



Pharmaceutical Agents for Targeting Autophagy and Their Applications in Clinics

 , , and
, , and
Abstract
:1. Introduction
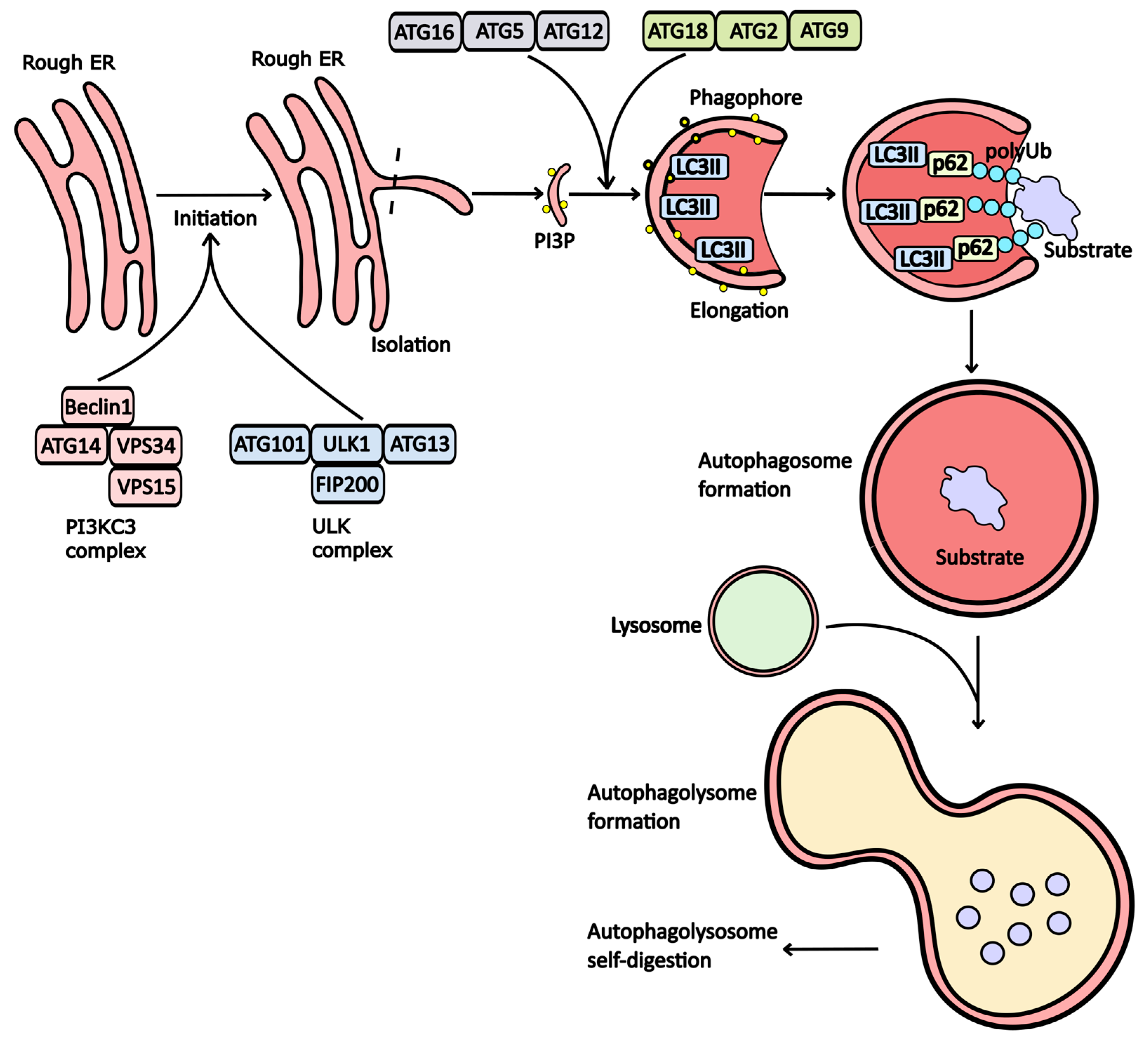
Mechanism of Autophagy
2. Drugs That Modulate Autophagy Activity
2.1. Inhibitors of Autophagy
2.1.1. PI3K Inhibitors
- -
- Class 1 PI3K (PI3KC1): Involved in the AKT signaling cascade and insulin receptor signaling.
- -
- Class 2 PI3K (PI3KC2): Plays a role in cell adhesion.
- -
- Class 3 PI3K (PI3KC3): Central to the formation of autophagosomes.
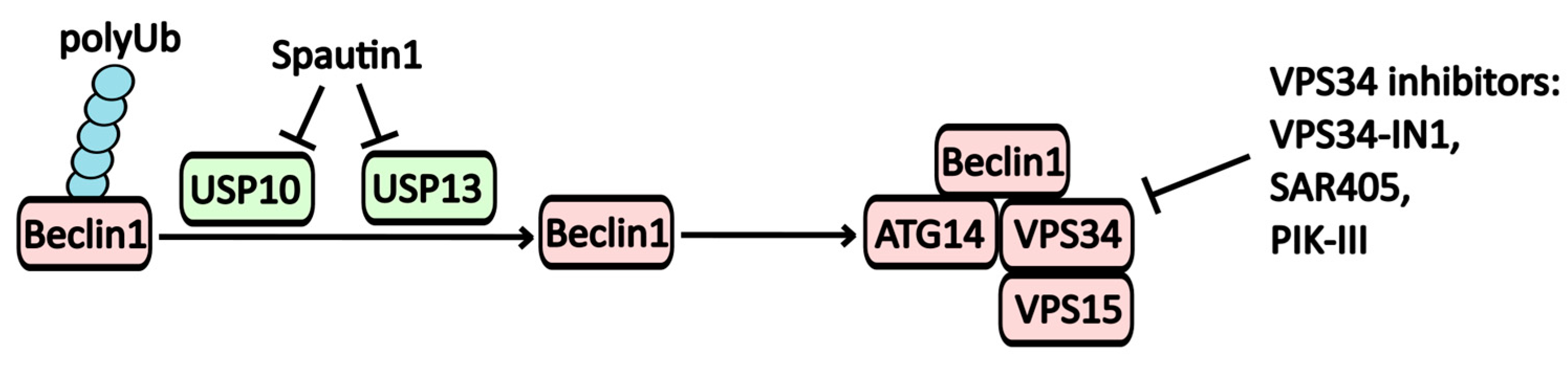
2.1.2. Inhibitors of the PI3KC3 Complex (Vps34)
2.1.3. ULK1 Inhibitors
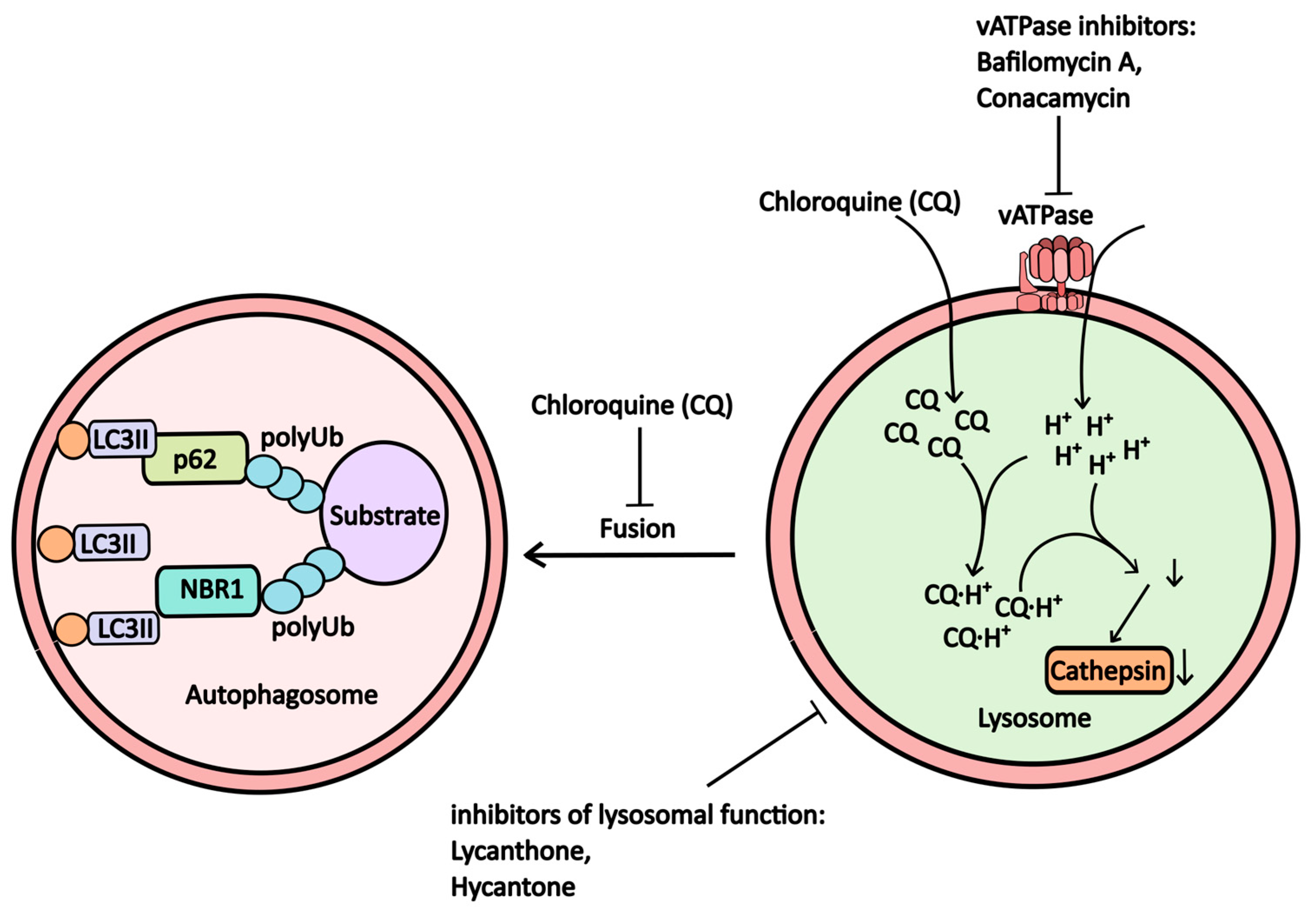
2.1.4. V-ATPase (Proton Pump) and Lysosomal Inhibitors
2.2. Inducers of Autophagy
2.2.1. mTOR Inhibitors
2.2.2. ER-Stress Inductors
2.2.3. Inhibitors of Inositol Monophosphatase (IMPase)
2.2.4. MTMR14 (Jumpy) Inhibitors
2.2.5. mTOR-Independent Inducers
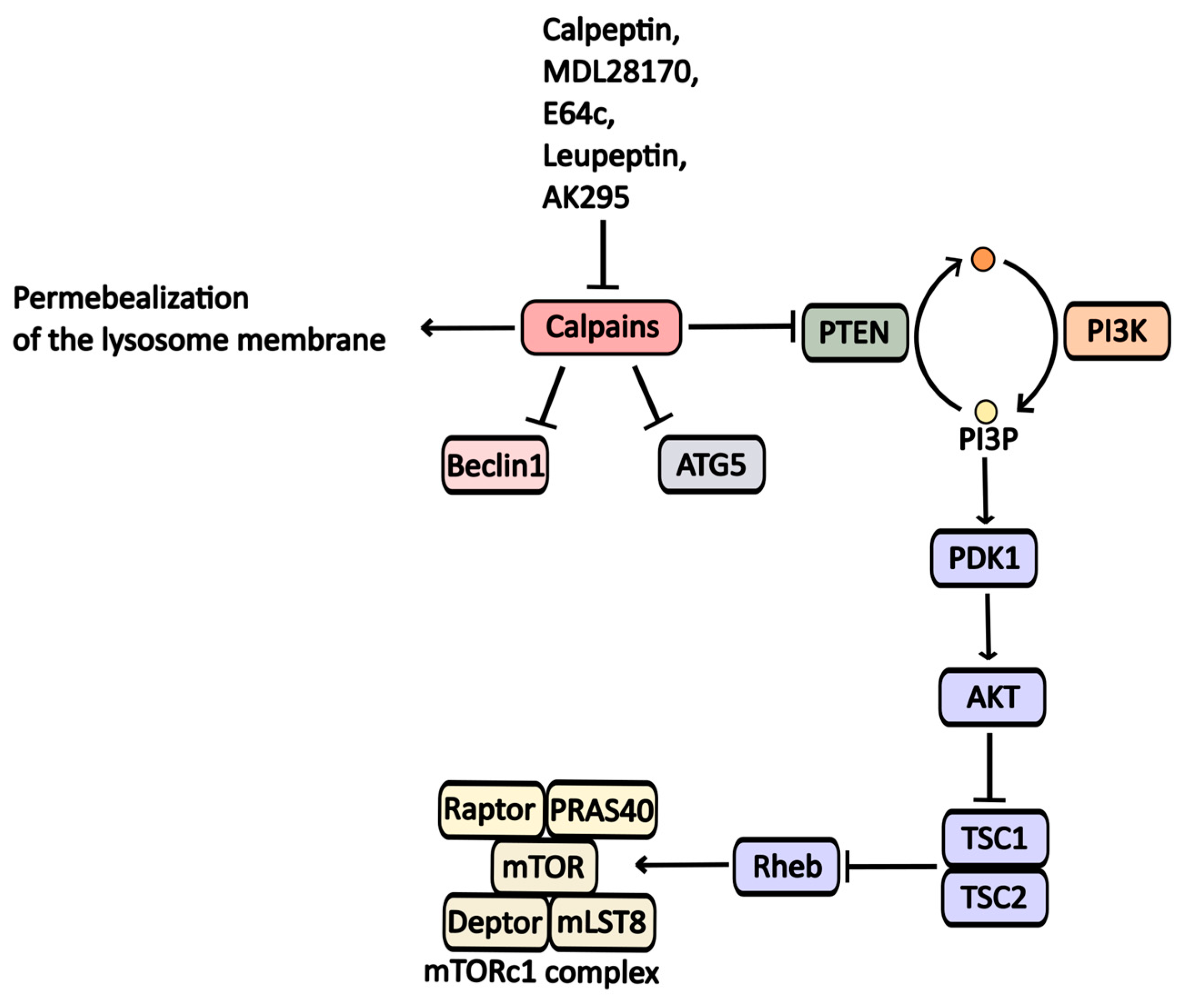
2.2.6. Calpain Inhibitors
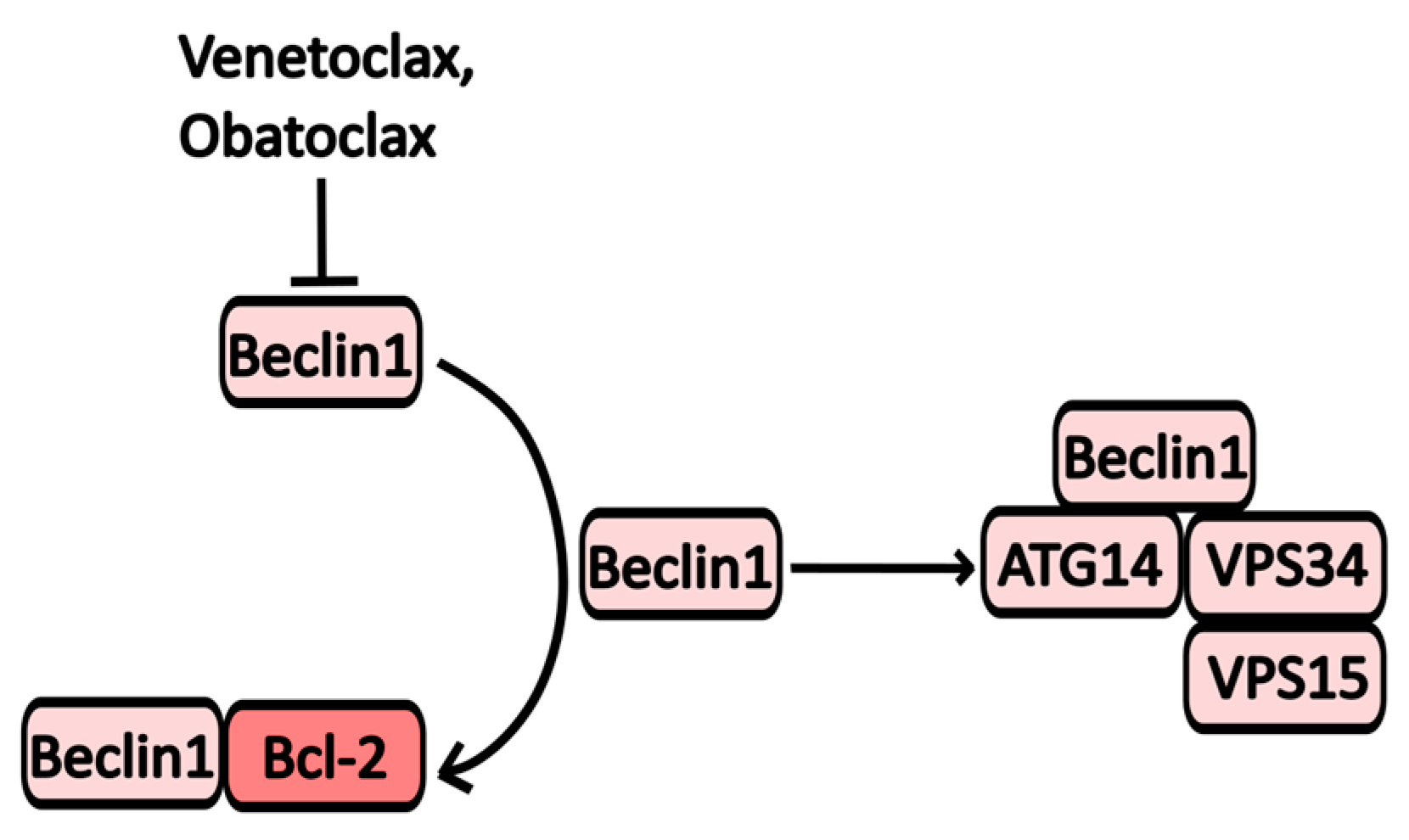
2.2.7. Bcl-2 Inhibitors
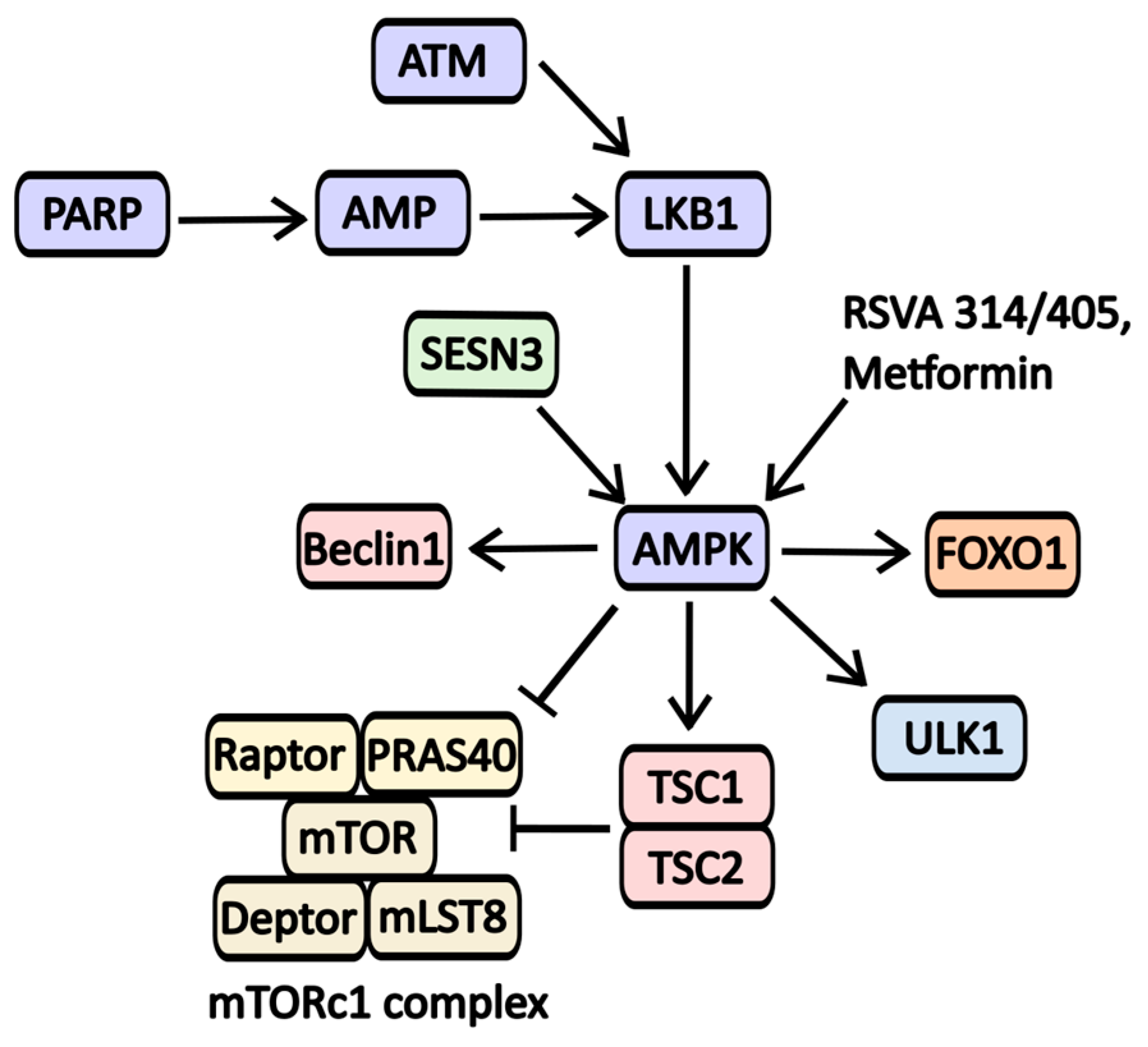
2.2.8. AMPK Activators
2.2.9. TFEB Activators
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy Pathway: Cellular and Molecular Mechanisms. Autophagy 2017, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Xia, H.G.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef]
- Kocak, M.; Ezazi Erdi, S.; Jorba, G.; Maestro, I.; Farrés, J.; Kirkin, V.; Martinez, A.; Pless, O. Targeting autophagy in disease: Established and new strategies. Autophagy 2022, 18, 473–495. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Joachim, J.; Tooze, S.A. Autophagosome Formation—The Role of ULK1 and Beclin1–PI3KC3 Complexes in Setting the Stage. Semin. Cancer Biol. 2013, 23, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Lystad, A.H.; Carlsson, S.R.; Simonsen, A. Toward the Function of Mammalian ATG12–ATG5-ATG16L1 Complex in Autophagy and Related Processes. Autophagy 2019, 15, 1485–1486. [Google Scholar] [CrossRef]
- Nagy, P.; Hegedűs, K.; Pircs, K.; Varga, Á.; Juhász, G. Different Effects of Atg2 and Atg18 Mutations on Atg8a and Atg9 Trafficking During Starvation in Drosophila. FEBS Lett. 2014, 588, 408–413. [Google Scholar] [CrossRef]
- Uluer, T.; Sonmez, P.K.; Akogullari, D.; Onal, M.; Tanriover, G.; Inan, S. Do Wortmannin and Thalidomide Induce Apoptosis by Autophagy Inhibition in 4T1 Breast Cancer Cells In Vitro and In Vivo? Am. J. Transl. Res. 2021, 13, 6236–6247. [Google Scholar]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.-M. Dual Role of 3-Methyladenine in Modulation of Autophagy via Different Temporal Patterns of Inhibition on Class I and III Phosphoinositide 3-Kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef]
- Feng, M.; Wang, J.; Sun, M.; Li, G.; Li, B.; Zhang, H. 3-Methyladenine but Not Antioxidants to Overcome BACH2-Mediated Bortezomib Resistance in Mantle Cell Lymphoma. Cancer Cell Int. 2021, 21, 279. [Google Scholar] [CrossRef]
- Blommaart, E.F.C.; Krause, U.; Schellens, J.P.M.; Vreeling-Sindelárová, H.; Meijer, A.J. The Phosphatidylinositol 3-Kinase Inhibitors Wortmannin and LY294002 Inhibit Autophagy in Isolated Rat Hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef]
- Xing, C.; Zhu, B.; Liu, H.; Yao, H.; Zhang, L. Class I Phosphatidylinositol 3-Kinase Inhibitor LY294002 Activates Autophagy and Induces Apoptosis Through p53 Pathway in Gastric Cancer Cell Line SGC7901. Acta Biochim. Biophys. Sin. 2008, 40, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Gao, Y.; Wang, D.; Xu, Z.; Sun, W.; Ren, P. Autophagy Inhibitor (LY294002) and 5-Fluorouracil (5-FU) Combination-Based Nanoliposome for Enhanced Efficacy Against Esophageal Squamous Cell Carcinoma. Nanoscale Res. Lett. 2018, 13, 325. [Google Scholar] [CrossRef]
- Liao, Y.; Guo, Z.; Xia, X.; Liu, Y.; Huang, C.; Jiang, L.; Wang, X.; Liu, J.; Huang, H. Inhibition of EGFR Signaling with Spautin-1 Represents a Novel Therapeutics for Prostate Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 157. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Vakifahmetoglu Norberg, H.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of p53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Jeung, H.-K.; Seol, S.-Y.; Chung, H.; Kim, Y.R.; Cheong, J.-W.; Min, Y.H. Inhibition of Unc-51-Like Kinase 1 (ULK1) with Novel Small Molecular Inhibitor MRT68921 Preferentially Induces Apoptosis and Autophagy in FLT3-ITD-Mutated Acute Myeloid Leukemia. Blood 2018, 132, 3499. [Google Scholar] [CrossRef]
- Avsec, D.; Jakoš Djordjevič, A.T.; Kandušer, M.; Podgornik, H.; Škerget, M.; Mlinarič-Raščan, I. Targeting Autophagy Triggers Apoptosis and Complements the Action of Venetoclax in Chronic Lymphocytic Leukemia Cells. Cancers 2021, 13, 4557. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.; Song, L.; Zhang, S.; Lin, W.; Cao, Y.; Xu, F.; Fang, Y.; Wang, Z.; Zhang, H.; Li, X.; et al. Bafilomycin A1 Targets Both Autophagy and Apoptosis Pathways in Pediatric B-Cell Acute Lymphoblastic Leukemia. Haematologica 2015, 100, 345–356. [Google Scholar] [CrossRef]
- Fedele, A.O.; Proud, C.G. Chloroquine and Bafilomycin A Mimic Lysosomal Storage Disorders and Impair mTORC1 Signalling. Biosci. Rep. 2020, 40, BSR20200905. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine Inhibits Autophagic Flux by Decreasing Autophagosome-Lysosome Fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Salata, C.; Calistri, A.; Parolin, C.; Baritussio, A.; Palù, G. Antiviral Activity of Cationic Amphiphilic Drugs. Expert Rev. Anti-Infective Ther. 2017, 15, 483–492. [Google Scholar] [CrossRef]
- Carew, J.S.; Espitia, C.M.; Esquivel, J.A.; Mahalingam, D.; Kelly, K.R.; Reddy, G.; Giles, F.J.; Nawrocki, S.T. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J. Biol. Chem. 2011, 286, 6602–6613. [Google Scholar] [CrossRef] [PubMed]
- Naidu, M.D.; Agarwal, R.; Pena, L.A.; Cunha, L.; Mezei, M.; Shen, M.; Wilson, D.M.; Liu, Y.; Sanchez, Z.; Chaudhary, P.; et al. Lucanthone and Its Derivative Hycanthone Inhibit Apurinic Endonuclease-1 (APE1) by Direct Protein Binding. PLoS ONE 2011, 6, e23679. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Lee, J.; Seo, H.-Y.; Lim, J.S.; Kim, E.K. Cathepsin Inhibition-Induced Lysosomal Dysfunction Enhances Pancreatic Beta-Cell Apoptosis in High Glucose. PLoS ONE 2015, 10, e0116972. [Google Scholar] [CrossRef] [PubMed]
- Mugume, Y.; Kazibwe, Z.; Bassham, D.C. Target of Mycin in Control of Autophagy: Puppet Master and Signal Integrator. Int. J. Mol. Sci. 2020, 21, 8259. [Google Scholar] [CrossRef]
- Smolewski, P. Recent Developments in Targeting the Mammalian Target of Rapamycin (mTOR) Kinase Pathway. Anti-Cancer Drugs 2006, 17, 487–494. [Google Scholar] [CrossRef]
- Niu, T.-K.; Pfeifer, A.C.; Lippincott-Schwartz, J.; Jackson, C.L. Dynamics of GBF1, a Brefeldin A-Sensitive Arf1 Exchange Factor at the Golgi. Mol. Biol. Cell 2005, 16, 1213–1222. [Google Scholar] [CrossRef]
- Li, D.; Li, M.; Li, X.; Liu, X.; Gao, W.; Yu, H.; Bai, J. Study of Brefeldin A on Induction of Autophagy in HepG2 Cells. Environ. Chem. 2022, 41, 3345–3352. [Google Scholar] [CrossRef]
- Zhu, K.; Dunner, K.; McConkey, D. Proteasome Inhibitors Activate Autophagy as a Cytoprotective Response in Human Prostate Cancer Cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, R.; Jiang, X.; Li, Z.; Zhang, B. Progress on the Application of Bortezomib and Bortezomib-Based Nanoformulations. Biomolecules 2021, 12, 51. [Google Scholar] [CrossRef]
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R D 2019, 19, 73–92. [Google Scholar] [CrossRef]
- Cusack, J.C., Jr.; Liu, R.; Xia, L.; Chao, T.H.; Pien, C.; Niu, W.; Palombella, V.J.; Neuteboom, S.T.; Palladino, M.A. NPI-0052 Enhances Tumoricidal Response to Conventional Cancer Therapy in a Colon Cancer Model. Clin. Cancer Res. 2006, 12, 6758–6764. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Rubinsztein, D.C. Inositol and IP3 Levels Regulate Autophagy—Biology and Therapeutic Speculations. Autophagy 2006, 2, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-W.; Choi, H.; Cotman, S.L.; Jung, Y.-K. Lithium Rescues the Impaired Autophagy Process in CbCln3Δex7/8/Δex7/8 Cerebellar Cells and Reduces Neuronal Vulnerability to Cell Death via IMPase Inhibition. J. Neurochem. 2011, 116, 659–668. [Google Scholar] [CrossRef]
- Vicencio, J.; Ortiz, C.; Criollo, A.; Jones, A.W.E.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The Inositol 1,4,5-Trisphosphate Receptor Regulates Autophagy through Its Interaction with Beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Billes, V.; Komlós, M.; Hotzi, B.; Manzéger, A.; Tarnóci, A.; Papp, D.; Szikszai, F.; Szinyákovics, J.; Rácz, Á.; et al. The Small Molecule AUTEN-99 (Autophagy Enhancer-99) Prevents the Progression of Neurodegenerative Symptoms. Sci. Rep. 2017, 7, 42014. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Szinyákovics, J.; Billes, V.; Murányi, G.; Varga, V.B.; Bjelik, A.; Légrádi, Á.; Szabó, M.; Sándor, S.; Kubinyi, E.; et al. A Conserved MTMR Lipid Phosphatase Increasingly Suppresses Autophagy in Brain Neurons during Aging. Sci. Rep. 2022, 12, 21817. [Google Scholar] [CrossRef]
- Zhu, Z.; Liu, L.F.; Su, C.F.; Liu, J.; Tong, B.C.-K.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Guan, X.-J.; Kan, Y.-X.; et al. Corynoxine B Derivative CB6 Prevents Parkinsonian Toxicity in Mice by Inducing PIK3C3 Complex-Dependent Autophagy. Acta Pharmacol. Sin. 2022, 43, 2511–2526. [Google Scholar] [CrossRef]
- Chen, L.-L.; Song, J.-X.; Lu, J.-H.; Yuan, Z.-W.; Liu, L.-F.; Durairajan, S.S.K.; Li, M. Corynoxine, a Natural Autophagy Enhancer, Promotes the Clearance of Alpha-Synuclein via Akt/mTOR Pathway. J. Neuroimmune Pharmacol. 2014, 9, 380–387. [Google Scholar] [CrossRef]
- Zhang, P.; Zheng, Z.; Ling, L.; Yang, X.; Zhang, N.; Wang, X.; Hu, M.; Xia, Y.; Ma, Y.; Yang, H.; et al. w09, a Novel Autophagy Enhancer, Induces Autophagy-Dependent Cell Apoptosis via Activation of the EGFR-Mediated RAS-RAF1-MAP2K-MAPK1/3 Pathway. Autophagy 2017, 13, 1093–1112. [Google Scholar] [CrossRef]
- Briz, V.; Hsu, Y.-T.; Li, Y.; Lee, E.; Bi, X.; Baudry, M. Calpain-2-Mediated PTEN Degradation Contributes to BDNF-Induced Stimulation of Dendritic Protein Synthesis. J. Neurosci. 2013, 33, 4317–4328. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, T.; Sun, L.; Luo, Y.; Liu, D.-H.; Xie, S.-T.; Song, X.-Y.; Wang, G.-F.; Chen, X.-L.; Zhou, B.-C.; et al. Calpain, Atg5 and Bak Play Important Roles in the Crosstalk between Apoptosis and Autophagy Induced by Influx of Extracellular Calcium. Apoptosis 2013, 18, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Garg, N.; Singh, T.G.; Kaur, A.; Thapa, K. Calpain Inhibitors as Potential Therapeutic Modulators in Neurodegenerative Diseases. Neurochem. Res. 2022, 47, 1125–1149. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Liu, E.; Wang, J.; Chen, Y.; Yu, J.; Baker, A.J. Calpain Inhibitor MDL-28170 Reduces the Functional and Structural Deterioration of Corpus Callosum Following Fluid Percussion Injury. J. Neurotrauma 2007, 24, 960–978. [Google Scholar] [CrossRef]
- Çolak, A.; Karaoğlan, A.; Kaya, M.; Sağmanligil, A.; Akdemir, O.; Şahan, E.; Çelik, Ö. Calpain Inhibitor AK 295 Inhibits Calpain-Induced Apoptosis and Improves Neurologic Function After Traumatic Spinal Cord Injury in Rats. Neurocirugía 2009, 20, 245–254. [Google Scholar] [CrossRef]
- Moldoveanu, T.; Campbell, R.L.; Cuerrier, D.; Davies, P.L. Crystal Structures of Calpain–E64 and –Leupeptin Inhibitor Complexes Reveal Mobile Loops Gating the Active Site. J. Mol. Biol. 2004, 343, 1313–1326. [Google Scholar] [CrossRef]
- Peng, S.; Kuang, Z.; Zhang, Y.; Xu, H.; Cheng, Q. The Protective Effects and Potential Mechanism of Calpain Inhibitor Calpeptin Against Focal Cerebral Ischemia–Reperfusion Injury in Rats. Mol. Biol. Rep. 2011, 38, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Joudeh, J.; Claxton, D. Obatoclax Mesylate: Pharmacology and Potential for Therapy of Hematological Neoplasms. Expert Opin. Investig. Drugs 2012, 21, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.-Z.; Ma, B.; Liang, Y.-J.; Liu, H.-C.; Zhang, T.-H.; Zheng, G.S.; Su, Y.-X.; Liao, G.-Q. Obatoclax Induces Beclin 1- and ATG5-Dependent Apoptosis and Autophagy in Adenoid Cystic Carcinoma Cells. Oral Dis. 2015, 21, 470–477. [Google Scholar] [CrossRef]
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15-070) Triggers Necroptosis by Promoting the Assembly of the Necrosome on Autophagosomal Membranes. Cell Death Differ. 2013, 20, 1161–1173. [Google Scholar] [CrossRef]
- Koehler, B.C.; Jassowicz, A.; Scherr, A.L.; Lorenz, S.; Radhakrishnan, P.; Kautz, N.; Elssner, C.; Weiss, J.; Jaeger, D.; Schneider, M.; et al. Pan-Bcl-2 Inhibitor Obatoclax is a Potent Late Stage Autophagy Inhibitor in Colorectal Cancer Cells Independent of Canonical Autophagy Signaling. BMC Cancer 2015, 15, 919. [Google Scholar] [CrossRef]
- Fulda, S. Autophagy in Cancer Therapy. Front. Oncol. 2017, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The Effects of Metformin on Autophagy. Biomed. Pharmacother. 2021, 137, 111286. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Lim, Y.M.; Kim, K.H.; Jeon, Y.E.; Park, K.; Kim, J.; Hwang, H.Y.; Lee, D.J.; Pagire, H.; Kwon, H.J.; et al. A Novel Autophagy Enhancer as a Therapeutic Agent Against Metabolic Syndrome and Diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef]
- Kim, J.; Park, K.; Kim, M.J.; Lim, H.; Kim, K.H.; Kim, S.W.; Lee, E.S.; Kim, H.; Kim, S.J.; Hur, K.Y.; et al. An Autophagy Enhancer Ameliorates Diabetes of Human IAPP-Transgenic Mice Through Clearance of Amyloidogenic Oligomer. Nat. Commun. 2021, 12, 183. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | NCT Number | Phase of Clinical Trial | Drugs That Were Used in Combination | Subtypes of Disease |
|---|---|---|---|---|
| Small cell lung cancer | NCT01575782 | I-III (Canceled) | -----//----- | -----//----- |
| Breast cancer | NCT02333890 | II | -----//----- | Breast cancer, Invasive breast cancer |
| Metastatic breast cancer | NCT01446016 | II (Completed) | (a) Chloroquine + Paclitaxel (b) Chloroquine + Docetaxel (c) Chloroquine + Abraxane (d) Chloroquine + Ixabepilon | Breast neoplasia, Breast cancer |
| Stage 4 small cell lung cancer | NCT00969306 | I (Canceled) | -----//----- | -----//----- |
| Dense tumors | NCT02071537 | I (Completed) | (a) Chloroquine + Carboplatin (b) Chloroquine + Gemcitabine | -----//----- |
| Pancreatic cancer | NCT01777477 | I (Completed) | Chloroquine + Gemcitabine | ----//----- |
| Intraductal carcinoma | NCT01023477 | I-II (Completed) | -----//----- | Intraductal non-filtering carcinoma, Ductal carcinoma |
| Dense tumors with IDH1/2 mutation | NCT02496741 | I-II | Chloroquine + Metformin | Glioma, Cholangiocarcinoma, Chondrosarcoma |
| Brain metastases from dense tumors | NCT01894633 | II (Canceled) | Chloroquine + radiotherapy | -----//----- |
| Brain metastases with IDO2 genetic status | NCT01727531 | (Canceled) | Chloroquine + radiotherapy | -----//----- |
| Brain cancer | NCT04397679 | Recruitment for clinical trials | Chloroquine, intensity modulated radiation therapy (IMRT), Temolozomide, tumor treating field therapy (TTF) | Glioblastoma, Gliosarcoma |
| Recurrent and refractory multiple myeloma | NCT01438177 | II (Completed) | Chloroquine + Bortezomib + Cyclophosphamide | Multiple myelomas |
| Melanoma | NCT01469455 | I | DT01 + Chloroquine + radiotherapy | -----//----- |
| Glioblastoma | NCT00224978 | III (Completed) | -----//----- | Glioblastoma multiform |
| Glioblastoma | NCT02432417 | II (Recruitment for clinical trials) | Chloroquine + Temolozomide + radiotherapy | -----//----- |
| Drug | Group | Targets | Application in Medicine | Clinical Trial |
|---|---|---|---|---|
| Wortmannin | PI3K inhibitor | PI3KC1, PI3KC2, PI3KC3 | -----//----- | -----//----- |
| 3-Methyladenine (3-MA) | PI3K inhibitor | PI3KC1, PI3KC3 | -----//----- | -----//----- |
| LY294002 | PI3K inhibitor | PI3KC1, p53, LC3, Caspase-3, PUMA | -----//----- | NCT02337309 (SF1126 for Patients with Neuroblastoma) |
| ZSTK474 | PI3K inhibitor | PI3KC1 | -----//----- | NCT01682473 (A Study of ZSTK474 in Japanese Patients with Advanced Solid Malignancies) |
| GSK-2126458 | PI3K inhibitor | PI3KC1, PI3KC3 | -----//----- | NCT00972686 (Dose-Escalation Study of GSK2126458), NCT01725139 (A Proof of Mechanism Study with GSK2126458 in Patients with Idiopathic Pulmonary Fibrosis), NCT01248858 (Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics, and Clinical Activity of GSK2126458 and GSK1120212 Combination Therapy in Subjects with Advanced Solid Tumors). |
| PT210 | PI3K inhibitor | PI3KC1, PI3KC3 | -----//----- | -----//----- |
| Spautin-1 | PI3KC3-C1 (Vps34) inhibitor | USP10, USP13 | -----//----- | -----//----- |
| VPS34-IN-1 | PI3KC3-C1 (Vps34) inhibitor | Vps34 | -----//----- | -----//----- |
| Compound-31 | PI3KC3-C1 (Vps34) inhibitor | Vps34 | -----//----- | -----//----- |
| SAR405 | PI3KC3-C1 (Vps34) inhibitor | Vps34 | -----//----- | -----//----- |
| PIK-III | PI3KC3-C1 (Vps34) inhibitor | Vps34 | -----//----- | -----//----- |
| MRT68921 | ULK1 inhibitor | ULK1, Atg13 | -----//----- | -----//----- |
| Bafilomycin A1 | vATPase inhibitor | vATPase | -----//----- | NCT04389580 (Combination Therapy with Isotretinoin and Tamoxifen Expected to Provide Complete Protection Against Severe Acute Respiratory Syndrome Coronavirus (Combination) NCT05711810 (Medicine-induced Cardiac Hemodialysis on COVID-19) |
| Lys05 | Lysosome | Lysosome | -----//----- | -----//----- |
| ARN 5187 | Lysosome | Lysosome, REV-ERBβ | -----//----- | -----//----- |
| Lucanthone, Hycanthone | Lysosome | Lysosomal membrane, APE1 | Helmints | NCT01587144 (Safety and Efficacy Study of Lucanthone When Used in Combination with Temozolomide (TMZ) and Radiation to Treat Glioblastoma Multiforme) NCT02014545 (Evaluation of Lucanthone to Whole Brain Radiation Therapy in Patients with Brain Metastases From Non-Small Cell Lung Cancer) |
| Rapamycin | mTOR inhibitor | mTOR | Immunosuppressant | NCT00411788 (Study of Rapamycin and Trastuzumab for Patients With HER-2 Receptor-Positive Metastatic Breast Cancer) NCT00409994 (Safety Study of Rapamycin Administered Before and During Radiotherapy to Treat Rectum Cancer) |
| Temsirolimus | mTOR inhibitor | mTOR | Renal cell carcinoma | NCT01827943 (Phase II Evaluating Efficacy of Temsirolimus in 2 Line Therapy for Patients with Advanced Bladder Cancer) NCT00919035 (Single Agent Temsirolimus in Chemotherapy-naïve Castration-Resistant Prostate Cancer Patients) |
| Everolimus | mTOR inhibitor | mTOR | Immunosuppressant, Kidney cancer | NCT02479490 (Prednisone Plus Everolimus in Patients with Metastatic Renal Cell Cancer After Failure of VEGFR -TKI) |
| Brefeldin A | ER-stress inductor | GBF1 | -----//----- | NCT03044509 (Diagnosis of Tuberculosis in Swiss Children) |
| Bortezomib | ER-stress inductor | 26S proteasome | Multiple myeloma, mantle cell lymphoma | NCT01328236 (Bortezomib in Combination with Liposomal Doxorubicin and Dexamethasone to Treat Plasma Cell Leukemia) NCT00183937 (Study of Bortezomib and Docetaxel for Patients with Hormone Refractory Prostate Cancer) |
| NPI-0052 | ER-stress inductor | 26S proteasome | -----//----- | NCT00461045 (Phase 2 Clinical Trial of NPI-0052 in Patients with Relapsed or Relapsed/Refractory Multiple Myeloma) NCT00396864 (Phase 1 Clinical Trial of NPI-0052 in Patients with Advanced Solid Tumor Malignancies or Refractory Lymphoma) |
| L-690/330 | IMPase inhibitors | IMPase | -----//----- | -----//----- |
| lithium carbonate, lithium chloride | IMPase inhibitors | IMPase | Bipolar disorder major depressive disorder | NCT02862210 (Low-Dose Lithium for the Treatment of Behavioral Symptoms in Frontotemporal Dementia) NCT01108068 (Trial of Lithium Carbonate for Treatment of Osteoporosis-pseudoglioma Syndrome) NCT01096082 (Safety and Efficacy of Lithium Carbonate in Patients with Spinocerebellar Ataxia Type 3) |
| Carbamazepine | IMPase inhibitors | Inositol | Epilepsy | NCT00203567 (Carbamazepine Extended-Release for the Treatment of Bipolar Depression) NCT01379469 (Carbamazepine in Severe Liver Disease Due to Alpha-1 Antitrypsin Deficiency) |
| Valproic acid | IMPase inhibitors | Inositol | Epilepsy | NCT04940572 (Efficacy Study of Daily Administration of VPA in Patients Affected by Wolfram Syndrome) NCT03919292 (Neratinib + Valproate in Advanced Solid Tumors, w/Expansion Cohort in Ras-Mutated Ca) |
| AUTEN99 AUTEN67 | MTMR14 inhibitor | MTMR14/Jumpy | -----//----- | -----//----- |
| Corynoxine | mTOR independent inducers | PI3KC3, mTOR, AKT, p70S6K | -----//----- | -----//----- |
| W09 | mTOR independent inducers | PARP, P62 | -----//----- | -----//----- |
| MDL-28170 | Calpains inhibitor | Calpains | -----//----- | -----//----- |
| AK-295 | Calpains inhibitor | Calpains | -----//----- | -----//----- |
| Leupeptin | Calpains inhibitor | Calpain 5, cathepsins | -----//----- | -----//----- |
| Obatoclax | Bcl2 inhibitor | Bcl-XL, Mcl-1, Bcl-2 | Various types of cancer | NCT00918931 (Obatoclax for Systemic Mastocytosis) NCT00684918 (Study of Obatoclax in Previously Untreated Acute Myeloid Leukemia) |
| Venetoclax | Bcl2 inhibitor | Bcl-2 | Chronic lymphocytic leukemia | NCT04501939 (Cirmtuzumab Consolidation for Treatment of Patients with Detectable CLL on Venetoclax) NCT04171791 (A Study of ABT-199 (Venetoclax) for Cutaneous T Cell Lymphoma) |
| Metformin | AMPK activator | AMPK | Type 2 Diabetes | NCT02978547 (The Effects of Neoadjuvant Metformin on Tumor Cell Proliferation and Tumor Progression in Pancreatic Ductal Adenocarcinoma) NCT05607316 (Evaluating the Efficacy and Safety of Metformin in Vitiligo) NCT05426044 (Metformin as a Neuroprotective Therapy for Glaucoma—A Randomized Controlled Trial) |
| RSVA 314/405 | AMPK activator | AMPK | -----//----- | -----//----- |
| MSL-7 | TFEB activator | TFEB | -----//----- | -----//----- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kench, U.; Sologova, S.; Smolyarchuk, E.; Prassolov, V.; Spirin, P. Pharmaceutical Agents for Targeting Autophagy and Their Applications in Clinics. Pharmaceuticals 2024, 17, 1355. https://doi.org/10.3390/ph17101355
Kench U, Sologova S, Smolyarchuk E, Prassolov V, Spirin P. Pharmaceutical Agents for Targeting Autophagy and Their Applications in Clinics. Pharmaceuticals. 2024; 17(10):1355. https://doi.org/10.3390/ph17101355
Chicago/Turabian StyleKench, Ulash, Susanna Sologova, Elena Smolyarchuk, Vladimir Prassolov, and Pavel Spirin. 2024. "Pharmaceutical Agents for Targeting Autophagy and Their Applications in Clinics" Pharmaceuticals 17, no. 10: 1355. https://doi.org/10.3390/ph17101355
APA StyleKench, U., Sologova, S., Smolyarchuk, E., Prassolov, V., & Spirin, P. (2024). Pharmaceutical Agents for Targeting Autophagy and Their Applications in Clinics. Pharmaceuticals, 17(10), 1355. https://doi.org/10.3390/ph17101355