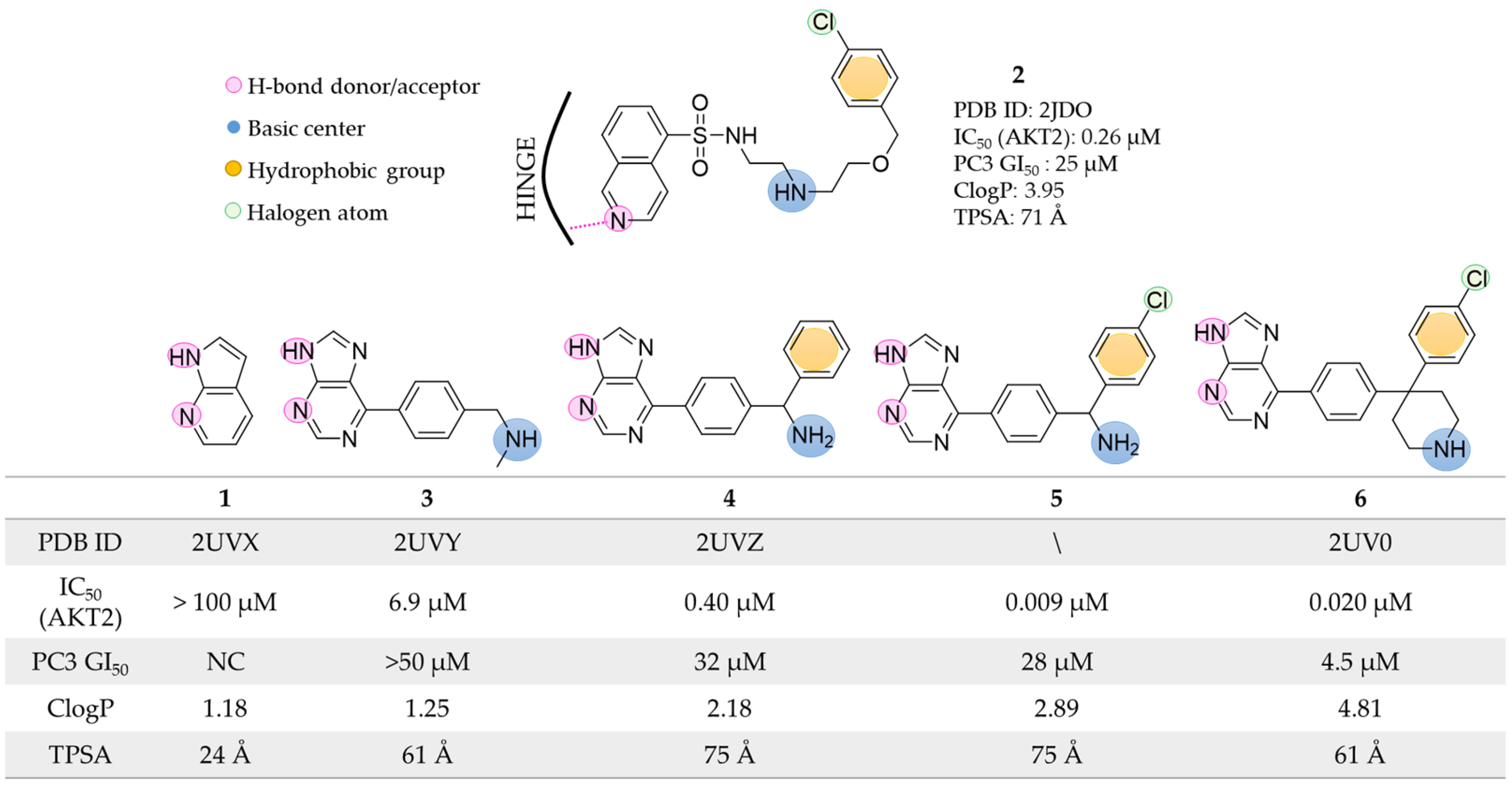
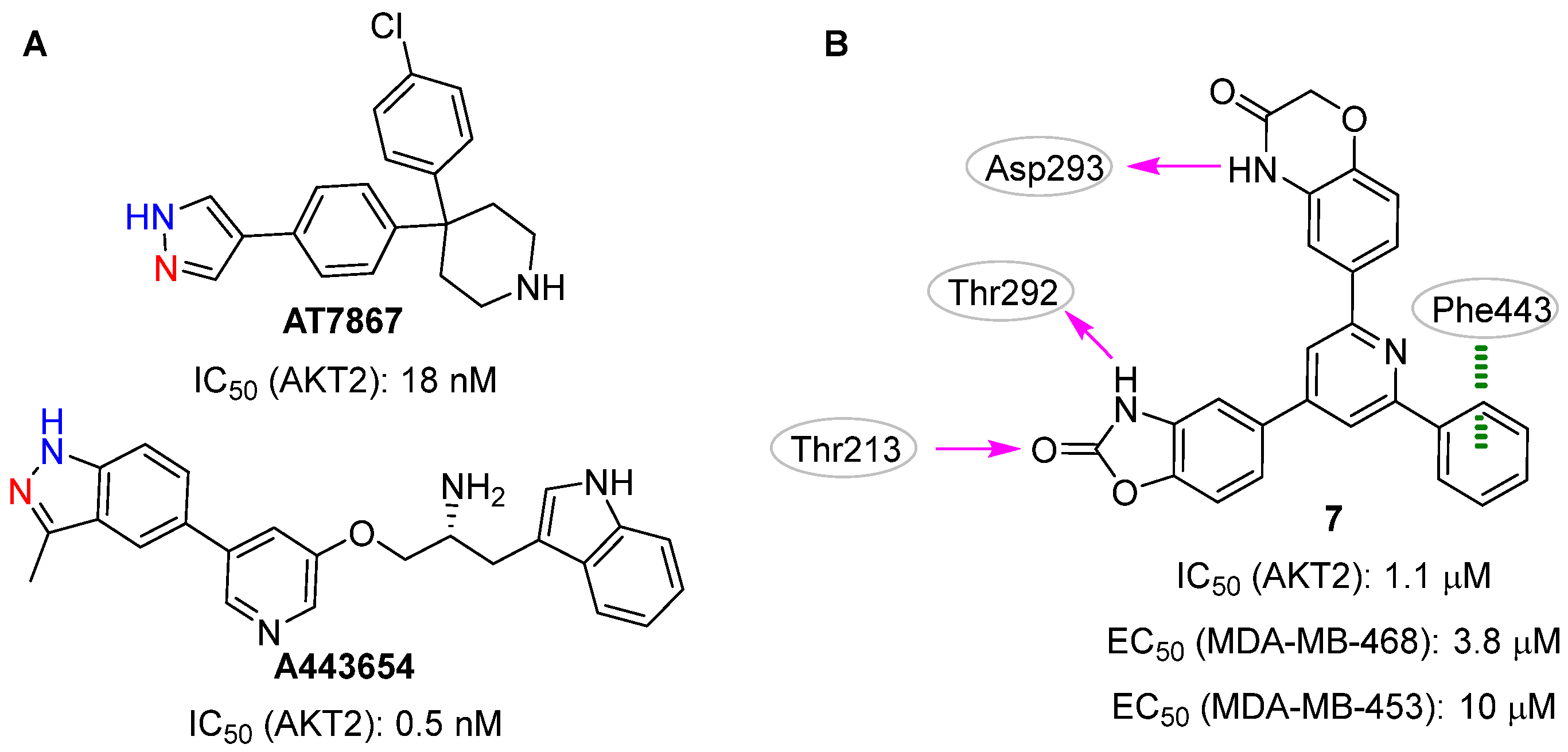
2. ATP-Binding Site
2.1. Docking-Based Approaches
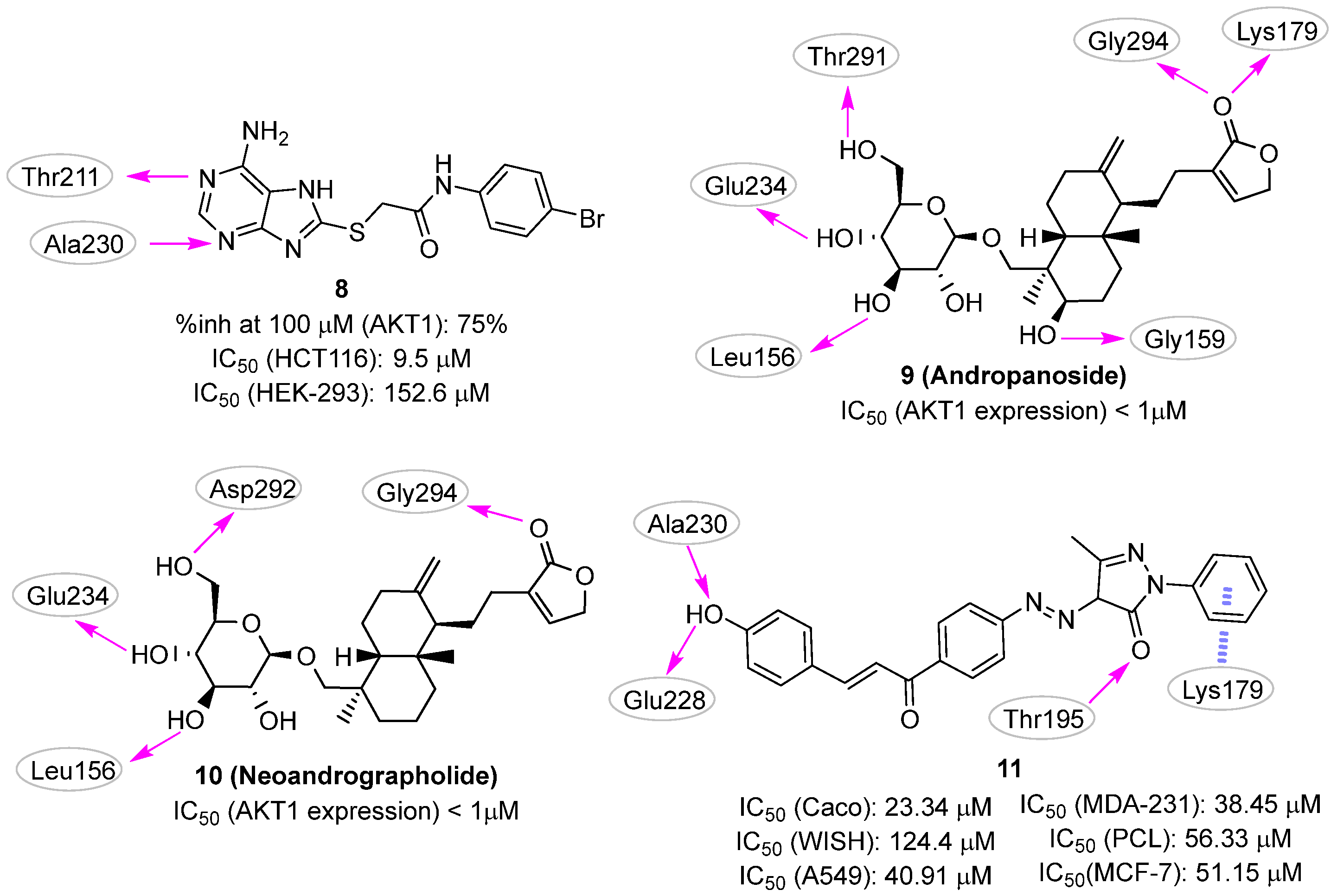
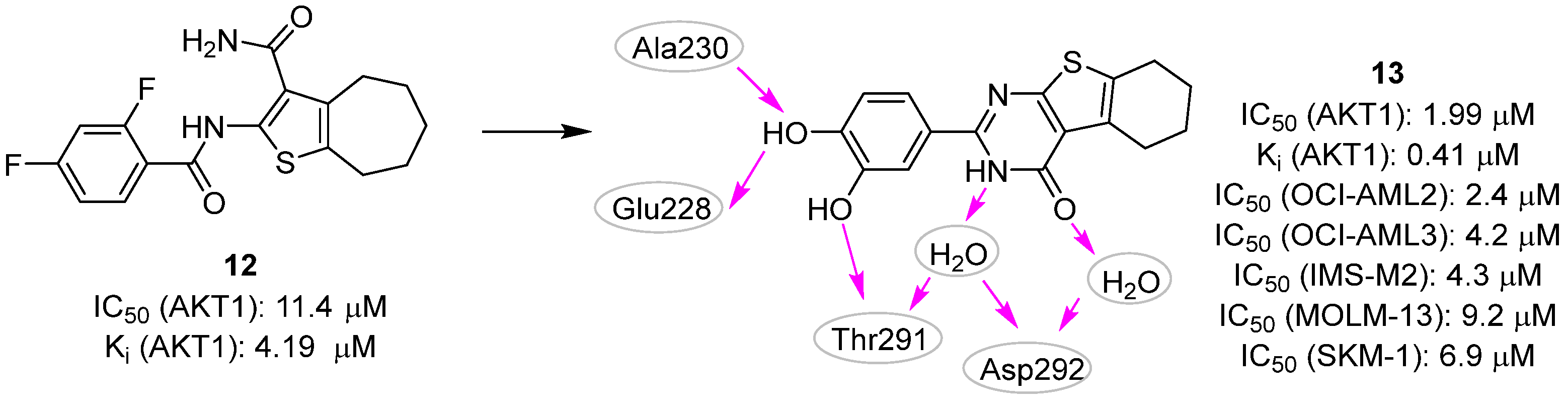
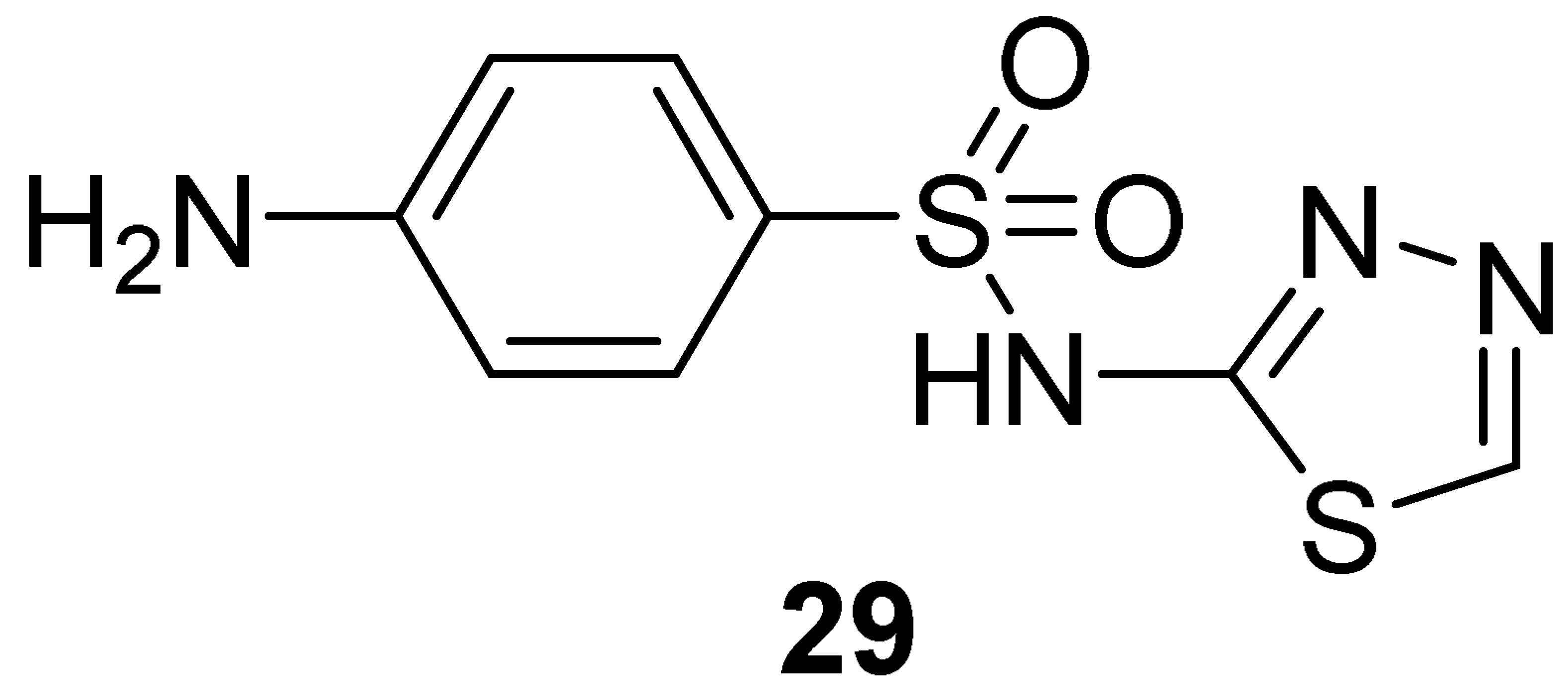
2.2. Pharmacophore-Based Approaches
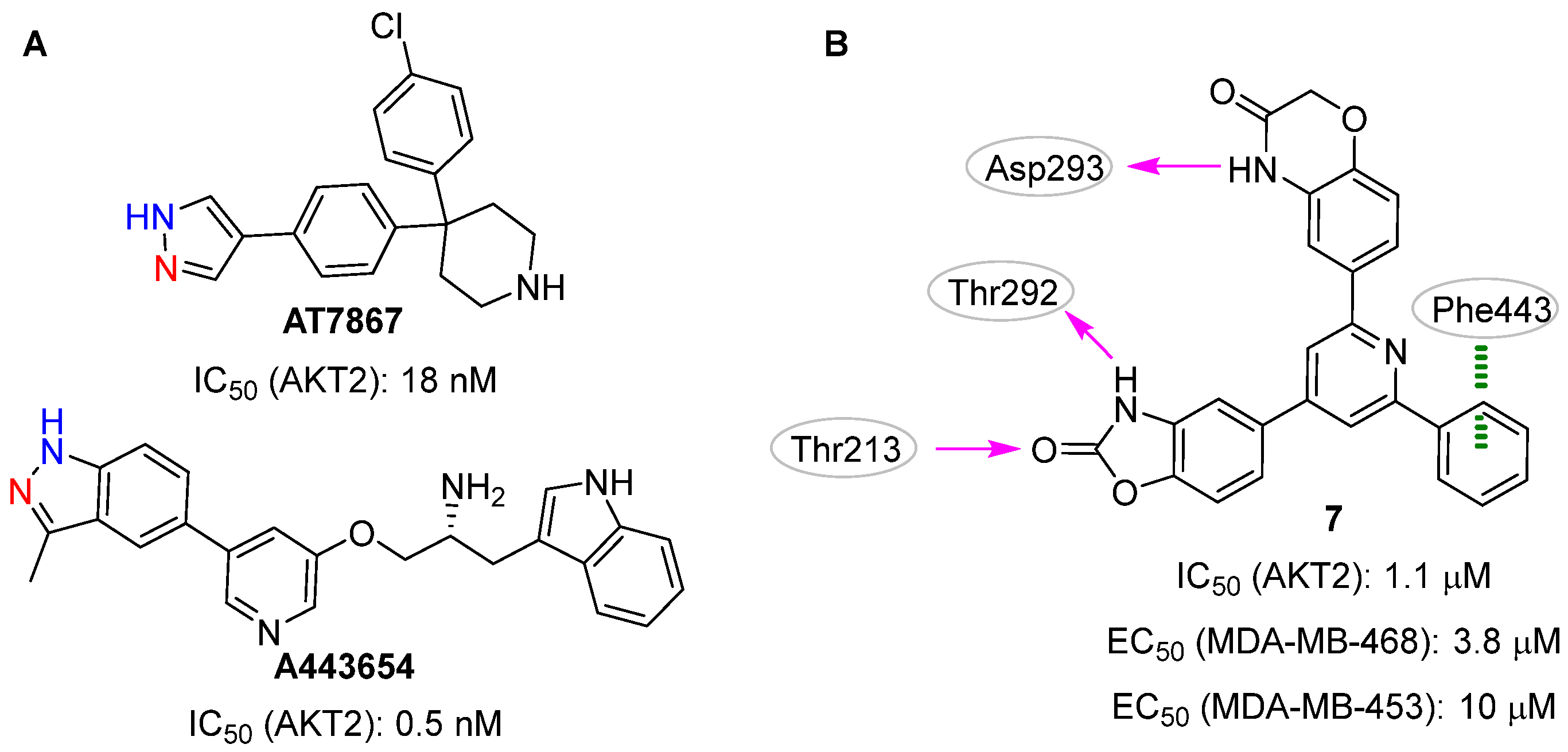
2.3. Machine Learning Approaches
2.4. QSAR Modelling
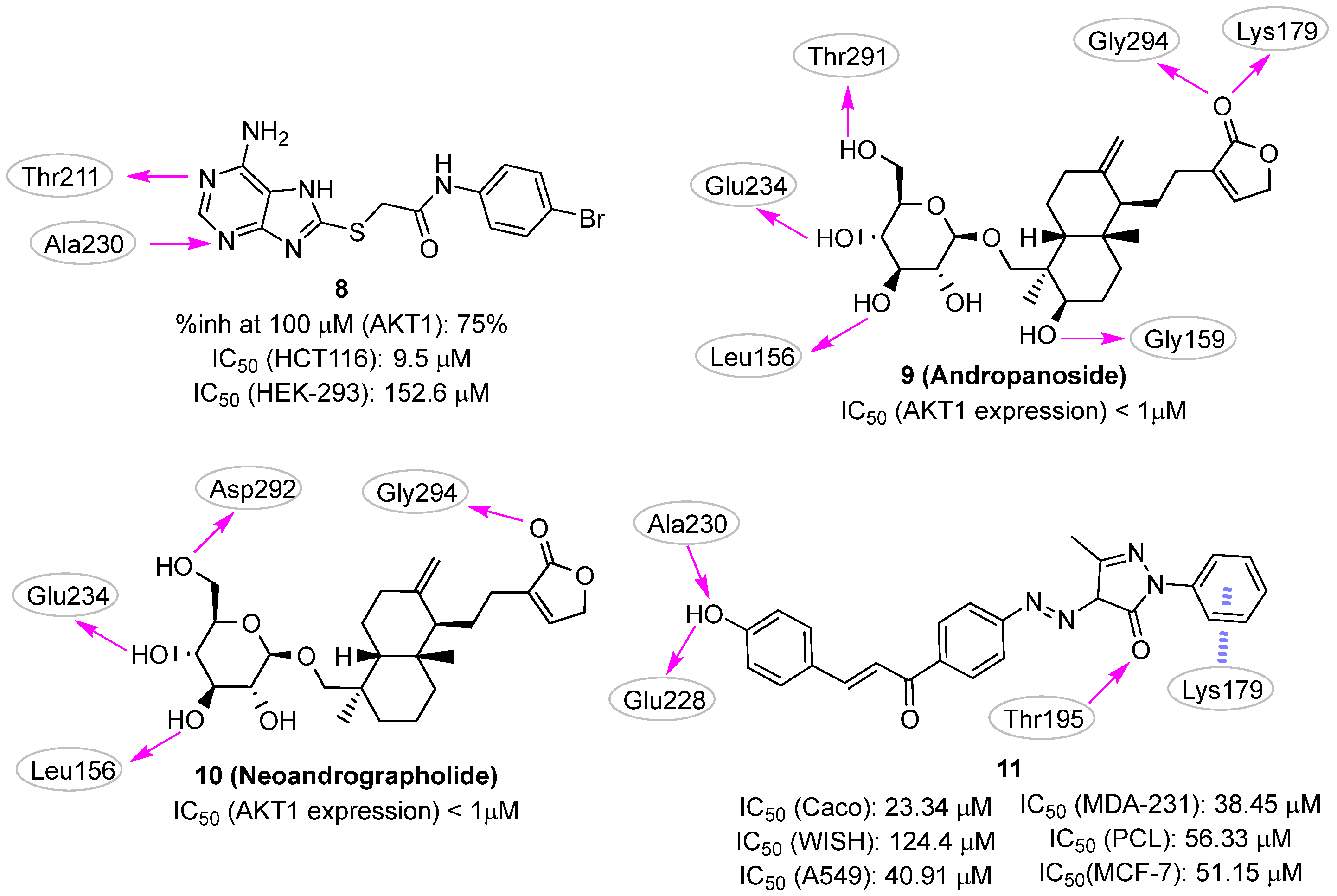
3. Allosteric Site
3.1. Docking-Based Approaches
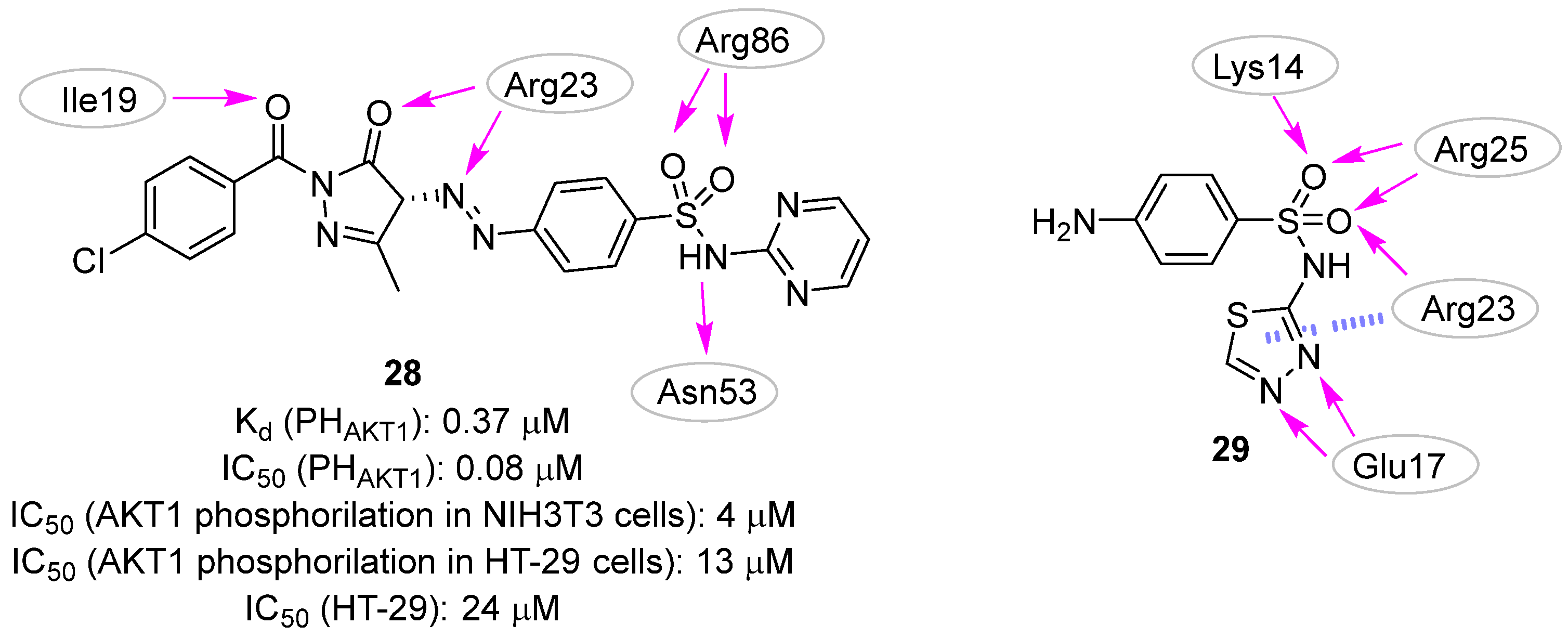
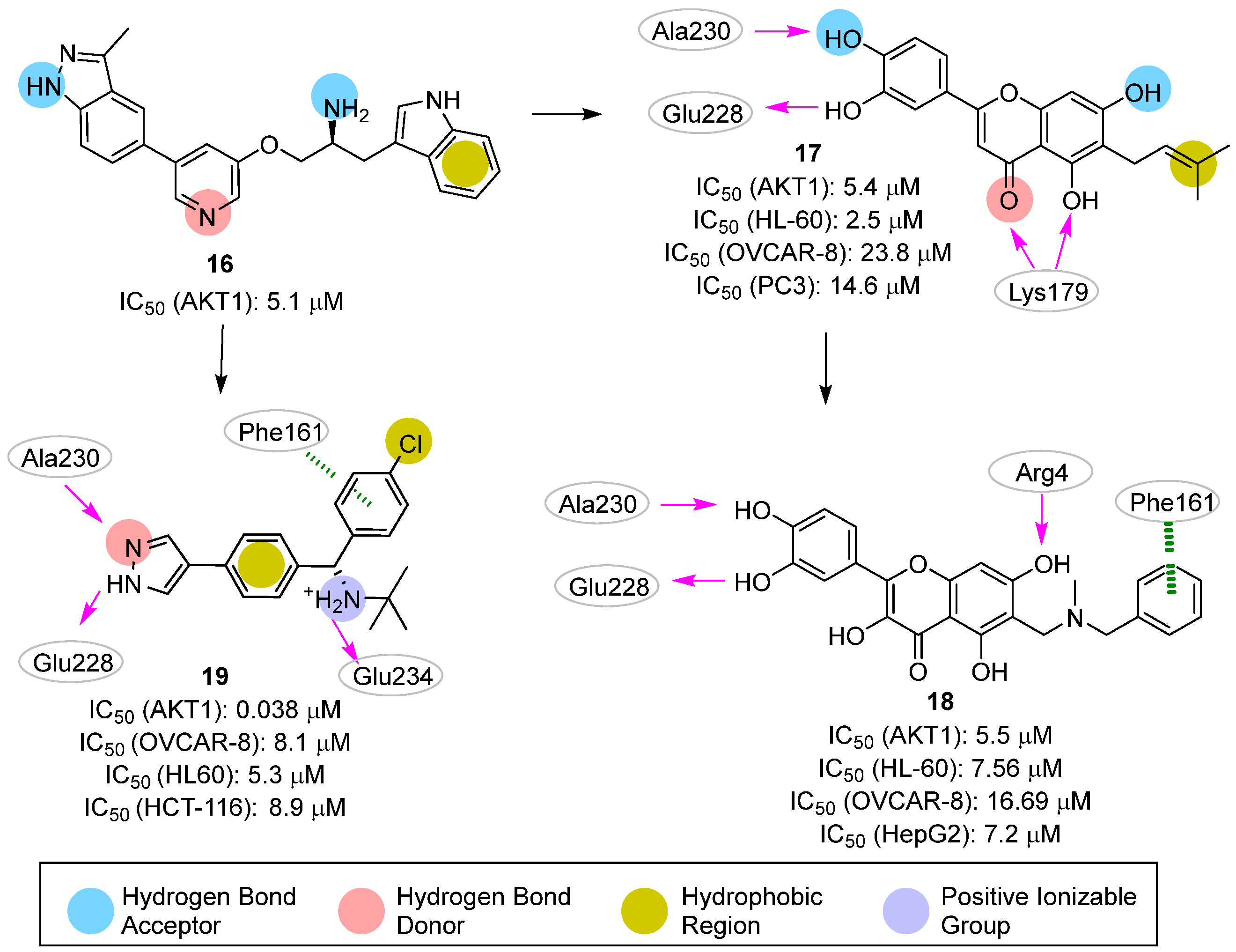
3.2. Pharmacophore-Based Approaches
4. PI3P-Binding Site
Pharmacophore-Based Approaches
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Duggal, S.; Jailkhani, N.; Midha, M.K.; Agrawal, N.; Rao, K.V.S.; Kumar, A. Defining the Akt1 interactome and its role in regulating the cell cycle. Sci. Rep. 2018, 8, 1303. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Testa, J.R.; Staal, S.P.; Tsichlis, P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 1991, 254, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Godwin, A.K.; Bellacosa, A.; Taguchi, T.; Franke, T.F.; Hamilton, T.C.; Tsichlis, P.N.; Testa, J.R. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc. Natl. Acad. Sci. USA 1992, 89, 9267–9271. [Google Scholar] [CrossRef]
- Konishi, H.; Kuroda, S.; Tanaka, M.; Matsuzaki, H.; Ono, Y.; Kameyama, K.; Haga, T.; Kikkawa, U. Molecular cloning and characterization of a new member of the RAC protein kinase family: Association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteins. Biochem. Biophys. Res. Commun. 1995, 216, 526–534. [Google Scholar] [CrossRef]
- Quambusch, L.; Depta, L.; Landel, I.; Lubeck, M.; Kirschner, T.; Nabert, J.; Uhlenbrock, N.; Weisner, J.; Kostka, M.; Levy, L.M.; et al. Cellular model system to dissect the isoform-selectivity of Akt inhibitors. Nat. Commun. 2021, 12, 5297. [Google Scholar] [CrossRef]
- Chin, Y.R.; Toker, A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Mol. Cell 2010, 38, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, W.; Guo, H.; Fang, Y.; Stockman, S.E.; Bai, S.; Ng, P.K.; Li, Y.; Yu, Q.; Lu, Y.; et al. AKT isoform-specific expression and activation across cancer lineages. BMC Cancer 2018, 18, 742. [Google Scholar] [CrossRef] [PubMed]
- Mattmann, M.E.; Stoops, S.L.; Lindsley, C.W. Inhibition of Akt with small molecules and biologics: Historical perspective and current status of the patent landscape. Expert. Opin. Ther. Pat. 2011, 21, 1309–1338. [Google Scholar] [CrossRef]
- Garcia-Echeverria, C.; Sellers, W.R. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 2008, 27, 5511–5526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.Q.; Lindsley, C.W.; Cheng, G.Z.; Yang, H.; Nicosia, S.V. The Akt/PKB pathway: Molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Lin, J.; Wu, W.I.; Ballard, J.; Lee, B.B.; Gloor, S.L.; Vigers, G.P.; Morales, T.H.; Friedman, L.S.; Skelton, N.; et al. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal 2012, 5, ra37. [Google Scholar] [CrossRef]
- Milburn, C.C.; Deak, M.; Kelly, S.M.; Price, N.C.; Alessi, D.R.; Van Aalten, D.M. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem. J. 2003, 375, 531–538. [Google Scholar] [CrossRef]
- Astolfi, A.; Iraci, N.; Manfroni, G.; Barreca, M.L.; Cecchetti, V. A Comprehensive Structural Overview of p38alpha MAPK in Complex with Type I Inhibitors. ChemMedChem 2015, 10, 957–969. [Google Scholar] [CrossRef]
- Astolfi, A.; Kudolo, M.; Brea, J.; Manni, G.; Manfroni, G.; Palazzotti, D.; Sabatini, S.; Cecchetti, F.; Felicetti, T.; Cannalire, R.; et al. Discovery of potent p38alpha MAPK inhibitors through a funnel like workflow combining in silico screening and in vitro validation. Eur. J. Med. Chem. 2019, 182, 111624. [Google Scholar] [CrossRef]
- Astolfi, A.; Milano, F.; Palazzotti, D.; Brea, J.; Pismataro, M.C.; Morlando, M.; Tabarrini, O.; Loza, M.I.; Massari, S.; Martelli, M.P.; et al. From Serendipity to Rational Identification of the 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one Core as a New Chemotype of AKT1 Inhibitors for Acute Myeloid Leukemia. Pharmaceutics 2022, 14, 2295. [Google Scholar] [CrossRef]
- Uko, N.E.; Guner, O.F.; Matesic, D.F.; Bowen, J.P. Akt Pathway Inhibitors. Curr. Top. Med. Chem. 2020, 20, 883–900. [Google Scholar] [CrossRef]
- Dotolo, S.; Cervellera, C.; Russo, M.; Russo, G.L.; Facchiano, A. Virtual Screening of Natural Compounds as Potential PI(3)K-AKT1 Signaling Pathway Inhibitors and Experimental Validation. Molecules 2021, 26, 492. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Giulianotti, M.A.; Yu, Y.; Shen, L.; Yao, L.; Singh, N. Discovery of a novel protein kinase B inhibitor by structure-based virtual screening. Bioorg. Med. Chem. Lett. 2009, 19, 4634–4638. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.H.; Cheng, T.C.; Leu, Y.L.; Chuang, K.H.; Tzou, S.C.; Chen, C.S. Discovery of Akt kinase inhibitors through structure-based virtual screening and their evaluation as potential anticancer agents. Int. J. Mol. Sci. 2015, 16, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Noser, A.A.; Shehadi, I.A.; Abdelmonsef, A.H.; Salem, M.M. Newly Synthesized Pyrazolinone Chalcones as Anticancer Agents via Inhibiting the PI3K/Akt/ERK1/2 Signaling Pathway. ACS Omega 2022, 7, 25265–25277. [Google Scholar] [CrossRef]
- Zhong, S.; Zhang, Z.; Guo, Z.; Yang, W.; Dou, G.; Lv, X.; Wang, X.; Ge, J.; Wu, B.; Pan, X.; et al. Identification of novel natural inhibitors targeting AKT Serine/Threonine Kinase 1 (AKT1) by computational study. Bioengineered 2022, 13, 12003–12020. [Google Scholar] [CrossRef]
- Zhu, C.L.; Luo, X.; Tian, T.; Rao, Z.; Wang, H.; Zhou, Z.; Mi, T.; Chen, D.; Xu, Y.; Wu, Y.; et al. Structure-based rational design enables efficient discovery of a new selective and potent AKT PROTAC degrader. Eur. J. Med. Chem. 2022, 238, 114459. [Google Scholar] [CrossRef]
- Dong, X.; Zhou, X.; Jing, H.; Chen, J.; Liu, T.; Yang, B.; He, Q.; Hu, Y. Pharmacophore identification, virtual screening and biological evaluation of prenylated flavonoids derivatives as PKB/Akt1 inhibitors. Eur. J. Med. Chem. 2011, 46, 5949–5958. [Google Scholar] [CrossRef]
- Fratev, F.; Gutierrez, D.A.; Aguilera, R.J.; Tyagi, A.; Damodaran, C.; Sirimulla, S. Discovery of new AKT1 inhibitors by combination of in silico structure based virtual screening approaches and biological evaluations. J. Biomol. Struct. Dyn. 2021, 39, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhan, W.; Wang, Y.; Zhang, L.; Yang, B.; Dong, X.; Hu, Y. Structure-based design, synthesis and biological evaluation of diphenylmethylamine derivatives as novel Akt1 inhibitors. Eur. J. Med. Chem. 2014, 73, 167–176. [Google Scholar] [CrossRef]
- Zhan, W.; Lin, S.; Chen, J.; Dong, X.; Chu, J.; Du, W. Design, synthesis, biological evaluation, and molecular docking of novel benzopyran and phenylpyrazole derivatives as Akt inhibitors. Chem. Biol. Drug Des. 2015, 85, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ran, T.; Xu, F.; Wen, C.; Song, S.; Zhou, Y.; Chen, H.; Lu, X. Deep learning-driven scaffold hopping in the discovery of Akt kinase inhibitors. Chem. Commun. 2021, 57, 10588–10591. [Google Scholar] [CrossRef]
- Zhan, W.; Li, D.; Che, J.; Zhang, L.; Yang, B.; Hu, Y.; Liu, T.; Dong, X. Integrating docking scores, interaction profiles and molecular descriptors to improve the accuracy of molecular docking: Toward the discovery of novel Akt1 inhibitors. Eur. J. Med. Chem. 2014, 75, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Abohassan, M.; Alshahrani, M.; Alshahrani, M.Y.; Rajagopalan, P. Insilco and Invitro approaches identify novel dual PI3K/AKT pathway inhibitors to control acute myeloid leukemia cell proliferations. Med. Oncol. 2022, 39, 249. [Google Scholar] [CrossRef] [PubMed]
- Al Shahrani, M.; Rajagopalan, P.; Abohassan, M.; Alshahrani, M.; Alraey, Y. CB-RAF600E-1 exerts efficacy in vemurafenib-resistant and non-resistant-melanoma cells via dual inhibition of RAS/RAF/MEK/ERK and PI3K/Akt signaling pathways. Saudi J. Biol. Sci. 2022, 29, 103285. [Google Scholar] [CrossRef]
- Pragna Lakshmi, T.; Kumar, A.; Vijaykumar, V.; Natarajan, S.; Krishna, R. Identification of natural allosteric inhibitor for Akt1 protein through computational approaches and in vitro evaluation. Int. J. Biol. Macromol. 2017, 96, 200–213. [Google Scholar] [CrossRef]
- Mahadevan, D.; Powis, G.; Mash, E.A.; George, B.; Gokhale, V.M.; Zhang, S.; Shakalya, K.; Du-Cuny, L.; Berggren, M.; Ali, M.A.; et al. Discovery of a novel class of AKT pleckstrin homology domain inhibitors. Mol. Cancer Ther. 2008, 7, 2621–2632. [Google Scholar] [CrossRef] [Green Version]
- Moses, S.A.; Ali, M.A.; Zuohe, S.; Du-Cuny, L.; Zhou, L.L.; Lemos, R.; Ihle, N.; Skillman, A.G.; Zhang, S.; Mash, E.A.; et al. In vitro and in vivo activity of novel small-molecule inhibitors targeting the pleckstrin homology domain of protein kinase B/AKT. Cancer Res. 2009, 69, 5073–5081. [Google Scholar] [CrossRef] [Green Version]
- Donald, A.; McHardy, T.; Rowlands, M.G.; Hunter, L.J.; Davies, T.G.; Berdini, V.; Boyle, R.G.; Aherne, G.W.; Garrett, M.D.; Collins, I. Rapid evolution of 6-phenylpurine inhibitors of protein kinase B through structure-based design. J. Med. Chem. 2007, 50, 2289–2292. [Google Scholar] [CrossRef]
- Gassel, M.; Breitenlechner, C.B.; Ruger, P.; Jucknischke, U.; Schneider, T.; Huber, R.; Bossemeyer, D.; Engh, R.A. Mutants of protein kinase a that mimic the ATP-binding site of protein kinase B (AKT). J. Mol. Biol. 2003, 329, 1021–1034. [Google Scholar] [CrossRef]
- Davies, T.G.; Verdonk, M.L.; Graham, B.; Saalau-Bethell, S.; Hamlett, C.C.; McHardy, T.; Collins, I.; Garrett, M.D.; Workman, P.; Woodhead, S.J.; et al. A structural comparison of inhibitor binding to PKB, PKA and PKA-PKB chimera. J. Mol. Biol. 2007, 367, 882–894. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Saxty, G.; Woodhead, S.J.; Berdini, V.; Davies, T.G.; Verdonk, M.L.; Wyatt, P.G.; Boyle, R.G.; Barford, D.; Downham, R.; Garrett, M.D.; et al. Identification of inhibitors of protein kinase B using fragment-based lead discovery. J. Med. Chem. 2007, 50, 2293–2296. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Jabeen, I. Pharmacoinformatic Approaches to Design Novel Inhibitors of Protein Kinase B Pathways in Cancer. Curr. Cancer Drug Targets 2018, 18, 830–846. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Gupta, M. Designing of kinase hinge binders: A medicinal chemistry perspective. Chem. Biol. Drug Des. 2022, 100, 968–980. [Google Scholar] [CrossRef]
- Xing, L.; Klug-Mcleod, J.; Rai, B.; Lunney, E.A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 2015, 23, 6520–6527. [Google Scholar] [CrossRef]
- Freeman-Cook, K.D.; Autry, C.; Borzillo, G.; Gordon, D.; Barbacci-Tobin, E.; Bernardo, V.; Briere, D.; Clark, T.; Corbett, M.; Jakubczak, J.; et al. Design of selective, ATP-competitive inhibitors of Akt. J. Med. Chem. 2010, 53, 4615–4622. [Google Scholar] [CrossRef]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated Docking with Grid-Based Energy Evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Rao, S.N.; Head, M.S.; Kulkarni, A.; LaLonde, J.M. Validation studies of the site-directed docking program LibDock. J. Chem. Inf. Model. 2007, 47, 2159–2171. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Rarey, M.; Dixon, J.S. Feature trees: A new molecular similarity measure based on tree matching. J. Comput. Aided Mol. Des. 1998, 12, 471–490. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Addie, M.; Ballard, P.; Buttar, D.; Crafter, C.; Currie, G.; Davies, B.R.; Debreczeni, J.; Dry, H.; Dudley, P.; Greenwood, R.; et al. Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J. Med. Chem. 2013, 56, 2059–2073. [Google Scholar] [CrossRef]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Drummond, M.L.; Henry, A.; Li, H.; Williams, C.I. Improved Accuracy for Modeling PROTAC-Mediated Ternary Complex Formation and Targeted Protein Degradation via New In Silico Methodologies. J. Chem. Inf. Model. 2020, 60, 5234–5254. [Google Scholar] [CrossRef]
- Li, H.; Sutter, J.; Hoffmann, R. HypoGen: An automated system for generating 3D predictive pharmacophore models. In Pharmacophore Perception, Development, and Use in Drug Design; Li, H., Sutter, J., Hoffmann, R., Eds.; Tsigelny, I. San Diego Supercomputer Center, University of California: San Diego, CA, USA, 2000; p. 171. [Google Scholar]
- Lippa, B.; Pan, G.; Corbett, M.; Li, C.; Kauffman, G.S.; Pandit, J.; Robinson, S.; Wei, L.; Kozina, E.; Marr, E.S.; et al. Synthesis and structure based optimization of novel Akt inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3359–3363. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Bencsik, J.R.; Xiao, D.; Blake, J.F.; Kallan, N.C.; Mitchell, I.S.; Spencer, K.L.; Xu, R.; Gloor, S.L.; Martinson, M.; Risom, T.; et al. Discovery of dihydrothieno- and dihydrofuropyrimidines as potent pan Akt inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 7037–7041. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieth, M.; Hirst, J.D.; Dominy, B.N.; Daigler, H.; Brooks, C.L. Assessing search strategies for flexible docking. J. Comput. Chem. 1998, 19, 1623–1631. [Google Scholar] [CrossRef]
- flexiDOCK, Sybyl 6.9 (2002), Molecular Modelling System, Tripos Associates. Available online: https://www.certara.com/ (accessed on 10 July 2023).
- Salam, N.K.; Nuti, R.; Sherman, W. Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef]
- Kallan, N.C.; Spencer, K.L.; Blake, J.F.; Xu, R.; Heizer, J.; Bencsik, J.R.; Mitchell, I.S.; Gloor, S.L.; Martinson, M.; Risom, T.; et al. Discovery and SAR of spirochromane Akt inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2410–2414. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, S.; Su, S.; Zhao, C.; Xu, J.; Chen, H. SyntaLinker: Automatic fragment linking with deep conditional transformer neural networks. Chem. Sci. 2020, 11, 8312–8322. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Uhlenbrock, N.; Smith, S.; Weisner, J.; Landel, I.; Lindemann, M.; Le, T.A.; Hardick, J.; Gontla, R.; Scheinpflug, R.; Czodrowski, P.; et al. Structural and chemical insights into the covalent-allosteric inhibition of the protein kinase Akt. Chem. Sci. 2019, 10, 3573–3585. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Wu, W.I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.C.; Deak, M.; Alessi, D.R.; van Aalten, D.M. High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr. Biol. 2002, 12, 1256–1262. [Google Scholar] [CrossRef]
- Certara; UNITY, SYBYL-X.: St. Louis, MO, USA, 2013.
- Moingeon, P.; Kuenemann, M.; Guedj, M. Artificial intelligence-enhanced drug design and development: Toward a computational precision medicine. Drug Discov. Today 2022, 27, 215–222. [Google Scholar] [CrossRef]
- Urbina, F.; Lentzos, F.; Invernizzi, C.; Ekins, S. Dual Use of Artificial Intelligence-powered Drug Discovery. Nat. Mach. Intell. 2022, 4, 189–191. [Google Scholar] [CrossRef]
- Palazzotti, D.; Fiorelli, M.; Sabatini, S.; Massari, S.; Barreca, M.L.; Astolfi, A. Q-raKtion: A Semiautomated KNIME Workflow for Bioactivity Data Points Curation. J. Chem. Inf. Model. 2022, 62, 6309–6315. [Google Scholar] [CrossRef]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Quambusch, L.; Landel, I.; Depta, L.; Weisner, J.; Uhlenbrock, N.; Muller, M.P.; Glanemann, F.; Althoff, K.; Siveke, J.T.; Rauh, D. Covalent-Allosteric Inhibitors to Achieve Akt Isoform-Selectivity. Angew. Chem. Int. Ed. Engl. 2019, 58, 18823–18829. [Google Scholar] [CrossRef] [Green Version]
- van der Westhuizen, L.; Weisner, J.; Taher, A.; Landel, I.; Quambusch, L.; Lindemann, M.; Uhlenbrock, N.; Muller, M.P.; Green, I.R.; Pelly, S.C.; et al. Covalent Allosteric Inhibitors of Akt Generated Using a Click Fragment Approach. ChemMedChem 2022, 17, e202100776. [Google Scholar] [CrossRef] [PubMed]
- Weisner, J.; Landel, I.; Reintjes, C.; Uhlenbrock, N.; Trajkovic-Arsic, M.; Dienstbier, N.; Hardick, J.; Ladigan, S.; Lindemann, M.; Smith, S.; et al. Preclinical Efficacy of Covalent-Allosteric AKT Inhibitor Borussertib in Combination with Trametinib in KRAS-Mutant Pancreatic and Colorectal Cancer. Cancer Res. 2019, 79, 2367–2378. [Google Scholar] [CrossRef] [Green Version]
- Landel, I.; Quambusch, L.; Depta, L.; Rauh, D. Spotlight on AKT: Current Therapeutic Challenges. ACS Med. Chem. Lett. 2020, 11, 225–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaro, G.; Kostaras, E.; Vivanco, I. Inhibitors in AKTion: ATP-competitive vs allosteric. Biochem. Soc. Trans. 2020, 48, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W.; Zhao, Z.; Leister, W.H.; Robinson, R.G.; Barnett, S.F.; Defeo-Jones, D.; Jones, R.E.; Hartman, G.D.; Huff, J.R.; Huber, H.E.; et al. Allosteric Akt (PKB) inhibitors: Discovery and SAR of isozyme selective inhibitors. Bioorg Med. Chem. Lett. 2005, 15, 761–764. [Google Scholar] [CrossRef]
- DeFeo-Jones, D.; Barnett, S.F.; Fu, S.; Hancock, P.J.; Haskell, K.M.; Leander, K.R.; McAvoy, E.; Robinson, R.G.; Duggan, M.E.; Lindsley, C.W.; et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol. Cancer Ther. 2005, 4, 271–279. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef]
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16, 1199–1207. [Google Scholar] [CrossRef]
- Spagnolli, G.; Massignan, T.; Astolfi, A.; Biggi, S.; Rigoli, M.; Brunelli, P.; Libergoli, M.; Ianeselli, A.; Orioli, S.; Boldrini, A.; et al. Pharmacological inactivation of the prion protein by targeting a folding intermediate. Commun. Biol. 2021, 4, 62. [Google Scholar] [CrossRef]
- Henning, R.K.; Varghese, J.O.; Das, S.; Nag, A.; Tang, G.; Tang, K.; Sutherland, A.M.; Heath, J.R. Degradation of Akt using protein-catalyzed capture agents. J. Pept. Sci. 2016, 22, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Xu, J.; Shen, Y.; Cahuzac, K.M.; Park, K.S.; Dale, B.; Liu, J.; Parsons, R.E.; Jin, J. Discovery of Potent, Selective, and In Vivo Efficacious AKT Kinase Protein Degraders via Structure-Activity Relationship Studies. J. Med. Chem. 2022, 65, 3644–3666. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Xu, J.; Xie, L.; Wang, L.; Shen, Y.; Cahuzac, K.M.; Chen, X.; Liu, J.; Parsons, R.E.; Jin, J. Design, Synthesis, and Evaluation of Potent, Selective, and Bioavailable AKT Kinase Degraders. J. Med. Chem. 2021, 64, 18054–18081. [Google Scholar] [CrossRef]
- Yu, X.; Xu, J.; Cahuzac, K.M.; Xie, L.; Shen, Y.; Chen, X.; Liu, J.; Parsons, R.E.; Jin, J. Novel Allosteric Inhibitor-Derived AKT Proteolysis Targeting Chimeras (PROTACs) Enable Potent and Selective AKT Degradation in KRAS/BRAF Mutant Cells. J. Med. Chem. 2022, 65, 14237–14260. [Google Scholar] [CrossRef]

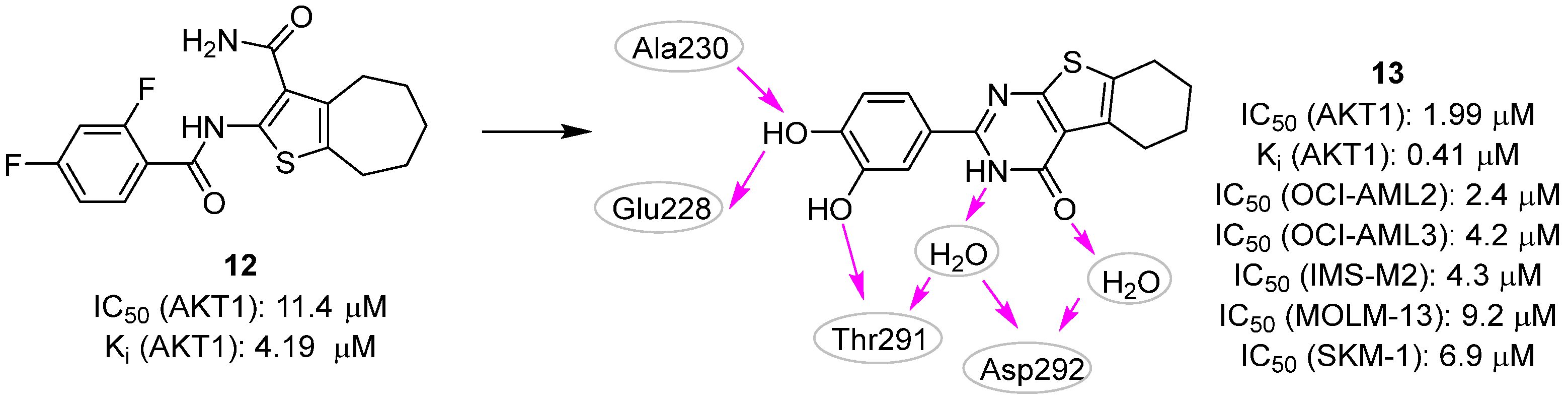
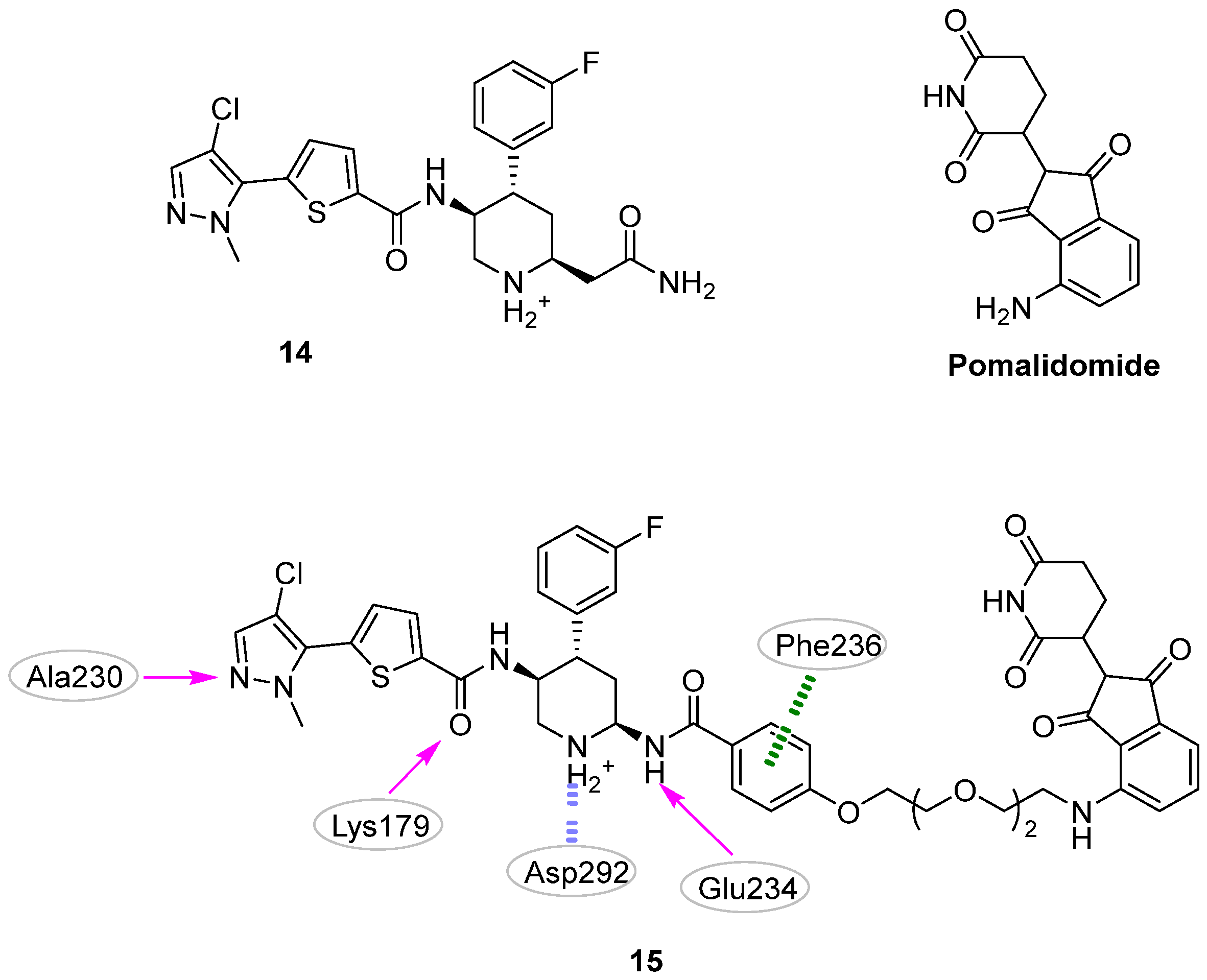
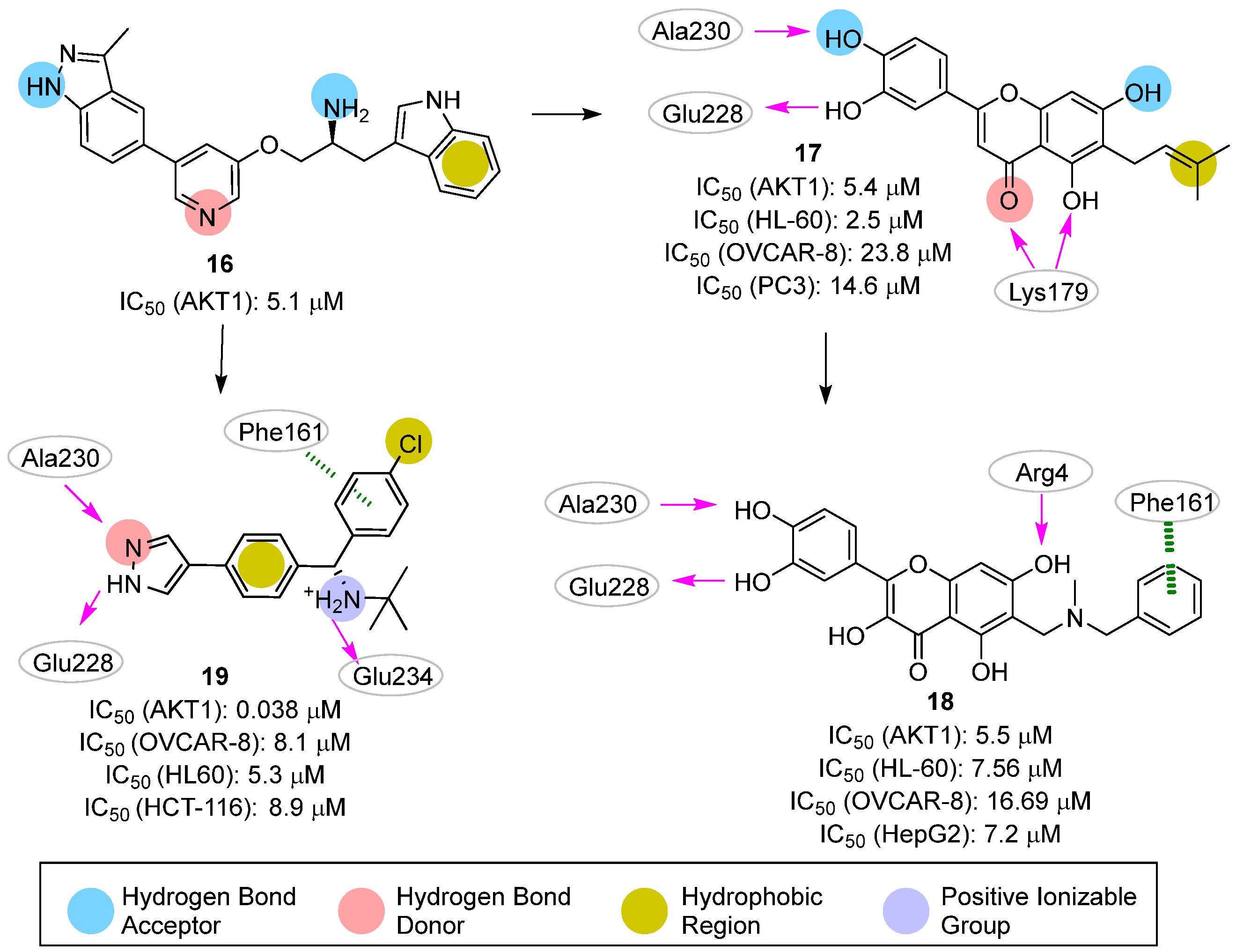
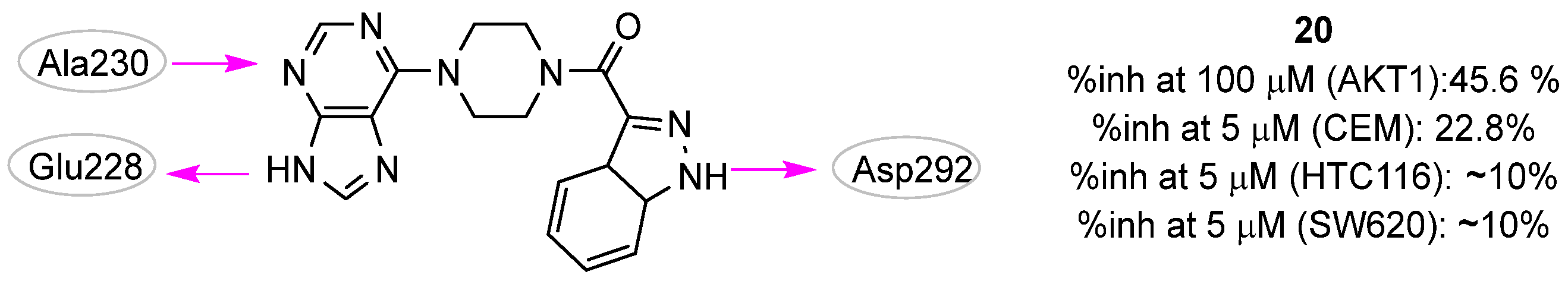
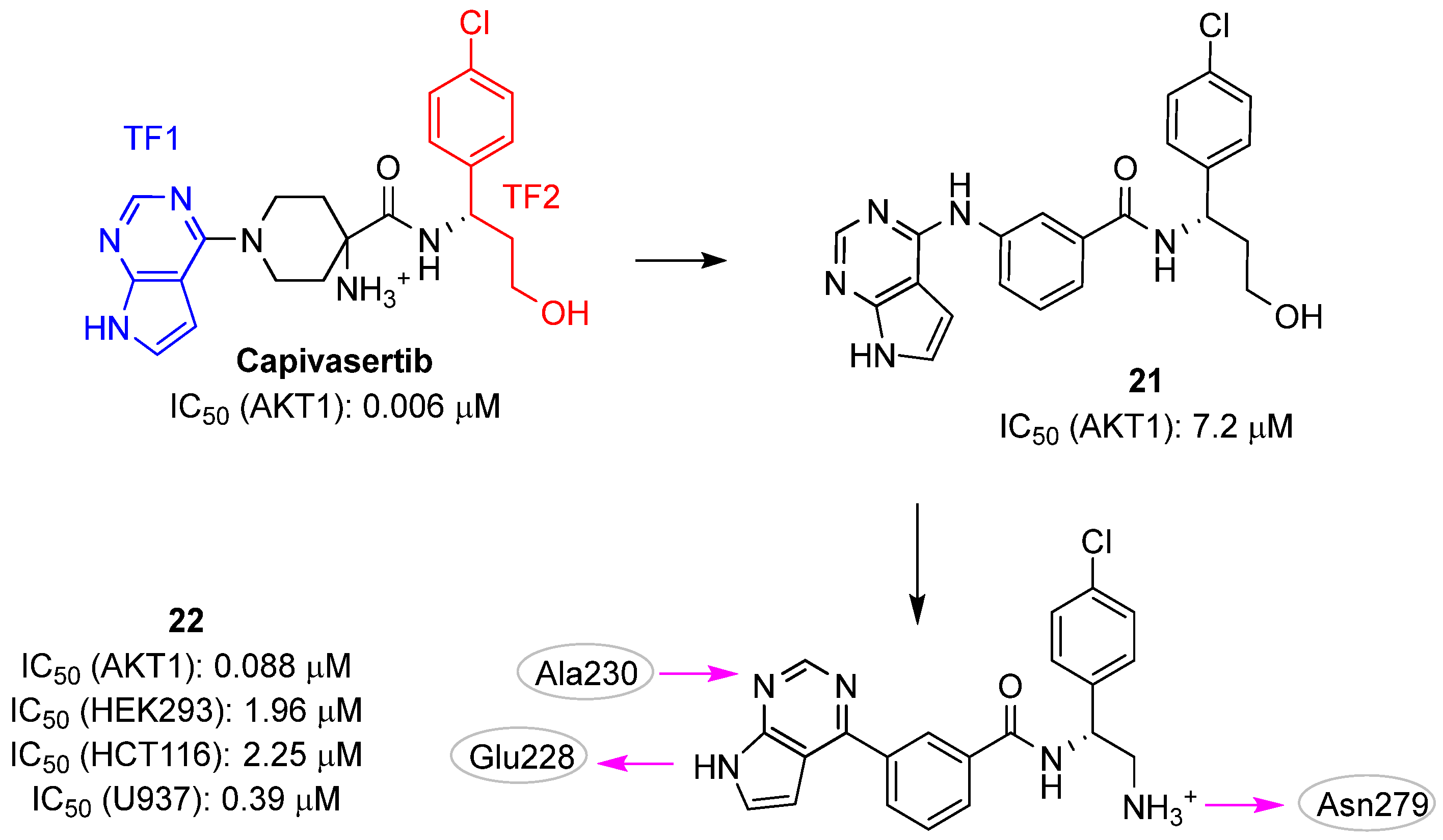
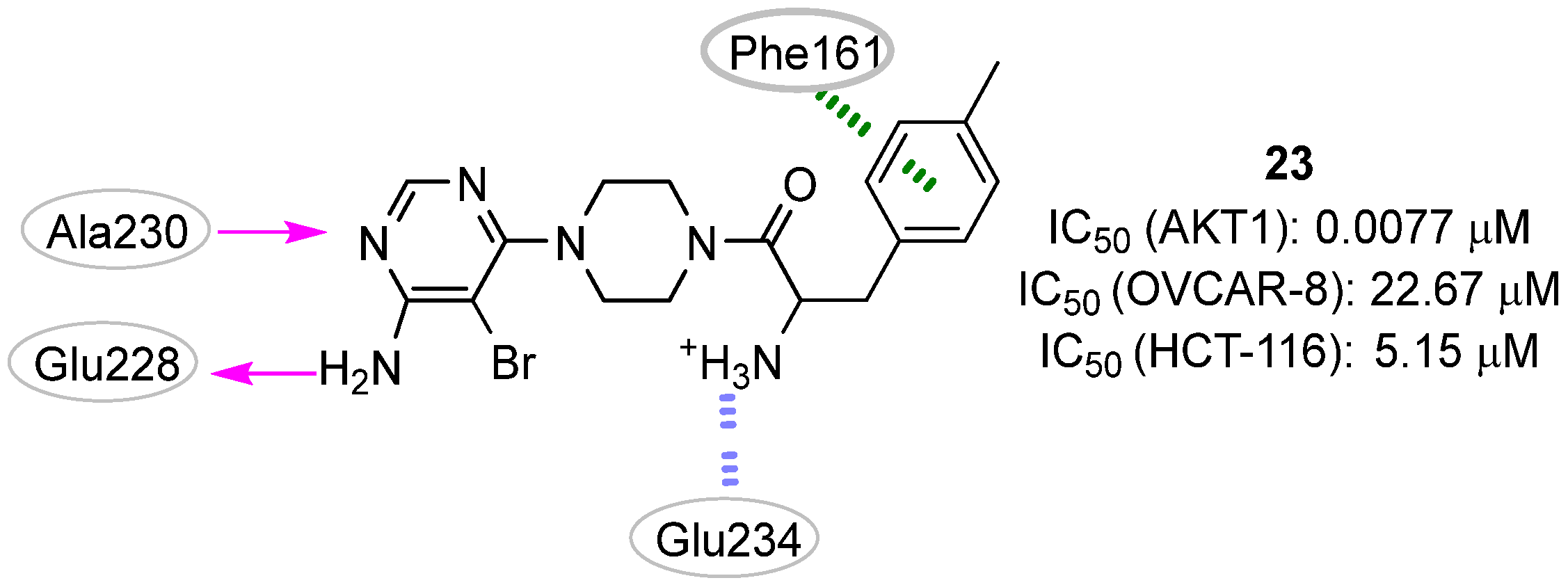
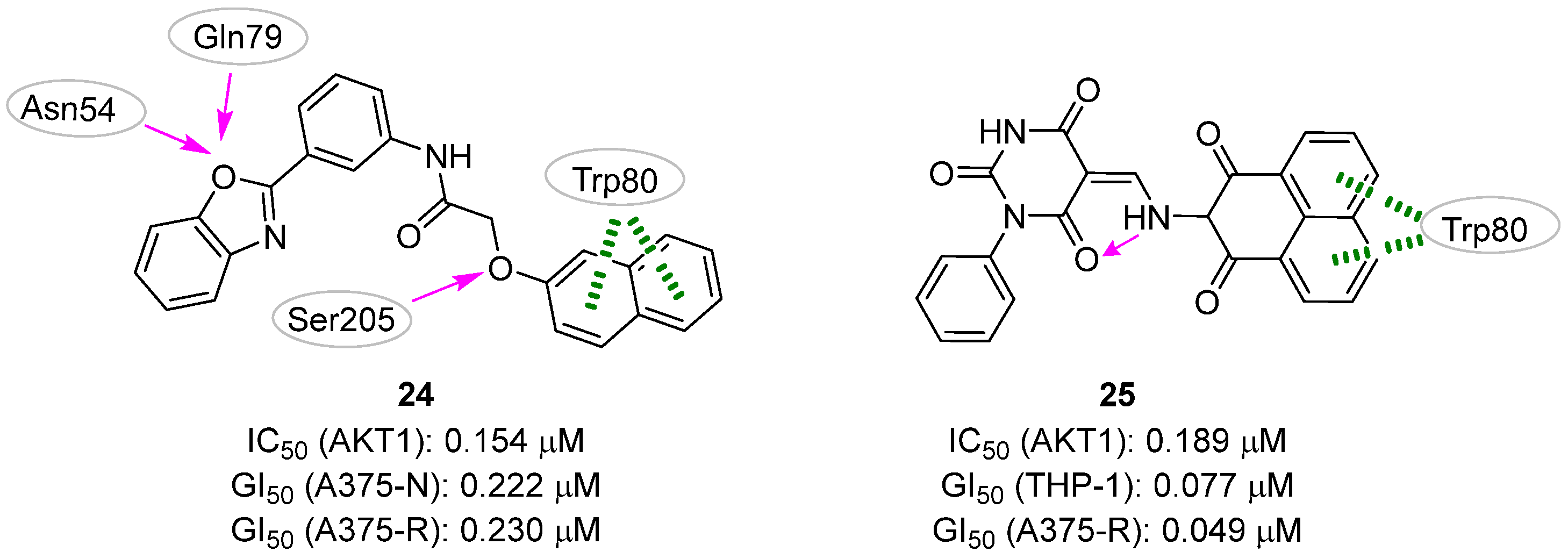
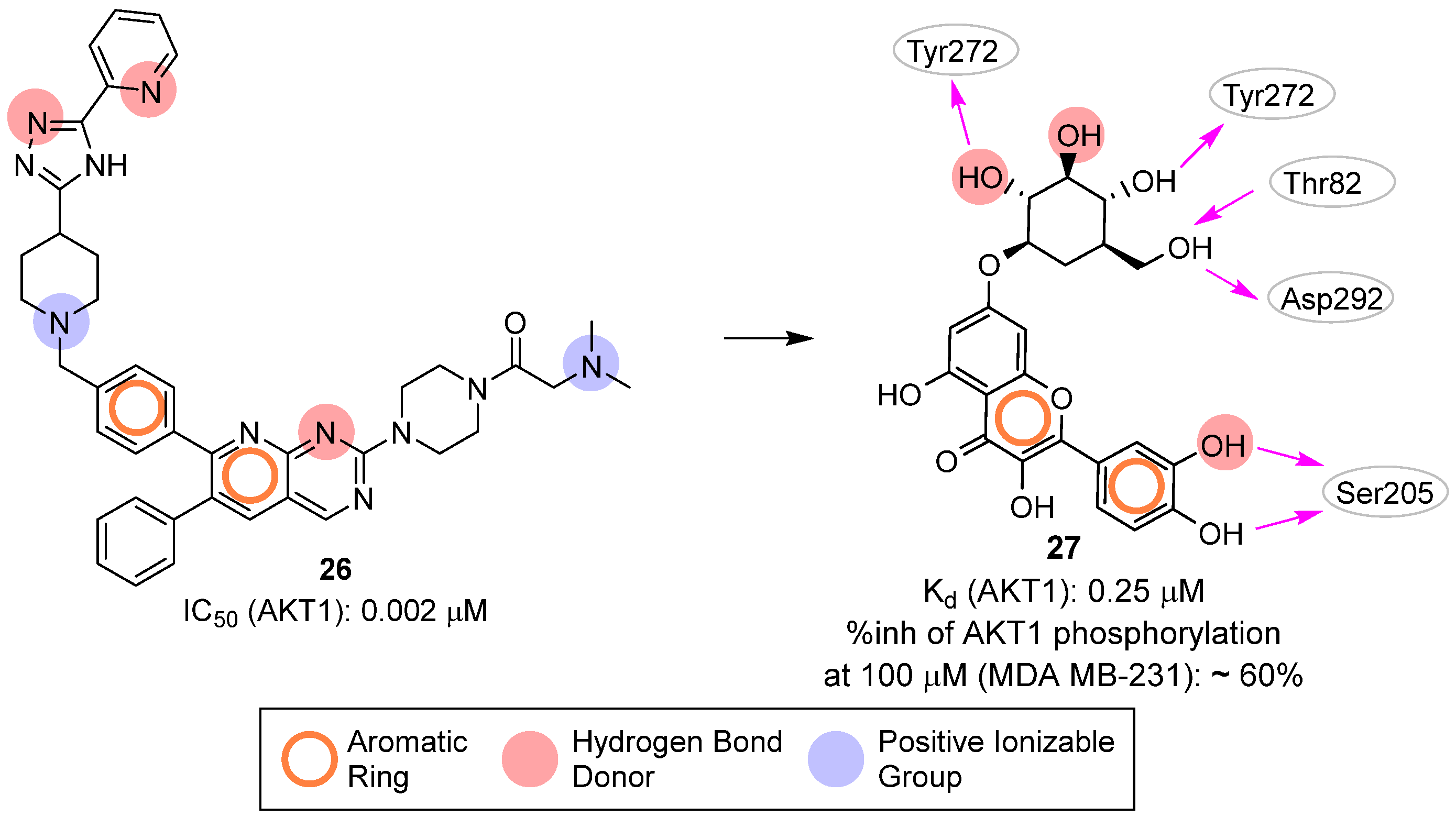

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Computational Methodology | Refs |
|---|---|---|
| ATP-binding site | docking-based | [20,22,23,24,25,26,27] |
| pharmacophore-based | [28,29,30,31] | |
| ML | [32] | |
| QSAR | [33] | |
| Allosteric site | docking-based | [34,35] |
| pharmacophore-based | [36] | |
| PI3P-binding site | pharmacophore-based | [37,38] |
| Compound | LogP a | Caco-2 a | Kd (μM) | Ki (μM) | pAKT1 Inhibition (IC50, μM) | Cellular Growth Inhibition (IC50, μM) |
|---|---|---|---|---|---|---|
 | 0.13 | 0.3 | 0.45 ± 0.1 | >50.0 | 20 b/25 c | NI b/NI c |
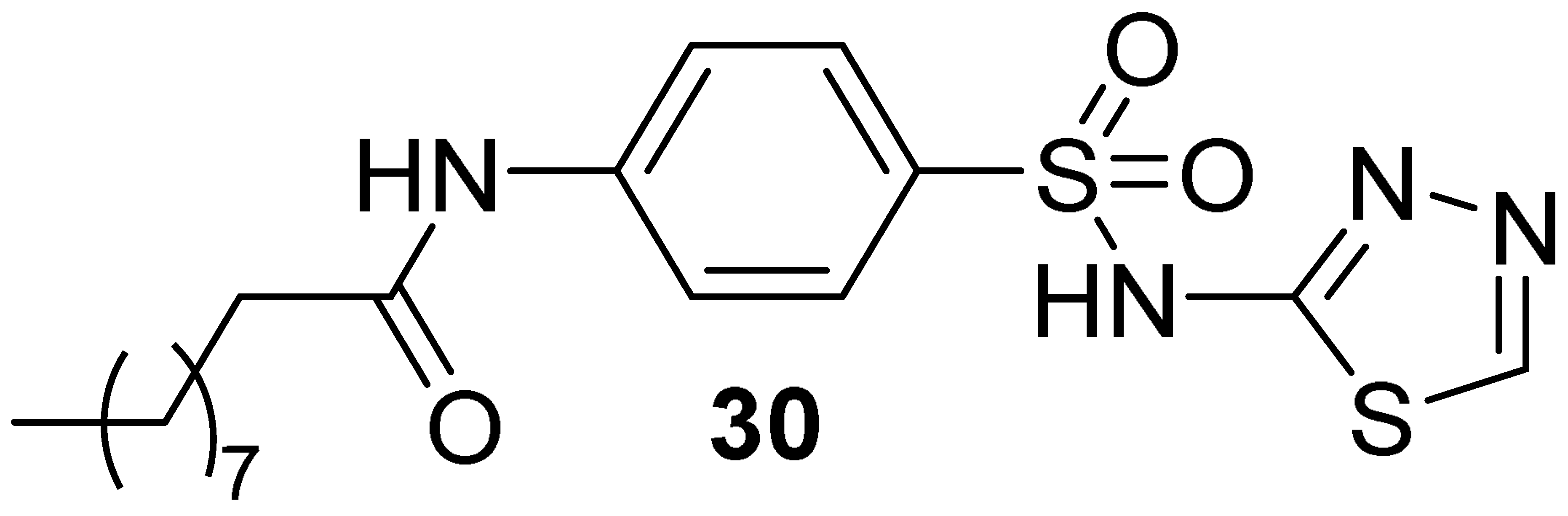 | 4.93 | 10.1 | 19.6 ± 4.9 | 21.8 ± 1.8 | 10 b/15 c | 127 b/90 c |
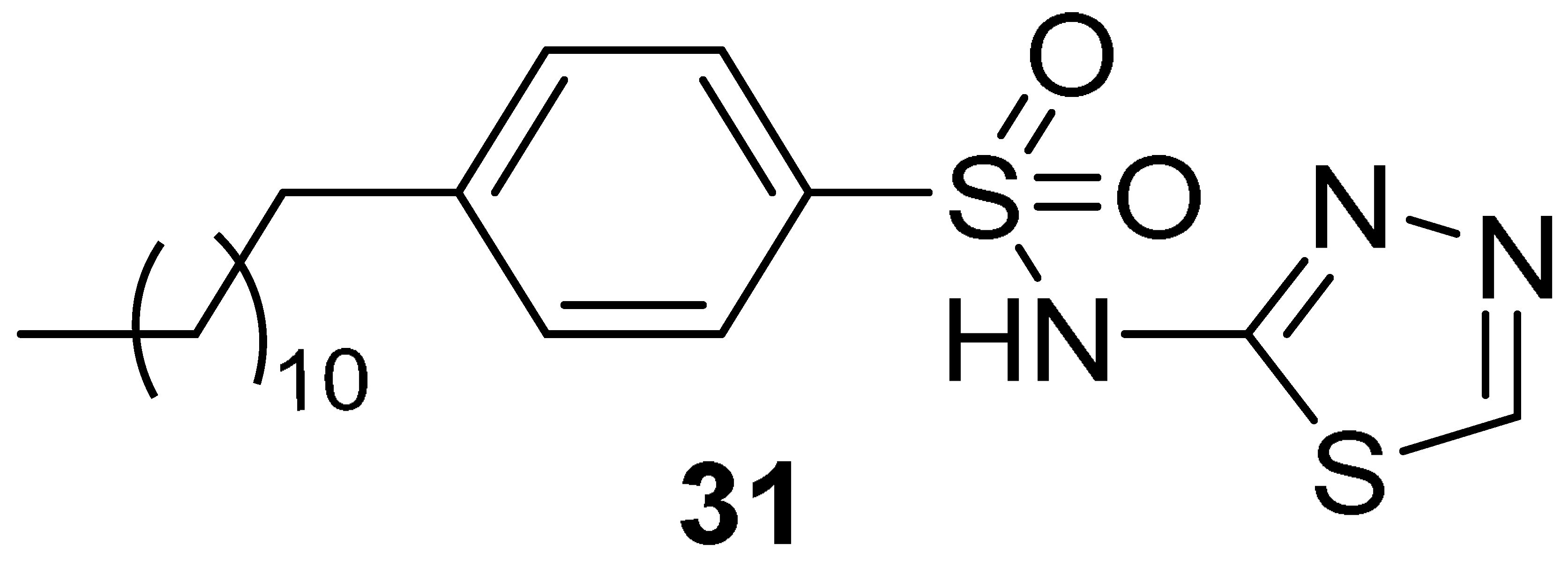 | 7.54 | 0.1 | 40.8 ± 2.5 | 2.4 ± 0.6 | 6 b/10 c | 65 b/30 c |
| Compound | Figure | IC50 | Ligand Binding Site | Clinical Phase | NCT Number |
|---|---|---|---|---|---|
| Capivasertib (AZD5363) | Figure 10 | 6 nM (AKT1) | ATP-binding site | Phase I | NCT01226316, NCT04556773 |
| Phase I/II | NCT01992952, NCT02208375, NCT03742102 | ||||
| Phase II | NCT02117167, NCT02299999, NCT02465060, NCT02664935, NCT04439123 | ||||
| Phase III | NCT03997123, NCT04305496 | ||||
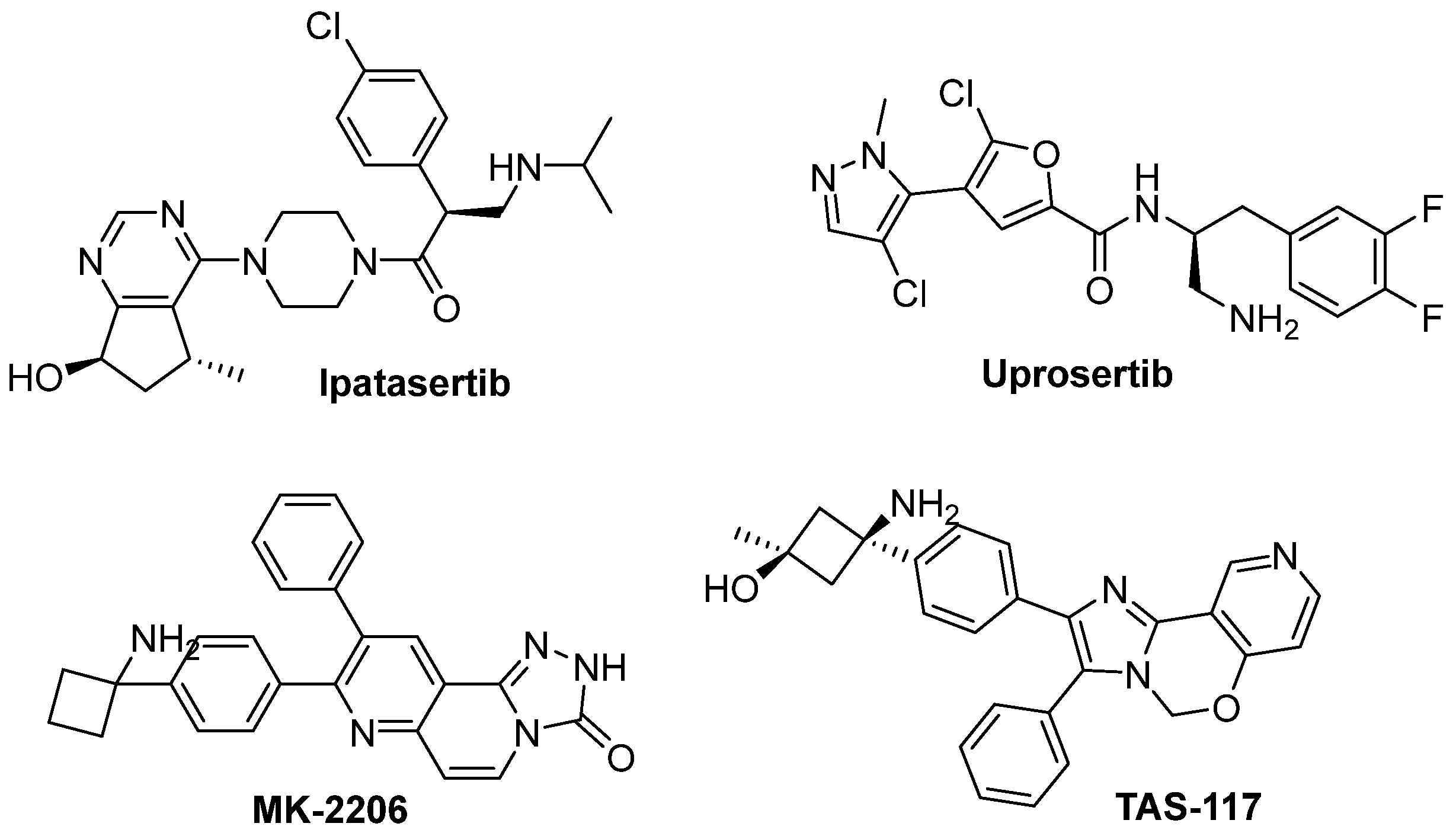
| Ipatasertib (GDC-0068) | Figure 15 | 5 nM (AKT1) | ATP-binding site | Phase I | NCT03959891 |
| Phase I/II | NCT03280563, NCT03424005, NCT03853707 | ||||
| Phase II | NCT02465060, NCT03395899, NCT03498521, NCT04464174, NCT04591431, NCT04632992, NCT05498896 | ||||
| Phase III | NCT03072238, NCT04060862 | ||||
| Uprosertib (GSK2141795) | Figure 15 | 9.6 nM (AKT1) | ATP-binding site | Phase I/II | NCT01902173 |
| MK-2206 | Figure 15 | 8 nM (AKT1) | Allosteric site | Phase I | NCT01480154 |
| Phase II | NCT01251861, NCT01306045 | ||||
| TAS-117 | Figure 15 | 0.55 nM (AKT1) | Allosteric site | Phase II | NCT04770246 |
| Domain | PDB ID | Release Date | Resolution (Å) | Exp. IC50 (nM) | Notes | |
|---|---|---|---|---|---|---|
| AKT1 | ||||||
| no ligands | PH | 1UNP | 2004 | 1.65 | ||
| PH | 1UNR | 2004 | 1.25 | |||
| PH | 2UZR | 2007 | 1.94 | |||
| Kinase | 6BUU | 2018 | 2.4 | |||
| Kinase | 6NPZ | 2019 | 2.12 | |||
| Full-length | 7APJ | 2021 | 2.05 | Complexed with antibody | ||
| PH | 7MYX | 2022 | 1.39 | |||
| ATP-binding site | Kinase | 3CQU | 2008 | 2.2 | 151 | |
| Kinase | 3CQW | 2008 | 2 | 318 | ||
| Kinase | 3MV5 | 2010 | 2.47 | 180 | ||
| Kinase | 3MVH | 2010 | 2.01 | 0.5 | ||
| Kinase | 3OCB | 2010 | 2.7 | 5 | ||
| Kinase | 3OW4 | 2010 | 2.6 | 22 | ||
| Kinase | 3QKK | 2011 | 2.3 | 300 | ||
| Kinase | 3QKL | 2011 | 1.9 | 9 | ||
| Kinase | 3QKM | 2011 | 2.2 | 38 | ||
| Kinase | 4EKK | 2012 | 2.8 | NR a | AMP-PNP | |
| Kinase | 4EKL | 2012 | 2 | 36.9 | Clinical candidate for cancer (Ipatasertib) | |
| Kinase | 4GV1 | 2013 | 1.49 | 4 | Clinical candidate for cancer (Capivasertib) | |
| Kinase | 6CCY | 2018 | 2.18 | 3 | ||
| Allosteric site | Full-length | 3O96 | 2010 | 2.7 | 58 | |
| Full-length | 4EJN | 2012 | 2.19 | 5 | ||
| Full-length | 5KCV | 2016 | 2.7 | 8 | Clinical candidate Proteus syndrome (Miransertib) | |
| Full-length | 6HHF | 2019 | 2.9 | 0.5 | Covalent binder | |
| Full-length | 6HHG | 2019 | 2.3 | 9.1 | Covalent binder | |
| Full-length | 6HHH | 2019 | 2.7 | 126 | Covalent binder | |
| Full-length | 6HHI | 2019 | 2.7 | 3.6 | Covalent binder | |
| Full-length | 6HHJ | 2019 | 2.3 | 3 | Covalent binder | |
| Full-length | 6S9W | 2019 | 2.3 | 39 | Covalent binder | |
| Full-length | 6S9X | 2019 | 2.6 | 381 | Covalent binder | |
| Full-length | 7NH4 | 2021 | 2.3 | 44 | Covalent binder | |
| Full-length | 7NH5 | 2021 | 1.9 | 112 | Covalent binder | |
| PI3P-binding site | PH | 1H10 | 2003 | 1.4 | NR a | 4IP |
| PH | 1UNQ | 2004 | 0.98 | NR a | 4IP | |
| PH | 2UVM | 2007 | 1.94 | Ki = 80 nM | ||
| PH | 2UZS | 2007 | 2.46 | NR a | 4IP, E17K mutation | |
| AKT2 | ||||||
| no ligands | Kinase | 1GZK | 2003 | 2.3 | ||
| Kinase | 1GZN | 2003 | 2.5 | |||
| Kinase | 1GZO | 2003 | 2.75 | |||
| Kinase | 1MRV | 2003 | 2.8 | |||
| Kinase | 1MRY | 2003 | 2.8 | |||
| PH | 1P6S | 2004 | NMR | |||
| ATP-binding site | Kinase | 1O6K | 2002 | 1.7 | NR a | ANP |
| Kinase | 1O6L | 2002 | 1.6 | NR a | ANP | |
| Kinase | 2JDO | 2007 | 1.8 | 230 | ||
| Kinase | 2JDR | 2007 | 2.3 | 0.5 | ||
| Kinase | 2UW9 | 2007 | 2.1 | 18 | ||
| Kinase | 2X39 | 2010 | 1.93 | 6 | ||
| Kinase | 2XH5 | 2010 | 2.72 | 27 | ||
| Kinase | 3D0E | 2008 | 2 | Ki: 4 nM | ||
| Kinase | 3E87 | 2008 | 2.3 | NR a | ||
| Kinase | 3E88 | 2008 | 2.5 | 0.6 | ||
| Kinase | 3E8D | 2008 | 2.7 | 2 | ||
| AKT3 | ||||||
| no ligands | PH | 2X18 | 2010 | 1.46 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primavera, E.; Palazzotti, D.; Barreca, M.L.; Astolfi, A. Computer-Aided Identification of Kinase-Targeted Small Molecules for Cancer: A Review on AKT Protein. Pharmaceuticals 2023, 16, 993. https://doi.org/10.3390/ph16070993
Primavera E, Palazzotti D, Barreca ML, Astolfi A. Computer-Aided Identification of Kinase-Targeted Small Molecules for Cancer: A Review on AKT Protein. Pharmaceuticals. 2023; 16(7):993. https://doi.org/10.3390/ph16070993
Chicago/Turabian StylePrimavera, Erika, Deborah Palazzotti, Maria Letizia Barreca, and Andrea Astolfi. 2023. "Computer-Aided Identification of Kinase-Targeted Small Molecules for Cancer: A Review on AKT Protein" Pharmaceuticals 16, no. 7: 993. https://doi.org/10.3390/ph16070993
APA StylePrimavera, E., Palazzotti, D., Barreca, M. L., & Astolfi, A. (2023). Computer-Aided Identification of Kinase-Targeted Small Molecules for Cancer: A Review on AKT Protein. Pharmaceuticals, 16(7), 993. https://doi.org/10.3390/ph16070993