

Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues
Abstract
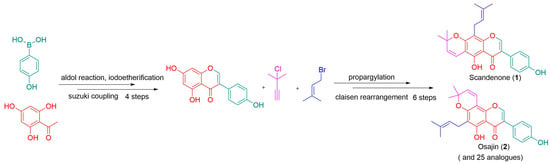
:
1. Introduction
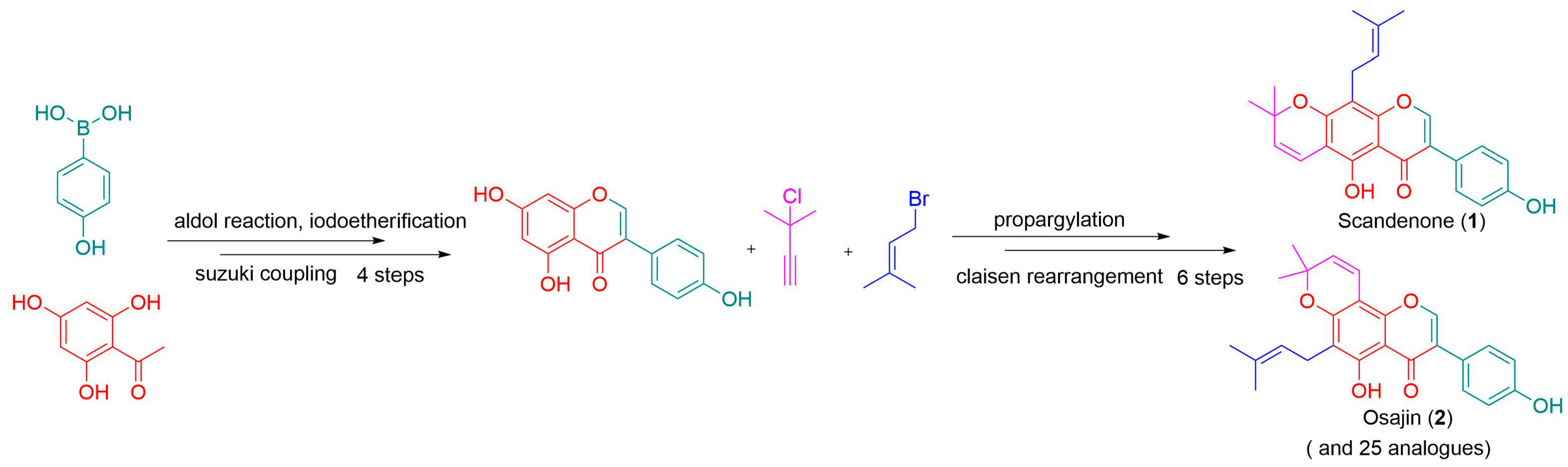
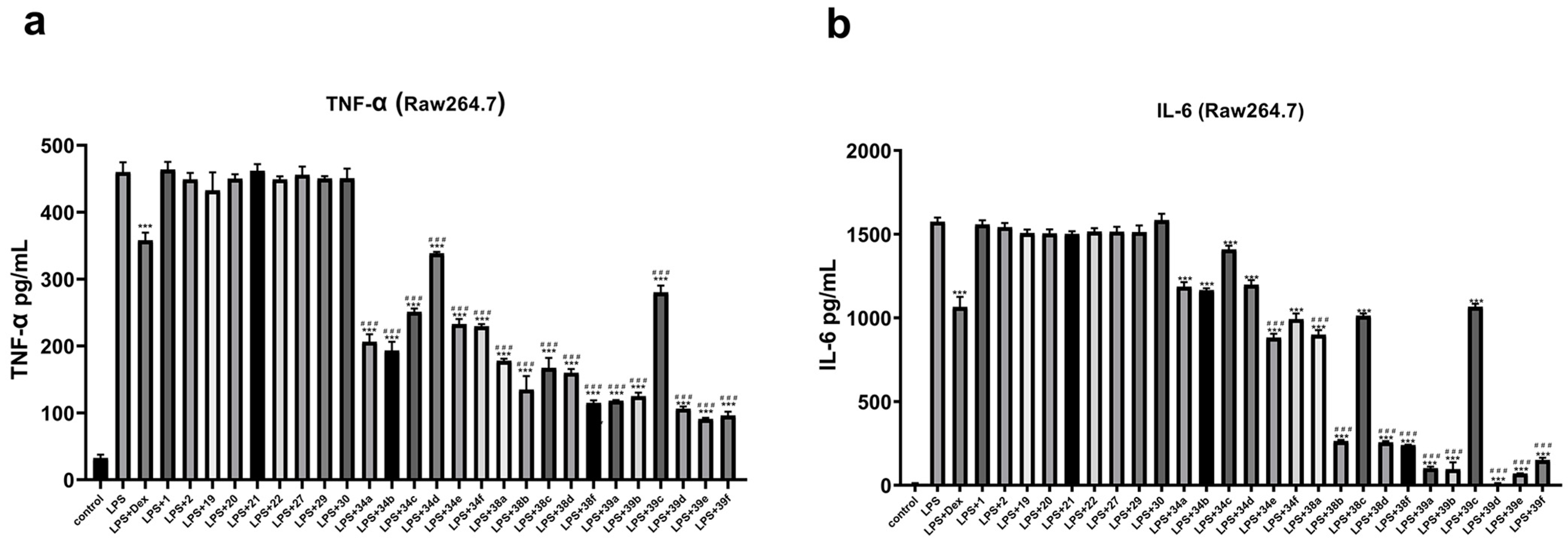
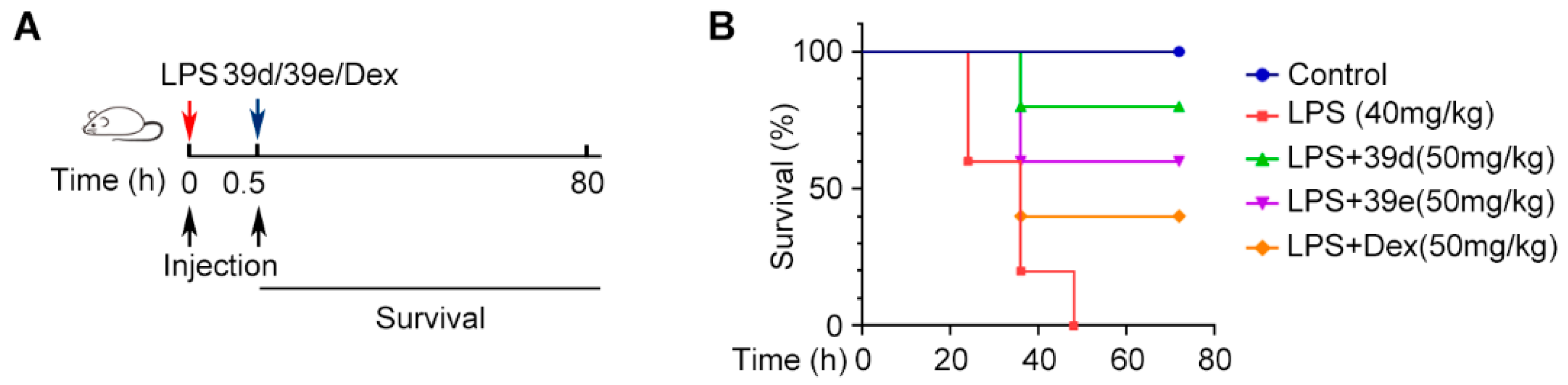
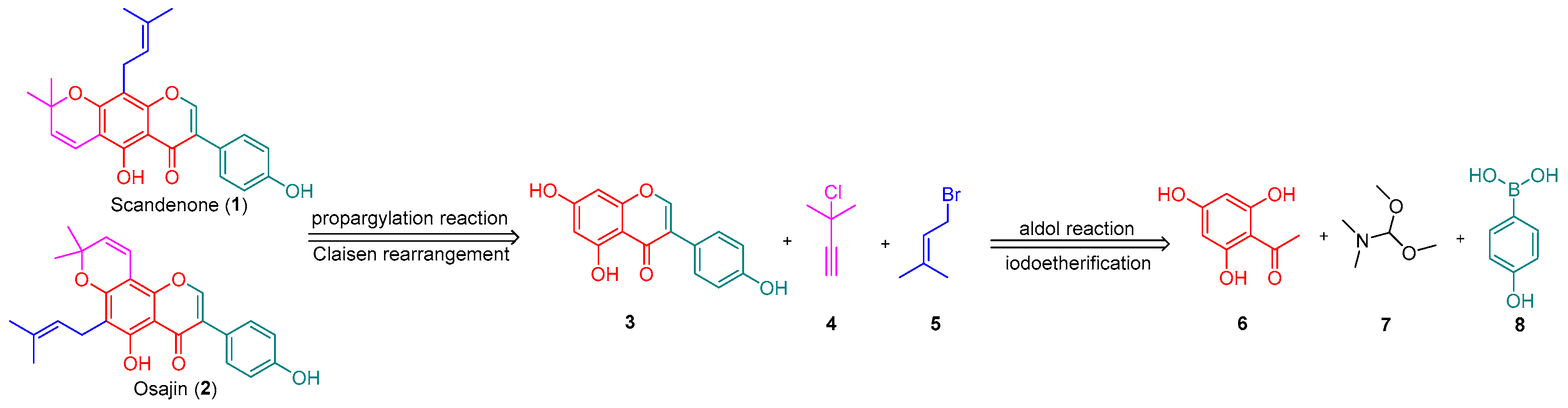
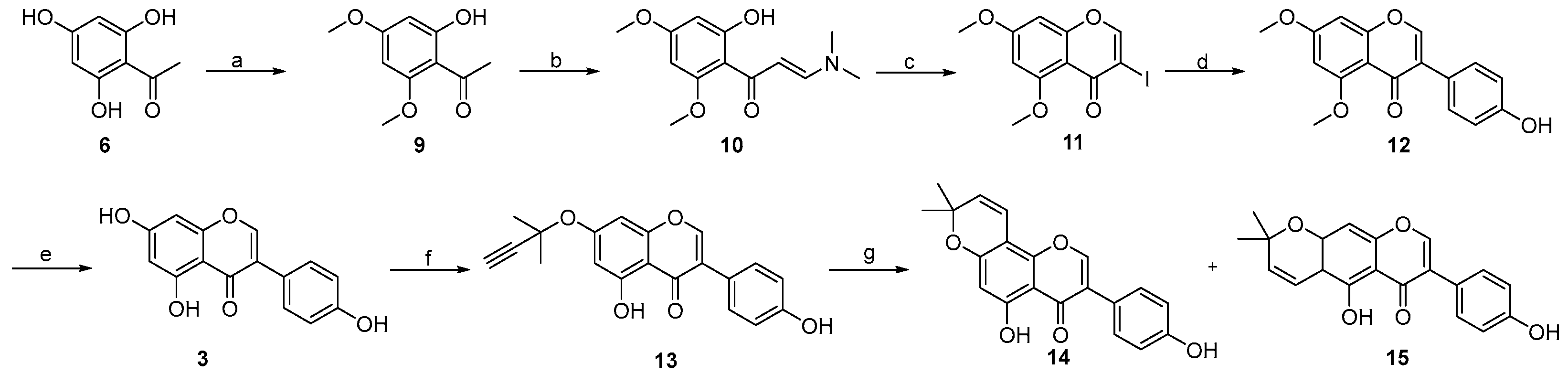
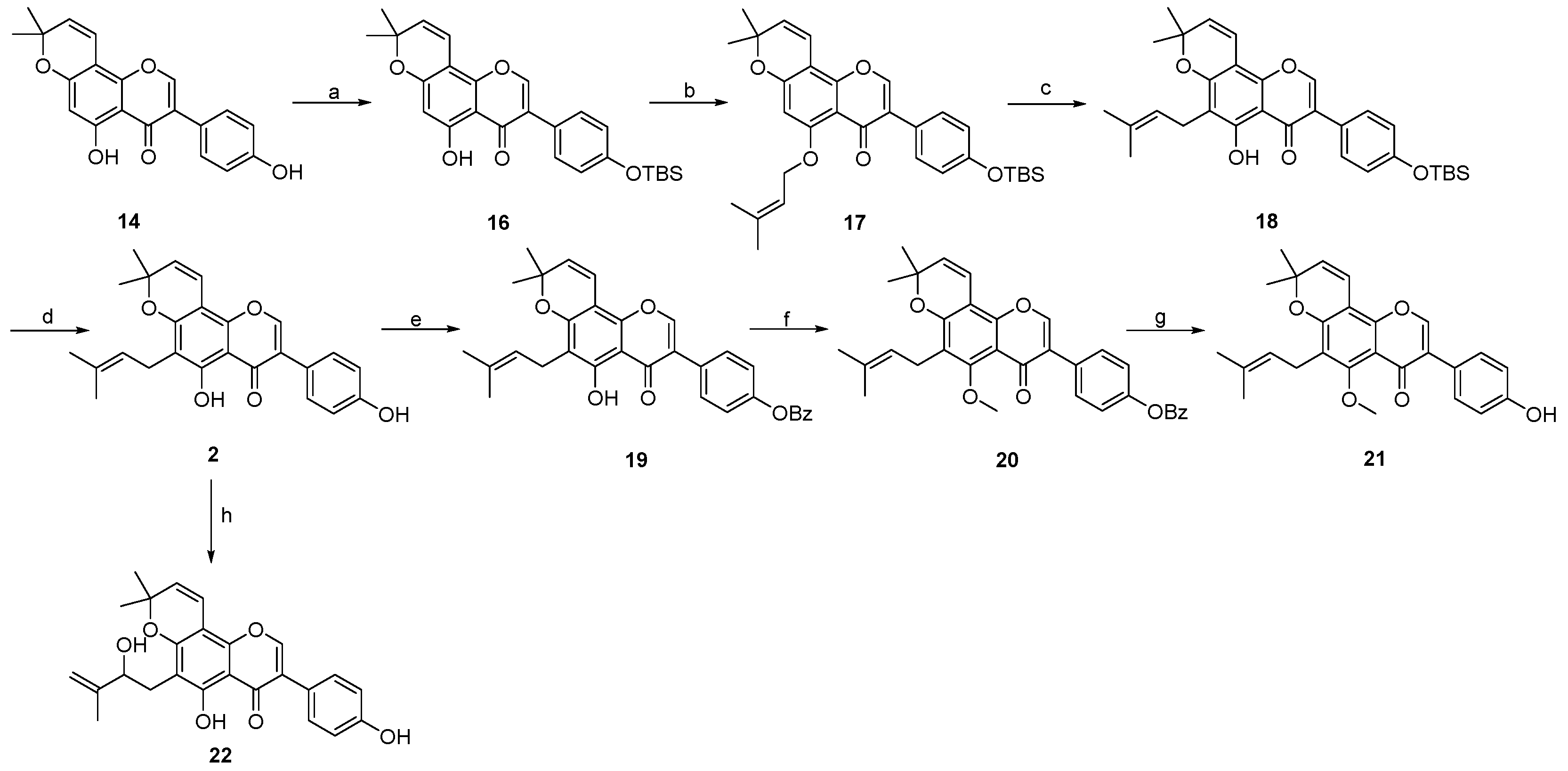
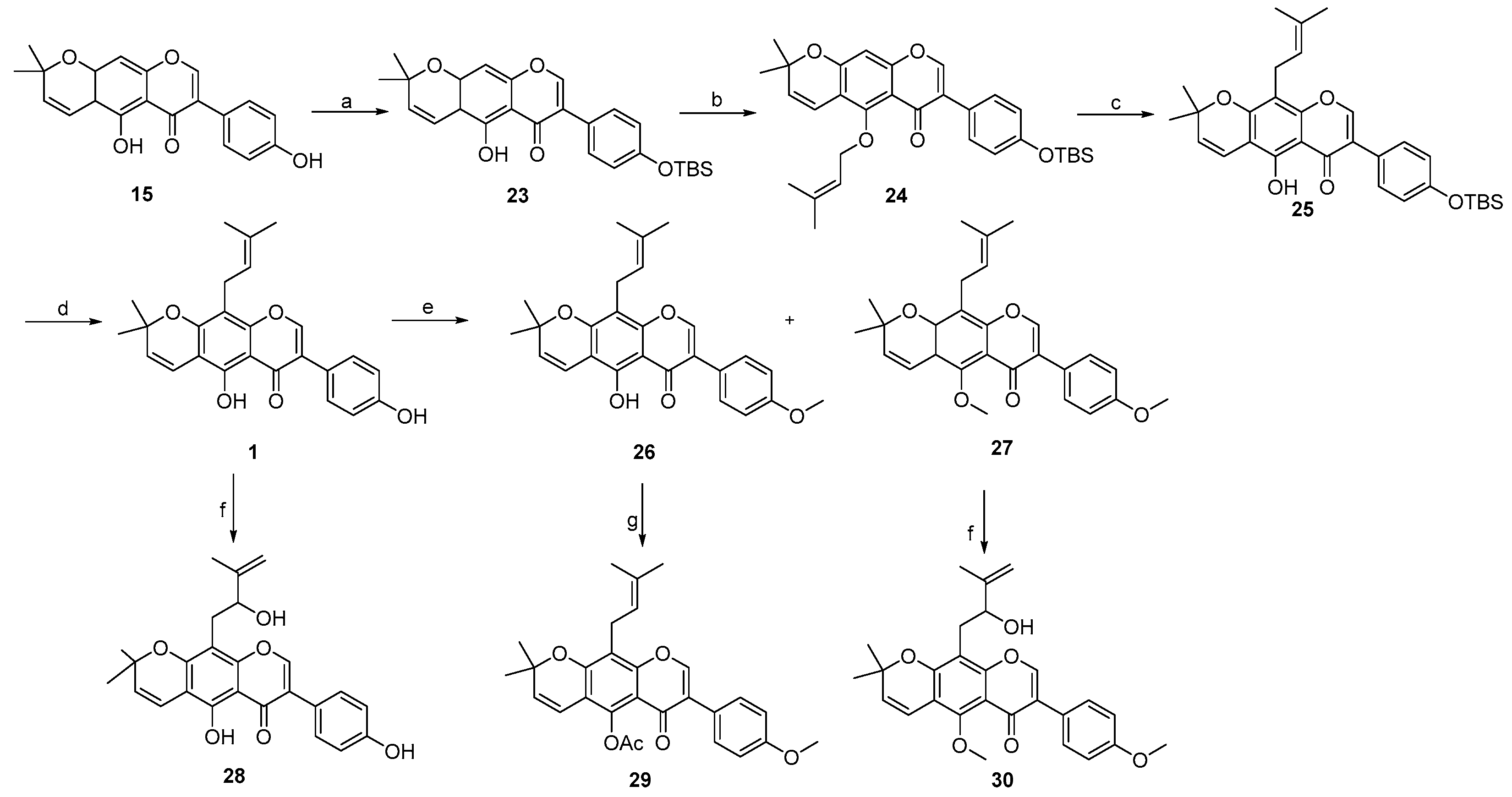
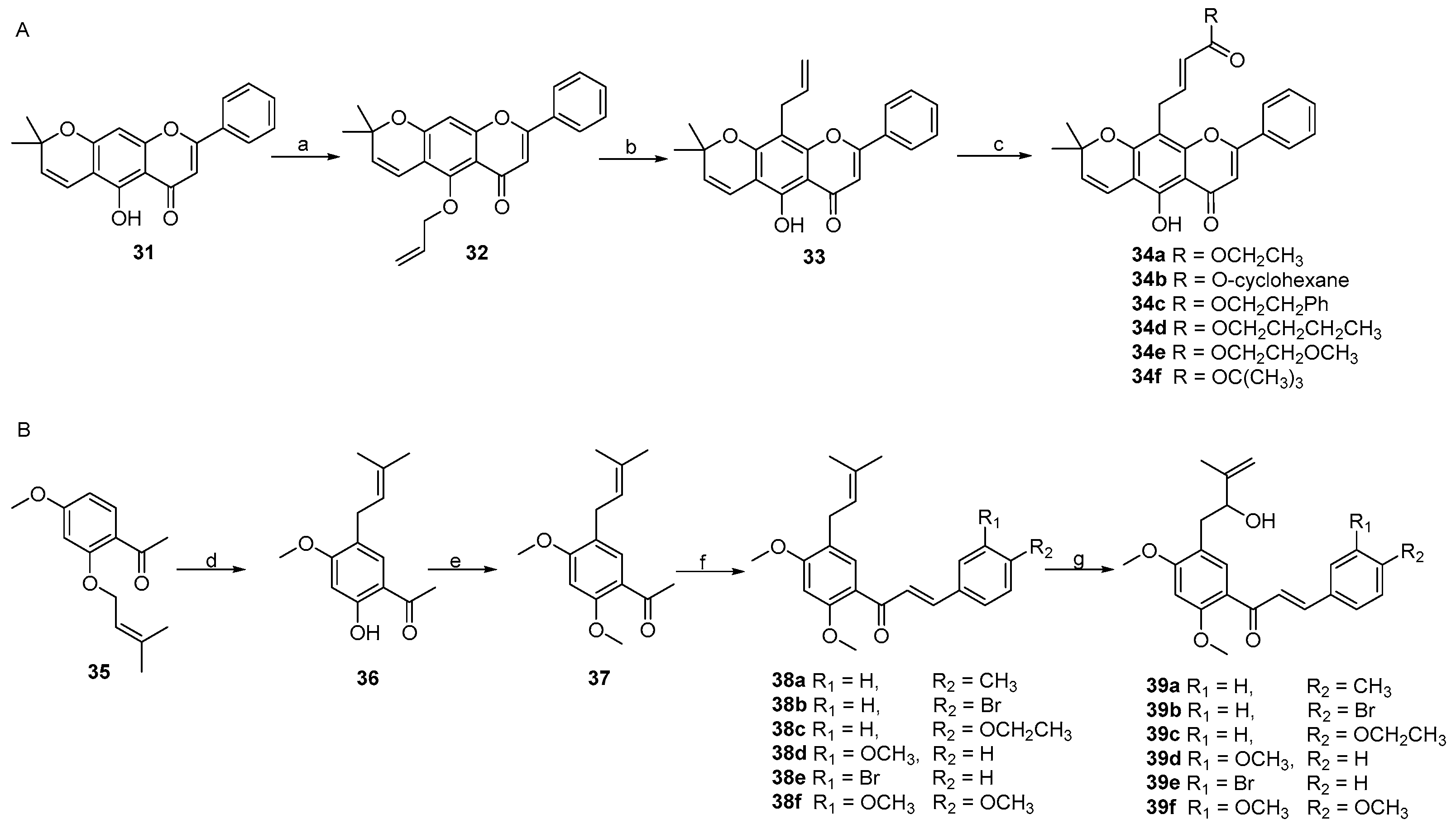
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Materials
3.3. Procedure for the Synthesis of Osajin and Scandenone
- 1-(2-hydroxy-4,6-dimethoxyphenyl)ethan-1-one 9:
- (E)-3-(dimethylamino)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one 10:
- 3-iodo-5,7-dimethoxy-4H-chromen-4-one 11:
- 3-(4-hydroxyphenyl)-5,7-dimethoxy-4H-chromen-4-one 12:
- 5,7-dihydroxy-3-(4-hydroxyphenyl)-4H-chromen-4-one 3:
- 5-hydroxy-3-(4-hydroxyphenyl)-7-((2-methylbut-3-yn-2-yl)oxy)-4H-chromen-4-one 13:
- 5-hydroxy-3-(4-hydroxyphenyl)-8,8-dimethyl-4H,8H-pyrano [2,3-f]chromen-4-one 14:
- 5-hydroxy-7-(4-hydroxyphenyl)-2,2-dimethyl-2H,6H-pyrano [3,2-g]chromen-6-one 15:
- 7-(4-((tert-butyldimethylsilyl)oxy)phenyl)-5-hydroxy-2,2-dimethyl-2H,6H-pyrano [3,2-g]chromen-6-one 23:
- 7-(4-((tert-butyldimethylsilyl)oxy)phenyl)-2,2-dimethyl-5-((3-methylbut-2-en-1-yl)oxy)-2H,6H-pyrano [3,2-g]chromen-6-one 24:
- 7-(4-((tert-butyldimethylsilyl)oxy)phenyl)-5-hydroxy-2,2-dimethyl-10-(3-methylbut-2-en-1-yl)-2H,6H-pyrano [3,2-g]chromen-6-one 25:
- 3-(4-((tert-butyldimethylsilyl)oxy)phenyl)-5-hydroxy-8,8-dimethyl-4H,8H-pyrano [2,3-f]chromen-4-one 16: Compound 16 was synthesized by following a similar procedure as that of 23.
- 3-(4-((tert-butyldimethylsilyl)oxy)phenyl)-8,8-dimethyl-5-((3-methylbut-2-en-1-yl)oxy)-4H,8H-pyrano [2,3-f]chromen-4-one 17: Compound 17 was synthesized by following a similar procedure as that of 24.
- 3-(4-((tert-butyldimethylsilyl)oxy)phenyl)-5-hydroxy-8,8-dimethyl-6-(3-methylbut-2-en-1-yl)-4H,8H-pyrano [2,3-f]chromen-4-one 18:
- 5-hydroxy-3-(4-hydroxyphenyl)-8,8-dimethyl-6-(3-methylbut-2-en-1-yl)-4H,8H-pyrano [2,3-f]chromen-4-one 2: Compound 2 was synthesized by following a similar procedure as that of 1.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 125124. [Google Scholar] [CrossRef]
- Sánchez, M.; Romero, M.; Gómez-Guzmán, M.; Tamargo, J.; Pérez-Vizcaino, F.; Duarte, J. Cardiovascular Effects of Flavonoids. Curr. Med. Chem. 2019, 26, 6991–7034. [Google Scholar] [CrossRef]
- Li, X.; Yin, M.; Yang, X.; Yang, G.; Gao, X. Flavonoids from Mirabilis himalaica. Fitoterapia 2018, 127, 89–95. [Google Scholar] [CrossRef]
- Liskova, A.; Koklesova, L.; Samec, M.; Smejkal, K.; Samuel, S.M.; Varghese, E.; Abotaleb, M.; Biringer, K.; Kudela, E.; Danko, J.; et al. Flavonoids in Cancer Metastasis. Cancers 2020, 12, 1498. [Google Scholar] [CrossRef]
- Lv, Y.; Marsafari, M.; Koffas, M.; Zhou, J.; Xu, P. Optimizing Oleaginous Yeast Cell Factories for Flavonoids and Hydroxylated Flavonoids Biosynthesis. ACS Synth. Biol. 2019, 8, 2514–2523. [Google Scholar] [CrossRef]
- Raffa, D.; Maggio, B.; Raimondi, M.V.; Plescia, F.; Daidone, G. Recent discoveries of anticancer flavonoids. Eur. J. Med. Chem. 2017, 142, 213–228. [Google Scholar] [CrossRef]
- Oh, T.W.; Do, H.J.; Jeon, J.-H.; Kim, K. Quercitrin inhibits platelet activation in arterial thrombosis. Phytomedicine 2021, 80, 153363. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, X.; Xiao, L.; Lu, Y.; Sang, S.; Lv, L.; Dong, W. Mechanistic studies of inhibition on acrolein by myricetin. Food Chem. 2020, 323, 126788. [Google Scholar] [CrossRef]
- Sreelatha, T.; Hymavathi, A.; Rama Subba Rao, V.; Devanand, P.; Usha Rani, P.; Madhusudana Rao, J.; Suresh Babu, K. A new benzil derivative from Derris scandens: Structure-insecticidal activity study. Bioorg. Med. Chem. Lett. 2010, 20, 549–553. [Google Scholar] [CrossRef]
- Wang, Y.; Curtis-Long, M.J.; Yuk, H.J.; Kim, D.W.; Tan, X.F.; Park, K.H. Bacterial neuraminidase inhibitory effects of prenylated isoflavones from roots of Flemingia philippinensis. Bioorg. Med. Chem. 2013, 21, 6398–6404. [Google Scholar] [CrossRef]
- Cheenpracha, S.; Chokchaisiri, R.; Laphookhieo, S.; Limtharakul, T.; Thepmalee, C. Rare prenylated isoflavonoids from the young twigs of Millettia extensa and their cytotoxic activities. RSC Adv. 2022, 12, 30359–30364. [Google Scholar] [CrossRef]
- Ribaudo, G.; Coghi, P.; Zanforlin, E.; Law, B.Y.K.; Wu, Y.Y.J.; Han, Y.; Qiu, A.C.; Qu, Y.Q.; Zagotto, G.; Wong, V.K.W. Semi-synthetic isoflavones as BACE-1 inhibitors against Alzheimer’s disease. Bioorg. Chem. 2019, 87, 474–483. [Google Scholar] [CrossRef]
- Adem, F.A.; Mbaveng, A.T.; Kuete, V.; Heydenreich, M.; Ndakala, A.; Irungu, B.; Yenesew, A.; Efferth, T. Cytotoxicity of isoflavones and biflavonoids from Ormocarpum kirkii towards multi-factorial drug resistant cancer. Phytomedicine 2019, 58, 152853. [Google Scholar] [CrossRef]
- Xue, L.-L.; Wu, W.-S.; Ma, X.; Pei, H.-Y.; Tang, M.-H.; Kuang, S.; Cai, X.-Y.; Wang, L.; Li, Y.; Zhang, R.-J.; et al. Modulation of LPS-induced inflammation in RAW264.7 murine cells by novel isoflavonoids from Millettia pulchra. Bioorg. Chem. 2020, 97, 103693. [Google Scholar] [CrossRef]
- Ito, S.; Kitamura, T.; Arulmozhiraja, S.; Manabe, K.; Tokiwa, H.; Suzuki, Y. Total Synthesis of Termicalcicolanone A via Organocatalysis and Regioselective Claisen Rearrangement. Org. Lett. 2019, 21, 2777–2781. [Google Scholar] [CrossRef]
- Boruah, J.J.; Das, S.P. Palladium (Pd)-based Photocatalysts for Suzuki Coupling Reactions: An Overview. Mini-Rev. Org. Chem. 2023, 20, 687–699. [Google Scholar] [CrossRef]
- Yi, J.; Du, G.; Zhao, Y.; Zhang, L.; Li, B.; Zhu, W.; Huang, C.; Li, Y.; Guo, F. Bavachinin analogues as agonists of pan-peroxisome proliferator-activated receptors. Med. Chem. Res. 2018, 27, 1851–1862. [Google Scholar] [CrossRef]
- Orsi, D.L.; Easley, B.J.; Lick, A.M.; Altman, R.A. Base Catalysis Enables Access to α,α-Difluoroalkylthioethers. Org. Lett. 2017, 19, 1570–1573. [Google Scholar] [CrossRef]
- Bensinger, D.; Stubba, D.; Cremer, A.; Kohl, V.; Waßmer, T.; Stuckert, J.; Engemann, V.; Stegmaier, K.; Schmitz, K.; Schmidt, B. Virtual Screening Identifies Irreversible FMS-like Tyrosine Kinase 3 Inhibitors with Activity toward Resistance-Conferring Mutations. J. Med. Chem. 2019, 62, 2428–2446. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Chan, Y.; Noor Armylisas, A.H.; Alahmdi, M. Asymmetric epoxidation of chromenes mediated by iminium salts: Synthesis of mollugin and (3S,4R)-trans-3,4-dihydroxy-3,4-dihydromollugin. Tetrahedron 2016, 72, 8406–8416. [Google Scholar] [CrossRef]
- Amolak, C.J.; Bhola, N.S. Synthesis of Alpinum Isoflavone, Osajin, and Warangalone. J. Org. Chem. 1974, 39, 2215–2217. [Google Scholar]
- Helesbeux, J.-J.; Duval, O.; Guilet, D.; Séraphin, D.; Rondeau, D.; Richomme, P. Regioselectivity in the ene reaction of singlet oxygen with ortho-prenylphenol derivatives. Tetrahedron 2003, 59, 5091–5104. [Google Scholar] [CrossRef]
- Parmar, V.S.; Jain, S.C.; Bisht, K.S.; Sharma, N.K.; Gupta, S.; Prasad, A.K.; Jha, A.; Malhotra, S.; Sharma, S.K.; Bracke, M.E. Synthesis and anti-invasive activity of novel 1,3-diarylpropenones. Indian J. Chem. 1998, 37B, 628–643. [Google Scholar]
- Dong, T.; Li, C.; Wang, X.; Dian, L.; Zhang, X.; Li, L.; Chen, S.; Cao, R.; Li, L.; Huang, N.; et al. Ainsliadimer A selectively inhibits IKKα/β by covalently binding a conserved cysteine. Nat. Commun. 2015, 6, 6522. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Solvent | Base | Temperature (°C) | Yield (%) c |
|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | MeOH | Na2CO3 | rt b | 40 |
| 2 | Pd(OAc)2 | DMF | Na2CO3 | rt | 45 |
| 3 | Pd(OAc)2 | DMF | K2CO3 | rt | 43 |
| 4 | Pd(OAc)2 | DMF | K2CO3 | 50 | 60 |
| 5 | Pd(PPh3)4 | DMF | K2CO3 | rt | 46 |
| 6 | Pd(PPh3)4 | DMF | K2CO3 | 40 | 57 |
| 7 | Pd(PPh3)4 | DMF | K2CO3 | 60 | 67 |
| 8 | Pd(PPh3)4 | DMF | K2CO3 | 80 | 70 |
| 9 | PdCl2(dppf) | 1,4-Dioxane/H2O d | K2CO3 | 50 | 84 |
| 10 | PdCl2(dppf) | 1,4-Dioxane/H2O | K2CO3 | 80 | 86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Ma, R.; Feng, K.; Lu, H.; Zhao, W.; Jin, H. Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues. Pharmaceuticals 2024, 17, 86. https://doi.org/10.3390/ph17010086
Wang R, Ma R, Feng K, Lu H, Zhao W, Jin H. Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues. Pharmaceuticals. 2024; 17(1):86. https://doi.org/10.3390/ph17010086
Chicago/Turabian StyleWang, Rui, Ran Ma, Ke Feng, Hongchen Lu, Wei Zhao, and Hongzhen Jin. 2024. "Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues" Pharmaceuticals 17, no. 1: 86. https://doi.org/10.3390/ph17010086
APA StyleWang, R., Ma, R., Feng, K., Lu, H., Zhao, W., & Jin, H. (2024). Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues. Pharmaceuticals, 17(1), 86. https://doi.org/10.3390/ph17010086