

Targeting Iron Responsive Elements (IREs) of APP mRNA into Novel Therapeutics to Control the Translation of Amyloid-β Precursor Protein in Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Iron Homeostasis in Brain
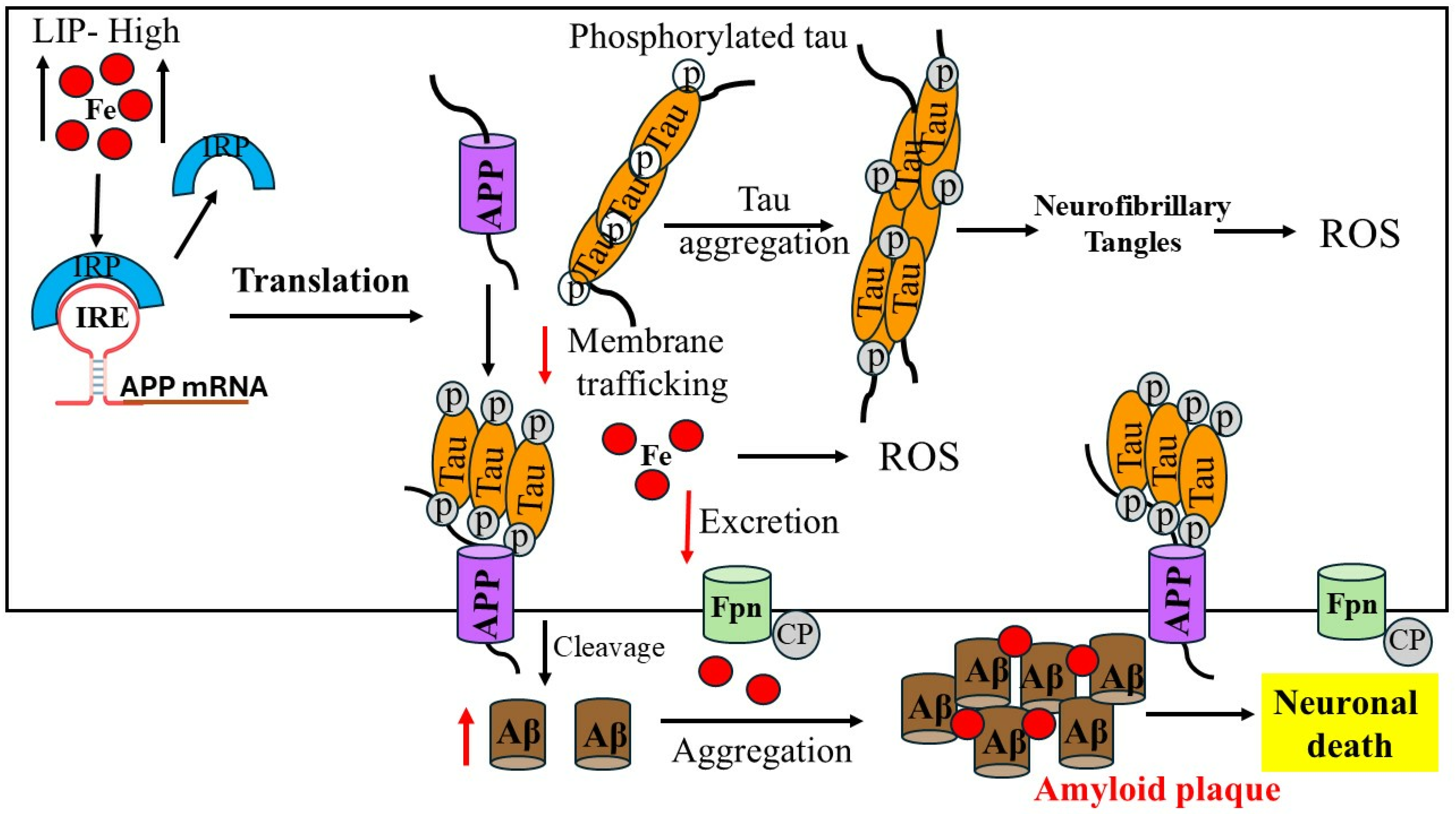
3. Iron Dysregulation in Alzheimer’s Disease
4. Iron-Induced Amyloid-β Aggregation

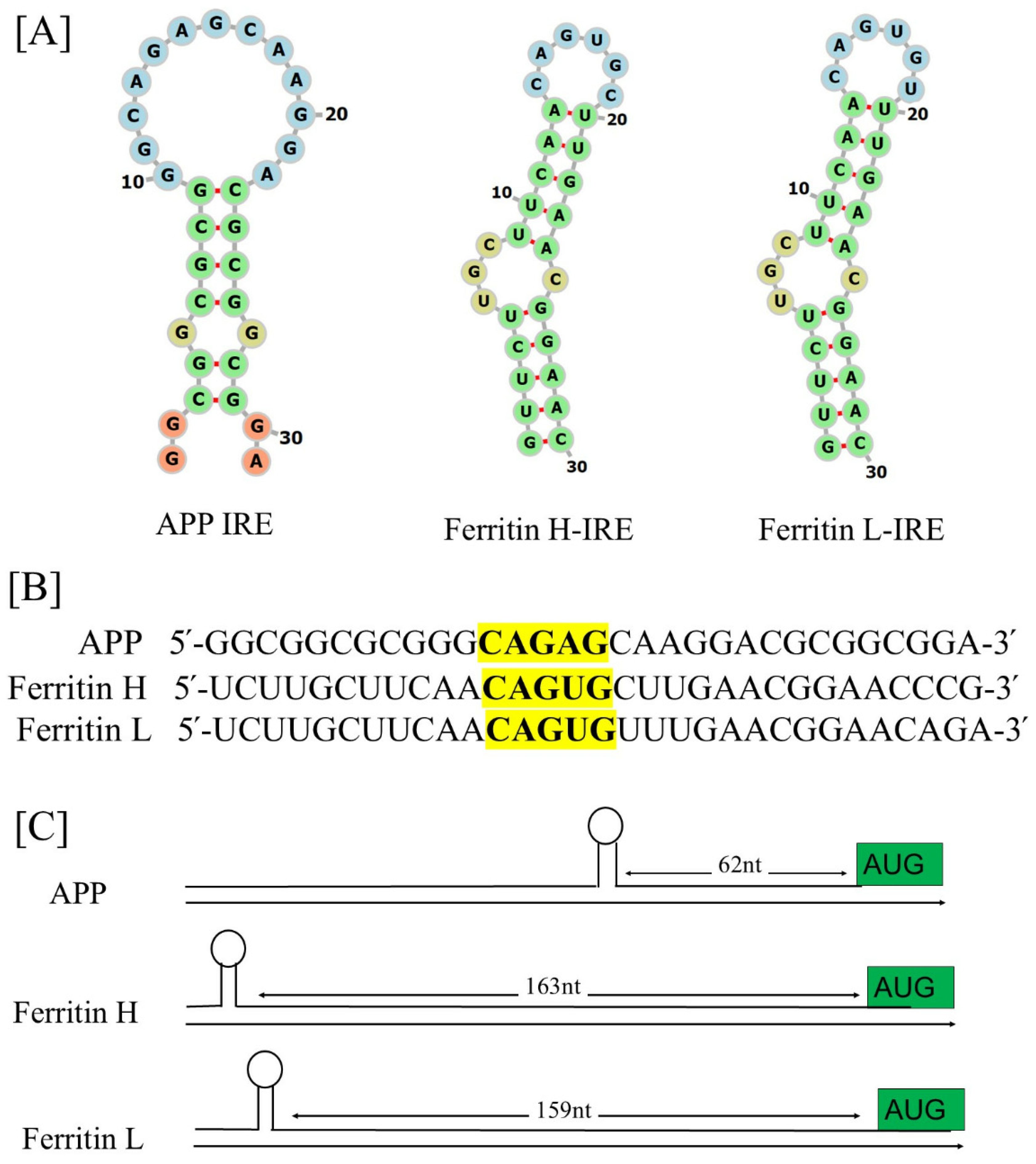
5. Alzheimer’s Amyloid-β Transcript Encodes Functional IRE mRNA
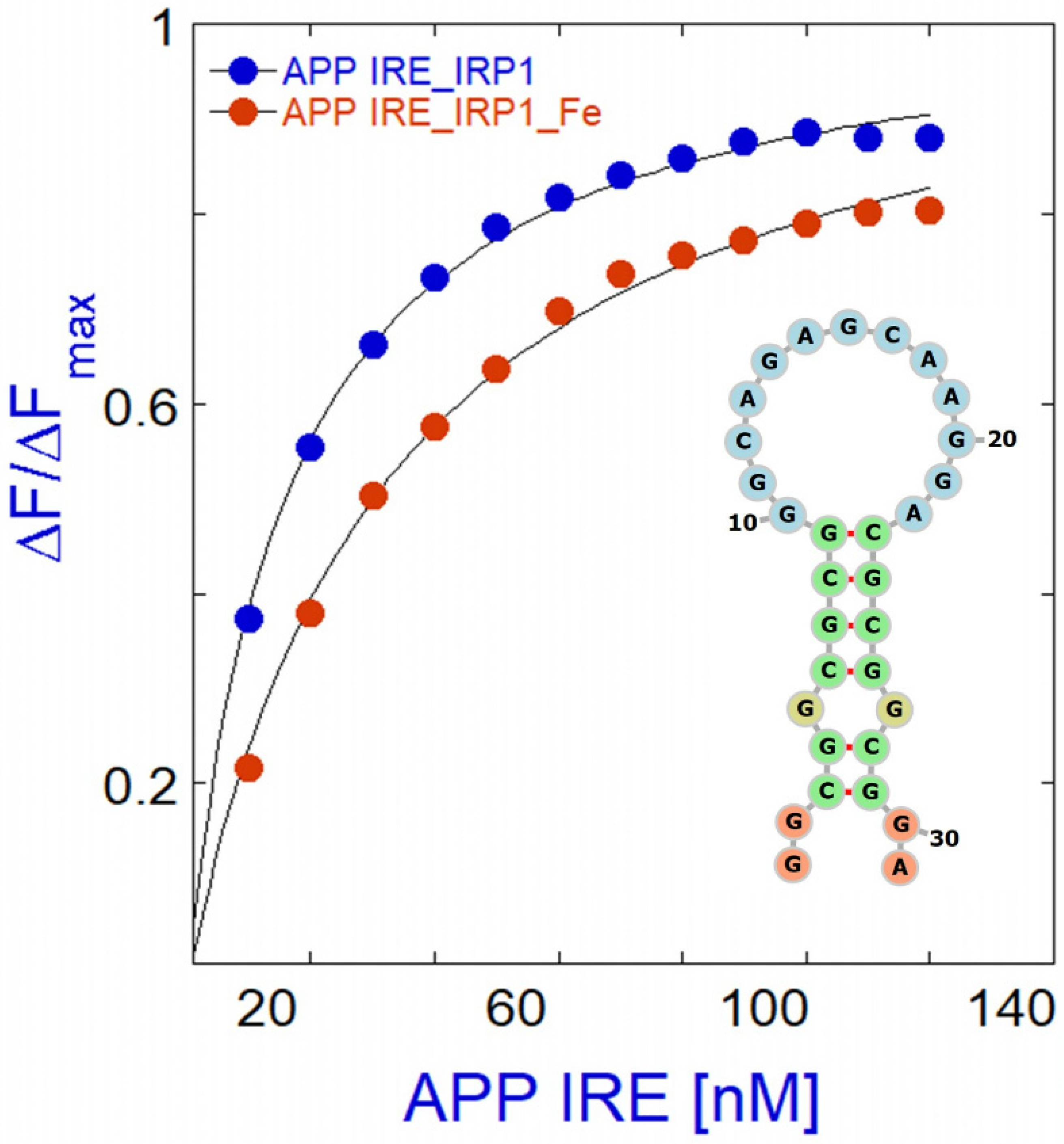
6. The Functional Interaction of Alzheimer’s APP mRNA with IRP1
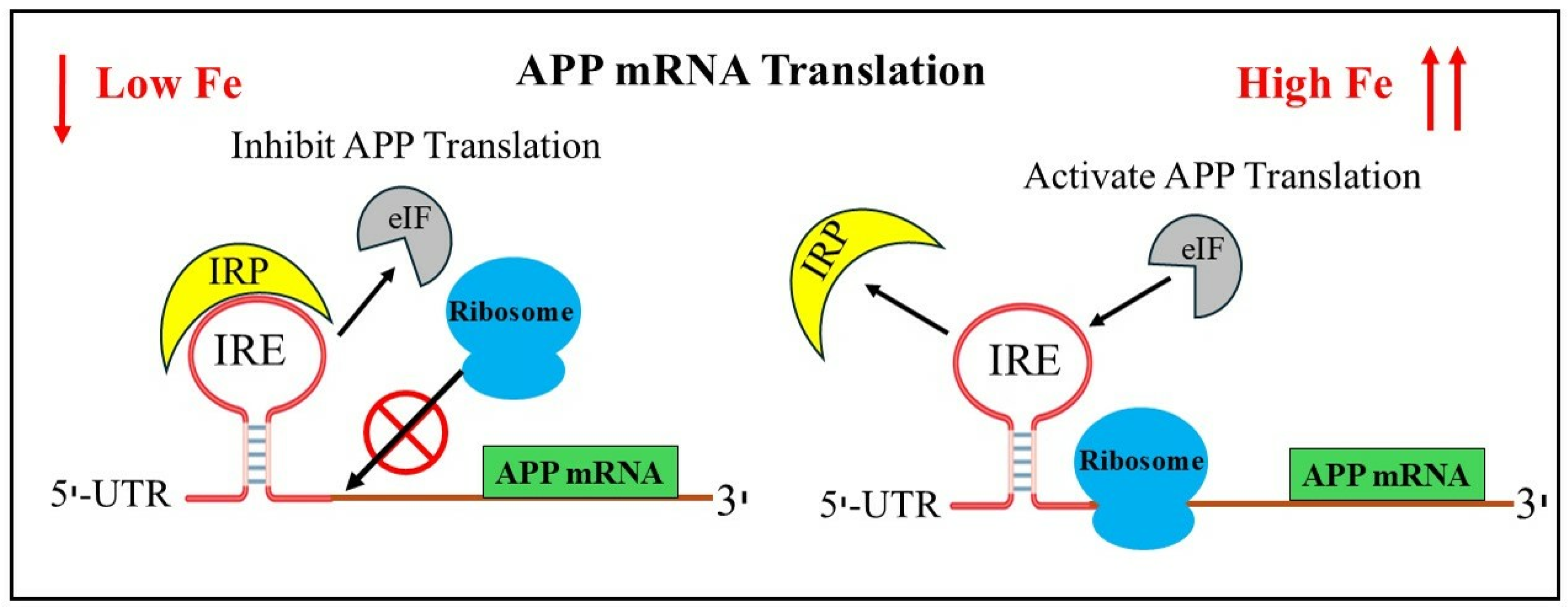
7. Translational Control of Alzheimer’s Amyloid Precursor mRNA
8. Therapies for Amyloid-β Control That Target APP IRE mRNA
9. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pardo, J.; Ventura, S. Cryo-EM structures of functional and pathological amyloid ribonucleoprotein assemblies. Trends Biochem. Sci. 2024, 49, 119–133. [Google Scholar] [CrossRef] [PubMed]
- van der Kant, R.; Louros, N.; Schymkowitz, J.; Rousseau, F. Thermodynamic analysis of amyloid fibril structures reveals a common framework for stability in amyloid polymorphs. Structure 2022, 30, 1178–1189.e3. [Google Scholar] [CrossRef] [PubMed]
- Phinney, A.L.; Drisaldi, B.; Schmidt, S.D.; Lugowski, S.; Coronado, V.; Liang, Y.; Horne, P.; Yang, J.; Sekoulidis, J.; Coomaraswamy, J.; et al. In Vivo reduction of amyloid-β by a mutant copper transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 14193–14198. [Google Scholar] [CrossRef]
- Upadhyay, A.; Chhangani, D.; Rao, N.R.; Kofler, J.; Vassar, R.; Rincon-Limas, D.E.; Savas, J.N. Amyloid fibril proteomics of AD brains reveals modifiers of aggregation and toxicity. Mol. Neurodegener. 2023, 18, 61. [Google Scholar] [CrossRef]
- Wolfe, M.S. Shutting down Alzheimer’s. Sci. Am. 2006, 294, 72–79. [Google Scholar] [CrossRef]
- Chiou, B.; Neely, E.B.; Mcdevitt, D.S.; Simpson, I.A.; Connor, J.R. Transferrin and H-ferritin involvement in brain iron acquisition during postnatal development: Impact of sex and genotype. J. Neurochem. 2020, 152, 381–396. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Zhang, L.; Meng, R.; Gao, J.; Jin, M.; Li, M.; Wang, X. Effect of metal ions on Alzheimer’s disease. Brain Behav. 2022, 12, e2527. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; I Giasson, B.; Trojanowski, J.Q. More than just two peas in a pod: Common amyloidogenic properties of tau and α-synuclein in neurodegenerative diseases. Trends Neurosci. 2004, 27, 129–134. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Holsinger, R.M.D. Roles of Non-Coding RNA in Alzheimer’s Disease Pathophysiology. Int. J. Mol. Sci. 2023, 24, 12498. [Google Scholar] [CrossRef]
- Zhang, N.; Yu, X.; Xie, J.; Xu, H. New Insights into the Role of Ferritin in Iron Homeostasis and Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Fleming, R.E.; Ponka, P. Iron overload in human disease. New Engl. J. Med. 2012, 366, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.M.; Shen, X. Brain iron transport and neurodegeneration. Trends Mol. Med. 2001, 7, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ming-Qian, Z. Iron misregulation in the brain: A primary cause of neurodegenerative disorders. Lancet Neurol. 2003, 2, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Baringer, S.L.; Lukacher, A.S.; Palsa, K.; Kim, H.; Lippmann, E.S.; Spiegelman, V.S.; Simpson, I.A.; Connor, J.R. Amyloid-β exposed astrocytes induce iron transport from endothelial cells at the blood–brain barrier by altering the ratio of apo- and holo-transferrin. J. Neurochem. 2023, 167, 248–261. [Google Scholar] [CrossRef]
- Baringer, S.L.; Simpson, I.A.; Connor, J.R. Brain iron acquisition: An overview of homeostatic regulation and disease dysregulation. J. Neurochem. 2023, 165, 625–642. [Google Scholar] [CrossRef]
- Paul, A.; Adlard, P.A.; Ashley, I.; Bush, A.I. Metals and Alzheimer’s Disease: How Far Have We Come in the Clinic? J. Alzheimer’s Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef]
- Jiang, H.; Song, N.; Jiao, Q.; Shi, L.; Du, X. Iron Pathophysiology in Parkinson Diseases. Adv. Exp. Med. Biol. 2019, 1173, 45–66. [Google Scholar] [CrossRef]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.X.; Tsatsanis, A.; Lim, L.Q.; Adlard, P.A.; Bush, A.I.; Duce, J.A. β-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 2014, 9, e114174. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Tan, E.-K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I. The metal theory of Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33, S277–S281. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pinero, D.J.; Hu, J.; Connor, J.R. Alterations in the interaction between iron regulatory proteins and their iron responsive element in normal and Alzheimer’s diseased brains. Cell Mol. Biol. 2000, 46, 761–776. [Google Scholar]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An Iron-responsive Element Type II in the 5′-Untranslated Region of the Alzheimer’s Amyloid Precursor Protein Transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Huang, X.; Cho, H.; Greig, N.H.; Youdim, M.B.; Rogers, J.T. Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5′untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J. Neural Transm. Suppl. 2006, 71, 237–247. [Google Scholar] [CrossRef]
- Piccinelli, P.; Samuelsson, T. Evolution of the iron-responsive element. RNA 2007, 13, 952–966. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pantopoulos, K. Iron metabolism and the IRE/IRP regulatory system: An update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-H.; Cahill, C.M.; Vanderburg, C.R.; Scherzer, C.R.; Wang, B.; Huang, X.; Rogers, J.T. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 2010, 285, 31217–31232. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2012, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Selezneva, A.I.; Cavigiolio, G.; Theil, E.C.; Walden, W.E.; Volz, K. Crystallization and preliminary X-ray diffraction analysis of iron regulatory protein 1 in complex with ferritin IRE RNA. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 249–252. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brazzolotto, X.; Timmins, P.; Dupont, Y.; Moulis, J.M. Structural changes associated with switching activities of human iron regulatory protein 1. J. Biol. Chem. 2002, 277, 11995–12000. [Google Scholar] [CrossRef] [PubMed]
- Volpon, L.O.M.; Capul, A.A.; de la Torre, J.C.; Borden, K.L. Cap-free structure of eIF4E suggests a basis for conformational regulation by its ligands. EMBO J. 2006, 25, 5138–5149. [Google Scholar] [CrossRef]
- Khan, M.A.; Malik, A.; Domashevskiy, A.V.; San, A.; Khan, J.A. Interaction of ferritin iron responsive element (IRE) mRNA with translation initiation factor eIF4F. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 243, 118776. [Google Scholar] [CrossRef]
- Khan, M.A.; Walden, W.E.; Theil, E.C.; Goss, D.J. Thermodynamic and kinetic analyses of iron response element (IRE)-mRNA binding to iron regulatory protein, IRP1. Sci. Rep. 2017, 7, 8532. [Google Scholar] [CrossRef]
- Khan, M.A.; Mohammad, T.; Malik, A.; Hassan, M.I.; Domashevskiy, A.V. Iron response elements (IREs)-mRNA of Alzheimer’s amyloid precursor protein binding to iron regulatory protein (IRP1): A combined molecular docking and spectroscopic approach. Sci. Rep. 2023, 13, 5073. [Google Scholar] [CrossRef]
- Khan, M.A.; Walden, W.E.; Goss, D.J.; Theil, E.C. Direct Fe2+ sensing by iron-responsive messenger RNA: Repressor complexes weakens binding. J. Biol. Chem. 2009, 284, 30122–30128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khan, M.A.; Yumak, S.; Miyoshi, H. Poly(A)-binding protein promotes VPg-dependent translation of potyvirus through enhanced binding of phosphorylated eIFiso4F and eIFiso4F∙eIF4B. PLoS ONE 2024, 19, e0300287. [Google Scholar] [CrossRef]
- Zhang, P.; Park, H.-J.; Zhang, J.; Junn, E.; Andrews, R.J.; Velagapudi, S.P.; Abegg, D.; Vishnu, K.; Costales, M.G.; Childs-Disney, J.L.; et al. Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 1457–1467. [Google Scholar] [CrossRef]
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar] [CrossRef]
- Moos, T.; Morgan, E.H. The significance of the mutated divalent metal transporter (DMT1) on iron transport into the Belgrade rat brain. J. Neurochem. 2004, 88, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, A.; Clark, M.; So, P.-W. The Aging of Iron Man. Front. Aging Neurosci. 2018, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Pelizzoni, I.; Zacchetti, D.; Campanella, A.; Grohovaz, F.; Codazzi, F. Iron uptake in quiescent and inflammation-activated astrocytes: A potentially neuroprotective control of iron burden. Biochim. Biophys. Acta. 2013, 1832, 1326–1333. [Google Scholar] [CrossRef]
- Rouault, T.A.; Cooperman, S. Brain iron metabolism. Semin. Pediatr. Neurol. 2006, 13, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P. Transferrin receptor 1. Int. J. Biochem. Cell Biol. 2004, 36, 2137–2143. [Google Scholar] [CrossRef]
- De Domenico, I.; McVey-Ward, D.; Kaplan, J. Regulation of iron acquisition and storage: Consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81. [Google Scholar] [CrossRef]
- Ingrassia, R.; Garavaglia, B.; Memo, M. DMT1 Expression and Iron Levels at the Crossroads Between Aging and Neurodegeneration. Front. Neurosci. 2019, 13, 575. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, S.L.; Kosman, D.J. Aberrant Cerebral Iron Trafficking Co-morbid With Chronic Inflammation: Molecular Mechanisms and Pharmacologic Intervention. Front. Neurol. 2022, 13, 855751. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef]
- Ganz, T. Cellular iron: Ferroportin is the only way out. Cell Metab. 2005, 1, 155–157. [Google Scholar] [CrossRef]
- Madsen, E.; Gitlin, J.D. Copper and iron disorders of the brain. Annu. Rev. Neurosci. 2007, 30, 317–337. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W. Toxicology of choroid plexus: Special reference to metal-induced neurotoxicities. Microsc. Res. Tech. 2001, 52, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Aschner, M.; Ghersi-Egea, J.F. Brain barrier systems: A new frontier in metal neurotoxicological research. Toxicol. Appl. Pharmacol. 2003, 192, 1–11. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Torres-Jardón, R.; Kulesza, R.J.; Mansour, Y.; González-González, L.O.; Gónzalez-Maciel, A.; Reynoso-Robles, R.; Mukherjee, P.S. Alzheimer disease starts in childhood in polluted Metropolitan Mexico City. A major health crisis in progress. Environ. Res. 2020, 183, 109137. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Ehrenberg, A.J.; Nguy, A.K.; Theofilas, P.; Dunlop, S.; Suemoto, C.K.; Alho, A.T.D.L.; Leite, R.P.; Rodriguez, R.D.; Mejia, M.B.; Rüb, U.; et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: The pathological building blocks of early Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Perry, G. What are the facts and artifacts of the pathogenesis and etiology of Alzheimer disease? J. Chem. Neuroanat. 1998, 16, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.; Perry, G. Nucleic acid oxidative damage in Alzheimer’s disease—explained by the hepcidin-ferroportin neuronal iron overload hypothesis? J. Trace Elem. Med. Biol. 2016, 38, 1–9. [Google Scholar] [CrossRef]
- Ma, L.; Azad, M.G.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Bórquez, D.A.; Núñez, M.T. Inflaming the Brain with Iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef]
- Hinarejos, I.; Machuca-Arellano, C.; Sancho, P.; Carmen Espinós, C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Review Antioxidants 2020, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Chebrolu, H.; Wekstein, D.R.; Schmitt, F.A.; Jicha, G.A.; Cooper, G.; Markesbery, W.R. Brain structural alterations before mild cognitive impairment. Neurology 2007, 68, 1268–1273. [Google Scholar] [CrossRef]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, L.; Zhao, Q.; Du, X.; Bi, M.; Li, Y.; Jiao, Q.; Jiang, H. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free. Radic. Biol. Med. 2020, 152, 227–234. [Google Scholar] [CrossRef]
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Cahill, C.M. Iron-responsive-like elements and neurodegenerative ferroptosis. Learn. Mem. 2020, 27, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-C.; Olsthoorn, R.C. Relevance of the iron-responsive element (IRE) pseudotriloop structure for IRP1/2 binding and validation of IRE-like structures using the yeast three-hybrid system. Gene 2019, 710, 399–405. [Google Scholar] [CrossRef]
- Yin, X.; Qiu, Y.; Zhao, C.; Zhou, Z.; Bao, J.; Qian, W. The Role of Amyloid-Beta and Tau in the Early Pathogenesis of Alzheimer’s Disease. Med. Sci. Monit. 2021, 27, e933084. [Google Scholar] [CrossRef]
- Pan, R.; Initiative, F.T.A.D.N.; Luo, S.; Huang, Q.; Li, W.; Cai, T.; Lai, K.; Shi, X. The Associations of Cerebrospinal Fluid Ferritin with Neurodegeneration and Neuroinflammation Along the Alzheimer’s Disease Continuum. J. Alzheimer’s Dis. 2022, 88, 1115–1125. [Google Scholar] [CrossRef]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Rogers, J.T. Alzheimer’s disease therapeutics targeted to the control of amyloid precursor protein translation: Maintenance of brain iron homeostasis. Biochem. Pharmacol. 2014, 88, 486–494. [Google Scholar] [CrossRef]
- Peng, Y.; Chang, X.; Lang, M. Iron Homeostasis Disorder and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12442. [Google Scholar] [CrossRef]
- Kagerer, S.M.; Bergen, J.M.G.; Li, X.; Quevenco, F.C.; Gietl, A.F.; Studer, S.; Treyer, V.; Meyer, R.; Kaufmann, P.A.; Nitsch, R.M.; et al. APOE4 moderates effects of cortical iron on synchronized default mode network activity in cognitively healthy old-aged adults. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12002. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Gunn, A.P.; Wong, B.X.; Ayton, S.; Appukuttan, A.T.; Roberts, B.R.; Duce, J.A.; Bush, A.I. Marked Age-Related Changes in Brain Iron Homeostasis in Amyloid Protein Precursor Knockout Mice. Neurotherapeutics 2018, 15, 1055–1062. [Google Scholar] [CrossRef]
- Wan, W.; Cao, L.; Kalionis, B.; Murthi, P.; Xia, S.; Guan, Y. Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling. Front. Neurol. 2019, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- LeVine, S.M. Exploring Potential Mechanisms Accounting for Iron Accumulation in the Central Nervous System of Patients with Alzheimer’s Disease. Cells 2024, 13, 689. [Google Scholar] [CrossRef] [PubMed]
- LeVine, S.M.; Tsau, S.S.G. Exploring Whether Iron Sequestration within the CNS of Patients with Alzheimer’s Disease Causes a Functional Iron Deficiency That Advances Neurodegeneration. Brain Sci. 2023, 13, 511. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.H.; Klevanski, M.; Saar, M.; Müller, U.C. Roles of the amyloid precursor protein family in the peripheral nervous system. Mech. Dev. 2013, 130, 433–446. [Google Scholar] [CrossRef]
- Wan, L.; Nie, G.; Zhang, J.; Zhao, B. Overexpression of human wild-type amyloid-β protein precursor decreases the iron content and increases the oxidative stress of neuroblastoma SH-SY5Y cells. J. Alzheimer’s Dis. 2012, 30, 523–530. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Park, Y.H.; Kosman, D.J. sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. Embo Rep. 2014, 15, 809–815. [Google Scholar] [CrossRef]
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1790, 615–628. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 2021, 296, 100105. [Google Scholar] [CrossRef]
- Hin, N.; Newman, M.; Pederson, S.; Lardelli, M. Iron Responsive Element-Mediated Responses to Iron Dyshomeostasis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 84, 1597–1630. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Li, Y.; Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Weiner, M.W.; Shaw, L.M.; Masters, C.L.; Fowler, C.J.; Trojanowski, J.Q.; et al. Validation of Plasma Amyloid-β 42/40 for Detecting Alzheimer Disease Amyloid Plaques. Neurology 2022, 98, e688–e699. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Fawzi, N.L. The C-terminal threonine of Aβ43 nucleates toxic aggregation via structural and dynamical changes in monomers and protofibrils. Biochemistry 2014, 53, 3095–3105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef]
- Härd, T. Amyloid Fibrils: Formation, Polymorphism, and Inhibition. J. Phys. Chem. Lett. 2014, 5, 607–614. [Google Scholar] [CrossRef]
- Gurry, T.; Stultz, C.M. Mechanism of amyloid-β fibril elongation. Biochemistry 2014, 53, 6981–6991. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Chakrabarti, M.; Govindaraju, T. Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem. Commun. 2015, 51, 13434–13450. [Google Scholar] [CrossRef] [PubMed]
- Teplyakov, A.; Obmolova, G.; Gilliland, G.L. A coiled conformation of amyloid-beta recognized by antibody C706. Alzheimer’s Res. Ther. 2017, 9, 66. [Google Scholar] [CrossRef]
- Tran, L.; Basdevant, N.; Prévost, C.; Ha-Duong, T. Structure of ring-shaped Aβ42 oligomers determined by conformational selection. Sci. Rep. 2016, 6, 21429. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 2001, 276, 20466–20473. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef]
- Lövestam, S.; Koh, F.A.; van Knippenberg, B.; Kotecha, A.; Murzin, A.G.; Goedert, M.; Scheres, S.H. Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. eLife 2022, 11, e76494. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Volz, K. Conservation in the Iron Responsive Element Family. Genes 2021, 12, 1365. [Google Scholar] [CrossRef] [PubMed]
- Leipuviene, R.; Theil, E.C. The family of iron responsive RNA structures regulated by changes in cellular iron and oxygen. Cell. Mol. Life Sci. 2007, 64, 2945–2955. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Volz, K. The functional duality of iron regulatory protein 1. Curr. Opin. Struct. Biol. 2008, 18, 106–111. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shen, M.; Goforth, J.B.; Eisenstein, R.S. Iron-dependent post transcriptional control of mitochondrial aconitase expression. Metallomics 2023, 15, mfac099. [Google Scholar] [CrossRef] [PubMed]
- Mikkilineni, S.; Cantuti-Castelvetri, I.; Cahill, C.M.; Balliedier, A.; Greig, N.H.; Rogers, J.T. The anticholinesterase phenserine and its enantiomer posiphen as 5′untranslated-region-directed translation blockers of the Parkinson’s alpha synuclein expression. Park. Dis. 2012, 2012, 142372. [Google Scholar] [CrossRef] [PubMed]
- Goforth, J.B.; Anderson, S.A.; Nizzi, C.P.; Eisenstein, R.S. Multiple determinants within iron-responsive elements dictate iron regulatory protein binding and regulatory hierarchy. RNA 2010, 16, 154–169. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Theil, E.C.; Goss, D.J. Living with iron (and oxygen): Questions and answers about iron homeostasis. Chem. Rev. 2009, 109, 4568–4579. [Google Scholar] [CrossRef]
- Walden, W.E.; Selezneva, A.I.; Dupuy, J.; Volbeda, A.; Fontecilla-Camps, J.C.; Theil, E.C.; Volz, K. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science 2006, 314, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Schalinske, K.L.; Chen, O.S.; Eisenstein, R.S. Iron differentially stimulates translation of mitochondrial aconitase and ferritin mRNAs in mammalian cells. Implications for iron regulatory proteins as regulators of mitochondrial citrate utilization. J. Biol. Chem. 1998, 273, 3740–3746. [Google Scholar] [CrossRef]
- Khan, M.A.; Ma, J.; Walden, W.E.; Merrick, W.C.; Theil, E.C.; Goss, D.J. Rapid kinetics of iron responsive element (IRE) RNA/iron regulatory protein 1 and IRE-RNA/eIF4F complexes respond differently to metal ions. Nucleic Acids Res. 2014, 42, 6567–6577. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ke, Y.; Wu, J.; Leibold, E.A.; Walden, W.E.; Theil, E.C. Loops and bulge/loops in iron-responsive element isoforms influence iron regulatory protein binding. Fine-tuning of mRNA regulation? J. Biol. Chem. 1998, 273, 23637–23640. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Zhang, D.L.; Ghosh, M.C.; Jain, A.; SantaMaria, A.M.; Rouault, T.A. Mechanisms of cellular iron sensing, regulation of erythropoiesis and mitochondrial iron utilization. Semin. Hematol. 2021, 58, 161–174. [Google Scholar] [CrossRef]
- Venkataramani, V.; Doeppner, T.R.; Willkommen, D.; Cahill, C.M.; Xin, Y.; Ye, G.; Liu, Y.; Southon, A.; Aron, A.; Au-Yeung, H.Y.; et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 2018, 147, 831–848. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Bush, A.I.; Cho, H.-H.; Smith, D.H.; Thomson, A.M.; Friedlich, A.L.; Lahiri, D.K.; Leedman, P.J.; Huang, X.; Cahill, C.M. Iron and the translation of the amyloid precursor protein (APP) and ferritin mRNAs: Riboregulation against neural oxidative damage in Alzheimer’s disease. Biochem. Soc. Trans. 2008, 36, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.; Galy, B.; Schwanhaeusser, B.; Blake, J.; Bähr-Ivacevic, T.; Benes, V.; Selbach, M.; Muckenthaler, M.U.; Hentze, M.W. Iron regulatory protein-1 and -2: Transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood 2011, 118, e168–e179. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Gu, M.B. Advances in aptamer screening and small molecule aptasensors. Adv. Biochem. Eng. Biotechnol. 2014, 140, 29–67. [Google Scholar] [CrossRef]
- Walden, W.E.; Selezneva, A.; Volz, K. Accomodating variety in iron-responsive elements: Crystal structure of transferrin receptor 1 B IRE bound to iron regulatory protein 1. FEBS Lett. 2012, 586, 32–35. [Google Scholar] [CrossRef]
- Gunshin, H.; Allerson, C.R.; Polycarpou-Schwarz, M.; Rofts, A.; Rogers, J.T.; Kishi, F.; Hentze, M.W.; Rouault, T.A.; Andrews, N.C.; Hediger, M.A. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001, 509, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Preiss, T. The REM phase of gene regulation. Trends Biochem. Sci. 2010, 35, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Sierzputowska-Gracz, H.; Gdaniec, Z.; Theil, E.C. Internal loop/bulge and hairpin loop of the iron-responsive element of ferritin mRNA contribute to maximal iron regulatory protein 2 binding and translational regulation in the iso-iron-responsive element/iso-iron regulatory protein family. Biochemistry 2000, 39, 6235–6242. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A. Ferritin iron responsive elements (IREs) mRNA interacts with eIF4G and activates in vitro translation. Front. Biosci. 2022, 14, 17. [Google Scholar] [CrossRef]
- Khan, M.A. Analysis of ion and pH effects on iron response element (IRE) and mRNA-iroin regulatory protein (IRP1) interactions. Curr. Chem. Biol. 2020, 14, 88–99. [Google Scholar] [CrossRef]
- Ma, J.; Haldar, S.; Khan, M.A.; Das Sharma, S.; Merrick, W.C.; Theil, E.C.; Goss, D.J. Fe2+ binds iron responsive element-RNA, selectively changing protein-binding affinities and regulating mRNA repression and activation. Proc. Natl. Acad. Sci. USA 2012, 109, 8417–8422. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rogers, J.T.; Lahiri, D.K. Metal and inflammatory targets for Alzheimer’s disease. Curr. Drug Targets 2004, 5, 535–551. [Google Scholar] [CrossRef]
- Netz, D.J.; Mascarenhas, J.; Stehling, O.; Pierik, A.J.; Lill, R. Maturation of cytosolic and nuclear iron-sulfur proteins. Trends Cell Biol. 2014, 24, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.-W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Deck, K.M.; Vasanthakumar, A.; Anderson, S.A.; Goforth, J.B.; Kennedy, M.C.; Antholine, W.E.; Eisenstein, R.S. Evidence that phosphorylation of iron regulatory protein 1 at Serine 138 destabilizes the [4Fe-4S] cluster in cytosolic aconitase by enhancing 4Fe-3Fe cycling. J. Biol. Chem. 2009, 284, 12701–12709. [Google Scholar] [CrossRef]
- Pantopoulos, K. Iron regulation of hepcidin through Hfe and Hjv: Common or distinct pathways? Hepatology 2015, 62, 1922–1923. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rogers, J.T.; Leiter, L.M.; McPhee, J.; Cahill, C.M.; Zhan, S.-S.; Potter, H.; Nilsson, L.N.G. Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J. Biol. Chem. 1999, 274, 6421–6431. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Cahill, C.; Balleidier, A.; Huang, C.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Novel 5′ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: Implications for down syndrome and Alzheimer’s disease. PLoS ONE 2013, 8, e65978. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Eder, P.S.; Huang, X.; Bush, A.I.; Tanzi, R.E.; Venti, A.; Payton, S.M.; Giordano, T.; Nagano, S.; et al. Alzheimer’s disease drug discovery targeted to the APP mRNA 5′Untranslated region. J. Mol. Neurosci. 2002, 19, 77–82. [Google Scholar] [CrossRef]
- Shaw, K.T.Y.; Utsuki, T.; Rogers, J.; Yu, Q.-S.; Sambamurti, K.; Brossi, A.; Ge, Y.-W.; Lahiri, D.K.; Greig, N.H. Phenserine regulates translation of β-amyloid precursor protein mRNA by a putative interleukin-1 responsive element, a target for drug development. Proc. Natl. Acad. Sci. USA 2001, 98, 7605–7610. [Google Scholar] [CrossRef] [PubMed]
- Venti, A.; Giordano, T.; Eder, P.; Bush, A.I.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T. The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5′-untranslated region. Ann. N. Y. Acad. Sci. 2004, 1035, 34–48. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Huang, X.; Lahiri, D.K.; Rogers, J.T. Novel drug targets based on metallobiology of Alzheimer’s disease. Expert Opin. Ther. Targets 2010, 14, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Ahl, M.; Cho, H.-H.; Bandyopadhyay, S.; Cuny, G.D.; Bush, A.I.; Goldstein, L.E.; Westaway, D.; Huang, X.; Rogers, J.T. RNA therapeutics directed to the non coding regions of APP mRNA, in vivo anti-amyloid efficacy of paroxetine, erythromycin, and N-acetyl cysteine. Curr. Alzheimer Res. 2006, 3, 221–227. [Google Scholar] [CrossRef]
- Chen, X.; Salehi, A.; Pearn, M.L.; Overk, C.; Nguyen, P.D.; Kleschevnikov, A.M.; Maccecchini, M.; Mobley, W.C. Targeting increased levels of APP in Down syndrome: Posiphen-mediated reductions in APP and its products reverse endosomal phenotypes in the Ts65Dn mouse model. Alzheimers Dement. 2020, 17, 271–292. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Chen, D.; Maloney, B.; Holloway, H.W.; Yu, Q.-S.; Utsuki, T.; Giordano, T.; Sambamurti, K.; Greig, N.H. The Experimental Alzheimer’s Disease Drug Posiphen [(+)-Phenserine] Lowers Amyloid-β Peptide Levels in Cell Culture and Mice. J. Pharmacol. Exp. Ther. 2007, 320, 386–396. [Google Scholar] [CrossRef]
- Maccecchini, M.; Roffman, M.; Greig, N.H. Posiphen lowers amyloid precursor protein and amyloid β as well as acetylcholinesterase levels in culture, animals and humans. Alzheimer’s Dementia 2009, 5, 247–248. [Google Scholar] [CrossRef]
- Avramovich-Tirosh, Y.; Amit, T.; Bar-Am, O.; Zheng, H.; Fridkin, M.; Youdim, M.B. Therapeutic targets and potential of the novel brain- permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer’s disease. J. Neurochem. 2007, 100, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Theil, E.C. An mRNA loop/bulge in the ferritin iron-responsive element forms in vivo and was detected by radical probing with Cu-1,10-phenantholine and iron regulatory protein footprinting. J. Biol. Chem. 2002, 277, 2373–2376. [Google Scholar] [CrossRef]
- Tibodeau, J.D.; Fox, P.M.; Ropp, P.A.; Theil, E.C.; Thorp, H.H. The up-regulation of ferritin expression using a small-molecule ligand to the native mRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 253–257. [Google Scholar] [CrossRef]
- Yang, X.; Childs-Disney, J.L.; Disney, M.D. A meditation on accelerating the development of small molecule medicines targeting RNA. Expert Opin. Drug Discov. 2023, 18, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.A. Targeting Iron Responsive Elements (IREs) of APP mRNA into Novel Therapeutics to Control the Translation of Amyloid-β Precursor Protein in Alzheimer’s Disease. Pharmaceuticals 2024, 17, 1669. https://doi.org/10.3390/ph17121669
Khan MA. Targeting Iron Responsive Elements (IREs) of APP mRNA into Novel Therapeutics to Control the Translation of Amyloid-β Precursor Protein in Alzheimer’s Disease. Pharmaceuticals. 2024; 17(12):1669. https://doi.org/10.3390/ph17121669
Chicago/Turabian StyleKhan, Mateen A. 2024. "Targeting Iron Responsive Elements (IREs) of APP mRNA into Novel Therapeutics to Control the Translation of Amyloid-β Precursor Protein in Alzheimer’s Disease" Pharmaceuticals 17, no. 12: 1669. https://doi.org/10.3390/ph17121669
APA StyleKhan, M. A. (2024). Targeting Iron Responsive Elements (IREs) of APP mRNA into Novel Therapeutics to Control the Translation of Amyloid-β Precursor Protein in Alzheimer’s Disease. Pharmaceuticals, 17(12), 1669. https://doi.org/10.3390/ph17121669