
Predicting Structural Consequences of Antibody Light Chain N-Glycosylation in AL Amyloidosis

 , , and
, , and
Abstract

1. Introduction
2. Results
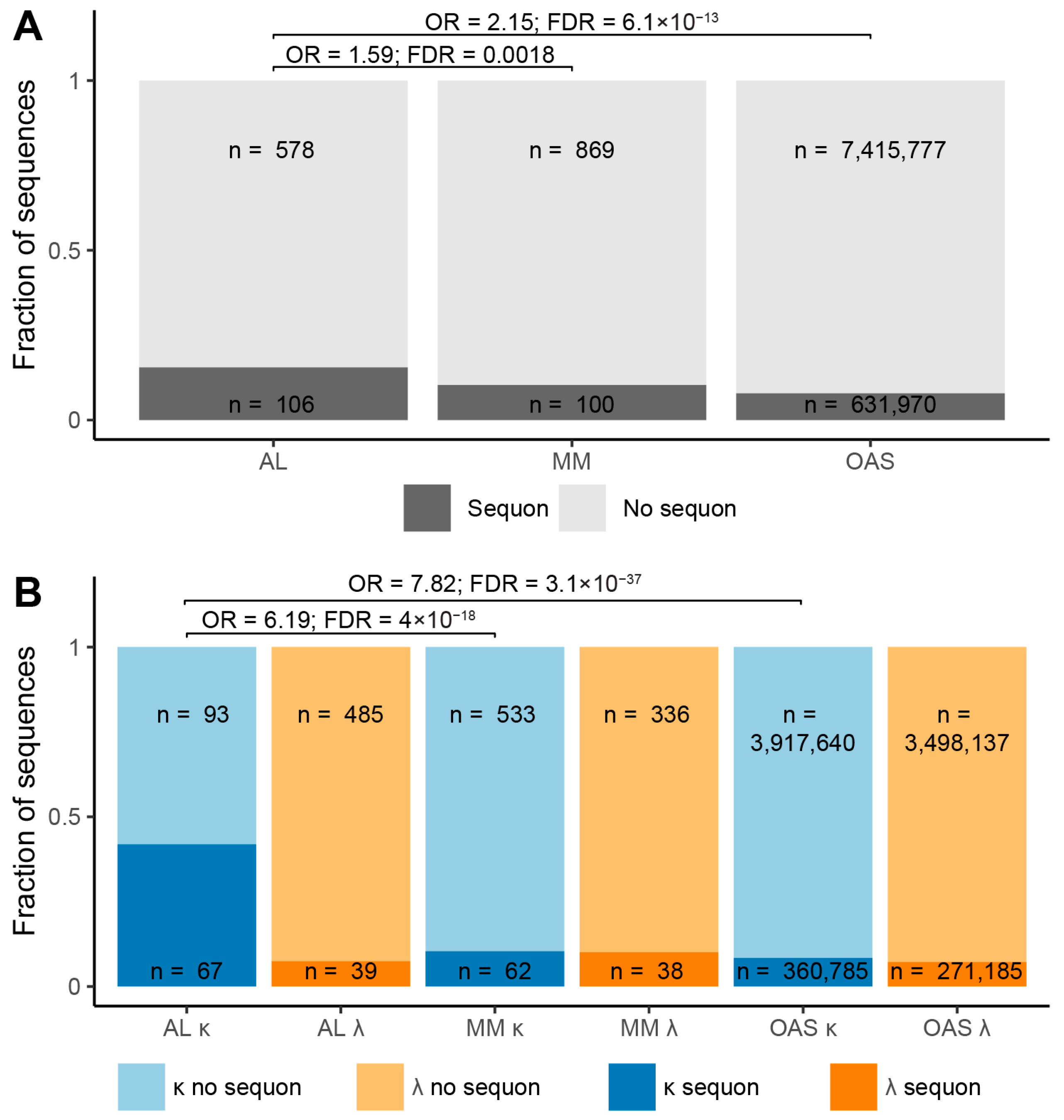
2.1. N-Glycosylation Sequons Are Enriched in AL-Associated κ LCs
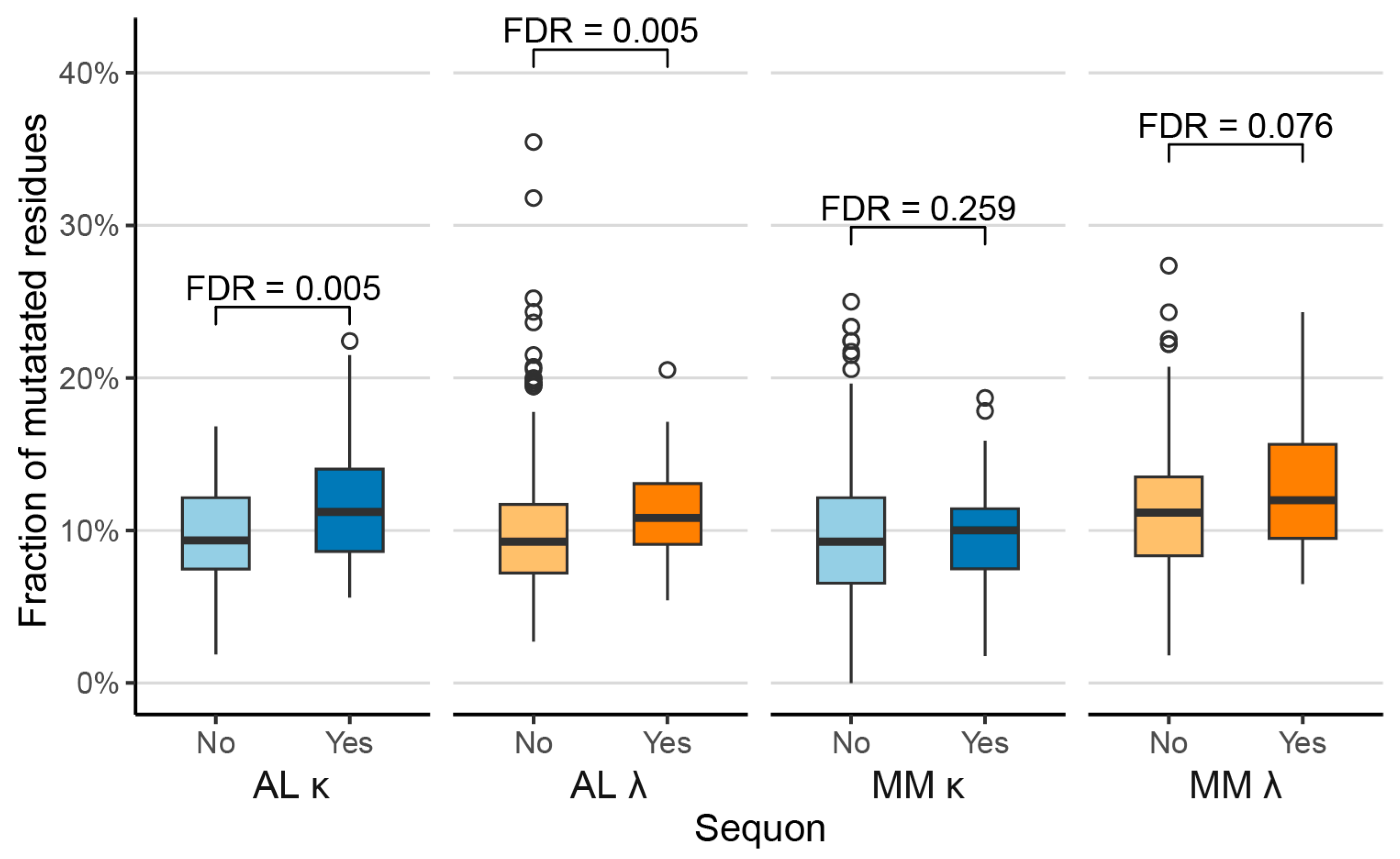
2.2. Sequons Are Associated with Increased Proportions of Mutated Residues
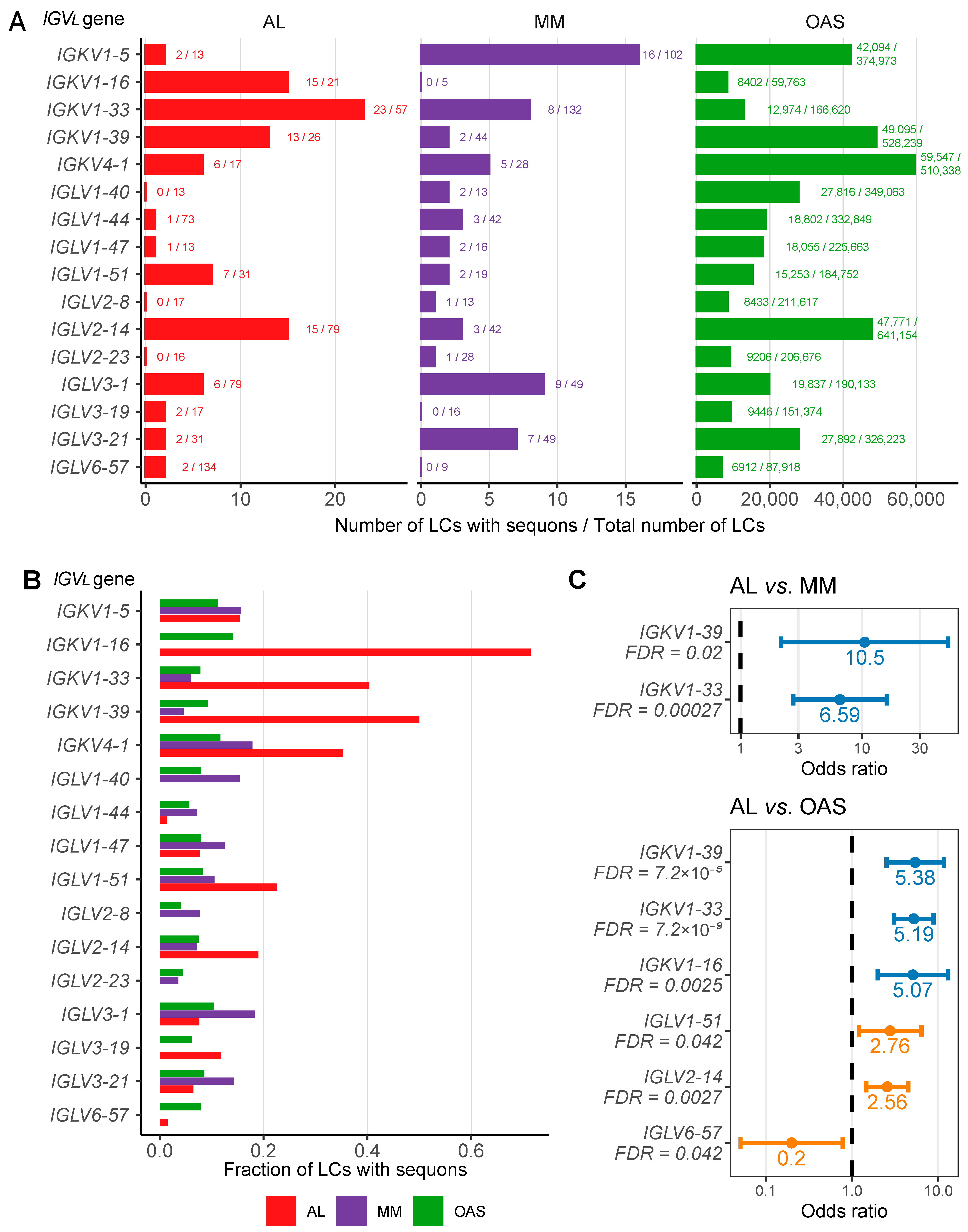
2.3. AL LCs Derived from a Subset of Precursor Genes Are Enriched in Sequons
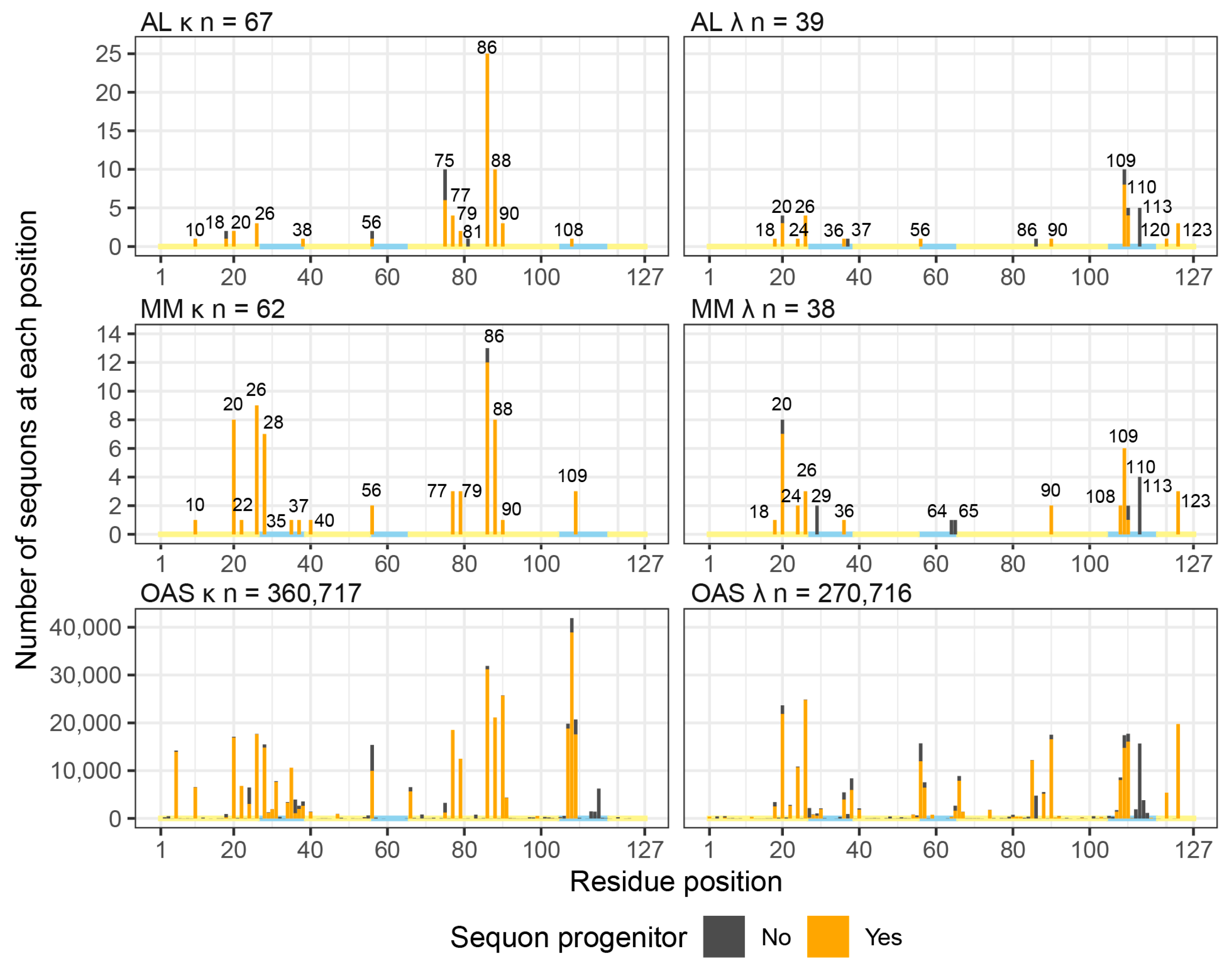
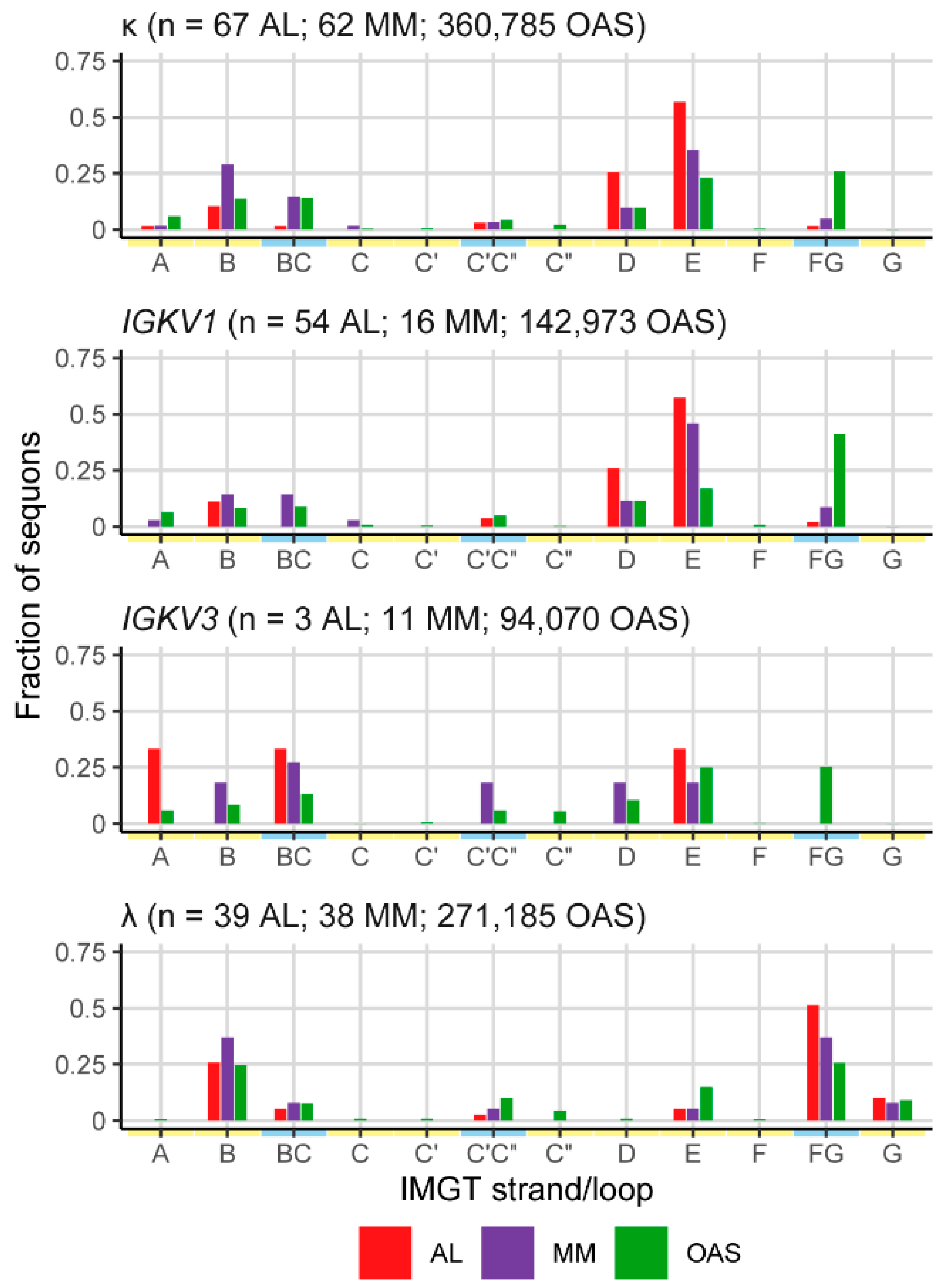
2.4. Sequons in AL and MM LCs Occur in Similar Positions
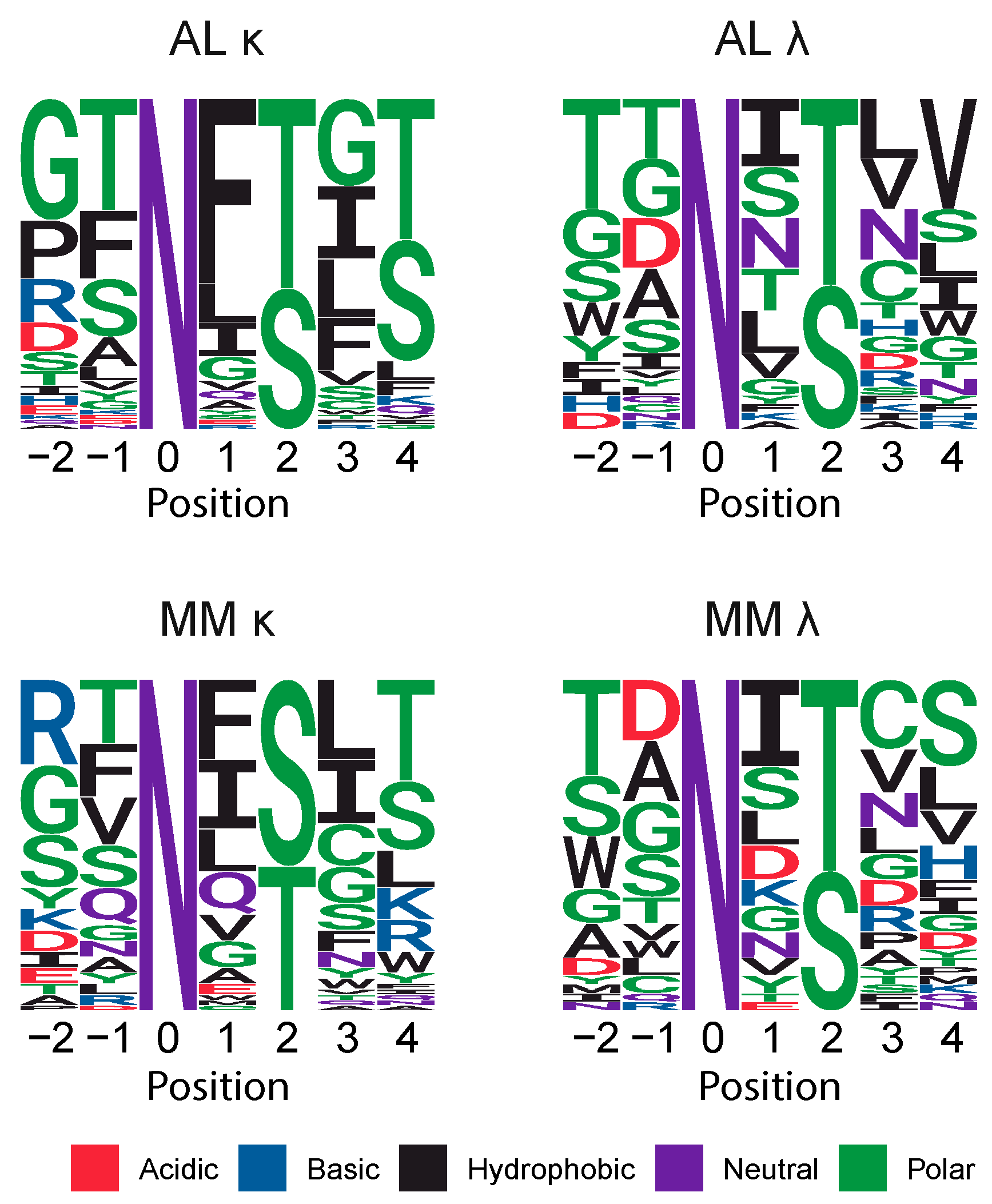
2.5. Sequons Occur in Similar Sequence Contexts
2.6. NxC Sequons Occur in LC CDRs
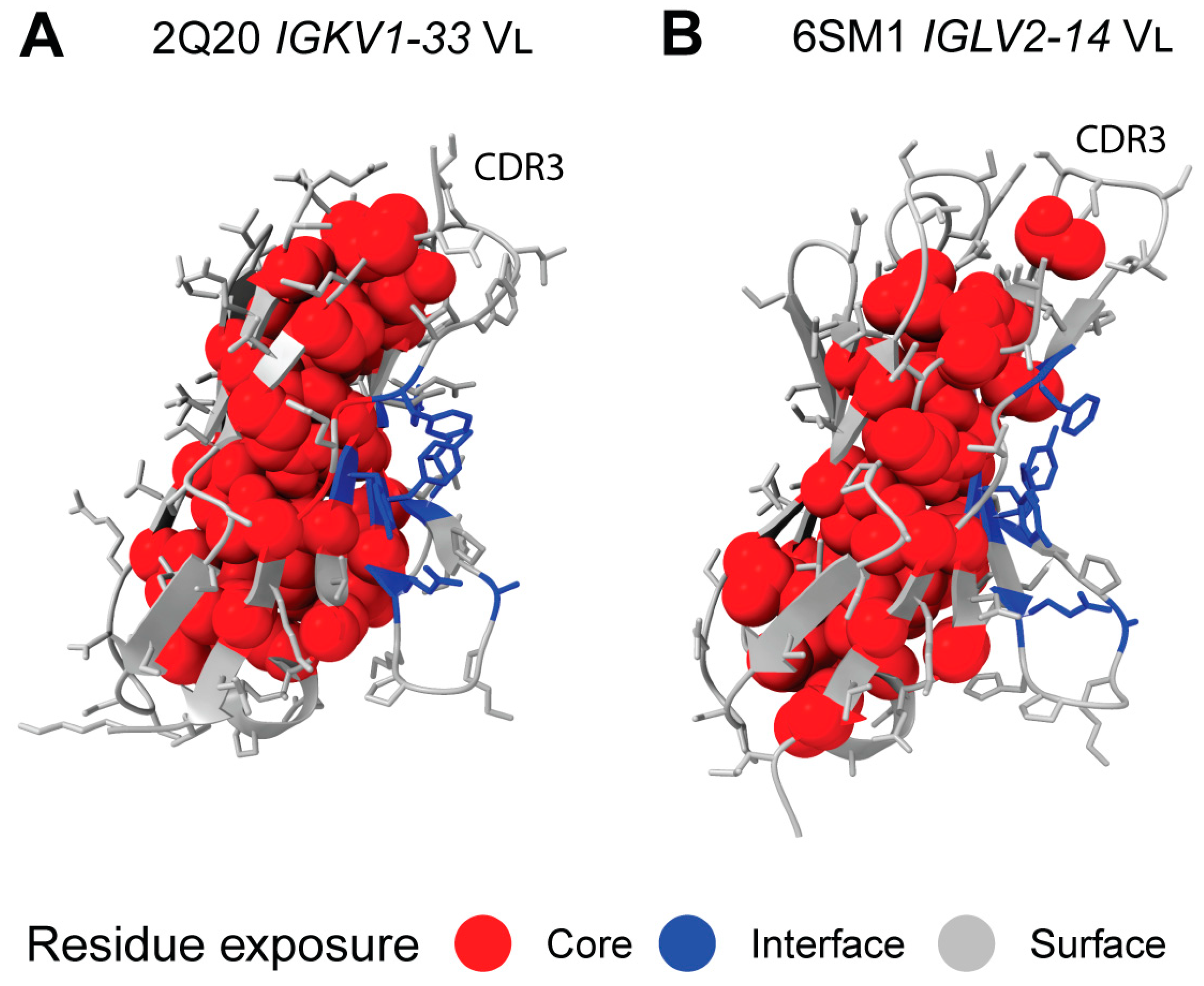
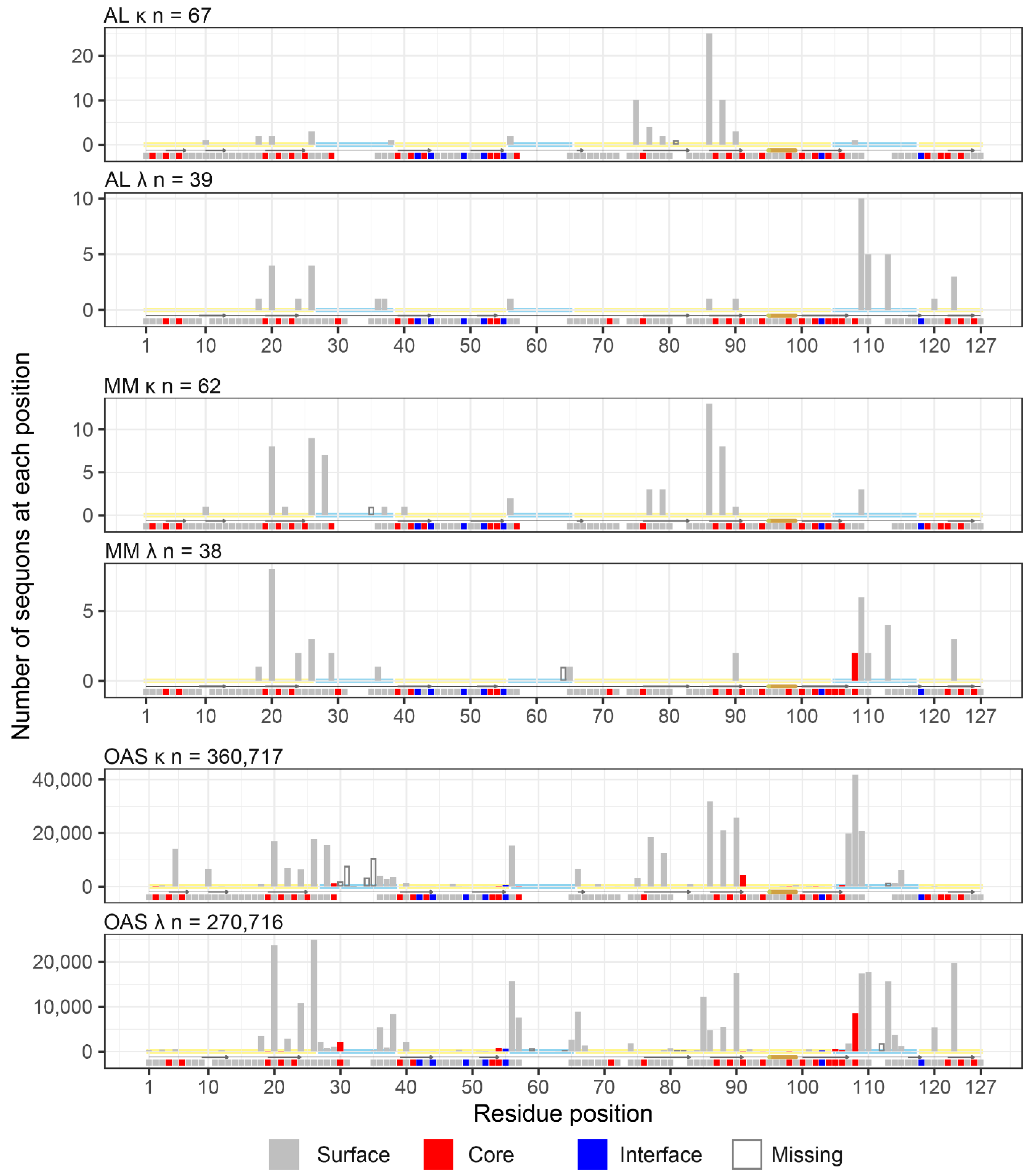
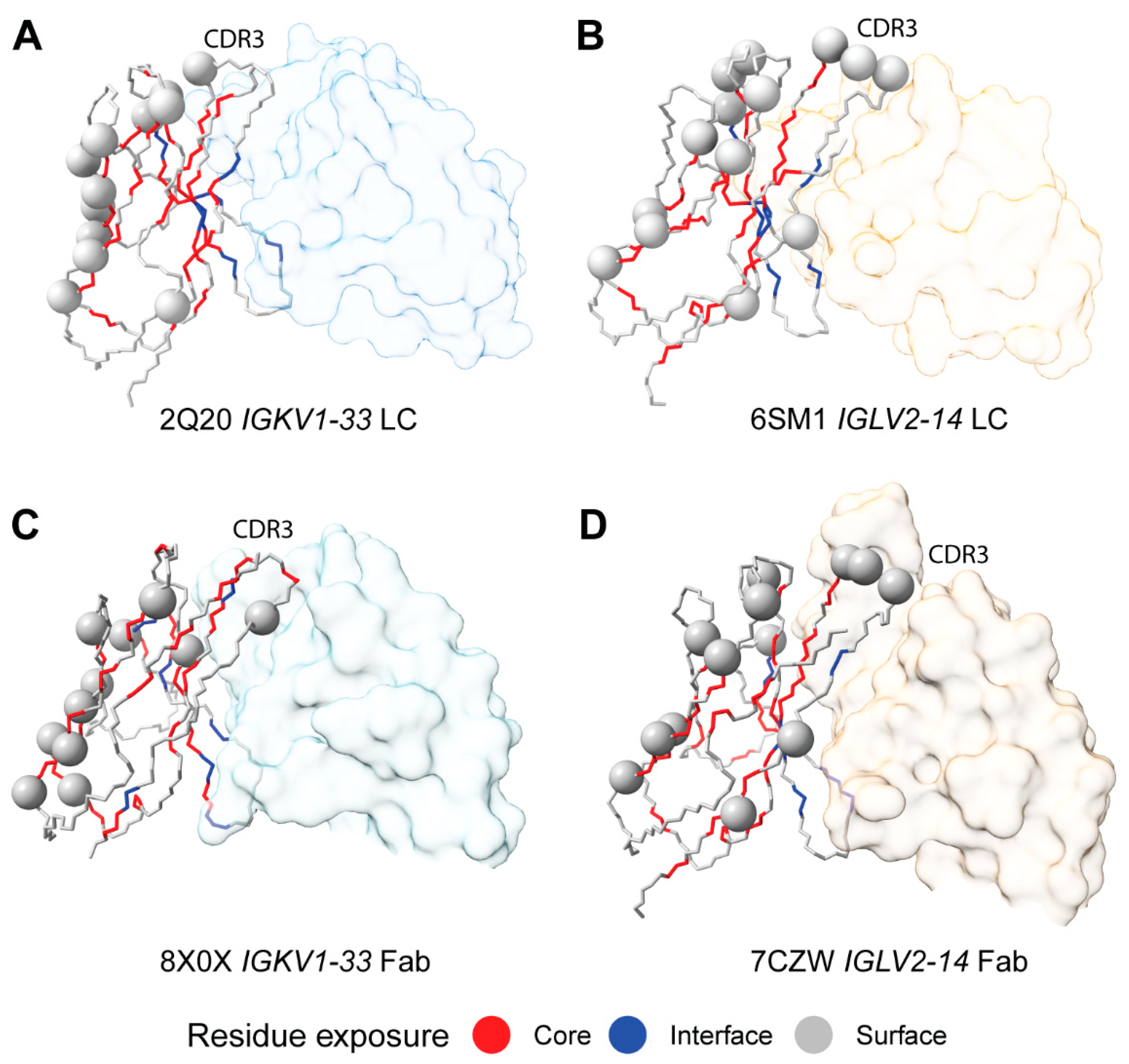
2.7. Sequon Positions Are Compatible with Native Antibody Structures
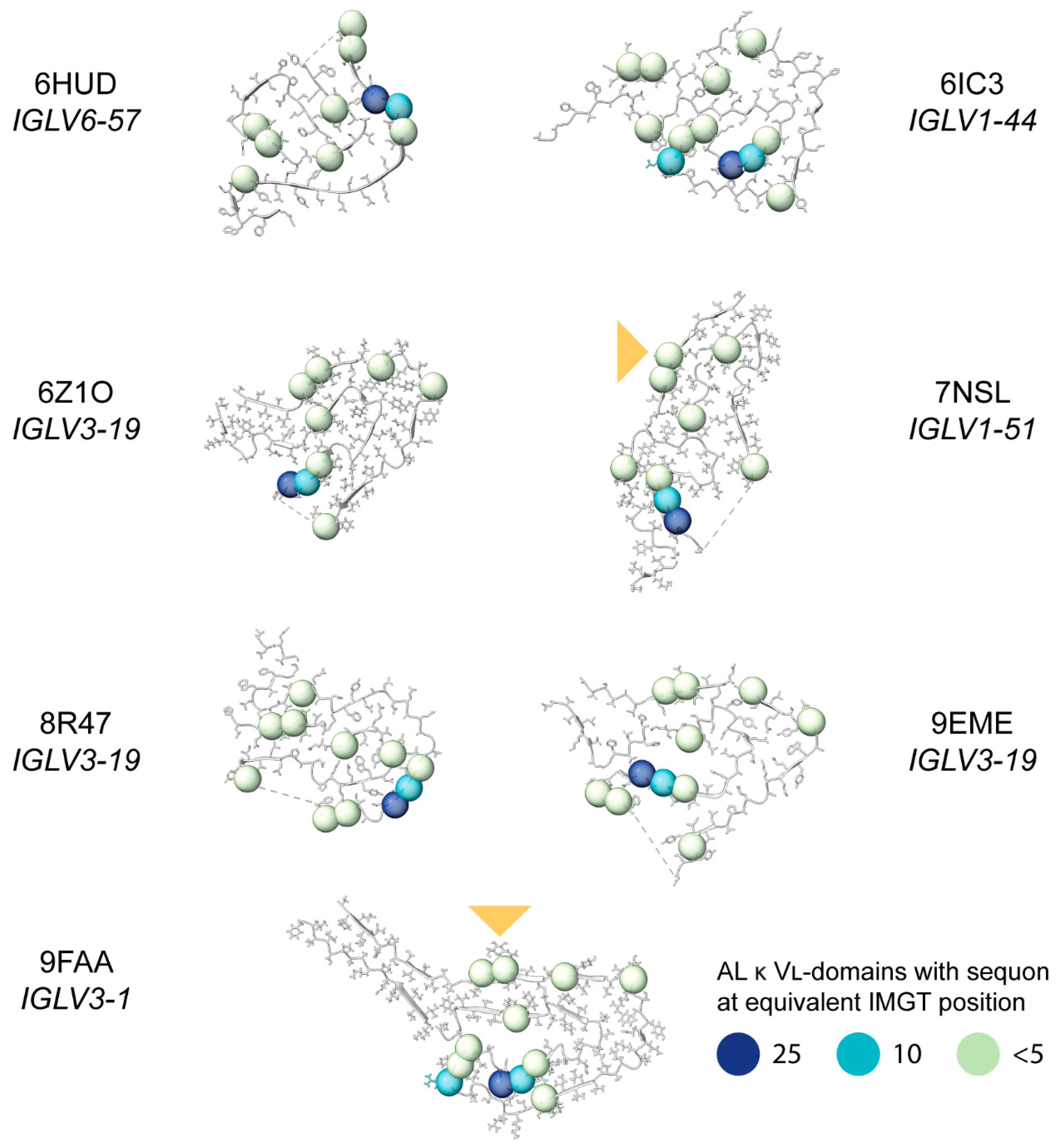
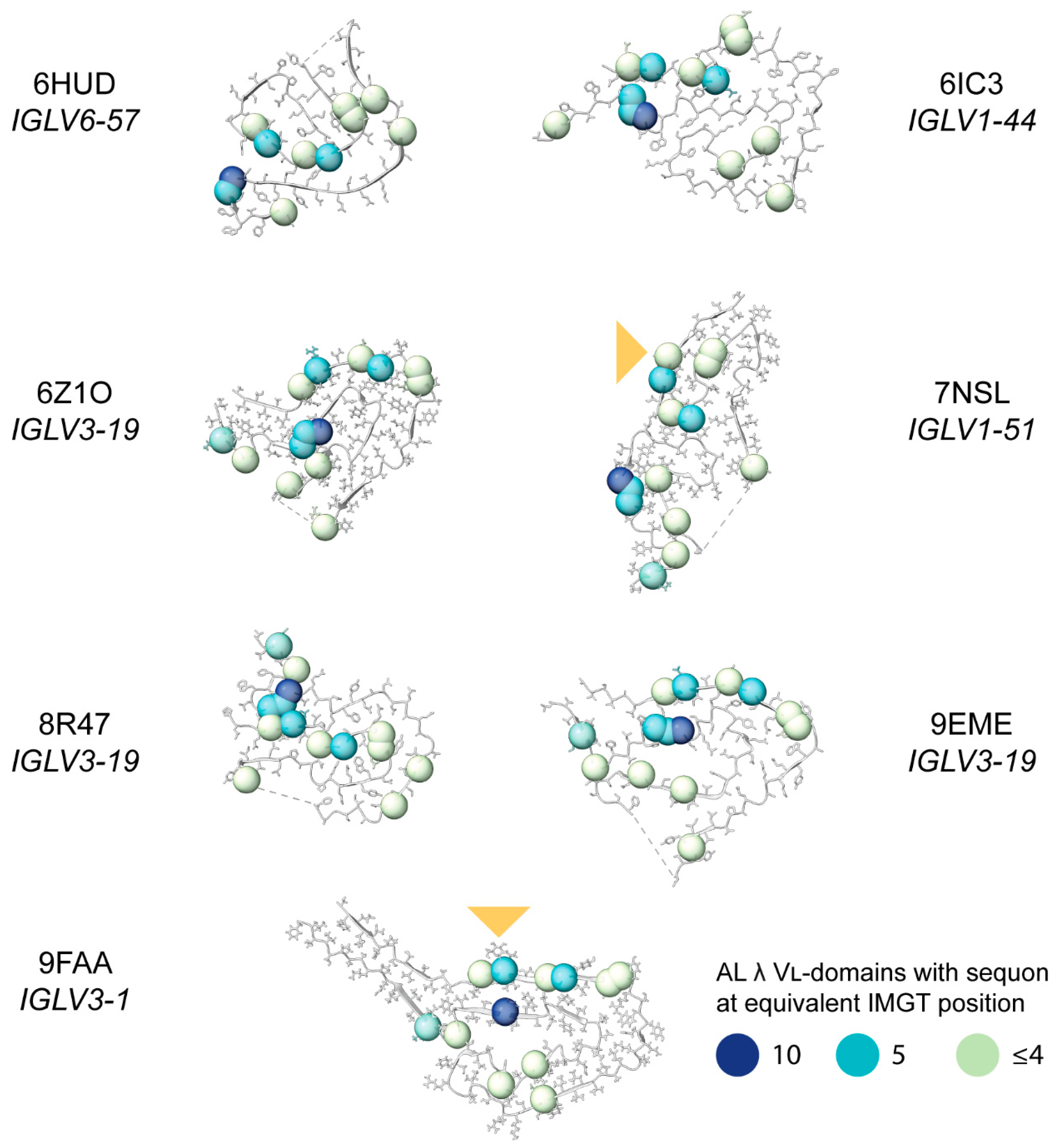
2.8. Sequon Positions Differ in Their Environment Among AL Fibril Structures
3. Discussion
4. Materials and Methods
4.1. LC Sequences
4.2. Statistics
4.3. Structural Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fändrich, M.; et al. Half a Century of Amyloids: Past, Present and Future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Mejía, L.M.; Ramirez-Alvarado, M. Systemic Amyloidoses. Annu. Rev. Biochem. 2013, 82, 745–774. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Eisenberg, D.S.; Fändrich, M.; McPhail, E.D.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Westermark, P. Amyloid Nomenclature 2024: Update, Novel Proteins, and Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2024. [Google Scholar] [CrossRef] [PubMed]
- Radamaker, L.; Karimi-Farsijani, S.; Andreotti, G.; Baur, J.; Neumann, M.; Schreiner, S.; Berghaus, N.; Motika, R.; Haupt, C.; Walther, P.; et al. Role of Mutations and Post-Translational Modifications in Systemic AL Amyloidosis Studied by Cryo-EM. Nat. Commun. 2021, 12, 6434. [Google Scholar] [CrossRef]
- Schulte, T.; Chaves-Sanjuan, A.; Speranzini, V.; Sicking, K.; Milazzo, M.; Mazzini, G.; Rognoni, P.; Caminito, S.; Milani, P.; Marabelli, C.; et al. Helical Superstructures between Amyloid and Collagen in Cardiac Fibrils from a Patient with AL Amyloidosis. Nat. Commun. 2024, 15, 6359. [Google Scholar] [CrossRef]
- Braakman, I.; Bulleid, N.J. Protein Folding and Modification in the Mammalian Endoplasmic Reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Sanchorawala, V. Systemic Light Chain Amyloidosis. N. Engl. J. Med. 2024, 390, 2295–2307. [Google Scholar] [CrossRef]
- Del Pozo-Yauner, L.; Herrera, G.A.; Perez Carreon, J.I.; Turbat-Herrera, E.A.; Rodriguez-Alvarez, F.J.; Ruiz Zamora, R.A. Role of the Mechanisms for Antibody Repertoire Diversification in Monoclonal Light Chain Deposition Disorders: When a Friend Becomes Foe. Front. Immunol. 2023, 14, 1203425. [Google Scholar] [CrossRef]
- Morgan, G.J.; Wall, J.S. The Process of Amyloid Formation Due to Monoclonal Immunoglobulins. Hematol. Oncol. Clin. N. Am. 2020, 34, 1041–1054. [Google Scholar] [CrossRef]
- Feige, M.J.; Hendershot, L.M.; Buchner, J. How Antibodies Fold. Trends Biochem. Sci. 2010, 35, 189–198. [Google Scholar] [CrossRef]
- Desikan, K.R.; Dhodapkar, M.V.; Hough, A.; Waldron, T.; Jagannath, S.; Siegel, D.; Barlogie, B.; Tricot, G. Incidence and Impact of Light Chain Associated (AL) Amyloidosis on the Prognosis of Patients with Multiple Myeloma Treated with Autologous Transplantation. Leuk. Lymphoma 1997, 27, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Madan, S.; Dispenzieri, A.; Lacy, M.Q.; Buadi, F.; Hayman, S.R.; Zeldenrust, S.R.; Rajkumar, S.V.; Gertz, M.A.; Kumar, S.K. Clinical Features and Treatment Response of Light Chain (AL) Amyloidosis Diagnosed in Patients with Previous Diagnosis of Multiple Myeloma. Mayo Clin. Proc. 2010, 85, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Bodi, K.; Prokaeva, T.; Spencer, B.; Eberhard, M.; Connors, L.H.; Seldin, D.C. AL-Base: A Visual Platform Analysis Tool for the Study of Amyloidogenic Immunoglobulin Light Chain Sequences. Amyloid 2009, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying Immunoglobulin Gene Usage in Systemic and Localized Immunoglobulin Light-Chain Amyloidosis by Mass Spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Morgan, G.; Nau, A.N.; Wong, S.; Spencer, B.H.; Shen, Y.; Hua, A.; Bullard, M.J.; Sanchorawala, V.; Prokaeva, T. An Updated AL-Base Reveals Ranked Enrichment of Immunoglobulin Light Chain Variable Genes in AL Amyloidosis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Radamaker, L.; Baur, J.; Huhn, S.; Haupt, C.; Hegenbart, U.; Schönland, S.; Bansal, A.; Schmidt, M.; Fändrich, M. Cryo-EM Reveals Structural Breaks in a Patient-Derived Amyloid Fibril from Systemic AL Amyloidosis. Nat. Commun. 2021, 12, 875. [Google Scholar] [CrossRef]
- Radamaker, L.; Lin, Y.-H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schönland, S.O.; Fritz, G.; Schmidt, M.; Fändrich, M. Cryo-EM Structure of a Light Chain-Derived Amyloid Fibril from a Patient with Systemic AL Amyloidosis. Nat. Commun. 2019, 10, 1103. [Google Scholar] [CrossRef]
- Karimi-Farsijani, S.; Pfeiffer, P.B.; Banerjee, S.; Baur, J.; Kuhn, L.; Kupfer, N.; Hegenbart, U.; Schönland, S.O.; Wiese, S.; Haupt, C.; et al. Light Chain Mutations Contribute to Defining the Fibril Morphology in Systemic AL Amyloidosis. Nat. Commun. 2024, 15, 5121. [Google Scholar] [CrossRef]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM Structure of Cardiac Amyloid Fibrils from an Immunoglobulin Light Chain AL Amyloidosis Patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef]
- Puri, S.; Schulte, T.; Chaves-Sanjuan, A.; Mazzini, G.; Caminito, S.; Pappone, C.; Anastasia, L.; Milani, P.; Merlini, G.; Bolognesi, M.; et al. The Cryo-EM Structure of Renal Amyloid Fibril Suggests Structurally Homogeneous Multiorgan Aggregation in AL Amyloidosis. J. Mol. Biol. 2023, 435, 168215. [Google Scholar] [CrossRef]
- Morgan, G.J. Transient Disorder along Pathways to Amyloid. Biophys. Chem. 2022, 281, 106711. [Google Scholar] [CrossRef] [PubMed]
- Kazman, P.; Vielberg, M.-T.; Pulido Cendales, M.D.; Hunziger, L.; Weber, B.; Hegenbart, U.; Zacharias, M.; Köhler, R.; Schönland, S.; Groll, M.; et al. Fatal Amyloid Formation in a Patient’s Antibody Light Chain Is Caused by a Single Point Mutation. Elife 2020, 9, e52300. [Google Scholar] [CrossRef] [PubMed]
- Baden, E.M.; Randles, E.G.; Aboagye, A.K.; Thompson, J.R.; Ramirez-Alvarado, M. Structural Insights into the Role of Mutations in Amyloidogenesis. J. Biol. Chem. 2008, 283, 30950–30956. [Google Scholar] [CrossRef] [PubMed]
- Hurle, M.R.; Helms, L.R.; Li, L.; Chan, W.; Wetzel, R. A Role for Destabilizing Amino Acid Replacements in Light-Chain Amyloidosis. Proc. Natl. Acad. Sci. USA 1994, 91, 5446–5450. [Google Scholar] [CrossRef]
- Morgan, G.J.; Kelly, J.W. The Kinetic Stability of a Full-Length Antibody Light Chain Dimer Determines Whether Endoproteolysis Can Release Amyloidogenic Variable Domains. J. Mol. Biol. 2016, 428, 4280–4297. [Google Scholar] [CrossRef]
- Klimtchuk, E.S.; Gursky, O.; Patel, R.S.; Laporte, K.L.; Connors, L.H.; Skinner, M.; Seldin, D.C. The Critical Role of the Constant Region in Thermal Stability and Aggregation of Amyloidogenic Immunoglobulin Light Chain. Biochemistry 2010, 49, 9848–9857. [Google Scholar] [CrossRef]
- Buxbaum, J. Mechanisms of Disease: Monoclonal Immunoglobulin Deposition. Amyloidosis, Light Chain Deposition Disease, and Light and Heavy Chain Deposition Disease. Hematol. Oncol. Clin. N. Am. 1992, 6, 323–346. [Google Scholar] [CrossRef]
- Lavatelli, F.; Natalello, A.; Marchese, L.; Ami, D.; Corazza, A.; Raimondi, S.; Mimmi, M.C.; Malinverni, S.; Mangione, P.P.; Palmer, M.T.; et al. Truncation of the Constant Domain Drives Amyloid Formation by Immunoglobulin Light Chains. J. Biol. Chem. 2024, 300, 107174. [Google Scholar] [CrossRef]
- Mazzini, G.; Ricagno, S.; Caminito, S.; Rognoni, P.; Milani, P.; Nuvolone, M.; Basset, M.; Foli, A.; Russo, R.; Merlini, G.; et al. Protease-Sensitive Regions in Amyloid Light Chains: What a Common Pattern of Fragmentation across Organs Suggests about Aggregation. FEBS J. 2022, 289, 494–506. [Google Scholar] [CrossRef]
- Lavatelli, F.; Mazzini, G.; Ricagno, S.; Iavarone, F.; Rognoni, P.; Milani, P.; Nuvolone, M.; Swuec, P.; Caminito, S.; Tasaki, M.; et al. Mass Spectrometry Characterization of Light Chain Fragmentation Sites in Cardiac AL Amyloidosis: Insights into the Timing of Proteolysis. J. Biol. Chem. 2020, 295, 16572–16584. [Google Scholar] [CrossRef]
- Lu, Y.; Jiang, Y.; Prokaeva, T.; Connors, L.H.; Costello, C.E. Oxidative Post-Translational Modifications of an Amyloidogenic Immunoglobulin Light Chain Protein. Int. J. Mass Spectrom. 2017, 416, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Connors, L.H.; Jiang, Y.; Budnik, M.; Théberge, R.; Prokaeva, T.; Bodi, K.L.; Seldin, D.C.; Costello, C.E.; Skinner, M. Heterogeneity in Primary Structure, Post-Translational Modifications, and Germline Gene Usage of Nine Full-Length Amyloidogenic Kappa1 Immunoglobulin Light Chains. Biochemistry 2007, 46, 14259–14271. [Google Scholar] [CrossRef]
- Lim, A.; Wally, J.; Walsh, M.T.; Skinner, M.; Costello, C.E. Identification and Location of a Cysteinyl Posttranslational Modification in an Amyloidogenic Kappa1 Light Chain Protein by Electrospray Ionization and Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Anal. Biochem. 2001, 295, 45–56. [Google Scholar] [CrossRef]
- Omtvedt, L.A.; Bailey, D.; Renouf, D.V.; Davies, M.J.; Paramonov, N.A.; Haavik, S.; Husby, G.; Sletten, K.; Hounsell, E.F. Glycosylation of Immunoglobulin Light Chains Associated with Amyloidosis. Amyloid 2000, 7, 227–244. [Google Scholar] [CrossRef]
- Tveteraas, T.; Sletten, K.; Westermark, P. The Amino Acid Sequence of a Carbohydrate-Containing Immunoglobulin-Light-Chain-Type Amyloid-Fibril Protein. Biochem. J 1985, 232, 183–190. [Google Scholar] [CrossRef]
- Fykse, E.M.; Sletten, K.; Husby, G.; Cornwell, G.G., 3rd. The Primary Structure of the Variable Region of an Immunoglobin IV Light-Chain Amyloid-Fibril Protein (AL GIL). Biochem. J 1988, 256, 973–980. [Google Scholar] [CrossRef]
- Stevens, F.J. Four Structural Risk Factors Identify Most Fibril-Forming Kappa Light Chains. Amyloid 2000, 7, 200–211. [Google Scholar] [CrossRef]
- Stevens, F.J.; Weiss, D.T.; Solomon, A. Structural Bases of Light Chain-Related Pathology. In The Antibodies; CRC Press: Boca Raton, FL, USA, 1999; pp. 175–208. ISBN 9780429180620. [Google Scholar]
- Dispenzieri, A.; Larson, D.R.; Rajkumar, S.V.; Kyle, R.A.; Kumar, S.K.; Kourelis, T.; Arendt, B.; Willrcih, M.; Dasari, S.; Murray, D. N-Glycosylation of Monoclonal Light Chains on Routine MASS-FIX Testing Is a Risk Factor for MGUS Progression. Leukemia 2020, 34, 2749–2753. [Google Scholar] [CrossRef]
- Nevone, A.; Girelli, M.; Mangiacavalli, S.; Paiva, B.; Milani, P.; Cascino, P.; Piscitelli, M.; Speranzini, V.; Cartia, C.S.; Benvenuti, P.; et al. An N-Glycosylation Hotspot in Immunoglobulin κ Light Chains Is Associated with AL Amyloidosis. Leukemia 2022, 36, 2076–2085. [Google Scholar] [CrossRef]
- Kumar, S.; Murray, D.; Dasari, S.; Milani, P.; Barnidge, D.; Madden, B.; Kourelis, T.; Arendt, B.; Merlini, G.; Ramirez-Alvarado, M.; et al. Assay to Rapidly Screen for Immunoglobulin Light Chain Glycosylation: A Potential Path to Earlier AL Diagnosis for a Subset of Patients. Leukemia 2019, 33, 254–257. [Google Scholar] [CrossRef]
- Mellors, P.W.; Dasari, S.; Kohlhagen, M.C.; Kourelis, T.; Go, R.S.; Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Buadi, F.K.; Willrich, M.A.V.; et al. MASS-FIX for the Detection of Monoclonal Proteins and Light Chain N-Glycosylation in Routine Clinical Practice: A Cross-Sectional Study of 6315 Patients. Blood Cancer J. 2021, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.D.; Kohlhagen, M.C.; Ladwig, P.M.; Dasari, S.; Kumar, S.; Dispenzieri, A.; Willrich, M.A.V.; Murray, D.L. Characterizing M-Protein Light Chain Glycosylation via Mass Spectrometry. Clin. Biochem. 2022, 109–110, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef]
- van de Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196, 1435–1441. [Google Scholar] [CrossRef]
- van de Bovenkamp, F.S.; Derksen, N.I.L.; Ooijevaar-de Heer, P.; van Schie, K.A.; Kruithof, S.; Berkowska, M.A.; van der Schoot, C.E.; IJspeert, H.; van der Burg, M.; Gils, A.; et al. Adaptive Antibody Diversification through N-Linked Glycosylation of the Immunoglobulin Variable Region. Proc. Natl. Acad. Sci. USA 2018, 115, 1901–1906. [Google Scholar] [CrossRef]
- Parodi, A.J. Role of N-Oligosaccharide Endoplasmic Reticulum Processing Reactions in Glycoprotein Folding and Degradation. Biochem. J. 2000, 348 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Varki, A. Biological Roles of Oligosaccharides: All of the Theories Are Correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-Resolution Structure and Strain Comparison of Infectious Mammalian Prions. Mol. Cell 2021, 81, 4540–4551.e6. [Google Scholar] [CrossRef]
- Culyba, E.K.; Price, J.L.; Hanson, S.R.; Dhar, A.; Wong, C.-H.; Gruebele, M.; Powers, E.T.; Kelly, J.W. Protein Native-State Stabilization by Placing Aromatic Side Chains in N-Glycosylated Reverse Turns. Science 2011, 331, 571–575. [Google Scholar] [CrossRef]
- Hanson, S.R.; Culyba, E.K.; Hsu, T.-L.; Wong, C.-H.; Kelly, J.W.; Powers, E.T. The Core Trisaccharide of an N-Linked Glycoprotein Intrinsically Accelerates Folding and Enhances Stability. Proc. Natl. Acad. Sci. USA 2009, 106, 3131–3136. [Google Scholar] [CrossRef]
- Chen, W.; Enck, S.; Price, J.L.; Powers, D.L.; Powers, E.T.; Wong, C.-H.; Dyson, H.J.; Kelly, J.W. Structural and Energetic Basis of Carbohydrate–Aromatic Packing Interactions in Proteins. J. Am. Chem. Soc. 2013, 135, 9877–9884. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.S.; et al. Structure-Based Engineering of a Monoclonal Antibody for Improved Solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef] [PubMed]
- van de Bovenkamp, F.S.; Derksen, N.I.L.; van Breemen, M.J.; de Taeye, S.W.; Ooijevaar-de Heer, P.; Sanders, R.W.; Rispens, T. Variable Domain N-Linked Glycans Acquired During Antigen-Specific Immune Responses Can Contribute to Immunoglobulin G Antibody Stability. Front. Immunol. 2018, 9, 740. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.H.; Boyles, F.; Deane, C.M. Observed Antibody Space: A Diverse Database of Cleaned, Annotated, and Translated Unpaired and Paired Antibody Sequences. Protein Sci. 2022, 31, 141–146. [Google Scholar] [CrossRef]
- Kovaltsuk, A.; Leem, J.; Kelm, S.; Snowden, J.; Deane, C.M.; Krawczyk, K. Observed Antibody Space: A Resource for Data Mining Next-Generation Sequencing of Antibody Repertoires. J. Immunol. 2018, 201, 2502–2509. [Google Scholar] [CrossRef]
- Petrescu, A.-J.; Milac, A.-L.; Petrescu, S.M.; Dwek, R.A.; Wormald, M.R. Statistical Analysis of the Protein Environment of N-Glycosylation Sites: Implications for Occupancy, Structure, and Folding. Glycobiology 2004, 14, 103–114. [Google Scholar] [CrossRef]
- Gupta, R.; Brunak, S. Prediction of Glycosylation across the Human Proteome and the Correlation to Protein Function. Pac. Symp. Biocomput. 2002, 7, 310–322. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Prokaeva, T.; Spencer, B.; Kaut, M.; Ozonoff, A.; Doros, G.; Connors, L.H.; Skinner, M.; Seldin, D.C. Soft Tissue, Joint, and Bone Manifestations of AL Amyloidosis: Clinical Presentation, Molecular Features, and Survival. Arthritis Rheum. 2007, 56, 3858–3868. [Google Scholar] [CrossRef]
- Lefranc, M.-P.; Lefranc, G. Immunoglobulins or Antibodies: IMGT® Bridging Genes, Structures and Functions. Biomedicines 2020, 8, 319. [Google Scholar] [CrossRef]
- Murray, A.N.; Chen, W.; Antonopoulos, A.; Hanson, S.R.; Wiseman, R.L.; Dell, A.; Haslam, S.M.; Powers, D.L.; Powers, E.T.; Kelly, J.W. Enhanced Aromatic Sequons Increase Oligosaccharyltransferase Glycosylation Efficiency and Glycan Homogeneity. Chem. Biol. 2015, 22, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.D.; Stephens, R.M. Sequence Logos: A New Way to Display Consensus Sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Lowenthal, M.S.; Davis, K.S.; Formolo, T.; Kilpatrick, L.E.; Phinney, K.W. Identification of Novel N-Glycosylation Sites at Noncanonical Protein Consensus Motifs. J. Proteome Res. 2016, 15, 2087–2101. [Google Scholar] [CrossRef]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Sternke-Hoffmann, R.; Boquoi, A.; Lopez, Y.; Niedenhoff, D.; Platten, F.; Fenk, R.; Haas, R.; Buell, A.K. Biochemical and Biophysical Characterisation of Immunoglobulin Free Light Chains Derived from an Initially Unbiased Population of Patients with Light Chain Disease. PeerJ 2020, 8, e8771. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Yan, N.L.; Mortenson, D.E.; Rennella, E.; Blundon, J.M.; Gwin, R.M.; Lin, C.-Y.; Stanfield, R.L.; Brown, S.J.; Rosen, H.; et al. Stabilization of Amyloidogenic Immunoglobulin Light Chains by Small Molecules. Proc. Natl. Acad. Sci. USA 2019, 116, 8360–8369. [Google Scholar] [CrossRef]
- Rennella, E.; Morgan, G.J.; Kelly, J.W.; Kay, L.E. Role of Domain Interactions in the Aggregation of Full-Length Immunoglobulin Light Chains. Proc. Natl. Acad. Sci. USA 2019, 116, 854–863. [Google Scholar] [CrossRef]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The Biological and Chemical Basis for Tissue-Selective Amyloid Disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef]
- Gertz, M.A.; Cohen, A.D.; Comenzo, R.L.; Kastritis, E.; Landau, H.J.; Libby, E.N.; Liedtke, M.; Sanchorawala, V.; Schönland, S.; Wechalekar, A.; et al. Birtamimab plus Standard of Care in Light-Chain Amyloidosis: The Phase 3 Randomized Placebo-Controlled VITAL Trial. Blood 2023, 142, 1208–1218. [Google Scholar] [CrossRef]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the Frequency of Protein Glycosylation, as Deduced from Analysis of the SWISS-PROT Database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Schanz, M.; Liechti, T.; Zagordi, O.; Miho, E.; Reddy, S.T.; Günthard, H.F.; Trkola, A.; Huber, M. High-Throughput Sequencing of Human Immunoglobulin Variable Regions with Subtype Identification. PLoS ONE 2014, 9, e111726. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.T.; Adams, K.D.; Briggs, A.W.; Timberlake, S.C.; Vigneault, F.; Kleinstein, S.H. Hierarchical Clustering Can Identify B Cell Clones with High Confidence in Ig Repertoire Sequencing Data. J. Immunol. 2017, 198, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Schramm, C.A.; Kong, R.; NISC Comparative Sequencing Program; Mullikin, J.C.; Mascola, J.R.; Kwong, P.D.; Shapiro, L. Gene-Specific Substitution Profiles Describe the Types and Frequencies of Amino Acid Changes during Antibody Somatic Hypermutation. Front. Immunol. 2017, 8, 537. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Bombardi, R.G.; Branchizio, A.; Kose, N.; Matta, P.; Sevy, A.M.; Sinkovits, R.S.; Gilchuk, P.; Finn, J.A.; Crowe, J.E., Jr. High Frequency of Shared Clonotypes in Human B Cell Receptor Repertoires. Nature 2019, 566, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Simonich, C.A.; Doepker, L.; Ralph, D.; Williams, J.A.; Dhar, A.; Yaffe, Z.; Gentles, L.; Small, C.T.; Oliver, B.; Vigdorovich, V.; et al. Kappa Chain Maturation Helps Drive Rapid Development of an Infant HIV-1 Broadly Neutralizing Antibody Lineage. Nat. Commun. 2019, 10, 2190. [Google Scholar] [CrossRef]
- Vander Heiden, J.A.; Stathopoulos, P.; Zhou, J.Q.; Chen, L.; Gilbert, T.J.; Bolen, C.R.; Barohn, R.J.; Dimachkie, M.M.; Ciafaloni, E.; Broering, T.J.; et al. Dysregulation of B Cell Repertoire Formation in Myasthenia Gravis Patients Revealed through Deep Sequencing. J. Immunol. 2017, 198, 1460–1473. [Google Scholar] [CrossRef]
- Lefranc, M.-P. Nomenclature of the Human Immunoglobulin Kappa (IGK) Genes. Exp. Clin. Immunogenet. 2001, 18, 161–174. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 16 October 2024).
- RStudio Team Posit. Available online: http://www.rstudio.com/ (accessed on 16 October 2024).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations. Available online: https://cran.r-project.org/package=stringr (accessed on 16 October 2024).
- Wagih, O. ggseqlogo: A ‘ggplot2’ Extension for Drawing Publication-Ready Sequence Logos. Available online: https://cran.r-project.org/package=ggseqlogo (accessed on 16 October 2024).
- Dunbar, J.; Deane, C.M. ANARCI: Antigen Receptor Numbering and Receptor Classification. Bioinformatics 2016, 32, 298–300. [Google Scholar] [CrossRef]
- Pagès, H.; Aboyoun, P.; Gentleman, R.; DebRoy, S. Biostrings: Efficient Manipulation of Biological Strings. Available online: https://bioconductor.org/packages/Biostrings (accessed on 16 October 2024).
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for Structure Building and Analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Topham, C.M.; Smith, J.C. Tri-Peptide Reference Structures for the Calculation of Relative Solvent Accessible Surface Area in Protein Amino Acid Residues. Comput. Biol. Chem. 2015, 54, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mohapatra, A.; Nguyen, H.T.V.; Schimanski, L.; Kit Tan, T.; Rijal, P.; Chen, C.-P.; Cheng, S.-H.; Lee, W.-H.; Chou, Y.-C.; et al. The Presence of Broadly Neutralizing Anti-SARS-CoV-2 RBD Antibodies Elicited by Primary Series and Booster Dose of COVID-19 Vaccine. PLoS Pathog. 2024, 20, e1012246. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Wang, R.; Ju, B.; Yu, J.; Zhang, Y.; Liu, N.; Wang, J.; Zhang, Q.; Chen, P.; Zhou, B.; et al. Structural Basis for Bivalent Binding and Inhibition of SARS-CoV-2 Infection by Human Potent Neutralizing Antibodies. Cell Res. 2021, 31, 517–525. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Origin | IGKV | IGLV | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | Sequon | No Sequon | Total | Sequon | No Sequon | Total | Sequon | No Sequon | |
| AL subcategory | 160 | 67 41.9% | 93 58.1% | 524 | 39 7.4% | 485 92.6% | 684 | 106 15.5% | 578 84.5% |
| MM subcategory | 595 | 62 10.4% | 533 89.6% | 374 | 38 10.2% | 336 89.8% | 969 | 100 10.3% | 869 89.7% |
| OAS repertoire | 4,278,425 | 360,785 8.4% | 3,917,640 91.6% | 3,769,322 | 271,185 7.2% | 3,498,137 92.8% | 8,047,747 | 631,970 7.9% | 7,415,777 92.1% |
| AL-Base Subcategory | IGVL Gene | Region | Asn Position (IMGT) | Sequence | Number of Sequences |
|---|---|---|---|---|---|
| AL | IGLV2-23 | CDR3 | 114 | NTC | 1 |
| AL | IGLV3-1 | CDR1 | 38 | NAC | 2 |
| AL | IGLV3-1 | CDR1 | 38 | NVC | 2 |
| MM | IGKV1-39 | CDR1 | 36 | NTC | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, G.J.; Yung, Z.; Spencer, B.H.; Sanchorawala, V.; Prokaeva, T. Predicting Structural Consequences of Antibody Light Chain N-Glycosylation in AL Amyloidosis. Pharmaceuticals 2024, 17, 1542. https://doi.org/10.3390/ph17111542
Morgan GJ, Yung Z, Spencer BH, Sanchorawala V, Prokaeva T. Predicting Structural Consequences of Antibody Light Chain N-Glycosylation in AL Amyloidosis. Pharmaceuticals. 2024; 17(11):1542. https://doi.org/10.3390/ph17111542
Chicago/Turabian StyleMorgan, Gareth J., Zach Yung, Brian H. Spencer, Vaishali Sanchorawala, and Tatiana Prokaeva. 2024. "Predicting Structural Consequences of Antibody Light Chain N-Glycosylation in AL Amyloidosis" Pharmaceuticals 17, no. 11: 1542. https://doi.org/10.3390/ph17111542
APA StyleMorgan, G. J., Yung, Z., Spencer, B. H., Sanchorawala, V., & Prokaeva, T. (2024). Predicting Structural Consequences of Antibody Light Chain N-Glycosylation in AL Amyloidosis. Pharmaceuticals, 17(11), 1542. https://doi.org/10.3390/ph17111542