Abstract
This comprehensive review examines the therapeutic potential of metformin, a well-established diabetes medication, in treating neurodegenerative disorders. Originally used as a first-line treatment for type 2 diabetes, recent studies have begun investigating metformin’s effects beyond metabolic disorders, particularly its neuroprotective capabilities against conditions like Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and multiple sclerosis. Key findings demonstrate that metformin’s neuroprotective effects operate through multiple pathways: AMPK activation enhancing cellular energy metabolism and autophagy; upregulation of antioxidant defenses; suppression of inflammation; inhibition of protein aggregation; and improvement of mitochondrial function. These mechanisms collectively address common pathological features in neurodegeneration and neuroinflammation, including oxidative stress, protein accumulation, and mitochondrial dysfunction. Clinical and preclinical evidence supporting metformin’s association with improved cognitive performance, reduced risk of dementia, and modulation of pathological hallmarks of neurodegenerative diseases is critically evaluated. While metformin shows promise as a therapeutic agent, this review emphasizes the need for further investigation to fully understand its mechanisms and optimal therapeutic applications in neurodegenerative diseases.
1. Introduction
Metformin, a drug from the biguanide group, has garnered increasing attention for its robust potential in various disease treatments. During the Middle Ages, Galega officinalis (goat’s rue) was used for diabetes, which later led to the isolation of guanidine and discovery of its blood sugar-lowering effects in 1918. This ultimately resulted in the synthesis of metformin, published in Journal of the Chemical Society, Transactions in 1922, and its successful clinical trials by Jean Sterne in 1957 [1,2,3]. The original use of metformin was as a first-line treatment for type 2 diabetes. In the following years, additional benefits of metformin treatment were observed, such as the prevention of cancer development, reduction in the risk of cardiovascular diseases, life prolongation, and improving fertility in women with polycystic ovary syndrome (PCOS). Recent studies have begun to investigate its effects beyond metabolic disorders, with a focus on its protective potential against neurodegenerative diseases (NDDs) like Parkinson’s disease (PD) and Alzheimer’s disease (AD). Additionally, new research suggests that it may help treat multiple sclerosis by lowering oxidative stress and inflammation in the central nervous system [4,5,6]. Preliminary research suggests that metformin’s ability to modulate mitochondrial function and reduce oxidative stress may offer therapeutic potential in Huntington’s disease (HD), though clinical evidence remains limited. Clinical studies suggest that metformin may improve not only mortality, but also cognitive function and memory in diabetic patients, and is associated with a lower risk of developing dementia [7,8,9,10,11]. It exerts its beneficial effects through a multifaceted mechanism, including AMP-activated protein kinase (AMPK) activation, enhanced antioxidant defenses, and potent anti-inflammatory actions [4,12]. These effects, coupled with its potential to modulate the gut microbiome and promote neurogenesis, suggest a significant impact on cognitive function [13,14]. Clinical and preclinical evidence supports its association with improved cognitive performance, a reduced risk of dementia, and advancement of pathological hallmarks of neurodegenerative diseases. Overall, even though metformin shows promise, more investigation is required to completely understand its processes and therapeutic uses in neurology [15,16]. This review will delve into the multifaceted mechanisms of metformin’s neuroprotective effects and critically evaluate the existing evidence to inform future research directions.
This review article focuses on potential use of metformin in Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and multiple sclerosis (MS). These conditions collectively encompass key neurodegenerative processes including protein misfolding and aggregation (AD, PD, HD), mitochondrial dysfunction (particularly in PD), oxidative stress (common across all), and neuroinflammation (prominent in MS). Furthermore, these diseases represent different anatomical patterns of neurodegeneration: cortical (AD), subcortical (PD), both cortical and subcortical (HD), and demyelinating (MS). This selection provides a comprehensive framework to evaluate metformin’s multifaceted mechanisms across diverse neurodegenerative pathways, while maintaining a focused analysis of conditions with substantial clinical and preclinical research data.
2. Metformin: Basic Pharmacology and Mechanisms
Metformin (N, N-dimethylbiguanide) exhibits complex pharmacokinetics, characterized by slow absorption in the upper small intestine via organic cation transporters (OCT1/3, PMAT) [17,18]. Oral bioavailability ranges from 50 to 60%, with peak plasma concentrations reached within 1–3 h. The drug undergoes minimal metabolism and is primarily eliminated unchanged through renal tubular secretion, with a plasma half-life of 4–9 h [19,20,21]. At therapeutic doses (1500–2500 mg/day), plasma concentrations typically range 1–4 μg/mL. The drug accumulates preferentially in the liver through OCT1-mediated uptake, achieving hepatic concentrations 3–5 times higher than plasma. This tissue distribution pattern aligns with its primary site of action [19,22].
Metformin’s primary effect is mediated through inhibition of hepatic gluconeogenesis [23,24]. While it exhibits effects on cellular metabolism, its primary molecular target has been identified as mitochondrial glycerophosphate dehydrogenase (GPD2), rather than complex I inhibition or AMPK activation which only occur at supra-pharmacological concentrations [25,26,27]. The drug’s action involves modulation of the cellular redox balance, where GPD2 inhibition affects the shuttle of redox equivalents from cytosolic to mitochondrial nicotinamide adenine dinucleotide (NAD) + hydrogen (H) (NADH) [28,29,30]. This leads to a reduced supply of dihydroxyacetone phosphate from glycerol and lower adenosine triphosphate (ATP) levels due to a decreased redox equivalent transfer into the mitochondrial matrix [25,28,31]. Additionally, the elevated NADH/NAD+ ratio in the cytosol inhibits lactate dehydrogenase, reducing the conversion of lactate to the gluconeogenic precursor pyruvate [25,32]. While this mechanism effectively reduces hepatic glucose production, it may also impact lactate metabolism, potentially leading to lactate acidosis [33,34]. The drug’s effects on other hepatic metabolic functions, particularly regarding hepatic steatosis, remain under investigation with controversial results [35].
2.1. Chemical Structure and Properties
The chemical structure of metformin contains two linked guanidine groups forming a biguanide scaffold, whose planar structure enables specific molecular interactions, e.g., hydrogen bonding capacity and charge distribution [25,36]. Moreover, the characteristic formation of metformin molecules acquires resonance stabilization across N-C bonds and delocalizes positive charge distribution [37,38]. The structure of metformin is illustrated in Figure 1. Metformin’s effectiveness is primarily driven by its hydrophilic nature and positive charge at physiological pH, which enables efficient transport through organic cation transporters and cellular membranes. Its small molecular size (129.16 g/mol) and planar biguanide structure, combined with high chemical stability and minimal protein binding, allow for predictable pharmacokinetics and sustained therapeutic action [19,39]. Metformin exhibits pH-dependent solution behavior, existing primarily as a cation at physiological pH with multiple possible protonation states that critically affect its tissue distribution and interaction with membrane transporters [40,41,42]. The compound exhibits good photostability and a defined thermal stability range, but is sensitive to humidity. Therefore, specific storage conditions are required, including protection from heat and moisture, controlled relative humidity, and appropriate packaging to maintain potency [19,37,43]. These physicochemical properties fundamentally determine metformin’s pharmaceutical performance and therapeutic efficacy.
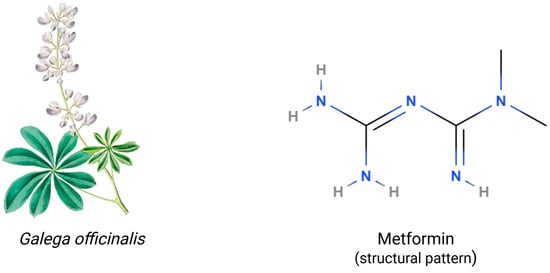
Figure 1.
Structural and botanical characterization of metformin (C4H11N5). Galega officinalis (Fabaceae), the plant from which metformin was originally derived, is characterized by palmately compound leaves and racemose inflorescences. The molecular structure of metformin shows a structural pattern that highlights the biguanide scaffold with terminal N-methylation. Image by rawpixel.com on Freepik. Created in BioRender. Kciuk, M. (2025) https://BioRender.com/o18q303 (accessed on 9 March 2025).
Metformin’s ability to cross the blood–brain barrier (BBB) is relatively limited, with only 10–20% of the drug reaching the central nervous system [44]. Levodopa, a known drug used in Parkinson’s disease, has effective penetration, but when administered alone its peripheral decarboxylation to dopamine by aromatic amino acid decarboxylase is rapid, reducing the amount of levodopa available for BBB transport. However, levodopa crosses the BBB effectively when combined with peripheral decarboxylase inhibitors like carbidopa or benserazide, which prevent its premature metabolism [45]. In contrast, anti-epileptic drugs (AEDs) like phenytoin and carbamazepine achieve significantly higher penetration, approximately 50–60% and 40–50%, respectively, due to their lipophilic properties. Valproic acid, another AED, shows moderate BBB penetration (similar to metformin and levodopa), at 10–20%. This highlights the variability in CNS drug delivery, with lipophilicity and specific transporter interactions playing crucial roles [46,47]. Metformin’s restricted penetration is primarily due to its hydrophilic nature and reliance on specific transport mechanisms. The drug primarily depends on organic cation transporters (OCTs), particularly OCT1 and OCT3, as well as the plasma membrane monoamine transporter (PMAT) to cross the BBB. However, these transporters have relatively low expression at the BBB compared to their abundance in peripheral tissues, such as the liver and kidneys [25,48]. Additionally, P-glycoprotein (P-gp) efflux transporters actively pump metformin back into the bloodstream, further limiting its brain accumulation [49,50]. Levodopa, utilizing the large neutral amino acids transporter small subunit 1 (LAT1) transporter, efficiently crosses the BBB, contrasting with metformin’s limited permeability via organic cation transporters [51,52]. Anti-epileptic drugs such as gabapentin can leverage specific transporters like LAT1, while others may be hindered by P-glycoprotein (P-gp)-mediated efflux. Conversely, lipophilic small molecules often achieve a wide CNS distribution through passive diffusion, circumventing the need for specific transporters. These observations underscore the importance of considering drug physicochemical properties and transporter affinities in the design and delivery of effective CNS therapeutics [53,54,55].
Metformin shows potential for treating neurodegenerative diseases, but its hydrophilic nature and limited blood–brain barrier penetration necessitate prolonged exposure and repeated dosing. Despite this, it has a well-established safety profile from decades of clinical use in T2DM. However, its long-term safety, particularly in patients with or without metabolic syndrome comorbidities, requires careful evaluation [56,57,58].
Metformin is generally well-tolerated, with transient gastrointestinal side effects like nausea, diarrhea, and abdominal discomfort, which can be managed through dose titration or extended-release formulations. It does not cause hypoglycemia when used alone, making it safer than many other antidiabetic drugs [59,60,61]. However, rare but serious complications like metformin-associated lactic acidosis (MALA) must be considered. MALA is extremely rare, occurring in 3–10 cases per 100,000 patient-years, primarily in those with renal impairment, hepatic dysfunction, or hypoxia-related conditions. Regular renal function monitoring is recommended for long-term use [34,62].
Another concern is vitamin B12 deficiency due to impaired absorption, which can lead to peripheral neuropathy or cognitive impairment, especially in those at risk of NDDs. Chronic metformin users have lower B12 levels than non-users, with deficiency rates of 5–10%. Regular monitoring and supplementation can help mitigate this risk [63,64].
Higher doses or longer treatment durations may be needed to achieve therapeutic CNS concentrations for NDDs like Alzheimer’s and Parkinson’s, raising concerns about systemic toxicity, particularly in elderly populations. While metformin is generally safe for long-term use in diabetics, its safety in non-diabetics for neuroprotection requires further study [65,66]. Emerging drug delivery strategies, such as nanocarrier-based formulations or intranasal delivery, aim to enhance BBB penetration while minimizing systemic exposure. These approaches could optimize therapeutic efficacy while reducing the risks associated with high-dose regimens [67,68].
Despite this limited BBB penetration, metformin still exhibits significant neuroprotective effects through both direct central nervous system actions (via the fraction that does cross the BBB) and indirect peripheral mechanisms that influence brain function through systemic metabolic and inflammatory pathways [69,70]. To overcome these limitations, current research focuses on developing modified formulations with enhanced BBB penetration, including lipophilic derivatives, nanocarrier delivery systems, and alternative administration routes such as intranasal delivery [71,72,73].
2.2. Traditional Use in Diabetes
The biguanide metformin represents the established first-line pharmacological intervention for type 2 diabetes mellitus (T2DM), with extensive clinical validation spanning several decades [74,75]. Clinical efficacy manifests primarily through significant reductions in glycated hemoglobin (HbA1c), typically achieving 1.0–2.0 percentage point decrements in controlled studies [76]. This therapeutic agent demonstrates particular utility in treatment-naive patients, especially those presenting with obesity or metabolic syndrome comorbidities [77]. The compound’s clinical profile is distinguished by several advantageous characteristics, e.g., neutral or favorable effects on body mass index, minimal risk of hypoglycemic events, and robust tolerability in most patient populations [78,79,80]. The medication’s extensive clinical history and favorable benefit–risk profile have established it as the cornerstone of T2DM pharmacological management. Recent investigations have suggested potential pleiotropic benefits beyond glycemic control [81,82].
2.3. Key Molecular Mechanisms
The molecular basis for metformin’s therapeutic effects centers on multiple interconnected pathways. While AMP-activated protein kinase activation serves as an important mediator, metformin also exerts significant AMPK-independent effects [42,83]. This complex network of molecular actions initiates a cascade of cellular responses that collectively improve glucose homeostasis [84]. The key mechanisms of metformin action include the suppression of hepatic glucose production, enhancement of peripheral insulin sensitivity, modulation of intestinal glucose metabolism, and regulation of cellular energy metabolism, as is summarized in Table 1.

Table 1.
Description of key mechanisms of metformin. Metformin enhances glucose metabolism through multiple mechanisms, including suppression of hepatic glucose production, increased peripheral insulin sensitivity via GLUT4 translocation, and modulation of intestinal glucose transport and gut microbiome. At the cellular level, it regulates energy metabolism by altering NADH/NAD+ ratios and activating AMPK pathways, while affecting mTOR signaling. It also provides anti-inflammatory benefits through reduction of pro-inflammatory cytokines and oxidative stress.
These mechanisms have therapeutic implications, especially for metformin timing and dosing. While metformin shows limited blood–brain barrier penetration, its beneficial effects on neurodegeneration appear to work through both direct and indirect pathways [44,104]. The indirect effects stem from its action on peripheral tissues—particularly liver and gut—where it reduces systemic inflammation, improves insulin sensitivity, and decreases oxidative stress, all of which positively impact brain health [13,105]. Additionally, metformin directly alleviates endoplasmic reticulum (ER) stress in neurons by activating the AMPK pathway and enhancing the unfolded protein response (UPR), which is particularly significant for neurodegenerative conditions where protein misfolding plays a central role [106,107,108]. These molecular mechanisms, along with metformin’s anti-inflammatory properties (which are particularly relevant in neurodegeneration), operate synergistically to improve glycemic control while simultaneously affecting multiple metabolic pathways involved in energy homeostasis [56,109]. The complex interplay between these mechanisms contributes to metformin’s broad therapeutic efficacy and emerging pleiotropic effects, including reduced cancer risk, improved cardiovascular outcomes, increased longevity, and neuroprotective properties [25,110,111,112].
Recent metformin research has significantly expanded our understanding of its mechanisms and therapeutic potential. Beyond traditional AMPK activation, studies reveal metformin’s influence on lysosomal pathways, redox states, and its interaction with specific genetic variants that impact individual responses. This has led to the identification of novel molecular targets and a deeper understanding of its glucose-lowering effects [113]. Metformin’s impact extends to modulating the gut microbiota, influencing bile acid metabolism, and affecting incretin secretion, further contributing to its multifaceted glucose-lowering effects. Specifically, the article reinforces metformin’s role in altering gut microbial composition, which in turn impacts glucose homeostasis. Furthermore, investigations into metformin’s benefits beyond diabetes, including cardiovascular protection and anti-inflammatory properties, are broadening its clinical applications. Ongoing research focuses on leveraging genetic profiling for personalized metformin therapy, exploring new drug targets based on its diverse actions, and even considering innovative approaches like mitochondrial transfer therapies to combat diabetic mitochondrial dysfunction [25,114,115].
3. Molecular Mechanism of Metformin Action in the Liver
There is a general consensus that the glucose-lowering impact in T2DM patients is primarily mediated by the inhibition of hepatic gluconeogenesis. However, after decades of research, the mechanism by which metformin suppresses this process remains contentious. Widely studied mechanisms of metformin action, such as complex I inhibition (reviewed by Fontaine in 2018) leading to 5′AMPK activation, have only been observed at supra-pharmacological (>1 mM) metformin concentrations, which do not occur in the clinical setting [23,116]. Furthermore, animal models imply that AMPK is not required for metformin’s anti-hyperglycemic actions, as mice with liver-specific genetic deletions of AMPK or the upstream kinase ‘liver kinase B1’ can still reduce hepatic gluconeogenesis in response to metformin [106,117,118].
Based on the discovery that metformin changes the cellular redox equilibrium, various groups have described a redox-dependent mode of action: Metformin inhibits the enzyme mitochondrial glycerophosphate dehydrogenase (GPD2), which, along with cytosolic glycerophosphate dehydrogenase (GPD1), catalyzes the transfer of redox equivalents from cytosolic to mitochondrial NADH during the reduction of glycerophosphate to dihydroxyacetone phosphate, an intermediate in the gluconeogenetic pathway. Reduced supply of dihydroxyacetone phosphate from glycerol, as well as a decrease in ATP levels caused by reduced transfer of redox equivalents into the mitochondrial matrix, limit gluconeogenesis [25,26,28].
Furthermore, an increased NADH/NAD+ cytosolic ratio inhibits the enzyme lactate dehydrogenase, which converts lactate to the gluconeogenetic precursor pyruvate. A possible negative effect of this method of action is impaired lactate recycling to glucose (Cori cycle), which increases the risk of lactic acidosis [33,119]. It is unknown whether metformin can have a significant temporary effect on other metabolic activities of the liver. In vitro and in vivo studies on metformin’s capacity to alleviate hepatic steatosis have yielded conflicting results [35].
4. Neurodegenerative Diseases: Common Pathological Mechanisms
While distinct in their primary manifestations, neurodegenerative conditions such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease share several key pathological mechanisms. Multiple sclerosis, while primarily an autoimmune inflammatory condition of the central nervous system, shares several of these same pathological mechanisms that ultimately contribute to neurodegeneration. Despite their different etiologies, all four conditions demonstrate overlapping features such as oxidative stress, mitochondrial dysfunction, neuroinflammation, and eventual neuronal loss [120,121,122].
Activated microglia and astrocytes release inflammatory mediators that can harm neurons and myelin in all four illnesses, a process known as neuroinflammation. There are key functions for protein misfolding and aggregation; amyloid beta and tau in Alzheimer’s disease, α-synuclein in Parkinson’s disease, mutant huntingtin in Huntington’s disease, and maybe misfolded proteins that contribute to autoimmune reactions in multiple sclerosis [123,124]. All of these disorders are characterized by oxidative stress and mitochondrial malfunction, which result in energy deficiencies and the generation of toxic reactive oxygen species that destroy cellular components [125,126]. Axonal transport disruption and synaptic dysfunction are also features of each illness, although they are caused by diverse molecular mechanisms: tau in Alzheimer’s, α-synuclein in Parkinson’s, huntingtin in Huntington’s, and inflammation in multiple sclerosis [127,128].
Each disease is characterized by the death of particular neuronal populations, such as oligodendrocytes with subsequent neuronal damage in multiple sclerosis, dopaminergic neurons in Parkinson’s, striatal and cortical neurons in Huntington’s, and hippocampus and cortical neurons in Alzheimer’s [129,130]. Lastly, all of these disorders exhibit signs of disturbed proteostasis and compromised cellular quality control systems, such as autophagy pathways and the ubiquitin–proteasome system [131].
4.1. Protein Aggregation
In neurodegenerative illnesses, protein aggregation is a key pathogenic process. These illnesses are typified by the build-up of misfolded proteins that congregate and cause neurodegeneration and cellular malfunction [132].
Usually, this process starts when proteins change from their original structural configuration for a variety of reasons, including oxidative stress, post-translational changes, or genetic abnormalities that impact protein homeostasis. The aggregation pathway typically occurs in phases: first, hydrophobic areas that are ordinarily hidden inside the protein’s tertiary structure are exposed by first protein misfolding, resulting in the production of oligomeric species that act as nucleation sites for additional aggregation [133,134,135]. After forming more ordered structures like protofibrils, these oligomers can develop into amyloid fibrils, which are characterized by their abundance of β-sheets. These protein aggregates may have prion-like characteristics, spreading sickness to nearby cells via methods of cell-to-cell transmission and accelerating the course of the illness. “Seeding” describes how misfolded protein “templates” spread between cells, converting normal proteins into the misfolded state and triggering a chain reaction of aggregation [136,137,138]. This spread occurs via exosomes, tunneling nanotubes, and synaptic transmission. The resulting aggregates disrupt key cellular processes—protein quality control, mitochondrial function, axonal transport, and synaptic transmission—ultimately causing neuronal dysfunction and death [139].
Notably, different neurodegenerative conditions exhibit disease-specific protein aggregates: α-synuclein in Parkinson’s disease forms Lewy bodies, amyloid beta and tau in Alzheimer’s disease produce extracellular plaques and intracellular neurofibrillary tangles, respectively, while the huntingtin protein with expanded polyglutamine repeats aggregates in Huntington’s disease. While the exact misfolded protein in multiple sclerosis (MS) is not yet identified, research indicates that protein misfolding might contribute to the disease’s development. Studies have found soluble oligomers in the brains and cerebrospinal fluid of MS patients, similar to those seen in Alzheimer’s disease [140,141,142,143,144].
Protein aggregation is caused by intricate and varied molecular processes that involve both environmental and cellular influences [145]. Under normal circumstances, cellular quality control systems—such as the autophagy–lysosomal pathway, the ubiquitin–proteasome system, molecular chaperones, and the endoplasmic reticulum stress response system, including the unfolded protein response—are essential in preventing protein aggregation and maintaining proteostasis [146,147]. However, when people age or experience sickness, these defenses may be overtaxed or stop working, leading to the accumulation of misfolded proteins in the ER lumen, which triggers ER stress, activates stress sensors (IRE1α, PERK, and ATF6), and initiates the UPR signaling cascade. If this stress persists and cannot be resolved, it can lead to chronic ER stress, further compromising protein homeostasis and potentially triggering cell death pathways [148,149]. Phosphorylation, ubiquitination, and acetylation are examples of post-translational changes that can have a major impact on the toxicity and propensity of proteins to aggregate. Moreover, oxidative stress induction and direct protein binding are two of the ways that metal ions—in particular, copper, zinc, and iron—have been linked to speeding up protein aggregation [150,151,152,153].
Protein aggregate toxicity arises from multiple mechanisms, which is illustrated in Figure 2. Oligomers, not fibrils, are the main culprits, disrupting cell membranes, calcium balance, and triggering inflammation. They also sequester essential proteins (e.g., transcription factors, chaperones), impairing cellular function [154,155,156]. Aggregates activate stress responses, causing chronic stress and cell death. Their accumulation fuels neuroinflammation via microglial and astrocyte activation, worsening neurodegeneration [157,158,159,160].
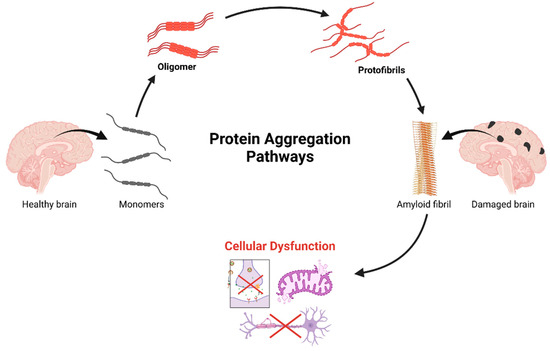
Figure 2.
A cascade of progressive protein aggregation in neurodegenerative pathogenesis. The process initiates in the healthy brain with native protein monomers, which undergo conformational changes leading to oligomerization. These oligomeric intermediates further aggregate into protofibrils, ultimately assembling into highly ordered amyloid fibrils. This terminal aggregation state correlates with observable neuropathology, characterized by distinct patterns of brain damage. The downstream of cellular consequences of protein aggregation, includes disruption of essential neuronal functions: protein quality control mechanisms, mitochondrial homeostasis, axonal transport efficiency, and synaptic transmission integrity. This cascade represents a crucial mechanistic framework in understanding the molecular basis of neurodegenerative disorders and identifies potential therapeutic intervention points along the aggregation pathway. Created in BioRender. Kciuk, M. (2025) https://BioRender.com/g29m614 (accessed on 19 February 2025).
4.2. Oxidative Stress
Oxidative stress (OS) occurs when there is an imbalance between the production of reactive oxygen species (ROS) and the body’s ability to neutralize them with antioxidants. ROS, which include free radicals like superoxide anion (O2−), hydroxyl radical (OH·), and non-free radicals such as hydrogen peroxide (H2O2), are produced during normal cellular metabolism, particularly in the mitochondria through the electron transport chain [161,162]. Under physiological conditions, ROS play crucial roles in cell signaling and homeostasis; however, excessive ROS can lead to cellular damage by oxidizing proteins, lipids, and nucleic acids [125,163].
Sources of ROS include mitochondrial respiration, inflammation, and exogenous factors like pollution and radiation, but interestingly, hyperglycemia as well [164,165]. In neurodegenerative conditions, mitochondrial dysfunction and neuroinflammation lead to increased ROS generation. These reactive species damage cellular components, including proteins, lipids, and DNA. For example, in Alzheimer’s disease, amyloid beta oligomers induce oxidative stress, leading to lipid peroxidation and neuronal membrane damage. Furthermore, ROS activate glial cells, triggering the release of pro-inflammatory cytokines that exacerbate neuronal injury. This creates a vicious cycle where inflammation amplifies oxidative stress, further promoting neuronal damage. Protein misfolding and aggregation, as seen in Parkinson’s disease with alpha-synuclein, are also exacerbated by oxidative modifications. The accumulation of oxidized DNA bases further impairs neuronal function and survival. Understanding these intricate mechanisms provides crucial insights for developing therapeutic strategies aimed at enhancing antioxidant defenses and targeting specific pathways involved in ROS-mediated neuronal damage, with the goal of mitigating disease progression [166,167].
4.3. Neuroinflammation
Neuroinflammation, a complex immune response in the central nervous system, is crucial in the development and progression of neurodegenerative diseases. It involves the activation of microglia and astrocytes, which undergo significant changes in form and function. Microglia, the brain’s immune cells, shift from a resting state to an active state, increasing their ability to engulf debris, producing pro-inflammatory molecules, and altering their gene expression. This activation is triggered by factors like misfolded proteins, cellular debris, and damage-associated molecular patterns (DAMPs), all linked to neurodegeneration [168,169,170,171].
Similarly, astrocytes experience reactive astrogliosis, which is characterized by pro- and anti-inflammatory factor production, cellular hypertrophy, and elevation of glial fibrillary acidic protein (GFAP) [172]. In the setting of neurodegeneration, the astrocytic response involves the creation of a glial scar, which can have both beneficial and harmful effects. Reactive oxygen species, chemokines, nitric oxide, pro-inflammatory cytokines (IL-1β, TNF-α, and IL-6), and other inflammatory mediators are released by these activated glial cells, resulting in a neuroinflammatory milieu that can have a major effect on neuronal survival and function [173,174].
An important factor in neuroinflammation is the blood–brain barrier, whose disruption frequently contributes to and results from inflammatory processes. Peripheral immune cells such as T lymphocytes, monocytes, and neutrophils can infiltrate when the BBB is compromised, which exacerbates the inflammatory response [157,175]. Through intricate signaling networks, these invading cells engage with native CNS cells, intensifying the inflammatory cascade and possibly worsening tissue damage. In addition to causing the extravasation of plasma proteins and the activation of complement cascades, the compromise of BBB integrity also contributes to the inflammatory environment [176].
Neuroinflammation features complex molecular pathways. Pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs), detect pathogen-associated molecular patterns (PAMPs), triggering the activation of inflammatory transcription factors like NF-κB and AP-1. This, in turn, leads to the increased expression of pro-inflammatory genes. Inflammasome complexes, particularly NLRP3, play a critical role in processing and releasing mature IL-1β and IL-18, further propagating the inflammatory cascade [177,178,179]. Chronic neuroinflammation establishes a self-sustaining cycle of cellular damage and immune activation. The persistent activation of glial cells, including microglia and astrocytes, results in the sustained production of neurotoxic factors, such as pro-inflammatory cytokines, reactive oxygen species, and excitotoxic molecules like glutamate. This chronic inflammatory state can significantly impair synaptic function, disrupt neuronal networks, and ultimately contribute to neurodegeneration [180,181,182]. Furthermore, the inflammatory environment can influence protein aggregation and clearance mechanisms, potentially exacerbating the accumulation of pathogenic protein species characteristic of various neurodegenerative disorders, such as amyloid beta in Alzheimer’s disease or alpha-synuclein in Parkinson’s disease. This interplay between inflammation and protein aggregation creates a vicious cycle that fuels disease progression [177,183]. This process is depicted in Figure 3.
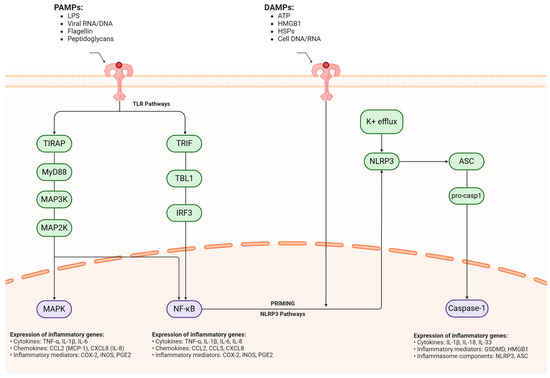
Figure 3.
Molecular mechanisms of neuroinflammatory signaling cascades in neurodegeneration. Converging molecular pathways mediating neuroinflammatory responses in neurodegenerative conditions have two primary inflammatory triggers: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), which initiate distinct but interconnected signaling cascades across the cellular membrane. PAMPs (pathogen-associated molecular patterns), including LPS, viral RNA/DNA, flagellin, and peptidoglycans, initiate inflammation through TLR-mediated signaling. This process branches into two main cascades: TIRAP–MyD88-MAP3K–MAP2K–MAPK and TRIF–TBL1–IRF3. Both pathways converge to activate NF-κB, triggering the expression of pro-inflammatory genes. These include critical cytokines (TNF-α, IL-1β, IL-6, IL-8), chemokines (CCL2, CXCL8), and inflammatory mediators (COX-2, iNOS, PGE2). Simultaneously, DAMPs (damage-associated molecular patterns) such as ATP, HMGB1, HSPs, and cellular DNA/RNA activate a separate inflammatory cascade through the NLRP3 inflammasome. This process begins with potassium efflux, which triggers NLRP3 activation, followed by ASC recruitment and pro-caspase-1 processing. The end result is active caspase-1, which promotes the expression of inflammatory mediators, particularly GSDMD and HMGB1. PAMPs = pathogen-associated molecular patterns, LPS = lipopolysaccharide, RNA = ribonucleic Acid, DNA = deoxyribonucleic acid, TLR = toll-like receptor, TIRAP = TIR domain-containing adaptor protein, MyD88 = myeloid differentiation primary response 88, MAP3K = mitogen-activated protein kinase kinase kinase, MAP2K = mitogen-activated protein kinase kinase, MAPK = mitogen-activated protein kinase, TRIF = TIR domain-containing adapter-inducing interferon-β, TBL1 = transducin beta-like 1, IRF3 = interferon regulatory factor 3, NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells, TNF-α = tumor necrosis factor alpha, IL-1β = interleukin-1 beta, IL-6 = interleukin-6, IL-8 = interleukin-8, CCL2 = C-C motif chemokine ligand 2, CXCL8 = C-X-C motif chemokine ligand 8, COX-2 = cyclooxygenase-2, iNOS = inducible nitric oxide synthase, PGE2 = prostaglandin E2, DAMPs = damage-associated molecular patterns, ATP = adenosine triphosphate, HMGB1 = high mobility group box 1, HSPs = heat shock proteins, NLRP3 = NOD-like receptor protein 3, ASC = apoptosis-associated speck-like protein containing a CARD, GSDMD = gasdermin D, K+ = potassium. Created in BioRender. Kciuk, M. (2025) https://BioRender.com/zb9c38p (accessed on 27 March 2025).
4.4. Mitochondrial Dysfunction
Given the high energy requirements of neurons and their dependence on oxidative phosphorylation for ATP synthesis, mitochondrial dysfunction is a key pathogenic factor in neurodegenerative diseases. In addition to their canonical function in bioenergetics, these organelles are key modulators of apoptotic pathways, redox signaling, and calcium homeostasis inside cells. Numerous interrelated pathways contribute to the reduced function of mitochondria in neurodegenerative diseases, resulting in a complex network of cellular disturbances that ultimately lead to neuronal death [184,185].
One of the main signs of mitochondrial impairment is the malfunction of the electron transport chain (ETC), which is typified by decreased activity of respiratory chain complexes, especially Complex I and IV. Reduced ATP synthesis and increased electron leakage are the results of this malfunction, which raises the creation of reactive oxygen species (ROS). A vicious loop is created by the increased generation of ROS, which can exacerbate the initial malfunction by further damaging proteins, lipids, and mitochondrial DNA (mtDNA). Because of its close proximity to the ETC and its lack of adequate repair mechanisms, mtDNA is especially susceptible to oxidative damage, which can result in accumulating mutations that further impair respiratory chain function [186,187,188].
Mitochondrial quality control is essential for neuronal health and becomes disrupted in neurodegenerative diseases. The balance between mitochondrial fusion and fission proteins becomes impaired, affecting mitochondrial morphology and function [189,190]. This disruption compromises mitophagy, the process that removes damaged mitochondria, as demonstrated by PINK1 and Parkin mutations in Parkinson’s disease. Additionally, mitochondrial calcium regulation becomes dysfunctional, leading to dangerous cytoplasmic calcium accumulation that triggers excitotoxicity and cell death pathways, particularly through the mitochondrial permeability transition pore [191,192].
Mitochondrial transport along neurons, crucial for synaptic function, is also compromised in these diseases. This disruption, affecting proteins like kinesin and dynein, causes energy deficits at synapses and contributes to synapse loss. Disease-specific protein aggregates like α-synuclein and amyloid beta can directly impair mitochondrial function, creating a destructive feedback loop [193,194]. These insights have led to therapeutic strategies including antioxidants, biogenesis enhancers, and modulators of mitochondrial dynamics. Early intervention and reliable biomarkers are crucial for treatment success. However, developing effective therapies remains challenging due to the complexity of mitochondrial dysfunction and diverse neuronal needs [195].
4.5. Insulin Resistance in the Brain
An increasingly acknowledged pathogenic mechanism causing neurodegenerative diseases, including Alzheimer’s disease, is insulin resistance in the brain. As a result, some researchers have proposed the moniker “type 3 diabetes”. Despite having enough insulin present, brain insulin resistance is defined by a decreased response to insulin signaling in neural tissues. This impairs cellular metabolism, synaptic plasticity, and cognitive function. Complex connections between cellular stress responses, inflammatory processes, and metabolic dysfunction underlie the molecular causes of central insulin resistance [5,196,197].
Brain insulin signaling, crucial for neuronal function, involves activating insulin receptors (IR) and IGF-1R, leading to IRS protein phosphorylation and downstream activation of PI3K/Akt and MAPK pathways, affecting glucose metabolism, synaptic plasticity, and cell survival. In insulin resistance, this cascade is compromised due to increased serine/threonine phosphorylation of IRS proteins, reduced tyrosine phosphorylation, and decreased pathway activation [198,199]. Several interconnected processes underlie brain insulin resistance. Inflammatory mediators (TNF-α, IL-6) interfere with signaling via JNK and IKK-β, promoting inhibitory IRS phosphorylation. Oxidative stress, often elevated in neurodegeneration, damages signaling components and creates AGEs, further impairing insulin sensitivity. Lipotoxicity, from aberrant lipid metabolism and ceramide/diacylglycerol accumulation, activates protein kinase C isoforms that disrupt insulin signaling [200,201].
Insulin resistance in the brain has wide-ranging and complex effects. Energy metabolism is compromised by reduced glucose transporter 4 (GLUT4) translocation and decreased glucose uptake in brain cells caused by impaired insulin signaling. Cellular repair processes, synaptic transmission, and neurotransmitter synthesis can all be impacted by this metabolic abnormality [202,203]. Additionally, by decreasing the activation of insulin-dependent protein phosphatases and increasing the activity of glycogen synthase kinase 3β (GSK-3β), insulin resistance can hinder the clearance of amyloid beta and promote tau hyperphosphorylation, directly connecting insulin resistance to the pathological hallmarks of Alzheimer’s disease [204,205].
Peripheral and central insulin resistance share a bidirectional relationship. Systemic metabolic disorders like obesity and type 2 diabetes promote brain insulin resistance through inflammation, oxidative stress, and disrupted adipokine signaling. The blood–brain barrier plays a crucial role, as metabolic stress compromises its function, affecting insulin transport and increasing inflammatory mediator infiltration. Brain insulin resistance impairs synaptic plasticity and cognitive function by disrupting NMDA receptors, AMPA receptor trafficking, and neurotrophic signaling, while also affecting key neurotransmitter systems [206,207].
Various therapeutic approaches have emerged, from insulin sensitizers to intranasal insulin delivery, while lifestyle changes may improve brain insulin signaling. Developing biomarkers for monitoring and understanding the complex interactions with other neurodegenerative mechanisms remains crucial for creating effective treatments [208] 39317178.
4.6. Diabetes Mellitus and Neurodegeneration: Shared Pathophysiological Mechanisms
The connection between diabetes mellitus and neurodegenerative diseases highlights shared pathophysiological mechanisms, explaining why antidiabetic drugs like metformin hold therapeutic promise for various neurodegenerative disorders [209,210]. It is also crucial to note that Alzheimer’s disease has been referred to as type 3 diabetes, which stems from shared molecular mechanisms and some risk factors. Chronic hyperglycemia in diabetes triggers neurotoxic processes such as oxidative stress, advanced glycation end product formation, and inflammation, which closely resemble pathological mechanisms in neurodegeneration [211,212]. These metabolic disruptions harm cellular components, accelerate protein misfolding and aggregation, and create a neurochemical environment favorable for neurodegeneration, as evidenced by the significantly higher risk of Alzheimer’s and Parkinson’s disease in diabetic patients. Central insulin resistance further strengthens the link to neurodegenerative pathology by impairing brain insulin signaling, which is crucial for glucose metabolism, synaptic plasticity, neuronal survival, and cognitive function [213,214,215]. The inflammatory interplay between these disorders is especially crucial, as systemic inflammation in diabetes activates microglia, fostering a neuroinflammatory environment similar to that seen in multiple neurodegenerative diseases [216,217]. Another key link is vascular pathology, where diabetic microangiopathy disrupts cerebral circulation, weakens the blood–brain barrier integrity, and accelerates neurodegenerative progression [4,218]. Clinically, these overlapping mechanisms result in faster cognitive decline and an earlier onset of neurodegenerative symptoms in diabetic individuals [219,220]. This biological convergence provides a mechanistic rationale for metformin’s potential to mitigate neurodegenerative processes by targeting multiple shared pathological pathways, paving the way for repurposing this well-established antidiabetic drug for neurodegenerative conditions.
5. Molecular Mechanisms of Metformin in Neuroprotection
5.1. Metformin in Specific Neurodegenerative Conditions
Metformin has emerged as a promising therapeutic agent in the field of neurodegenerative disorders [56]. Originally developed to control blood glucose levels, this biguanide drug has revealed unexpected neuroprotective properties that extend far beyond its traditional metabolic effects [221].
In recent years, researchers have uncovered compelling evidence suggesting that metformin’s mechanisms of action may offer significant benefits in various neurodegenerative conditions, sparking intense interest in its potential repurposing for brain health [222,223]. The intersection of metabolic regulation and neurodegeneration has become increasingly apparent as scientists uncover the complex relationships between cellular energy metabolism, inflammation, and neuronal health [224]. Metformin’s ability to modulate these fundamental biological processes, particularly through its activation of AMPK signaling pathways and regulation of mitochondrial function as we mentioned before, positions the drug as a valuable candidate for addressing the multifaceted nature of neurodegenerative diseases [84,225,226].
The neuroprotective effects of metformin can be attributed to both common pathogenic pathway-dependent mechanisms and disease-specific modes of action. Metformin exerts significant effects on protein homeostasis by inhibiting protein misfolding and aggregation. Studies have demonstrated that metformin reduces the formation of toxic protein aggregates including α-synuclein, amyloid-β, tau, and huntingtin [227,228]. Furthermore, metformin disrupts the prion-like spreading of misfolded proteins between neurons in mouse models [229]. Metformin also enhances autophagic and proteasomal degradation of misfolded proteins [230]. Neuroinflammation modulation constitutes a significant aspect of metformin’s neuroprotective profile. Metformin inhibits microglial activation and proliferation while reducing pro-inflammatory cytokine production, including TNF-α, IL-1β, and IL-6 [231]. Additionally, studies have found that metformin promotes anti-inflammatory phenotypes in glial cells and suppresses the NF-κB signaling pathway [231,232].
From Alzheimer’s and Parkinson’s disease to multiple sclerosis and Huntington’s disease, research indicates that metformin may influence key pathological processes common to these conditions [112,233,234]. Understanding metformin’s role in neurodegenerative disorders not only offers hope for new therapeutic strategies but also provides valuable insights into the underlying mechanisms of these devastating conditions [224,235]. As the global burden of neurodegenerative diseases continues to grow with an aging population, the exploration of established medications like metformin represents an efficient approach to drug development, potentially accelerating the path to effective treatments for patients in need [236,237].
5.2. Alzheimer’s Disease
Metformin has gained interest for its potential effects on Alzheimer’s disease (AD). It activates the AMPK, reducing amyloid beta production and inhibiting neuroinflammation. It also reduces oxidative stress by stabilizing the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway and enhancing antioxidant enzyme expression. Metformin also decreases neuronal apoptosis and improves mitochondrial function, potentially alleviating memory loss associated with AD. However, findings are inconclusive [220,238].
Based on the research literature, T2DM significantly increases the risk of neurodegenerative conditions, particularly Alzheimer’s disease (AD). Interestingly, while T2DM shows these strong connections to neurodegenerative diseases, recent Mendelian randomization studies suggest that type 1 diabetes mellitus (T1DM) may have a different relationship, showing reduced genetic susceptibility to Parkinson’s disease and no significant genetic connection to AD [239,240]. Studies have shown that individuals with T2DM experience a 45% faster decline in cognitive abilities, memory, and reasoning compared to non-diabetic individuals [241,242]. The connection between these conditions is driven by several shared risk factors. These include obesity (which promotes insulin resistance and neuroinflammation), hypertension (which damages blood vessels and impairs brain blood flow), oxidative stress (which affects both glucose metabolism and neuronal function), poor diet (particularly high-fat, high-sugar diets that contribute to insulin resistance and brain inflammation), and a sedentary lifestyle (which reduces insulin sensitivity and is associated with increased amyloid beta deposition). Genetics also plays a role, with certain genes affecting both conditions through shared pathways. Additionally, lifestyle factors such as smoking and excessive alcohol consumption can exacerbate both conditions by increasing oxidative stress and inflammation [5,207,243]. Hence, the analysis of metformin’s effect on neurodegenerative diseases is a subject of considerable current interest.
In a Veterans Administration cohort study comparing different diabetes medications, metformin monotherapy was associated with lower dementia risk compared to sulfonylureas [244]. On the other hand, a meta-analysis from 2018 found that metformin use was associated with a reduced risk of dementia compared to no pharmacological treatment, with longer treatment duration showing greater benefit [222]. Similarly, in trials of patients with mild cognitive impairment and early dementia, metformin demonstrated potential cognitive benefits, including improved executive functioning and trends toward improved learning/memory [236]. Recent Alzheimer’s Disease Neuroimaging Initiative (ADNI) data have shown that diabetic patients treated with metformin during early AD stages exhibited better cognitive performance and cerebrospinal fluid biomarker profiles, with treatment associated with higher amyloid beta and lower tau/phosphorylated tau levels [245]. However, some studies have found no protective effect or even suggested increased risk—a meta-analysis examining metformin use and AD risk found no clear overall association (odds ratio (OR) 1.17, 95% confidence interval (CI) 0.88–1.56), though subgroup analysis revealed increased AD risk among Asian populations [246]. The heterogeneous treatment effects may be explained by factors such as timing of intervention, presence of neuropsychiatric disorders, and concurrent medication use, highlighting the need for careful patient selection when considering metformin for cognitive protection [247]. Building on this mixed evidence, the ongoing MET-FINGER randomized controlled trial is evaluating a novel precision prevention approach combining lifestyle intervention with metformin in non-diabetic older adults at increased dementia risk, which may provide crucial evidence for implementing innovative combination strategies for AD prevention [248]. Taking into account the research published in ClinicalTrials.gov, there are two completed studies on this topic, one focusing on AD biomarkers and the second on the topic of cognitive impairment [249,250]. There are two other studies, one assessing the use of metformin in AD prevention (active, not recruiting), and the second being a cross-sectional pilot study of brain imaging biomarkers in the Diabetes Prevention Program (terminated) [9,251].
Current research directions intend to clarify contradictory findings by investigating distinct biological targets of metformin in the brain, the balance of positive and negative effects of this drug, as well as possible genetic variables that may alter individual reactions to metformin therapy [252].
5.3. Parkinson’s Disease
Metformin offers neuroprotection in Parkinson’s disease through several pathways. It primarily activates AMPK, a key energy sensor, boosting mitochondrial biogenesis (via PGC-1α), improving energy metabolism, and reducing oxidative stress in dopaminergic neurons. Metformin also has anti-inflammatory effects, suppressing microglial activation and reducing pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) [4,6]. It enhances autophagy (via mTOR inhibition and AMPK activation), clearing damaged components and protein aggregates. Furthermore, metformin modulates genes involved in cellular stress response (Nrf2), boosting antioxidant defenses [253,254,255].
Clinical evidence for metformin’s efficacy in Parkinson’s comes from observational studies and trials. Epidemiological studies suggest a reduced Parkinson’s incidence in diabetic metformin users [256,257]. A retrospective study found that metformin users have lower risk of developing Parkinson’s. Interventional trials show promising results in motor function improvement (based on the Unified Parkinson’s Disease Rating Scale), though evidence is somewhat mixed. Ongoing phase II trials are exploring metformin’s potential as a disease-modifying therapy, especially in early-stage Parkinson’s [11,258]. Clinicaltrials.gov also provides a study which analyzes the correlation between idiopathic Parkinson’s disease and diabetes mellitus in Egyptian elderly patients; however, its status is unknown [259].
Metformin’s impact on α-synuclein is crucial to its potential therapeutic benefits. It influences α-synuclein aggregation and clearance through multiple mechanisms. It can directly interact with α-synuclein monomers, potentially altering their conformation and reducing toxic oligomer formation [260,261]. Via AMPK activation, metformin enhances autophagic α-synuclein clearance (macroautophagy and chaperone-mediated autophagy). It also reduces α-synuclein phosphorylation at Ser129 (associated with aggregation and toxicity) and modulates α-synuclein expression (potentially epigenetically via AMPK). Metformin’s reduction of oxidative stress and inflammation indirectly affects α-synuclein aggregation [262,263,264].
Proteomic analyses show that metformin alters the α-synuclein interactome, potentially affecting its processing and degradation. Transgenic models show reduced α-synuclein aggregates and improved neuronal survival in metformin-treated animals. Its effect on α-synuclein propagation is under investigation, with preliminary evidence suggesting that it might affect pathogenic α-synuclein release and uptake. These findings support metformin’s potential to modify α-synuclein pathology in Parkinson’s disease [265,266].
These mechanisms have therapeutic implications, especially for metformin timing and dosing. Its multiple effects on α-synuclein suggest that early intervention might be most beneficial, as demonstrated in multiple preclinical studies [7,267]. In a rotenone-induced mouse model of Parkinson’s disease, metformin administration (300 mg/kg/day) attenuated the loss of tyrosine hydroxylase (TH+) neurons in the substantia nigra (SN), decreased cleaved caspase-3 and α-synuclein accumulation in the SN, and reduced the levels of malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), indicating decreased lipid peroxidation, compared to the rotenone-only group [268]. In C. elegans models of Parkinson’s disease, metformin treatment (10 mM) significantly decreased 6-OHDA-induced dopaminergic neurodegeneration, inhibited α-synuclein aggregation in transgenic worms expressing human α-synuclein, and upregulated expression of the antioxidant gene sod-3. Specifically, 10 mM metformin reduced α-synuclein aggregation and improved lipid deposition in the NL5901 strain expressing α-synuclein yellow fluorescent (YFP) fusion proteins [269].
5.4. Huntington’s Disease
Metformin has emerged as a promising therapeutic agent for Huntington’s disease (HD) through multiple mechanisms of action, especially its activation of AMP-activated protein kinase [270]. Metformin inhibits the MID1/PP2A/mTOR protein complex, reducing mutant huntingtin (mHtt) mRNA translation and reducing toxic protein levels [271]. It activates AMPK, improving cognitive function and reducing neuropsychiatric symptoms in HD models [272]. Metformin also influences autophagy and mitochondrial dynamics, mitigating toxic effects from mHtt aggregation. It enhances brain-derived neurotrophic factor levels and reduces inflammation-related glial activation [273,274].
In the Hdh150 mouse model, metformin administration during very far from disease onset (VFDO) stages reversed early cortical network dysfunction, including aberrant neuronal hyperactivity and enhanced synchronicity, while also normalizing anxiety-related behavioral changes [271]. These findings collectively indicate that metformin may have therapeutic potential for HD, particularly when administered early in the disease course, before substantial neuronal damage occurs. Preclinical studies have demonstrated that metformin reduces mHTT aggregation, prevents neuronal death, and improves neuropsychiatric phenotypes in HD models. In the zQ175 mouse model, metformin treatment reduced nuclear mHTT aggregates in the striatum, partially restored brain-derived neurotrophic factor (BDNF) levels, and attenuated glial activation [275]. These neuroprotective effects appear to be mediated through AMPK-dependent mechanisms and enhanced autophagy, as demonstrated in C. elegans models where metformin reduced polyQ aggregation and improved neuronal function in an AMPK- and lysosome-dependent manner. However, the timing of AMPK activation appears critical, as late-stage activation may have detrimental effects. A post hoc analysis of the Enroll-HD database revealed that HD patients taking metformin showed better cognitive performance compared to non-metformin users, particularly in verbal fluency and Stroop interference tests [276]. These findings have led to an ongoing phase III clinical trial (which can be found on Clinicaltrials.gov) evaluating metformin’s efficacy in treating cognitive decline in HD patients, with results that were expected in 2024; however, its status is currently unknown [10].
Metformin is being investigated not only as a treatment for existing symptoms but also as a preventive measure for those at risk of developing Huntington’s disease. The existing body of research supports its potential as a promising therapeutic option, particularly due to its established safety profile from long-term use in diabetes management.
5.5. Multiple Sclerosis
Multiple sclerosis treatment strategies continue to evolve, with increasing interest in repurposing established medications. The following section concentrates on the potential therapeutic applications of metformin in MS treatment, with emphasis on its molecular mechanisms, immunomodulatory effects, and clinical implications. Current evidence suggests that metformin’s activation of AMPK and modulation of the mTOR pathway may provide beneficial effects in MS through multiple mechanisms, including immunomodulation, neuroprotection, and metabolic regulation [277,278,279,280].
Research evidence from randomized controlled trial demonstrates metformin’s multifaceted therapeutic potential in multiple sclerosis through several key mechanisms, as reported by Keersmaecker et al. [281]. The drug exhibited significant remyelinating properties via the enhancement of oligodendrocyte precursor cell function and differentiation, mediated through AMPK pathway activation. The study has established metformin’s ability to rejuvenate aged oligodendrocyte precursor cells (OPCs) and restore their proliferative capacity, particularly in white matter regions. Metformin demonstrated considerable immunomodulatory effects, evidenced by reduction in inflammatory markers and T2 lesion formation. The drug’s neuroprotective capabilities were attributed to its antioxidant properties, mitochondrial function optimization, and downregulation of pro-inflammatory microglial activity through Mac-3 mRNA modulation [281]. Clinical observations support these mechanisms, with studies reporting reduced disability progression in MS patients receiving metformin, particularly notable in cohorts with comorbid type 2 diabetes. A pilot study (N = 50) demonstrated significant reduction in new T2 hyperintense lesions alongside favorable immunological changes. Although only a small percentage of metformin crosses the blood–brain barrier, this amount appears to be sufficient to potentially exert a beneficial effect in the treatment of multiple sclerosis, as suggested by similar results obtained by Gilbert and Livingstone et al. Their experimental study demonstrated that metformin administration at disease onset significantly reduced neurological impairment and protected myelin integrity in experimental autoimmune encephalomyelitis through microglial modulation, without affecting oligodendrocyte precursor dynamics. However, delayed treatment showed limited efficacy, suggesting metformin’s potential therapeutic value in multiple sclerosis may depend on early intervention. The drug’s established safety profile and demonstrated neuroprotective properties warrant further investigation for clinical translation [282].
While current disease-modifying therapies (DMTs) have shown significant efficacy, there remains a need for treatments that can address both the inflammatory and neurodegenerative aspects of MS [283,284]. Metformin has emerged as a promising candidate for MS treatment due to its diverse mechanisms of action and well-established safety profile. Metformin’s primary mechanism of action involves the activation of AMP-activated protein kinase, a fundamental regulator of cellular energy homeostasis [281,284]. In the context of MS pathophysiology, AMPK activation produces several beneficial effects. The pathway reduces inflammatory cytokine production while enhancing mitochondrial function and glucose metabolism in neural cells [285,286]. Additionally, AMPK activation modulates T-cell responses, potentially affecting the autoimmune component of MS [287,288]. The mammalian target of rapamycin pathway represents another crucial mechanism through which metformin may exert therapeutic effects in MS [289,290]. Metformin’s inhibition of mTOR leads to decreased inflammatory cell proliferation and regulated immune cell differentiation, as was shown in a recent study on Hiradentis suppurtiva, using the same mTOR pathway [216]. Furthermore, this modulation impacts myelin regeneration processes and influences neuronal survival mechanisms, both of which are critical in MS pathophysiology [290]. Metformin demonstrates significant effects on T-cell function, including the suppression of pro-inflammatory T-cell responses and promotion of regulatory T-cell development [291]. The drug modulates the Th17/Treg balance, which is crucial in autoimmune conditions [280]. The effects of metformin on B-cells include modulation of activation and differentiation processes, reduction in antibody production, and influence on B-cell-mediated inflammation. These actions may contribute to reducing the adaptive immune response involved in MS pathogenesis [292]. Metformin significantly impacts microglial cell function by reducing activation and decreasing pro-inflammatory cytokine production. It promotes an anti-inflammatory phenotype and enhances neuroprotective functions, potentially contributing to reduced neuroinflammation in MS [293,294]. The drug demonstrates positive effects on blood–brain barrier integrity, reducing immune cell infiltration and modulating endothelial cell function [44,295]. Current research suggests promising therapeutic potential for metformin in multiple sclerosis. Provided studies indicate that metformin’s effects may help limit central nervous system inflammation in MS. The drug’s metabolic effects, including improved glucose utilization, enhanced insulin sensitivity, and reduced oxidative stress, target key pathogenic mechanisms in MS. While early clinical observations suggest potential benefits in reducing relapse frequency and slowing disability progression, these findings primarily come from basic research [296].
Currently there are seven trials available at Clinicaltrials.gov. These studies explore metformin’s ability to promote remyelination, modulate immune responses, and impact neurodegeneration in various MS subtypes, with trials ranging from Phase I to Phase IIa and encompassing both pediatric and adult populations. Trial statuses include “recruiting”, “completed”, “unknown status”, and “not yet recruiting”, reflecting the ongoing and evolving nature of this research [297,298,299,300,301,302,303]. Larger clinical trials are needed to definitively establish metformin’s therapeutic value, particularly its potential to enhance existing MS treatments and provide neuroprotective effects. The metabolic benefits may be especially relevant for MS patients, who often show altered metabolic profiles, but human studies are required to confirm these promising preclinical results.
5.6. Metformin vs. Other Treatments
Metformin presents a multifaceted therapeutic approach for neurodegenerative disorders and multiple sclerosis, exhibiting distinct advantages over conventional treatments. In neurodegenerative diseases, while other insulin sensitizers like pioglitazone improve insulin sensitivity, metformin’s broader impact on oxidative stress, mitochondrial dysfunction, and protein aggregation, through AMPK activation and mTOR inhibition, offers a more comprehensive neuroprotective strategy [12,304]. Compared to antioxidants like vitamin E, which primarily target oxidative stress, metformin additionally modulates cellular aging and inflammatory pathways, potentially offering disease-modifying benefits beyond symptom management [305,306,307]. In MS, metformin’s capacity to promote remyelination and reduce neuroinflammation, through mechanisms targeting mTOR and Nrf2, distinguishes it from simple antioxidant therapies like N-acetylcysteine. While disease-modifying drugs like interferon-beta remain the cornerstone of MS treatment, metformin’s potential as an adjunct therapy, though still under investigation in trials like MACSiMiSE-BRAIN, suggests a valuable role in addressing metabolic comorbidities and potentially enhancing remyelination [281,282]. However, variability in clinical responses and the need for larger, well-controlled trials emphasize the importance of further research to fully elucidate metformin’s therapeutic potential in these complex neurological conditions.
5.7. Biomarkers for Predicting Metformin Response
5.7.1. Genetic Biomarkers
Polymorphisms in genes encoding transporters responsible for metformin pharmacokinetics have shown consistent associations with treatment response. The SLC22A1 gene, encoding the organic cation transporter 1 (OCT1), contains several reduced-function variants (R61C, G401S, M420del) that correlate with decreased metformin efficacy. In the Rotterdam study, carriers of two reduced-function SLC22A1 alleles demonstrated significantly smaller HbA1c reductions compared to carriers of functional alleles [308]. Similarly, variants in the SLC22A2 (OCT2) and SLC47A1/2 (MATE1/2) genes affect metformin excretion and plasma concentration. The common rs316019 variant in SLC22A2 has been linked to reduced metformin clearance, while the rs2289669 G>A polymorphism in SLC47A1 correlates with enhanced metformin efficacy, particularly in Asian populations [309].
Perhaps most significantly, a genome-wide association study identified the rs11212617 variant near the ATM gene as strongly associated with metformin response. This finding, replicated across multiple cohorts, suggests that carriers of the C allele achieve better glycemic control with metformin treatment. Mechanistic studies indicate ATM may influence AMPK signaling, a central pathway in metformin’s action [310].
5.7.2. Metabolic Biomarkers
Baseline glycemic parameters consistently predict treatment outcomes, with higher pre-treatment fasting glucose and HbA1c values associated with greater absolute reductions following metformin therapy. The trial demonstrated that baseline insulin sensitivity and beta-cell function measurements significantly predicted long-term glycemic response [311]. Metabolomic approaches have identified specific plasma markers with predictive potential. Elevated baseline levels of branched-chain amino acids (particularly valine, leucine, and isoleucine) and acylcarnitines correlate with suboptimal metformin response. Alpha-hydroxybutyrate, an early marker of insulin resistance, shows promise as a predictor of efficacy [312].
5.7.3. Clinical Biomarkers
Several readily accessible clinical parameters demonstrate associations with metformin response. Lower BMI consistently predicts better treatment outcomes [313]. Visceral adiposity, as measured by waist circumference or imaging techniques, may offer superior predictive value compared to BMI alone, potentially explaining observed gender differences in response [314]. Age and diabetes duration significantly influence treatment efficacy. Younger patients and those with shorter diabetes duration prior to metformin initiation typically achieve greater HbA1c reductions [315]. The Diabetes Prevention Program clearly demonstrated superior efficacy of metformin in preventing diabetes progression among younger individuals with higher BMI [316].
6. Research Gaps, Emerging Technologies, and Future Perspectives
Significant research gaps remain in understanding the complex pathophysiology of neurodegenerative diseases and optimizing therapeutic interventions, particularly regarding metformin’s potential preventive role [317]. A critical challenge lies in identifying patients at risk of developing neurodegenerative conditions or those in early disease stages, when therapeutic interventions like metformin might be most effective [236,237]. This is especially relevant given that type 2 diabetes mellitus (T2DM), a primary indication for metformin, is preceded by long-term insulin resistance, hyperglycemia, and oxidative stress—all key factors predisposing to neurodegenerative diseases [5,207,243]. Currently, we may be missing opportunities for metformin’s preventive benefits due to delayed implementation and incomplete understanding of the temporal relationship between metabolic dysfunction and brain pathology [200,201].
The complex interplay between protein aggregation, neuroinflammation, mitochondrial dysfunction, insulin resistance, and metabolic disruption makes it difficult to pinpoint primary pathogenic mechanisms and optimal intervention timing [318]. This is particularly relevant for metformin therapy, as its mechanisms of action intersect with multiple pathological pathways [221,222]. Translating preclinical findings to effective treatments remains challenging due to limitations in current animal models and high clinical trial failure rates. The lack of reliable early biomarkers and incomplete understanding of cell type-specific vulnerabilities hinder both research and clinical practice, potentially delaying the implementation of preventive strategies with metformin and other therapeutics [317].
Novel technologies are revolutionizing the field with potential to address these gaps. Single-cell RNA sequencing, spatial transcriptomics, and advanced proteomics offer unprecedented insights into cell type-specific responses and disease mechanisms, which could help identify early molecular signatures indicating the need for preventive metformin therapy [130,319]. Artificial intelligence and machine learning facilitate complex data analysis, drug response prediction, and multi-omics data integration, potentially enabling better patient stratification for metformin treatment [320]. These technologies could help identify which patients might benefit most from early metformin intervention, even before clinical manifestations of neurodegeneration.
Human brain organoids, microfluidic organ-on-chip platforms, and CRISPR-based genome editing are advancing disease modeling and therapeutic development, allowing better understanding of metformin’s effects on neural tissue [130,319,320]. Advanced imaging techniques are improving disease monitoring and could help track early pathological changes when metformin intervention might be most beneficial. These technologies are also being leveraged to enhance drug delivery across the blood–brain barrier, particularly relevant for medications like metformin where optimizing BBB penetration remains a crucial challenge [71,72,73].
The field is moving toward precision medicine based on genetic and biomarker profiles, which could help identify patients who might particularly benefit from metformin’s neuroprotective effects [321,322,323,324]. This individualized approach is essential given that metformin’s effectiveness may vary among different patient populations and disease stages. A critical challenge remains the timing of therapeutic intervention, as evidence suggests that metformin may be most effective in early disease stages or as a preventive measure, particularly in patients with metabolic dysfunction who are at higher risk for neurodegeneration [325,326,327].
Key research priorities should include:
- Developing early detection biomarkers specifically focused on identifying patients who might benefit from preventive metformin therapy [236,237],
- Understanding the temporal relationship between metabolic dysfunction and neurodegeneration to optimize metformin intervention timing [207],
- Investigating the link between systemic inflammation, insulin resistance, and neurodegeneration to better target metformin’s therapeutic effects [200,201],
- Establishing clear criteria for preventive metformin use in patients with metabolic dysfunction at risk for neurodegeneration [241,243],
- Developing more sensitive methods to monitor brain health in T2DM patients to guide metformin therapy optimization [324,325],
- Gene therapy advancements for neurodegenerative conditions or combined with stem cells [114],
- Personalized medicine approaches integrating these novel technologies with established treatments like metformin [114].
These advances require robust collaboration between clinicians, researchers, and industry partners, along with significant research investment. The focus should be on translating basic research findings into practical clinical applications, particularly regarding the preventive use of metformin in at-risk populations [324,325,326]. This approach could maximize the therapeutic potential of this well-established medication while developing new strategies for early intervention in neurodegenerative diseases.
7. Conclusions
Based on the extensive literature review, metformin shows promising potential in treating various neurodegenerative conditions through multiple mechanisms. Research demonstrates its ability to activate AMPK pathways, enhance antioxidant defenses, and provide anti-inflammatory effects as well as increase brain sensitivity to insulin [4,12].
The current clinical evidence base for metformin in neurodegenerative diseases is predominantly comprised of observational studies, which suggest a reduced risk of neurodegeneration in metformin-treated patients, though some findings remain mixed [7,8,246]. While these observational data are encouraging, there is a notable scarcity of interventional clinical trials. For Huntington’s disease and multiple sclerosis, current evidence largely relies on basic research and animal studies, highlighting the need for more robust clinical investigations.
The drug’s neuroprotective effects are particularly notable in Parkinson’s disease, where it influences α-synuclein aggregation and clearance, and in multiple sclerosis, where it modulates neuroinflammation and immune responses [260,261,277,278]. In Huntington’s disease, metformin shows promise through its effects on mutant huntingtin protein levels and mitochondrial function [270,271].
However, significant research gaps remain, particularly in understanding the optimal timing of intervention and patient-specific responses [317]. Emerging technologies, including single-cell sequencing and advanced imaging, are providing new insights into disease mechanisms and potential therapeutic approaches [319,320]. Moving forward, the field is trending toward precision medicine approaches and multi-modal therapies [325,326].
Notably, metformin holds several distinct advantages over newer therapeutic agents. As a long-established medication, it has well-characterized pharmacodynamic and pharmacokinetic properties, along with a thoroughly documented safety profile. Its demonstrated neuroprotective effects, combined with its low cost and extensive clinical experience, make it an attractive candidate for broader investigation. These factors strongly support the expansion of clinical trials examining metformin’s preventive and therapeutic potential in neurodegenerative diseases, particularly as part of comprehensive treatment strategies.
It is important to acknowledge that several studies have reported no significant benefits or even adverse effects of metformin in neurodegenerative contexts [328,329]. These contradictory findings may be attributed to various factors, including heterogeneity in study populations, differences in disease stages, varying comorbidity profiles, and inconsistent dosing protocols [222,330].
Methodological limitations in existing studies further complicate the interpretation of these mixed results. Many observational studies face challenges with confounding variables, inadequate control for diabetes severity, and selection bias that may mask or artificially enhance metformin’s apparent effects [56,331]. Additionally, genetic polymorphisms affecting metformin transporters and receptors may significantly influence the treatment response, potentially explaining inconsistent outcomes across different populations [332].
The drug’s relatively limited blood–brain barrier penetration presents another potential explanation for null findings, as insufficient central nervous system concentrations may fail to achieve therapeutic effects in some patients, particularly those with compromised blood–brain barrier integrity [169]. Future studies should address these limitations through more rigorous methodologies, careful patient stratification, standardized dosing regimens, and longer follow-up periods to better characterize metformin’s true potential in neurodegenerative disease management [224].
Author Contributions
W.K., J.G. and P.B. conceptualized the article; A.Ś. and E.P. supervised the article; W.K., J.G., P.B., M.K., A.Ś. and E.P. reviewed the literature; W.K., P.B., J.G. and M.K. visualized the figures and prepared the tables; W.K., J.G. and P.B. wrote the original draft; W.K., J.G., P.B., M.K., A.Ś. and E.P. reviewed and edited article. All authors have read and agreed to the published version of the manuscript.
Funding
This paper was supported by a grant from the Medical University of Lodz (No. 503/1-159-01/503-11-001; 503/0-149-04/503-01-001) and the Polish Society of Metabolic Disorders.
Data Availability Statement
Data sharing is not applicable, because no datasets were generated or analyzed during the review article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on mechanisms of action and repurposing potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef]
- Werner, E.A.; Bell, J. CCXIV.—The preparation of methylguanidine, and of ββ-dimethylguanidine by the interaction of dicyanodiamide, and methylammonium and dimethylammonium chlorides respectively. J. Chem. Soc. Trans. 1922, 121, 1790–1794. [Google Scholar] [CrossRef]
- Loan, A.; Syal, C.; Lui, M.; He, L.; Wang, J. Promising use of metformin in treating neurological disorders: Biomarker-guided therapies. Neural Regen. Res. 2024, 19, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Kruczkowska, W.; Galeziewska, J.; Wanke, K.; Kaluzinska-Kolat, Z.; Aleksandrowicz, M.; Kontek, R. Alzheimer’s Disease as Type 3 Diabetes: Understanding the Link and Implications. Int. J. Mol. Sci. 2024, 25, 11955. [Google Scholar] [CrossRef] [PubMed]
- Alrouji, M.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Ashour, N.A.; Jabir, M.S.; Negm, W.A.; Batiha, G.E. Metformin role in Parkinson’s disease: A double-sword effect. Mol. Cell. Biochem. 2024, 479, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Masato, A.; Bubacco, L.; Bisaglia, M. Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges. Int. J. Mol. Sci. 2021, 23, 398. [Google Scholar] [CrossRef]
- Battini, V.; Cirnigliaro, G.; Leuzzi, R.; Rissotto, E.; Mosini, G.; Benatti, B.; Pozzi, M.; Nobile, M.; Radice, S.; Carnovale, C.; et al. The potential effect of metformin on cognitive and other symptom dimensions in patients with schizophrenia and antipsychotic-induced weight gain: A systematic review, meta-analysis, and meta-regression. Front. Psychiatry 2023, 14, 1215807. [Google Scholar] [CrossRef]
- NCT04098666; Metformin in Alzheimer’s Dementia Prevention (MAP), José A. Luchsinger, Columbia University. 2025. Available online: https://clinicaltrials.gov/study/NCT04098666 (accessed on 13 March 2025).
- NCT04826692; TEsting METformin Against Cognitive Decline in HD, Instituto de Investigacion Sanitaria La Fe. 2022. Available online: https://clinicaltrials.gov/study/NCT04826692 (accessed on 13 March 2025).
- NCT05781711; Clinical Study to Evaluate the Possible Efficacy of Metformin in Patients with Parkinson’s Disease, Mostafa Bahaa, Tanta University. 2025. Available online: https://clinicaltrials.gov/study/NCT05781711 (accessed on 13 March 2025).
- Demare, S.; Kothari, A.; Calcutt, N.A.; Fernyhough, P. Metformin as a potential therapeutic for neurological disease: Mobilizing AMPK to repair the nervous system. Expert Rev. Neurother. 2021, 21, 45–63. [Google Scholar] [CrossRef]
- Rosell-Diaz, M.; Fernandez-Real, J.M. Metformin, Cognitive Function, and Changes in the Gut Microbiome. Endocr. Rev. 2024, 45, 210–226. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Therapeutic profile in the treatment of type 2 diabetes. Diabetes Obes. Metab. 2024, 26 (Suppl. S3), 3–19. [Google Scholar] [CrossRef] [PubMed]
- Samaras, K.; Makkar, S.; Crawford, J.D.; Kochan, N.A.; Wen, W.; Draper, B.; Trollor, J.N.; Brodaty, H.; Sachdev, P.S. Metformin Use Is Associated with Slowed Cognitive Decline and Reduced Incident Dementia in Older Adults with Type 2 Diabetes: The Sydney Memory and Ageing Study. Diabetes Care 2020, 43, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaei Malazy, O.; Bandarian, F.; Qorbani, M.; Mohseni, S.; Mirsadeghi, S.; Peimani, M.; Larijani, B. The effect of metformin on cognitive function: A systematic review and meta-analysis. J. Psychopharmacol. 2022, 36, 666–679. [Google Scholar] [CrossRef]
- Liang, X.; Giacomini, K.M. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J. Pharm. Sci. 2017, 106, 2245–2250. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathways: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Szymczak-Pajor, I.; Wenclewska, S.; Sliwinska, A. Metabolic Action of Metformin. Pharmaceuticals 2022, 15, 810. [Google Scholar] [CrossRef]
- Sun, M.L.; Liu, F.; Yan, P.; Chen, W.; Wang, X.H. Effects of food on pharmacokinetics and safety of metformin hydrochloride tablets: A meta-analysis of pharmacokinetic, bioavailability, or bioequivalence studies. Heliyon 2023, 9, e17906. [Google Scholar] [CrossRef]
- McCreight, L.J.; Stage, T.B.; Connelly, P.; Lonergan, M.; Nielsen, F.; Prehn, C.; Adamski, J.; Brosen, K.; Pearson, E.R. Pharmacokinetics of metformin in patients with gastrointestinal intolerance. Diabetes Obes. Metab. 2018, 20, 1593–1601. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Wrobel, M.P.; Marek, B.; Kajdaniuk, D.; Rokicka, D.; Szymborska-Kajanek, A.; Strojek, K. Metformin—A new old drug. Endokrynol. Pol. 2017, 68, 482–496. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Iannantuoni, F.; Gruevska, A.; Muntane, J.; Rocha, M.; Victor, V.M. Mechanisms of action of metformin in type 2 diabetes: Effects on mitochondria and leukocyte-endothelium interactions. Redox Biol. 2020, 34, 101517. [Google Scholar] [CrossRef] [PubMed]
- Vezza, T.; Luna-Marco, C.; Rovira-Llopis, S.; Víctor, V.M. Metformin and its redox-related mechanisms of action in type 2 diabetes. Redox Exp. Med. 2023, 2023, e230015. [Google Scholar] [CrossRef]
- Alshawi, A.; Agius, L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J. Biol. Chem. 2019, 294, 2839–2853. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Washio, J.; Sato, S.; Abiko, Y.; Shinohara, Y.; Kobayashi, Y.; Otani, H.; Sasaki, S.; Wang, X.; Takahashi, N. Rewired Cellular Metabolic Profiles in Response to Metformin under Different Oxygen and Nutrient Conditions. Int. J. Mol. Sci. 2022, 23, 989. [Google Scholar] [CrossRef]
- Salpeter, S.R.; Greyber, E.; Pasternak, G.A.; Salpeter, E.E. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2010, 2010, CD002967. [Google Scholar] [CrossRef]
- See, K.C. Metformin-associated lactic acidosis: A mini review of pathophysiology, diagnosis and management in critically ill patients. World J. Diabetes 2024, 15, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Pinyopornpanish, K.; Leerapun, A.; Pinyopornpanish, K.; Chattipakorn, N. Effects of Metformin on Hepatic Steatosis in Adults with Nonalcoholic Fatty Liver Disease and Diabetes: Insights from the Cellular to Patient Levels. Gut Liver 2021, 15, 827–840. [Google Scholar] [CrossRef]
- Rena, G.; Pearson, E.R.; Sakamoto, K. Molecular mechanism of action of metformin: Old or new insights? Diabetologia 2013, 56, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Gumieniczek, A.; Berecka-Rycerz, A.; Mroczek, T.; Wojtanowski, A.K. Determination of Chemical Stability of Two Oral Antidiabetics, Metformin and Repaglinide in the Solid State and Solutions Using LC-UV, LC-MS, and FT-IR Methods. Molecules 2019, 24, 4430. [Google Scholar] [CrossRef]
- Niranjana Devi, R.; Jelsch, C.; Israel, S.; Aubert, E.; Anzline, C.; Hosamani, A.A. Charge density analysis of metformin chloride, a biguanide anti-hyperglycemic agent. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 10–22. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Desai, D.; Wong, B.; Huang, Y.; Ye, Q.; Tang, D.; Guo, H.; Huang, M.; Timmins, P. Surfactant-mediated dissolution of metformin hydrochloride tablets: Wetting effects versus ion pairs diffusivity. J. Pharm. Sci. 2014, 103, 920–926. [Google Scholar] [CrossRef]
- Elezovic, A.; Maric, A.; Biscevic, A.; Hadziabdic, J.; Skrbo, S.; Spirtovic-Halilovic, S.; Rahic, O.; Vranic, E.; Elezovic, A. In vitro pH dependent passive transport of ketoprofen and metformin. ADMET DMPK 2021, 9, 57–68. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Mohamed, M.A.; Mersal, G.A.M.; Al-Juaid, S. Insulin-like action of novel metformin-containing vanadate as a new antidiabatic drug: Synthesis, characterization and crystal structure of [Metformin-H]2[V2O6]]·H2O. J. Mol. Struct. 2015, 1098, 92–100. [Google Scholar] [CrossRef]
- An, Q.; Xing, C.; Wang, Z.; Li, S.; Wang, W.; Yang, S.; Kong, L.; Yang, D.; Zhang, L.; Du, G.; et al. Metformin-Mediated Improvement in Solubility, Stability, and Permeability of Nonsteroidal Anti-Inflammatory Drugs. Pharmaceutics 2024, 16, 382. [Google Scholar] [CrossRef]
- Li, N.; Zhou, T.; Fei, E. Actions of Metformin in the Brain: A New Perspective of Metformin Treatments in Related Neurological Disorders. Int. J. Mol. Sci. 2022, 23, 8281. [Google Scholar] [CrossRef] [PubMed]
- Beckers, M.; Bloem, B.R.; Verbeek, M.M. Mechanisms of peripheral levodopa resistance in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Li, W.; Liu, Z.; Ding, Z.; Tuo, D. Towards Improvements for Penetrating the Blood-Brain Barrier-Recent Progress from a Material and Pharmaceutical Perspective. Cells 2018, 7, 24. [Google Scholar] [CrossRef]
- Chevalier, R. 2018. Available online: https://www.biorxiv.org/content/10.1101/362541v2.abstract (accessed on 13 March 2025).
- Sharma, S.; Zhang, Y.; Akter, K.A.; Nozohouri, S.; Archie, S.R.; Patel, D.; Villalba, H.; Abbruscato, T. Permeability of Metformin across an In Vitro Blood–Brain Barrier Model during Normoxia and Oxygen-Glucose Deprivation Conditions: Role of Organic Cation Transporters (Octs). Pharmaceutics 2023, 15, 1357. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Hien, T.T.; Han, E.H.; Hwang, Y.P.; Choi, J.H.; Kang, K.W.; Kwon, K.I.; Kim, B.H.; Kim, S.K.; Song, G.Y.; et al. Metformin inhibits P-glycoprotein expression via the NF-κB pathway and CRE transcriptional activity through AMPK activation. Br. J. Pharmacol. 2011, 162, 1096–1108. [Google Scholar] [CrossRef]
- Abbasi, M.M.; Valizadeh, H.; Hamishehkar, H.; Zakeri-Milani, P. Inhibition of P-glycoprotein expression and function by anti-diabetic drugs gliclazide, metformin, and pioglitazone in vitro and in situ. Res. Pharm. Sci. 2016, 11, 177–186. [Google Scholar]
- Huttunen, J.; Peltokangas, S.; Gynther, M.; Natunen, T.; Hiltunen, M.; Auriola, S.; Ruponen, M.; Vellonen, K.S.; Huttunen, K.M. L-Type Amino Acid Transporter 1 (LAT1/Lat1)-Utilizing Prodrugs Can Improve the Delivery of Drugs into Neurons, Astrocytes and Microglia. Sci. Rep. 2019, 9, 12860. [Google Scholar] [CrossRef]
- Singh, N.; Ecker, G.F. Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1. Int. J. Mol. Sci. 2018, 19, 1278. [Google Scholar] [CrossRef]
- Sato, R.; Ohmori, K.; Umetsu, M.; Takao, M.; Tano, M.; Grant, G.; Porter, B.; Bet, A.; Terasaki, T.; Uchida, Y. An Atlas of the Quantitative Protein Expression of Anti-Epileptic-Drug Transporters, Metabolizing Enzymes and Tight Junctions at the Blood-Brain Barrier in Epileptic Patients. Pharmaceutics 2021, 13, 2122. [Google Scholar] [CrossRef]
- Zhang, C.; Kwan, P.; Zuo, Z.; Baum, L. The transport of antiepileptic drugs by P-glycoprotein. Adv. Drug Deliv. Rev. 2012, 64, 930–942. [Google Scholar] [CrossRef]
- Ghosh, C.; Achar, A. Multiple hurdle mechanism and blood-brain barrier in epilepsy: Glucocorticoid receptor-heat shock proteins on drug regulation. Neural Regen. Res. 2021, 16, 2427–2428. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Sikora, J.; Szydlowska, A.; Skupien, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A Future Therapy for Neurodegenerative Diseases: Theme: Drug Discovery, Development and Delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Giaccari, A.; Solini, A.; Frontoni, S.; Del Prato, S. Metformin Benefits: Another Example for Alternative Energy Substrate Mechanism? Diabetes Care 2021, 44, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Crowley, M.J.; Diamantidis, C.J.; McDuffie, J.R.; Cameron, B.; Stanifer, J.; Mock, C.K.; Kosinski, A.; Wang, X.; Tang, S.; Williams, J.W., Jr. Metformin Use in Patients with Historical Contraindications or Precautions; Department of Veterans Affairs: Washington, DC, USA, 2016; Appendix A, FDA Safety Announcements For Metformin. Available online: https://www.ncbi.nlm.nih.gov/books/NBK409379/ (accessed on 13 March 2025).
- Flory, J.H.; Keating, S.; Guelce, D.; Mushlin, A.I. Overcoming barriers to the use of metformin: Patient and provider perspectives. Patient Prefer. Adherence 2019, 13, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Silverii, G.A. Optimizing metformin therapy in practice: Tailoring therapy in specific patient groups to improve tolerability, efficacy and outcomes. Diabetes Obes. Metab. 2024, 26 (Suppl. S3), 42–54. [Google Scholar] [CrossRef]
- Alibrahim, N.T.Y.; Chasib, M.G.; Hamadi, S.S.; Mansour, A.A. Predictors of Metformin Side Effects in Patients with Newly Diagnosed Type 2 Diabetes Mellitus. Ibnosina J. Med. Biomed. Sci. 2023, 15, 67–73. [Google Scholar] [CrossRef]
- Lalau, J.D. Lactic acidosis induced by metformin: Incidence, management and prevention. Drug Saf. 2010, 33, 727–740. [Google Scholar] [CrossRef]
- Sayedali, E.; Yalin, A.E.; Yalin, S. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes. World J. Diabetes 2023, 14, 585–593. [Google Scholar] [CrossRef]
- Bahl, G.; Hussain, M.S.; Saraswat, N.; Agrawal, M. Beyond Diabetes Management: Unraveling Metformin’s Long-Term Effects on Vitamin B12. Clin. Diabetol. 2023, 12, 279–282. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Saad, H.M.; Batiha, G.E. Long-term use of metformin and Alzheimer’s disease: Beneficial or detrimental effects. Inflammopharmacology 2023, 31, 1107–1115. [Google Scholar] [CrossRef]
- Abdelgadir, E.; Ali, R.; Rashid, F.; Bashier, A. Effect of Metformin on Different Non-Diabetes Related Conditions, a Special Focus on Malignant Conditions: Review of Literature. J. Clin. Med. Res. 2017, 9, 388–395. [Google Scholar] [CrossRef]
- Shen, D.; Ye, X.; Li, J.; Hao, X.; Jin, L.; Jin, Y.; Tong, L.; Gao, F. Metformin Preserves VE-Cadherin in Choroid Plexus and Attenuates Hydrocephalus via VEGF/VEGFR2/p-Src in an Intraventricular Hemorrhage Rat Model. Int. J. Mol. Sci. 2022, 23, 8552. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Heath, B.; McGinness, L. Advances in metformin-delivery systems for diabetes and obesity management. Diabetes Obes. Metab. 2024, 26, 3513–3529. [Google Scholar] [CrossRef]
- Du, M.R.; Gao, Q.Y.; Liu, C.L.; Bai, L.Y.; Li, T.; Wei, F.L. Exploring the Pharmacological Potential of Metformin for Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 838173. [Google Scholar] [CrossRef]
- Kodi, T.; Praveen, S.; Paka, S.K.; Sankhe, R.; Gopinathan, A.; Krishnadas, N.; Kishore, A. Neuroprotective Effects of Metformin and Berberine in Lipopolysaccharide-Induced Sickness-Like Behaviour in Mice. Adv. Pharmacol. Pharm. Sci. 2024, 2024, 8599268. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, H.; Kazem Nezhad, S.; Farmoudeh, A.; Babaei, A.; Ebrahimnejad, P.; Akbari, E.; Siahposht-Khachaki, A. Design and optimization of metformin-loaded solid lipid nanoparticles for neuroprotective effects in a rat model of diffuse traumatic brain injury: A biochemical, behavioral, and histological study. Eur. J. Pharm. Biopharm. 2022, 181, 122–135. [Google Scholar] [CrossRef]
- Kumar, H.; Chakrabarti, A.; Sarma, P.; Modi, M.; Banerjee, D.; Radotra, B.D.; Bhatia, A.; Medhi, B. Novel therapeutic mechanism of action of metformin and its nanoformulation in Alzheimer’s disease and role of AKT/ERK/GSK pathway. Eur. J. Pharm. Sci. 2023, 181, 106348. [Google Scholar] [CrossRef]
- Yuan, M.; Han, Z.; Somayaji, Y.; Nguyen, N.; Hu, H.; Madhu, L.N.; Attaluri, S.; Kodali, M.; Yang, Y.; Hsu, Y.C.; et al. Intranasal delivery of metformin using metal-organic framework (MOF)-74-Mg nanocarriers. Adv. Compos. Hybrid Mater. 2025, 8, 131. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45 (Suppl. S1), S17–S38. [Google Scholar] [CrossRef]
- Maruthur, N.M.; Tseng, E.; Hutfless, S.; Wilson, L.M.; Suarez-Cuervo, C.; Berger, Z.; Chu, Y.; Iyoha, E.; Segal, J.B.; Bolen, S. Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 Diabetes: A Systematic Review and Meta-analysis. Ann. Intern. Med. 2016, 164, 740–751. [Google Scholar] [CrossRef]
- Hirst, J.A.; Farmer, A.J.; Ali, R.; Roberts, N.W.; Stevens, R.J. Quantifying the effect of metformin treatment and dose on glycemic control. Diabetes Care 2012, 35, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Mavridis, D.; Nicolucci, A.; Johnson, D.W.; Tonelli, M.; Craig, J.C.; Maggo, J.; Gray, V.; De Berardis, G.; Ruospo, M.; et al. Comparison of Clinical Outcomes and Adverse Events Associated with Glucose-Lowering Drugs in Patients with Type 2 Diabetes: A Meta-analysis. JAMA 2016, 316, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhu, Y.J.; Zhou, Y.X.; Ding, J.; Liu, J.Y. Metformin in therapeutic applications in human diseases: Its mechanism of action and clinical study. Mol. Biomed. 2022, 3, 41. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R.; et al. Management of hyperglycemia in type 2 diabetes: A patient-centered approach: Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012, 35, 1364–1379. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, F.; Scheen, A. Understanding and overcoming metformin gastrointestinal intolerance. Diabetes Obes. Metab. 2017, 19, 473–481. [Google Scholar] [CrossRef]
- Larsen, A.H.; Wiggers, H.; Dollerup, O.L.; Jespersen, N.R.; Hansson, N.H.; Frokiaer, J.; Brosen, K.; Norrelund, H.; Botker, H.E.; Moller, N.; et al. Metformin Lowers Body Weight But Fails to Increase Insulin Sensitivity in Chronic Heart Failure Patients without Diabetes: A Randomized, Double-Blind, Placebo-Controlled Study. Cardiovasc. Drugs Ther. 2021, 35, 491–503. [Google Scholar] [CrossRef]
- Yuan, Y.; Fan, X.; Guo, Z.; Zhou, Z.; Gao, W. Metformin Protects against Spinal Cord Injury and Cell Pyroptosis via AMPK/NLRP3 Inflammasome Pathway. Anal. Cell. Pathol. 2022, 2022, 3634908. [Google Scholar] [CrossRef]
- Deschemin, J.C.; Foretz, M.; Viollet, B.; Vaulont, S. AMPK is not required for the effect of metformin on the inhibition of BMP6-induced hepcidin gene expression in hepatocytes. Sci. Rep. 2017, 7, 12679. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef]
- Ji, X.; Wang, S.; Tang, H.; Zhang, Y.; Zhou, F.; Zhang, L.; Zhu, Q.; Zhu, K.; Liu, Q.; Liu, Y.; et al. PPP1R3C mediates metformin-inhibited hepatic gluconeogenesis. Metabolism 2019, 98, 62–75. [Google Scholar] [CrossRef]
- Pavlovic, K.; Krako Jakovljevic, N.; Isakovic, A.M.; Ivanovic, T.; Markovic, I.; Lalic, N.M. Therapeutic vs. Suprapharmacological Metformin Concentrations: Different Effects on Energy Metabolism and Mitochondrial Function in Skeletal Muscle Cells in vitro. Front. Pharmacol. 2022, 13, 930308. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Woo, S.L.; Hu, X.; Botchlett, R.; Chen, L.; Huo, Y.; Wu, C. Metformin and metabolic diseases: A focus on hepatic aspects. Front. Med. 2015, 9, 173–186. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Detaille, D.; R-Villanueva, G.; Delgado-Esteban, M.; Guigas, B.; Attia, S.; Fontaine, E.; Almeida, A.; Leverve, X. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J. Mol. Neurosci. 2008, 34, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Del Guerra, S.; Marselli, L.; Lupi, R.; Masini, M.; Pollera, M.; Bugliani, M.; Boggi, U.; Vistoli, F.; Mosca, F.; et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J. Clin. Endocrinol. Metab. 2004, 89, 5535–5541. [Google Scholar] [CrossRef]
- Yang, J.; Holman, G.D. Long-term metformin treatment stimulates cardiomyocyte glucose transport through an AMP-activated protein kinase-dependent reduction in GLUT4 endocytosis. Endocrinology 2006, 147, 2728–2736. [Google Scholar] [CrossRef]
- Shaw, L.M. The insulin receptor substrate (IRS) proteins: At the intersection of metabolism and cancer. Cell Cycle 2011, 10, 1750–1756. [Google Scholar] [CrossRef]
- Pilon, M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis. 2016, 15, 167. [Google Scholar] [CrossRef]
- Ahyayauch, H. Relationship between obesity, insulin resistance and cell membrane properties. Eur. J. Clin. Exp. Med. 2023, 21, 357–364. [Google Scholar] [CrossRef]
- Lupi, R.; Del Guerra, S.; Fierabracci, V.; Marselli, L.; Novelli, M.; Patane, G.; Boggi, U.; Mosca, F.; Piro, S.; Del Prato, S.; et al. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes 2002, 51 (Suppl. S1), S134–S137. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Cheng, M.; Ren, L.; Jia, X.; Wang, J.; Cong, B. Understanding the action mechanisms of metformin in the gastrointestinal tract. Front. Pharmacol. 2024, 15, 1347047. [Google Scholar] [CrossRef]
- Yasuda, N.; Inoue, T.; Nagakura, T.; Yamazaki, K.; Kira, K.; Saeki, T.; Tanaka, I. Enhanced secretion of glucagon-like peptide 1 by biguanide compounds. Biochem. Biophys. Res. Commun. 2002, 298, 779–784. [Google Scholar] [CrossRef]
- Goel, S.; Singh, R.; Singh, V.; Singh, H.; Kumari, P.; Chopra, H.; Sharma, R.; Nepovimova, E.; Valis, M.; Kuca, K.; et al. Metformin: Activation of 5′ AMP-activated protein kinase and its emerging potential beyond anti-hyperglycemic action. Front. Genet. 2022, 13, 1022739. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Ao, H.; Guo, G.; Liu, M. The Role and Mechanism of Metformin in Inflammatory Diseases. J. Inflamm. Res. 2023, 16, 5545–5564. [Google Scholar] [CrossRef] [PubMed]
- Dehkordi, A.H.; Abbaszadeh, A.; Mir, S.; Hasanvand, A. Metformin and its anti-inflammatory and anti-oxidative effects; new concepts. J. Ren. Inj. Prev. 2018, 8, 54–61. [Google Scholar] [CrossRef]
- Jiang, Y.; Huang, W.; Wang, J.; Xu, Z.; He, J.; Lin, X.; Zhou, Z.; Zhang, J. Metformin plays a dual role in MIN6 pancreatic β cell function through AMPK-dependent autophagy. Int. J. Biol. Sci. 2014, 10, 268–277. [Google Scholar] [CrossRef]
- Cao, G.; Gong, T.; Du, Y.; Wang, Y.; Ge, T.; Liu, J. Mechanism of metformin regulation in central nervous system: Progression and future perspectives. Biomed. Pharmacother. 2022, 156, 113686. [Google Scholar] [CrossRef]
- Chele, D.; Sirbu, C.-A.; Mitrica, M.; Toma, M.; Vasiliu, O.; Sirbu, A.-M.; Authier, F.J.; Mischianu, D.; Munteanu, A.E. Metformin’s Effects on Cognitive Function from a Biovariance Perspective: A Narrative Review. Int. J. Mol. Sci. 2025, 26, 1783. [Google Scholar] [CrossRef]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Morales, N.; Iannantuoni, F.; Escribano-Lopez, I.; Banuls, C.; Rovira-Llopis, S.; Sola, E.; Rocha, M.; Hernandez-Mijares, A.; Victor, V.M. Does Metformin Modulate Endoplasmic Reticulum Stress and Autophagy in Type 2 Diabetic Peripheral Blood Mononuclear Cells? Antioxid. Redox Signal. 2018, 28, 1562–1569. [Google Scholar] [CrossRef]
- Yang, L.; Lu, P.; Qi, X.; Yang, Q.; Liu, L.; Dou, T.; Guan, Q.; Yu, C. Metformin inhibits inflammatory response and endoplasmic reticulum stress to improve hypothalamic aging in obese mice. iScience 2023, 26, 108082. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef]
- Griffin, S.J.; Leaver, J.K.; Irving, G.J. Impact of metformin on cardiovascular disease: A meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia 2017, 60, 1620–1629. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Zhu, H.; Jia, Z.; Li, Y.R.; Danelisen, I. Molecular mechanisms of action of metformin: Latest advances and therapeutic implications. Clin. Exp. Med. 2023, 23, 2941–2951. [Google Scholar] [CrossRef]
- Ricci, S.; Cacialli, P. Stem Cell Research Tools in Human Metabolic Disorders: An Overview. Cells 2021, 10, 2681. [Google Scholar] [CrossRef]
- Ezzamouri, B.; Rosario, D.; Bidkhori, G.; Lee, S.; Uhlen, M.; Shoaie, S. Metabolic modelling of the human gut microbiome in type 2 diabetes patients in response to metformin treatment. NPJ Syst. Biol. Appl. 2023, 9, 2. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, X.; Cong, B. Advances in the mechanism of metformin with wide-ranging effects on regulation of the intestinal microbiota. Front. Microbiol. 2024, 15, 1396031. [Google Scholar] [CrossRef]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef]
- An, H.; Wang, Y.; Qin, C.; Li, M.; Maheshwari, A.; He, L. The importance of the AMPK gamma 1 subunit in metformin suppression of liver glucose production. Sci. Rep. 2020, 10, 10482. [Google Scholar] [CrossRef]
- Shibata, S.; Sogabe, S.; Miwa, M.; Fujimoto, T.; Takakura, N.; Naotsuka, A.; Kitamura, S.; Kawamoto, T.; Soga, T. Identification of the first highly selective inhibitor of human lactate dehydrogenase B. Sci. Rep. 2021, 11, 21353. [Google Scholar] [CrossRef] [PubMed]
- Arneson, D.; Zhang, Y.; Yang, X.; Narayanan, M. Shared mechanisms among neurodegenerative diseases: From genetic factors to gene networks. J. Genet. 2018, 97, 795–806. [Google Scholar] [CrossRef]
- Li, P.; Nie, Y.; Yu, J. An Effective Method to Identify Shared Pathways and Common Factors among Neurodegenerative Diseases. PLoS ONE 2015, 10, e0143045. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Marrodan, M.; Ysrraelit, M.C. Mechanisms of Neurodegeneration and Axonal Dysfunction in Progressive Multiple Sclerosis. Biomedicines 2019, 7, 14. [Google Scholar] [CrossRef]
- Wingo, T.S.; Liu, Y.; Gerasimov, E.S.; Vattathil, S.M.; Wynne, M.E.; Liu, J.; Lori, A.; Faundez, V.; Bennett, D.A.; Seyfried, N.T.; et al. Shared mechanisms across the major psychiatric and neurodegenerative diseases. Nat. Commun. 2022, 13, 4314. [Google Scholar] [CrossRef]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Gerke-Duncan, M.B.; Holsinger, R.M.D. Oxidative Stress and Antioxidants in Neurodegenerative Disorders. Antioxidants 2023, 12, 517. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, X.; Liu, C.; Liu, Q.; Chai, H.; Luo, Y.; Li, S. Role of Mitochondria in Neurodegenerative Diseases: From an Epigenetic Perspective. Front. Cell Dev. Biol. 2021, 9, 688789. [Google Scholar] [CrossRef]
- Berth, S.H.; Lloyd, T.E. Disruption of axonal transport in neurodegeneration. J. Clin. Investig. 2023, 133, e168554. [Google Scholar] [CrossRef]
- Giri, P.M.; Banerjee, A.; Ghosal, A.; Layek, B. Neuroinflammation in Neurodegenerative Disorders: Current Knowledge and Therapeutic Implications. Int. J. Mol. Sci. 2024, 25, 3995. [Google Scholar] [CrossRef] [PubMed]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative Diseases—Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef]
- Amartumur, S.; Nguyen, H.; Huynh, T.; Kim, T.S.; Woo, R.S.; Oh, E.; Kim, K.K.; Lee, L.P.; Heo, C. Neuropathogenesis-on-chips for neurodegenerative diseases. Nat. Commun. 2024, 15, 2219. [Google Scholar] [CrossRef] [PubMed]
- Sonninen, T.M.; Goldsteins, G.; Laham-Karam, N.; Koistinaho, J.; Lehtonen, S. Proteostasis Disturbances and Inflammation in Neurodegenerative Diseases. Cells 2020, 9, 2183. [Google Scholar] [CrossRef] [PubMed]
- Koszla, O.; Solek, P. Misfolding and aggregation in neurodegenerative diseases: Protein quality control machinery as potential therapeutic clearance pathways. Cell Commun. Signal. 2024, 22, 421. [Google Scholar] [CrossRef]
- Tsuboyama, K.; Dauparas, J.; Chen, J.; Laine, E.; Mohseni Behbahani, Y.; Weinstein, J.J.; Mangan, N.M.; Ovchinnikov, S.; Rocklin, G.J. Mega-scale experimental analysis of protein folding stability in biology and design. Nature 2023, 620, 434–444. [Google Scholar] [CrossRef]
- Sanz-Hernandez, M.; Barritt, J.D.; Sobek, J.; Hornemann, S.; Aguzzi, A.; De Simone, A. Mechanism of misfolding of the human prion protein revealed by a pathological mutation. Proc. Natl. Acad. Sci. USA 2021, 118, e2019631118. [Google Scholar] [CrossRef]
- Wille, H.; Dorosh, L.; Amidian, S.; Schmitt-Ulms, G.; Stepanova, M. Combining molecular dynamics simulations and experimental analyses in protein misfolding. Adv. Protein Chem. Struct. Biol. 2019, 118, 33–110. [Google Scholar] [CrossRef]
- Diociaiuti, M.; Bonanni, R.; Cariati, I.; Frank, C.; D’Arcangelo, G. Amyloid Prefibrillar Oligomers: The Surprising Commonalities in Their Structure and Activity. Int. J. Mol. Sci. 2021, 22, 6435. [Google Scholar] [CrossRef] [PubMed]
- Alibhai, J.; Blanco, R.A.; Barria, M.A.; Piccardo, P.; Caughey, B.; Perry, V.H.; Freeman, T.C.; Manson, J.C. Distribution of Misfolded Prion Protein Seeding Activity Alone Does Not Predict Regions of Neurodegeneration. PLoS Biol. 2016, 14, e1002579. [Google Scholar] [CrossRef]
- Okuzumi, A.; Kurosawa, M.; Hatano, T.; Takanashi, M.; Nojiri, S.; Fukuhara, T.; Yamanaka, T.; Miyazaki, H.; Yoshinaga, S.; Furukawa, Y.; et al. Rapid dissemination of α-synuclein seeds through neural circuits in an in-vivo prion-like seeding experiment. Acta Neuropathol. Commun. 2018, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Belian, S.; Zurzolo, C. Hijacking intercellular trafficking for the spread of protein aggregates in neurodegenerative diseases: A focus on tunneling nanotubes (TNTs). Extracell. Vesicles Circ. Nucleic Acids 2023, 4, 27–43. [Google Scholar] [CrossRef]
- Moda, F.; Ciullini, A.; Dellarole, I.L.; Lombardo, A.; Campanella, N.; Bufano, G.; Cazzaniga, F.A.; Giaccone, G. Secondary Protein Aggregates in Neurodegenerative Diseases: Almost the Rule Rather than the Exception. Front. Biosci. 2023, 28, 255. [Google Scholar] [CrossRef]
- Prymaczok, N.C.; De Francesco, P.N.; Mazzetti, S.; Humbert-Claude, M.; Tenenbaum, L.; Cappelletti, G.; Masliah, E.; Perello, M.; Riek, R.; Gerez, J.A. Cell-to-cell transmitted α-synuclein recapitulates experimental Parkinson’s disease. NPJ Park. Dis. 2024, 10, 10. [Google Scholar] [CrossRef]
- Cai, Y.; Du, J.; Li, A.; Zhu, Y.; Xu, L.; Sun, K.; Ma, S.; Guo, T.; Alzheimer’s Disease Neuroimaging Initiative. Initial levels of β-amyloid and tau deposition have distinct effects on longitudinal tau accumulation in Alzheimer’s disease. Alzheimer’s Res. Ther. 2023, 15, 30. [Google Scholar] [CrossRef]
- Miguez, A.; Gomis, C.; Vila, C.; Monguio-Tortajada, M.; Fernandez-Garcia, S.; Bombau, G.; Galofre, M.; Garcia-Bravo, M.; Sanders, P.; Fernandez-Medina, H.; et al. Soluble mutant huntingtin drives early human pathogenesis in Huntington’s disease. Cell. Mol. Life Sci. 2023, 80, 238. [Google Scholar] [CrossRef]
- Mosleth, E.F.; Vedeler, C.A.; Liland, K.H.; McLeod, A.; Bringeland, G.H.; Kroondijk, L.; Berven, F.S.; Lysenko, A.; Rawlings, C.J.; Eid, K.E.; et al. Cerebrospinal fluid proteome shows disrupted neuronal development in multiple sclerosis. Sci. Rep. 2021, 11, 4087. [Google Scholar] [CrossRef]
- Weids, A.J.; Ibstedt, S.; Tamas, M.J.; Grant, C.M. Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 2016, 6, 24554. [Google Scholar] [CrossRef]
- Dong, Z.; Cui, H. The Autophagy-Lysosomal Pathways and Their Emerging Roles in Modulating Proteostasis in Tumors. Cells 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Shahheydari, H.; Ragagnin, A.; Walker, A.K.; Toth, R.P.; Vidal, M.; Jagaraj, C.J.; Perri, E.R.; Konopka, A.; Sultana, J.M.; Atkin, J.D. Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front. Mol. Neurosci. 2017, 10, 119. [Google Scholar] [CrossRef]
- Watanabe, Y.; Takeda, H.; Honda, N.; Hanajima, R. A bioinformatic investigation of proteasome and autophagy expression in the central nervous system. Heliyon 2023, 9, e18188. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Nahm, M.; Kim, S.H. Proteostasis and Ribostasis Impairment as Common Cell Death Mechanisms in Neurodegenerative Diseases. J. Clin. Neurol. 2023, 19, 101–114. [Google Scholar] [CrossRef]
- Feleciano, D.R.; Juenemann, K.; Iburg, M.; Bras, I.C.; Holmberg, C.I.; Kirstein, J. Crosstalk Between Chaperone-Mediated Protein Disaggregation and Proteolytic Pathways in Aging and Disease. Front. Aging Neurosci. 2019, 11, 9. [Google Scholar] [CrossRef]
- Stojowska-Swedrzynska, K.; Kuczynska-Wisnik, D.; Laskowska, E. Influence of Nε-Lysine Acetylation on the Formation of Protein Aggregates and Antibiotic Persistence in E. coli. Molecules 2024, 29, 383. [Google Scholar] [CrossRef]
- Posadas, Y.; Sanchez-Lopez, C.; Quintanar, L. Copper binding and protein aggregation: A journey from the brain to the human lens. RSC Chem. Biol. 2023, 4, 974–985. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef]
- Yao, Q.; Mascarenhas Dos Santos, A.C.; Zhang, H.; Manas, A.; Hussaini, A.; Kim, U.; Xu, C.; Basheer, S.; Tasaki, S.; Xiang, J. Unconventional Source of Neurotoxic Protein Aggregation from Organelle Off-Target Bax∆2 in Alzheimer’s Disease. Biomolecules 2023, 13, 970. [Google Scholar] [CrossRef]
- Bonfanti, S.; Lionetti, M.C.; Fumagalli, M.R.; Chirasani, V.R.; Tiana, G.; Dokholyan, N.V.; Zapperi, S.; La Porta, C.A.M. Molecular mechanisms of heterogeneous oligomerization of huntingtin proteins. Sci. Rep. 2019, 9, 7615. [Google Scholar] [CrossRef]
- Zhou, W.; Graner, M.; Paucek, P.; Beseler, C.; Boisen, M.; Bubak, A.; Asturias, F.; George, W.; Graner, A.; Ormond, D.; et al. Multiple sclerosis plasma IgG aggregates induce complement-dependent neuronal apoptosis. Cell Death Dis. 2023, 14, 254. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.B.; Ferrari, L.; Rudiger, S.G.D. How do protein aggregates escape quality control in neurodegeneration? Trends Neurosci. 2022, 45, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Bevan-Jones, W.R.; Cope, T.E.; Jones, P.S.; Kaalund, S.S.; Passamonti, L.; Allinson, K.; Green, O.; Hong, Y.T.; Fryer, T.D.; Arnold, R.; et al. Neuroinflammation and protein aggregation co-localize across the frontotemporal dementia spectrum. Brain 2020, 143, 1010–1026. [Google Scholar] [CrossRef]
- Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergstrom, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J. Neuroinflammation 2021, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Garcia-Sanchez, A.; Miranda-Diaz, A.G.; Cardona-Munoz, E.G. The Role of Oxidative Stress in Physiopathology and Pharmacological Treatment with Pro- and Antioxidant Properties in Chronic Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 2082145. [Google Scholar] [CrossRef]
- Albano, G.D.; Gagliardo, R.P.; Montalbano, A.M.; Profita, M. Overview of the Mechanisms of Oxidative Stress: Impact in Inflammation of the Airway Diseases. Antioxidants 2022, 11, 2237. [Google Scholar] [CrossRef]
- Afzal, S.; Abdul Manap, A.S.; Attiq, A.; Albokhadaim, I.; Kandeel, M.; Alhojaily, S.M. From imbalance to impairment: The central role of reactive oxygen species in oxidative stress-induced disorders and therapeutic exploration. Front. Pharmacol. 2023, 14, 1269581. [Google Scholar] [CrossRef]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells 2022, 11, 552. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Houldsworth, A. Role of oxidative stress in neurodegenerative disorders: A review of reactive oxygen species and prevention by antioxidants. Brain Commun. 2024, 6, fcad356. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, I.I.; Radu, C.I.; Vladacenco, O.; Roza, E.; Costachescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Ihnatovych, I.; Birkaya, B.; Notari, E.; Szigeti, K. iPSC-Derived Microglia for Modeling Human-Specific DAMP and PAMP Responses in the Context of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9668. [Google Scholar] [CrossRef]
- Chang, N.P.; DaPrano, E.M.; Lindman, M.; Estevez, I.; Chou, T.W.; Evans, W.R.; Nissenbaum, M.; McCourt, M.; Alzate, D.; Atkins, C.; et al. Neuronal DAMPs exacerbate neurodegeneration via astrocytic RIPK3 signaling. JCI Insight 2024, 9, e177002. [Google Scholar] [CrossRef]
- Li, X.; Tong, H.; Xu, S.; Zhou, G.; Yang, T.; Yin, S.; Yang, S.; Li, X.; Li, S. Neuroinflammatory Proteins in Huntington’s Disease: Insights into Mechanisms, Diagnosis, and Therapeutic Implications. Int. J. Mol. Sci. 2024, 25, 11787. [Google Scholar] [CrossRef] [PubMed]
- Djebar, M.; Anselme, I.; Pezeron, G.; Bardet, P.L.; Cantaut-Belarif, Y.; Eschstruth, A.; Lopez-Santos, D.; Le Ribeuz, H.; Jenett, A.; Khoury, H.; et al. Astrogliosis and neuroinflammation underlie scoliosis upon cilia dysfunction. eLife 2024, 13, RP96831. [Google Scholar] [CrossRef]
- Ishijima, T.; Nakajima, K. Inflammatory cytokines TNFα, IL-1β, and IL-6 are induced in endotoxin-stimulated microglia through different signaling cascades. Sci. Prog. 2021, 104, 368504211054985. [Google Scholar] [CrossRef]
- Güven, G.; Köseoğlu, P.; Lohmann, E.; Samancı, B.; Şahin, E.; Bilgiç, B.; Hanağası, H.A.; Gürvit, H.; Erginel-Ünaltuna, N. Peripheral Expression of IL-6, TNF-α and TGF-β1 in Alzheimer’s Disease Patients. Turk. J. Immunol. 2024, 12, 28–34. [Google Scholar] [CrossRef]
- Preis, L.; Villringer, K.; Brosseron, F.; Duzel, E.; Jessen, F.; Petzold, G.C.; Ramirez, A.; Spottke, A.; Fiebach, J.B.; Peters, O. Assessing blood-brain barrier dysfunction and its association with Alzheimer’s pathology, cognitive impairment and neuroinflammation. Alzheimer’s Res. Ther. 2024, 16, 172. [Google Scholar] [CrossRef]
- Mayer, M.G.; Fischer, T. Microglia at the blood brain barrier in health and disease. Front. Cell. Neurosci. 2024, 18, 1360195. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zeng, Q.; Baris, M.; Tezel, G. Transgenic inhibition of astroglial NF-κB restrains the neuroinflammatory and neurodegenerative outcomes of experimental mouse glaucoma. J. Neuroinflammation 2020, 17, 252. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, V.A.; Alameda, J.P.; Page, A.; Casanova, M.L. Role of NF-κB in Ageing and Age-Related Diseases: Lessons from Genetically Modified Mouse Models. Cells 2021, 10, 1906. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Basurco, L.; Abellanas, M.A.; Ayerra, L.; Conde, E.; Vinueza-Gavilanes, R.; Luquin, E.; Vales, A.; Vilas, A.; Martin-Uriz, P.S.; Tamayo, I.; et al. Microglia and astrocyte activation is region-dependent in the α-synuclein mouse model of Parkinson’s disease. Glia 2023, 71, 571–587. [Google Scholar] [CrossRef]
- Edler, M.K.; Munger, E.L.; Maycon, H.; Hopkins, W.D.; Hof, P.R.; Sherwood, C.C.; Raghanti, M.A. The association of astrogliosis and microglial activation with aging and Alzheimer’s disease pathology in the chimpanzee brain. J. Neurosci. Res. 2023, 101, 881–900. [Google Scholar] [CrossRef]
- Hohn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef]
- Peng, K.; Xiao, J.; Yang, L.; Ye, F.; Cao, J.; Sai, Y. Mutual Antagonism of PINK1/Parkin and PGC-1α Contributes to Maintenance of Mitochondrial Homeostasis in Rotenone-Induced Neurotoxicity. Neurotox. Res. 2019, 35, 331–343. [Google Scholar] [CrossRef]
- Van Acker, Z.P.; Leroy, T.; Annaert, W. Mitochondrial dysfunction, cause or consequence in neurodegenerative diseases? Bioessays 2025, 47, e2400023. [Google Scholar] [CrossRef]
- Tresse, E.; Marturia-Navarro, J.; Sew, W.Q.G.; Cisquella-Serra, M.; Jaberi, E.; Riera-Ponsati, L.; Fauerby, N.; Hu, E.; Kretz, O.; Aznar, S.; et al. Mitochondrial DNA damage triggers spread of Parkinson’s disease-like pathology. Mol. Psychiatry 2023, 28, 4902–4914. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants 2021, 10, 1917. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wan, Z.; Tang, Y.; Xu, J.; Laboret, B.; Nallamothu, S.; Yang, C.; Liu, B.; Lu, R.O.; Lu, B.; et al. Pantothenate kinase 2 interacts with PINK1 to regulate mitochondrial quality control via acetyl-CoA metabolism. Nat. Commun. 2022, 13, 2412. [Google Scholar] [CrossRef]
- Yan, X.; Wang, B.; Hu, Y.; Wang, S.; Zhang, X. Abnormal Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 138. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Brassard, R.; Arutyunova, E.; Takyi, E.; Espinoza-Fonseca, L.M.; Young, H.S.; Touret, N.; Lemieux, M.J. Transmembrane Parkinson’s disease mutation of PINK1 leads to altered mitochondrial anchoring. J. Biol. Chem. 2025, 301, 108253. [Google Scholar] [CrossRef]
- Canty, J.T.; Hensley, A.; Aslan, M.; Jack, A.; Yildiz, A. TRAK adaptors regulate the recruitment and activation of dynein and kinesin in mitochondrial transport. Nat. Commun. 2023, 14, 1376. [Google Scholar] [CrossRef] [PubMed]
- Fenton, A.R.; Jongens, T.A.; Holzbaur, E.L.F. Mitochondrial adaptor TRAK2 activates and functionally links opposing kinesin and dynein motors. Nat. Commun. 2021, 12, 4578. [Google Scholar] [CrossRef]
- Gu, Y.Y.; Zhao, X.R.; Zhang, N.; Yang, Y.; Yi, Y.; Shao, Q.H.; Liu, M.X.; Zhang, X.L. Mitochondrial dysfunction as a therapeutic strategy for neurodegenerative diseases: Current insights and future directions. Ageing Res. Rev. 2024, 102, 102577. [Google Scholar] [CrossRef]
- Holscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.J.O.; de Oliveira, L.M.; Bittencourt, A.M.V.; Lourenco, L.G.C.; de Oliveira, G.C.M. Brain insulin resistance and Alzheimer’s disease: A systematic review. Dement. Neuropsychol. 2024, 18, e20230032. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Al-Jarallah, A.; Babiker, F.A. Impaired Insulin Signaling Alters Mediators of Hippocampal Synaptic Dynamics/Plasticity: A Possible Mechanism of Hyperglycemia-Induced Cognitive Impairment. Cells 2023, 12, 1728. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A. Deciphering Brain Insulin Receptor and Insulin-Like Growth Factor 1 Receptor Signalling. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef]
- Pomytkin, I.; Pinelis, V. Brain Insulin Resistance: Focus on Insulin Receptor-Mitochondria Interactions. Life 2021, 11, 262. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Oliveira, L.T.; Leon, G.V.O.; Provance, D.W., Jr.; de Mello, F.G.; Sorenson, M.M.; Salerno, V.P. Exogenous β-amyloid peptide interferes with GLUT4 localization in neurons. Brain Res. 2015, 1615, 42–50. [Google Scholar] [CrossRef]
- Rebelos, E.; Bucci, M.; Karjalainen, T.; Oikonen, V.; Bertoldo, A.; Hannukainen, J.C.; Virtanen, K.A.; Latva-Rasku, A.; Hirvonen, J.; Heinonen, I.; et al. Insulin Resistance Is Associated with Enhanced Brain Glucose Uptake During Euglycemic Hyperinsulinemia: A Large-Scale PET Cohort. Diabetes Care 2021, 44, 788–794. [Google Scholar] [CrossRef]
- Xia, H.; Scholtes, C.; Dufour, C.R.; Ouellet, C.; Ghahremani, M.; Giguere, V. Insulin action and resistance are dependent on a GSK3β-FBXW7-ERRα transcriptional axis. Nat. Commun. 2022, 13, 2105. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef]
- Madhusudhanan, J.; Suresh, G.; Devanathan, V. Neurodegeneration in type 2 diabetes: Alzheimer’s as a case study. Brain Behav. 2020, 10, e01577. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Shi, X.; Han, J.; Lin, B.; Peng, W.; Mei, Z.; Lin, Y. Metformin and the risk of neurodegenerative diseases in patients with diabetes: A meta-analysis of population-based cohort studies. Diabet. Med. 2022, 39, e14821. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Liu, S.; Fonseca, V.A.; Thethi, T.K.; Shi, L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open 2019, 9, e024954. [Google Scholar] [CrossRef]
- Gupta, M.; Pandey, S.; Rumman, M.; Singh, B.; Mahdi, A.A. Molecular mechanisms underlying hyperglycemia associated cognitive decline. IBRO Neurosci. Rep. 2023, 14, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Violante-Cumpa, J.R.; Perez-Arredondo, L.A.; Gonzalez-Gonzalez, J.G.; Mancillas-Adame, L.G. Comment on Samara et al. Metformin Use Is Associated with Slowed Cognitive Decline and Reduced Incident Dementia in Older Adults with Type 2 Diabetes: The Sydney Memory and Ageing Study. Diabetes Care 2020;43:2691–2701. Diabetes Care 2021, 44, e73. [Google Scholar] [CrossRef]
- Khezri, M.R.; Yousefi, K.; Mahboubi, N.; Hodaei, D.; Ghasemnejad-Berenji, M. Metformin in Alzheimer’s disease: An overview of potential mechanisms, preclinical and clinical findings. Biochem. Pharmacol. 2022, 197, 114945. [Google Scholar] [CrossRef]
- Mendonca, I.P.; de Paiva, I.H.R.; Duarte-Silva, E.P.; de Melo, M.G.; da Silva, R.S.; do Nascimento, M.I.X.; Peixoto, C.A. Metformin improves depressive-like behavior in experimental Parkinson’s disease by inducing autophagy in the substantia nigra and hippocampus. Inflammopharmacology 2022, 30, 1705–1716. [Google Scholar] [CrossRef]
- Hurtado-Carneiro, V.; LeBaut-Ayuso, Y.; Velazquez, E.; Flores-Lamas, C.; Fernandez-de la Rosa, R.; Garcia-Garcia, L.; Gomez-Oliver, F.; Ruiz-Albusac, J.M.; Pozo, M.A. Effects of chronic treatment with metformin on brain glucose hypometabolism and central insulin actions in transgenic mice with tauopathy. Heliyon 2024, 10, e35752. [Google Scholar] [CrossRef]
- Petrasca, A.; Hambly, R.; Kearney, N.; Smith, C.M.; Pender, E.K.; Mac Mahon, J.; O’Rourke, A.M.; Ismaiel, M.; Boland, P.A.; Almeida, J.P.; et al. Metformin has anti-inflammatory effects and induces immunometabolic reprogramming via multiple mechanisms in hidradenitis suppurativa. Br. J. Dermatol. 2023, 189, 730–740. [Google Scholar] [CrossRef]
- Sakata, N. The anti-inflammatory effect of metformin: The molecular targets. Genes Cells 2024, 29, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Y.; Liu, S.; Gao, M.; Wang, W.; Chen, K.; Huang, L.; Liu, Y. Diabetic vascular diseases: Molecular mechanisms and therapeutic strategies. Signal Transduct. Target. Ther. 2023, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.; Yadav, D.; Patel, S.; Song, M. Effects of a Diabetic Microenvironment on Neurodegeneration: Special Focus on Neurological Cells. Brain Sci. 2024, 14, 284. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Lv, C.; Geng, P.; Fu, M.; Zhou, W.; Xiong, M.; Li, T. Novel targets and therapies of metformin in dementia: Old drug, new insights. Front. Pharmacol. 2024, 15, 1415740. [Google Scholar] [CrossRef]
- Ruan, C.; Guo, H.; Gao, J.; Wang, Y.; Liu, Z.; Yan, J.; Li, X.; Lv, H. Neuroprotective effects of metformin on cerebral ischemia-reperfusion injury by regulating PI3K/Akt pathway. Brain Behav. 2021, 11, e2335. [Google Scholar] [CrossRef]
- Campbell, J.M.; Stephenson, M.D.; de Courten, B.; Chapman, I.; Bellman, S.M.; Aromataris, E. Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2018, 65, 1225–1236. [Google Scholar] [CrossRef]
- Papini, N.; Giussani, P.; Tringali, C. Metformin Lysosomal Targeting: A Novel Aspect to Be Investigated for Metformin Repurposing in Neurodegenerative Diseases? Int. J. Mol. Sci. 2024, 25, 8884. [Google Scholar] [CrossRef]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, K.; Wang, R.; Liu, Y.; Kwak, Y.D.; Ma, T.; Thompson, R.C.; Zhao, Y.; Smith, L.; Gasparini, L.; et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc. Natl. Acad. Sci. USA 2009, 106, 3907–3912. [Google Scholar] [CrossRef]
- Son, S.M.; Shin, H.J.; Byun, J.; Kook, S.Y.; Moon, M.; Chang, Y.J.; Mook-Jung, I. Metformin Facilitates Amyloid-β Generation by β- and γ-Secretases via Autophagy Activation. J. Alzheimer’s Dis. 2016, 51, 1197–1208. [Google Scholar] [CrossRef]
- Abdelaziz, D.H.; Thapa, S.; Abdulrahman, B.; Vankuppeveld, L.; Schatzl, H.M. Metformin reduces prion infection in neuronal cells by enhancing autophagy. Biochem. Biophys. Res. Commun. 2020, 523, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, C.; Gao, K.; Zhao, H.; Zhou, Z.; Shen, Z.; Guo, Y.; Li, Z.; Yao, T.; Mei, X. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem. Biophys. Res. Commun. 2016, 477, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Tayara, K.; Espinosa-Oliva, A.M.; Garcia-Dominguez, I.; Ismaiel, A.A.; Boza-Serrano, A.; Deierborg, T.; Machado, A.; Herrera, A.J.; Venero, J.L.; de Pablos, R.M. Divergent Effects of Metformin on an Inflammatory Model of Parkinson’s Disease. Front. Cell. Neurosci. 2018, 12, 440. [Google Scholar] [CrossRef]
- Zhu, X.C.; Jiang, T.; Zhang, Q.Q.; Cao, L.; Tan, M.S.; Wang, H.F.; Ding, Z.Z.; Tan, L.; Yu, J.T. Chronic Metformin Preconditioning Provides Neuroprotection via Suppression of NF-κB-Mediated Inflammatory Pathway in Rats with Permanent Cerebral Ischemia. Mol. Neurobiol. 2015, 52, 375–385. [Google Scholar] [CrossRef]
- Sportelli, C.; Urso, D.; Jenner, P.; Chaudhuri, K.R. Metformin as a Potential Neuroprotective Agent in Prodromal Parkinson’s Disease-Viewpoint. Front. Neurol. 2020, 11, 556. [Google Scholar] [CrossRef]
- Taheri, M.; Roghani, M.; Sedaghat, R. Metformin Mitigates Trimethyltin-Induced Cognition Impairment and Hippocampal Neurodegeneration. Cell. Mol. Neurobiol. 2024, 44, 70. [Google Scholar] [CrossRef]
- Li, H.; Liu, R.; Liu, J.; Qu, Y. The Role and Mechanism of Metformin in the Treatment of Nervous System Diseases. Biomolecules 2024, 14, 1579. [Google Scholar] [CrossRef]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.S.; He, Y.; Kivisakk, P.; Arnold, S.E.; Das, S. Effect of Metformin on Plasma and Cerebrospinal Fluid Biomarkers in Non-Diabetic Older Adults with Mild Cognitive Impairment Related to Alzheimer’s Disease. J. Alzheimer’s Dis. 2024, 99 (Suppl. S2), S355–S365. [Google Scholar] [CrossRef]
- Poor, S.R.; Ettcheto, M.; Cano, A.; Sanchez-Lopez, E.; Manzine, P.R.; Olloquequi, J.; Camins, A.; Javan, M. Metformin a Potential Pharmacological Strategy in Late Onset Alzheimer’s Disease Treatment. Pharmaceuticals 2021, 14, 890. [Google Scholar] [CrossRef]
- Geng, C.; Meng, K.; Zhao, B.; Liu, X.; Tang, Y. Causal relationships between type 1 diabetes mellitus and Alzheimer’s disease and Parkinson’s disease: A bidirectional two-sample Mendelian randomization study. Eur. J. Med. Res. 2024, 29, 53. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Associations Between Diabetes Mellitus and Neurodegenerative Diseases. Int. J. Mol. Sci. 2025, 26, 542. [Google Scholar] [CrossRef]
- Tuligenga, R.H.; Dugravot, A.; Tabak, A.G.; Elbaz, A.; Brunner, E.J.; Kivimaki, M.; Singh-Manoux, A. Midlife type 2 diabetes and poor glycaemic control as risk factors for cognitive decline in early old age: A post-hoc analysis of the Whitehall II cohort study. Lancet Diabetes Endocrinol. 2014, 2, 228–235. [Google Scholar] [CrossRef]
- Wu, J.; Fang, Y.; Tan, X.; Kang, S.; Yue, X.; Rao, Y.; Huang, H.; Liu, M.; Qiu, S.; Yap, P.T. Detecting type 2 diabetes mellitus cognitive impairment using whole-brain functional connectivity. Sci. Rep. 2023, 13, 3940. [Google Scholar] [CrossRef]
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [PubMed]
- Orkaby, A.R.; Cho, K.; Cormack, J.; Gagnon, D.R.; Driver, J.A. Metformin vs sulfonylurea use and risk of dementia in US veterans aged ≥65 years with diabetes. Neurology 2017, 89, 1877–1885. [Google Scholar] [CrossRef]
- Pomilio, C.; Perez, N.G.; Calandri, I.; Crivelli, L.; Allegri, R.; Initiative, A.A.s.D.N.; Sevlever, G.; Saravia, F. Diabetic patients treated with metformin during early stages of Alzheimer’s disease show a better integral performance: Data from ADNI study. Geroscience 2022, 44, 1791–1805. [Google Scholar] [CrossRef]
- Luo, A.; Ning, P.; Lu, H.; Huang, H.; Shen, Q.; Zhang, D.; Xu, F.; Yang, L.; Xu, Y. Association Between Metformin and Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Clinical Observational Studies. J. Alzheimer’s Dis. 2022, 88, 1311–1323. [Google Scholar] [CrossRef]
- Tang, H.; Guo, J.; Shaaban, C.E.; Feng, Z.; Wu, Y.; Magoc, T.; Hu, X.; Donahoo, W.T.; DeKosky, S.T.; Bian, J. Heterogeneous treatment effects of metformin on risk of dementia in patients with type 2 diabetes: A longitudinal observational study. Alzheimer’s Dement. 2024, 20, 975–985. [Google Scholar] [CrossRef]
- Barbera, M.; Lehtisalo, J.; Perera, D.; Aspo, M.; Cross, M.; De Jager Loots, C.A.; Falaschetti, E.; Friel, N.; Luchsinger, J.A.; Gavelin, H.M.; et al. A multimodal precision-prevention approach combining lifestyle intervention with metformin repurposing to prevent cognitive impairment and disability: The MET-FINGER randomised controlled trial protocol. Alzheimer’s Res. Ther. 2024, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- NCT00620191, Metformin in Amnestic Mild Cognitive Impairment (MCI), José A. Luchsinger, Columbia University. 2020. Available online: https://clinicaltrials.gov/study/NCT00620191 (accessed on 13 March 2025).
- NCT01965756, Effect of Insulin Sensitizer Metformin on AD Biomarkers, University of Pennsylvania. 2017. Available online: https://clinicaltrials.gov/study/NCT01965756 (accessed on 13 March 2025).
- NCT03757910; Brain Imaging in the Diabetes Prevention Program Outcomes Study (DPPOS-Brain), José A. Luchsinger, Columbia University. 2024. Available online: https://clinicaltrials.gov/study/NCT03757910 (accessed on 13 March 2025).
- Isop, L.M.; Neculau, A.E.; Necula, R.D.; Kakucs, C.; Moga, M.A.; Dima, L. Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders. Pharmaceuticals 2023, 16, 1714. [Google Scholar] [CrossRef]
- El-Ghaiesh, S.H.; Bahr, H.I.; Ibrahiem, A.T.; Ghorab, D.; Alomar, S.Y.; Farag, N.E.; Zaitone, S.A. Metformin Protects From Rotenone-Induced Nigrostriatal Neuronal Death in Adult Mice by Activating AMPK-FOXO3 Signaling and Mitigation of Angiogenesis. Front. Mol. Neurosci. 2020, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.R.; Yin, X.; Kang, S.U.; Mitra, J.; Wang, H.; Ryu, T.; Brahmachari, S.; Karuppagounder, S.S.; Kimura, Y.; Jhaldiyal, A.; et al. Enhanced mTORC1 signaling and protein synthesis in pathologic α-synuclein cellular and animal models of Parkinson’s disease. Sci. Transl. Med. 2023, 15, eadd0499. [Google Scholar] [CrossRef]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin functionally interacts with PGC-1α to preserve mitochondria and protect dopaminergic neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar] [CrossRef]
- Newby, D.; Linden, A.B.; Fernandes, M.; Molero, Y.; Winchester, L.; Sproviero, W.; Ghose, U.; Li, Q.S.; Launer, L.J.; Duijn, C.M.V.; et al. Comparative effect of metformin versus sulfonylureas with dementia and Parkinson’s disease risk in US patients over 50 with type 2 diabetes mellitus. BMJ Open Diabetes Res. Care 2022, 10, e003036. [Google Scholar] [CrossRef]
- Huang, K.H.; Chang, Y.L.; Gau, S.Y.; Tsai, T.H.; Lee, C.Y. Dose-Response Association of Metformin with Parkinson’s Disease Odds in Type 2 Diabetes Mellitus. Pharmaceutics 2022, 14, 946. [Google Scholar] [CrossRef]
- Rozani, V.; Bezimianski, M.G.; Azuri, J.; Bitan, M.; Peretz, C. Anti-diabetic drug use and reduced risk of Parkinson’s disease: A community-based cohort study. Park. Relat. Disord. 2024, 128, 107132. [Google Scholar] [CrossRef]
- NCT03685357; Correlation Between Idiopathic Parkinson’s Disease and Diabetes Mellitus, Afnan Awad-Allah Elgnainy, Ain Shams University. 2018. Available online: https://www.clinicaltrials.gov/study/NCT03685357 (accessed on 13 March 2025).
- Boi, L.; Pisanu, A.; Palmas, M.F.; Fusco, G.; Carboni, E.; Casu, M.A.; Satta, V.; Scherma, M.; Janda, E.; Mocci, I.; et al. Modeling Parkinson’s Disease Neuropathology and Symptoms by Intranigral Inoculation of Preformed Human α-Synuclein Oligomers. Int. J. Mol. Sci. 2020, 21, 8535. [Google Scholar] [CrossRef]
- Zhang, J.B.; Wan, X.J.; Duan, W.X.; Dai, X.Q.; Xia, D.; Fu, X.; Hu, L.F.; Wang, F.; Liu, C.F. Circadian disruption promotes the neurotoxicity of oligomeric α-synuclein in mice. NPJ Park. Dis. 2024, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Perez-Revuelta, B.I.; Hettich, M.M.; Ciociaro, A.; Rotermund, C.; Kahle, P.J.; Krauss, S.; Di Monte, D.A. Metformin lowers Ser-129 phosphorylated α-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. 2014, 5, e1209. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.S.; Choi, D.Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]
- Park, J.M.; Lee, D.H.; Kim, D.H. Redefining the role of AMPK in autophagy and the energy stress response. Nat. Commun. 2023, 14, 2994. [Google Scholar] [CrossRef]
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447. [Google Scholar] [CrossRef] [PubMed]
- Norradee, C.; Khwanraj, K.; Balit, T.; Dharmasaroja, P. Evaluation of the Combination of Metformin and Rapamycin in an MPP(+)-Treated SH-SY5Y Model of Parkinson’s Disease. Adv. Pharmacol. Pharm. Sci. 2023, 2023, 3830861. [Google Scholar] [CrossRef]
- Ping, F.; Jiang, N.; Li, Y. Association between metformin and neurodegenerative diseases of observational studies: Systematic review and meta-analysis. BMJ Open Diabetes Res. Care 2020, 8, e001370. [Google Scholar] [CrossRef]
- Ozbey, G.; Nemutlu-Samur, D.; Parlak, H.; Yildirim, S.; Aslan, M.; Tanriover, G.; Agar, A. Metformin protects rotenone-induced dopaminergic neurodegeneration by reducing lipid peroxidation. Pharmacol. Rep. 2020, 72, 1397–1406. [Google Scholar] [CrossRef]
- Saewanee, N.; Praputpittaya, T.; Malaiwong, N.; Chalorak, P.; Meemon, K. Neuroprotective effect of metformin on dopaminergic neurodegeneration and α-synuclein aggregation in C. elegans model of Parkinson’s disease. Neurosci. Res. 2021, 162, 13–21. [Google Scholar] [CrossRef]
- Van de Roovaart, H.J.; Nguyen, N.; Veenstra, T.D. Huntington’s Disease Drug Development: A Phase 3 Pipeline Analysis. Pharmaceuticals 2023, 16, 1513. [Google Scholar] [CrossRef] [PubMed]
- Arnoux, I.; Willam, M.; Griesche, N.; Krummeich, J.; Watari, H.; Offermann, N.; Weber, S.; Narayan Dey, P.; Chen, C.; Monteiro, O.; et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. eLife 2018, 7, e38744. [Google Scholar] [CrossRef]
- Trujillo-Del Rio, C.; Tortajada-Perez, J.; Gomez-Escribano, A.P.; Castera, F.; Peiro, C.; Millan, J.M.; Herrero, M.J.; Vazquez-Manrique, R.P. Metformin to treat Huntington disease: A pleiotropic drug against a multi-system disorder. Mech. Ageing Dev. 2022, 204, 111670. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Gu, H.; Anders, N.M.; Ren, T.; Jiang, M.; Tao, M.; Peng, Q.; Rudek, M.A.; Duan, W. Metformin Protects Cells from Mutant Huntingtin Toxicity Through Activation of AMPK and Modulation of Mitochondrial Dynamics. Neuromol. Med. 2016, 18, 581–592. [Google Scholar] [CrossRef]
- Ameen, O.; Samaka, R.M.; Abo-Elsoud, R.A.A. Metformin alleviates neurocognitive impairment in aging via activation of AMPK/BDNF/PI3K pathway. Sci. Rep. 2022, 12, 17084. [Google Scholar] [CrossRef]
- Sanchis, A.; Garcia-Gimeno, M.A.; Canada-Martinez, A.J.; Sequedo, M.D.; Millan, J.M.; Sanz, P.; Vazquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Hervas, D.; Fornes-Ferrer, V.; Gomez-Escribano, A.P.; Sequedo, M.D.; Peiro, C.; Millan, J.M.; Vazquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef]
- Narine, M.; Azmi, M.A.; Umali, M.; Volz, A.; Colognato, H. The AMPK activator metformin improves recovery from demyelination by shifting oligodendrocyte bioenergetics and accelerating OPC differentiation. Front. Cell. Neurosci. 2023, 17, 1254303. [Google Scholar] [CrossRef]
- Mather, M.L.; Jeffries, M.A.; Wood, T.L. The mechanistic target of rapamycin as a regulator of metabolic function in oligodendroglia during remyelination. Curr. Opin. Pharmacol. 2022, 63, 102193. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Ursini, F.; Russo, E.; Pellino, G.; D’Angelo, S.; Chiaravalloti, A.; De Sarro, G.; Manfredini, R.; De Giorgio, R. Metformin and Autoimmunity: A “New Deal” of an Old Drug. Front. Immunol. 2018, 9, 1236. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, A.V.; Van Doninck, E.; Popescu, V.; Willem, L.; Cambron, M.; Laureys, G.; D’ Haeseleer, M.; Bjerke, M.; Roelant, E.; Lemmerling, M.; et al. A metformin add-on clinical study in multiple sclerosis to evaluate brain remyelination and neurodegeneration (MACSiMiSE-BRAIN): Study protocol for a multi-center randomized placebo controlled clinical trial. Front. Immunol. 2024, 15, 1362629. [Google Scholar] [CrossRef]
- Gilbert, E.A.B.; Livingston, J.; Flores, E.G.; Khan, M.; Kandavel, H.; Morshead, C.M. Metformin treatment reduces inflammation, dysmyelination and disease severity in a mouse model of multiple sclerosis, experimental autoimmune encephalomyelitis. Brain Res. 2024, 1822, 148648. [Google Scholar] [CrossRef] [PubMed]
- Samjoo, I.A.; Drudge, C.; Walsh, S.; Tiwari, S.; Brennan, R.; Boer, I.; Haring, D.A.; Klotz, L.; Adlard, N.; Banhazi, J. Comparative efficacy of therapies for relapsing multiple sclerosis: A systematic review and network meta-analysis. J. Comp. Eff. Res. 2023, 12, e230016. [Google Scholar] [CrossRef]
- Negrotto, L.; Farez, M.F.; Correale, J. Immunologic Effects of Metformin and Pioglitazone Treatment on Metabolic Syndrome and Multiple Sclerosis. JAMA Neurol. 2016, 73, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Askari, H.; Rabiei, F.; Lohrasbi, F.; Ghadir, S.; Mehdipour Arbastan, A.; Ghasemi-Kasman, M. AMP-activated protein kinase as a mediator of mitochondrial dysfunction of multiple sclerosis in animal models: A systematic review. J. Cell. Physiol. 2024, 239, e31230. [Google Scholar] [CrossRef]
- Muraleedharan, R.; Dasgupta, B. AMPK in the brain: Its roles in glucose and neural metabolism. FEBS J. 2022, 289, 2247–2262. [Google Scholar] [CrossRef]
- Lepez, A.; Pirnay, T.; Denanglaire, S.; Perez-Morga, D.; Vermeersch, M.; Leo, O.; Andris, F. Long-term T cell fitness and proliferation is driven by AMPK-dependent regulation of reactive oxygen species. Sci. Rep. 2020, 10, 21673. [Google Scholar] [CrossRef]
- Wei, J.; Raynor, J.; Nguyen, T.L.; Chi, H. Nutrient and Metabolic Sensing in T Cell Responses. Front. Immunol. 2017, 8, 247. [Google Scholar] [CrossRef]
- Vakrakou, A.G.; Alexaki, A.; Brinia, M.E.; Anagnostouli, M.; Stefanis, L.; Stathopoulos, P. The mTOR Signaling Pathway in Multiple Sclerosis; from Animal Models to Human Data. Int. J. Mol. Sci. 2022, 23, 8077. [Google Scholar] [CrossRef]
- Sanadgol, N.; Barati, M.; Houshmand, F.; Hassani, S.; Clarner, T.; Shahlaei, M.; Golab, F. Metformin accelerates myelin recovery and ameliorates behavioral deficits in the animal model of multiple sclerosis via adjustment of AMPK/Nrf2/mTOR signaling and maintenance of endogenous oligodendrogenesis during brain self-repairing period. Pharmacol. Rep. 2020, 72, 641–658. [Google Scholar] [CrossRef]
- Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. Adenosine-mono-phosphate-activated protein kinase-independent effects of metformin in T cells. PLoS ONE 2014, 9, e106710. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Metformin Enhances B Cell Function and Antibody Responses of Elderly Individuals with Type-2 Diabetes Mellitus. Front. Aging 2021, 2, 715981. [Google Scholar] [CrossRef]
- Wu, Y.Q.; Xiong, J.; He, Z.L.; Yuan, Y.; Wang, B.N.; Xu, J.Y.; Wu, M.; Zhang, S.S.; Cai, S.F.; Zhao, J.X.; et al. Metformin promotes microglial cells to facilitate myelin debris clearance and accelerate nerve repairment after spinal cord injury. Acta Pharmacol. Sin. 2022, 43, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-S.; Yeom, Y.-S.; Jang, J.-H.; Kim, Y.-H.; Im, J.I.; Kim, I.S.; Yang, S.-J. Anti-inflammatory Effects of Metformin on Neuro-inflammation and NLRP3 Inflammasome Activation in BV-2 Microglial Cells. Biomed. Sci. Lett. 2019, 25, 92–98. [Google Scholar] [CrossRef]
- Liao, W.; Xu, J.; Li, B.; Ruan, Y.; Li, T.; Liu, J. Deciphering the Roles of Metformin in Alzheimer’s Disease: A Snapshot. Front. Pharmacol. 2021, 12, 728315. [Google Scholar] [CrossRef] [PubMed]
- Abdelgaied, M.Y.; Rashad, M.H.; El-Tayebi, H.M.; Solayman, M.H. The impact of metformin use on the outcomes of relapse-remitting multiple sclerosis patients receiving interferon beta 1a: An exploratory prospective phase II open-label randomized controlled trial. J. Neurol. 2024, 271, 1124–1132. [Google Scholar] [CrossRef]
- NCT04121468; A Phase I Double Blind Study of Metformin Acting on Endogenous Neural Progenitor Cells in Children with Multiple Sclerosis, E. Ann Yeh, The Hospital for Sick Children. 2022. Available online: https://clinicaltrials.gov/study/NCT04121468?cond=Multiple%20Sclerosis&intr=Metformin&rank=1 (accessed on 13 March 2025).
- NCT06463743; Metformin as an Add-On or Monotherapy in Treatment of Aging People with Multiple Sclerosis (MS), Bianca Weinstock-Guttman, State University of New York at Buffalo. 2025. Available online: https://clinicaltrials.gov/study/NCT06463743?cond=Multiple%20Sclerosis&intr=Metformin&rank=2 (accessed on 13 March 2025).
- NCT05893225; Metformin Add-On Clinical Study in Multiple Sclerosis to Evaluate Brain Remyelination and Neurodegeneration, University Hospital, Antwerp. 2024. Available online: https://clinicaltrials.gov/study/NCT05893225?cond=Multiple%20Sclerosis&intr=Metformin&rank=3 (accessed on 13 March 2025).
- NCT06812585; The Effect of Metformin as an Adjuvant Therapy on Immunological Parameters in Egyptian Patients with RRMS: A Pilot Study, Mohamed Youssef Elsayed, German University in Cairo. 2025. Available online: https://clinicaltrials.gov/study/NCT06812585?cond=Multiple%20Sclerosis&intr=Metformin&rank=4 (accessed on 13 March 2025).
- NCT05131828; CCMR Two: A Phase IIa, Randomised, Double-Blind, Placebo-Controlled Trial of the Ability of the Combination of Metformin and Clemastine to Promote Remyelination in People with Relapsing-Remitting Multiple Sclerosis Already on Disease-modifying Therapy, Nicholas Cunniffe, Cambridge University Hospitals NHS Foundation Trust. 2023. Available online: https://clinicaltrials.gov/study/NCT05131828?cond=Multiple%20Sclerosis&intr=Metformin&rank=5 (accessed on 13 March 2025).
- NCT05298670; Drug Repurposing Using Metformin for Improving the Therapeutic Outcome in Multiple Sclerosis Patients, Mohamed Youssef Elsayed, German University in Cairo. 2023. Available online: https://clinicaltrials.gov/study/NCT05298670?cond=Multiple%20Sclerosis&intr=Metformin&rank=6 (accessed on 13 March 2025).
- NCT05349474; Metformin Treatment in Progressive Multiple Sclerosis, Kevin Patel, University of California, Los Angeles. 2023. Available online: https://clinicaltrials.gov/study/NCT05349474?cond=Multiple%20Sclerosis&intr=Metformin&rank=7 (accessed on 13 March 2025).
- Ale Mahmoud Mehraban, R.; Babaei, P.; Rohampour, K.; Jafari, A.; Golipoor, Z. Metformin improves memory via AMPK/mTOR-dependent route in a rat model of Alzheimer’s disease. Iran. J. Basic Med. Sci. 2024, 27, 360–365. [Google Scholar] [CrossRef]
- Rai, S.N.; Singh, P.; Steinbusch, H.W.M.; Vamanu, E.; Ashraf, G.; Singh, M.P. The Role of Vitamins in Neurodegenerative Disease: An Update. Biomedicines 2021, 9, 1284. [Google Scholar] [CrossRef]
- Ha, J.; Choi, D.W.; Kim, K.J.; Cho, S.Y.; Kim, H.; Kim, K.Y.; Koh, Y.; Nam, C.M.; Kim, E. Association of metformin use with Alzheimer’s disease in patients with newly diagnosed type 2 diabetes: A population-based nested case-control study. Sci. Rep. 2021, 11, 24069. [Google Scholar] [CrossRef]
- Gawryś, A.; Cholewa, M.M.; Kromer, A.; Wilk, J.; Cichoń, K.; Zapała, M.A.; Łapaj, M.; Chyćko, M.; Środoń, A.; Czarnota, J. The effect of antioxidants on the course and prevention of Alzheimer’s disease on the example of vitamin E. J. Educ. Health Sport 2023, 46, 216–232. [Google Scholar] [CrossRef]
- Becker, M.L.; Visser, L.E.; van Schaik, R.H.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H. Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: A preliminary study. Diabetes 2009, 58, 745–749. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Zhang, D.; Lu, W.; Zheng, T.; Wan, L.; Liu, F.; Jia, W. SLC47A1 gene rs2289669 G>A variants enhance the glucose-lowering effect of metformin via delaying its excretion in Chinese type 2 diabetes patients. Diabetes Res. Clin. Pract. 2015, 109, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Yee, S.W.; Chen, L.; Giacomini, K.M. The role of ATM in response to metformin treatment and activation of AMPK. Nat. Genet. 2012, 44, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Haffner, S.M.; Heise, M.A.; Herman, W.H.; Holman, R.R.; Jones, N.P.; Kravitz, B.G.; Lachin, J.M.; O’Neill, M.C.; Zinman, B.; et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443. [Google Scholar] [CrossRef]
- Cobb, J.; Gall, W.; Adam, K.P.; Nakhle, P.; Button, E.; Hathorn, J.; Lawton, K.; Milburn, M.; Perichon, R.; Mitchell, M.; et al. A novel fasting blood test for insulin resistance and prediabetes. J. Diabetes Sci. Technol. 2013, 7, 100–110. [Google Scholar] [CrossRef]
- Donnelly, L.A.; Doney, A.S.; Hattersley, A.T.; Morris, A.D.; Pearson, E.R. The effect of obesity on glycaemic response to metformin or sulphonylureas in Type 2 diabetes. Diabet. Med. 2006, 23, 128–133. [Google Scholar] [CrossRef]
- Vaag, A.; Lund, S.S. Non-obese patients with type 2 diabetes and prediabetic subjects: Distinct phenotypes requiring special diabetes treatment and (or) prevention? Appl. Physiol. Nutr. Metab. 2007, 32, 912–920. [Google Scholar] [CrossRef]
- Dennis, J.M.; Shields, B.M.; Hill, A.V.; Knight, B.A.; McDonald, T.J.; Rodgers, L.R.; Weedon, M.N.; Henley, W.E.; Sattar, N.; Holman, R.R.; et al. Precision Medicine in Type 2 Diabetes: Clinical Markers of Insulin Resistance Are Associated with Altered Short- and Long-term Glycemic Response to DPP-4 Inhibitor Therapy. Diabetes Care 2018, 41, 705–712. [Google Scholar] [CrossRef]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M.; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Cozzolino, M.; Apolloni, S. The Contribution of Non-Neuronal Cells in Neurodegeneration: From Molecular Pathogenesis to Therapeutic Challenges. Cells 2022, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, C.P.; Pei, J.; Alugoju, P.; Anthikapalli, N.V.A.; Jayaraman, S.; Veeraraghavan, V.P.; Gopathy, S.; Roy, J.R.; Janaki, C.S.; Thalamati, D.; et al. New strategies of neurodegenerative disease treatment with extracellular vesicles (EVs) derived from mesenchymal stem cells (MSCs). Theranostics 2023, 13, 4138–4165. [Google Scholar] [CrossRef]
- Nouri Nojadeh, J.; Bildiren Eryilmaz, N.S.; Erguder, B.I. CRISPR/Cas9 genome editing for neurodegenerative diseases. EXCLI J. 2023, 22, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Benchekroun, M.; Maramai, S. Multitarget-directed ligands for neurodegenerative diseases: Real opportunity or blurry mirage? Future Med. Chem. 2019, 11, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Roy, S. Gene-based therapies for neurodegenerative diseases. Nat. Neurosci. 2021, 24, 297–311. [Google Scholar] [CrossRef]
- Quilon, P.G.; Volpedo, G.; Cappato, S.; Ferrera, L.; Zara, F.; Bocciardi, R.; Riva, A.; Striano, P. Antisense oligonucleotides as a precision therapy for developmental and epileptic encephalopathies. CNS Neurosci. Ther. 2024, 30, e70050. [Google Scholar] [CrossRef]
- Temple, S. Advancing cell therapy for neurodegenerative diseases. Cell Stem Cell 2023, 30, 512–529. [Google Scholar] [CrossRef]
- Jafleh, E.A.; Alnaqbi, F.A.; Almaeeni, H.A.; Faqeeh, S.; Alzaabi, M.A.; Al Zaman, K. The Role of Wearable Devices in Chronic Disease Monitoring and Patient Care: A Comprehensive Review. Cureus 2024, 16, e68921. [Google Scholar] [CrossRef]
- Wang, Y. Editorial: Understanding the link between lifestyle and neurodegenerative diseases. Front. Neurosci. 2024, 18, 1365734. [Google Scholar] [CrossRef]
- Jain, A.; Madkan, S.; Patil, P. The Role of Gut Microbiota in Neurodegenerative Diseases: Current Insights and Therapeutic Implications. Cureus 2023, 15, e47861. [Google Scholar] [CrossRef] [PubMed]
- Picone, P.; Nuzzo, D.; Caruana, L.; Messina, E.; Barera, A.; Vasto, S.; Di Carlo, M. Metformin increases APP expression and processing via oxidative stress, mitochondrial dysfunction and NF-κB activation: Use of insulin to attenuate metformin’s effect. Biochim. Biophys. Acta 2015, 1853, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.M.; Mander, A.G.; Ames, D.; Kotowicz, M.A.; Carne, R.P.; Brodaty, H.; Woodward, M.; Boundy, K.; Ellis, K.A.; Bush, A.I.; et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 2013, 36, 2981–2987. [Google Scholar] [CrossRef]
- Chin-Hsiao, T. Metformin and the Risk of Dementia in Type 2 Diabetes Patients. Aging Dis. 2019, 10, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Kuan, Y.C.; Huang, K.W.; Lin, C.L.; Hu, C.J.; Kao, C.H. Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79 Pt B, 77–83. [Google Scholar] [CrossRef]
- Yarchoan, M.; Arnold, S.E. Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 2014, 63, 2253–2261. [Google Scholar] [CrossRef]
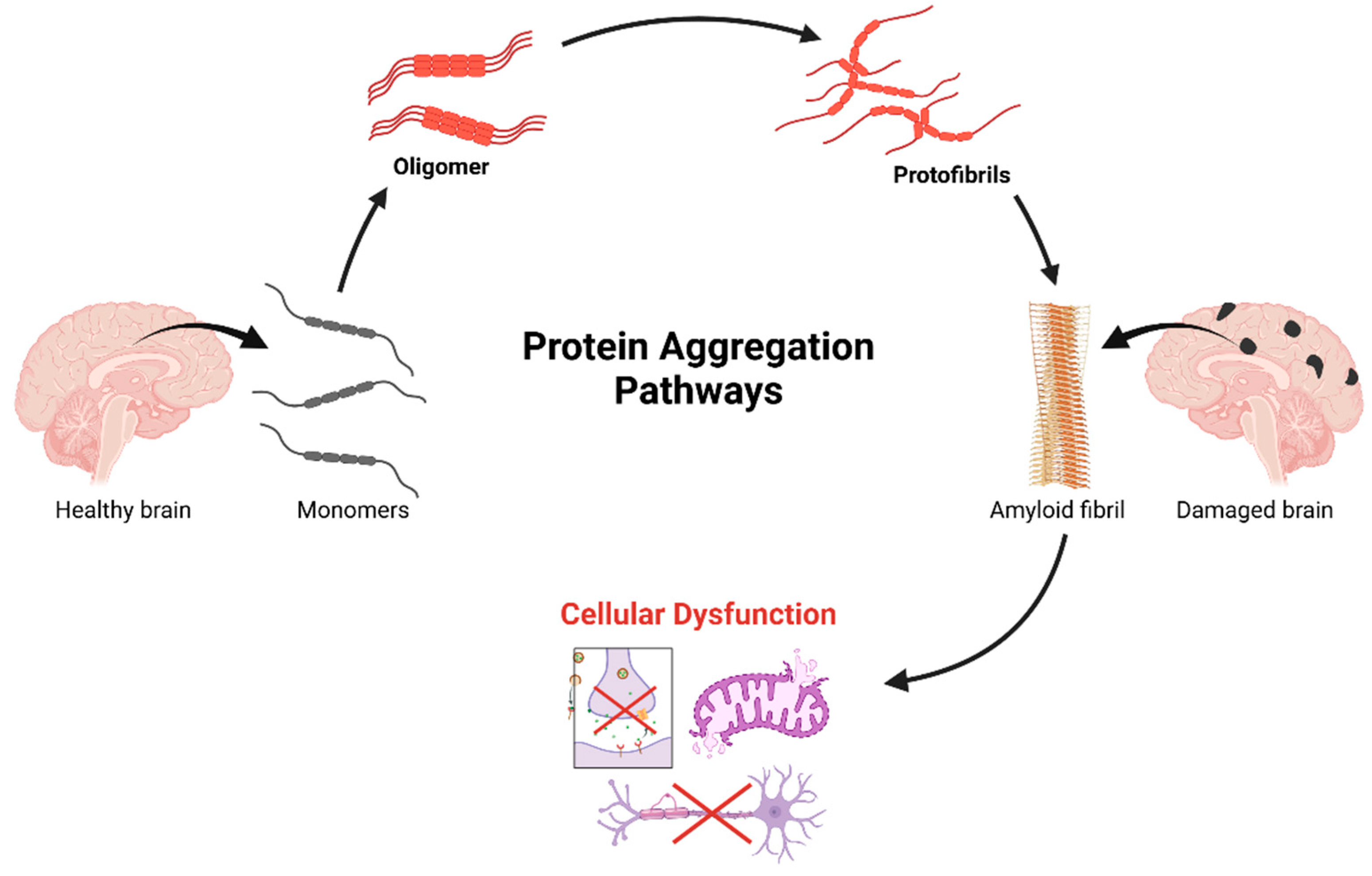
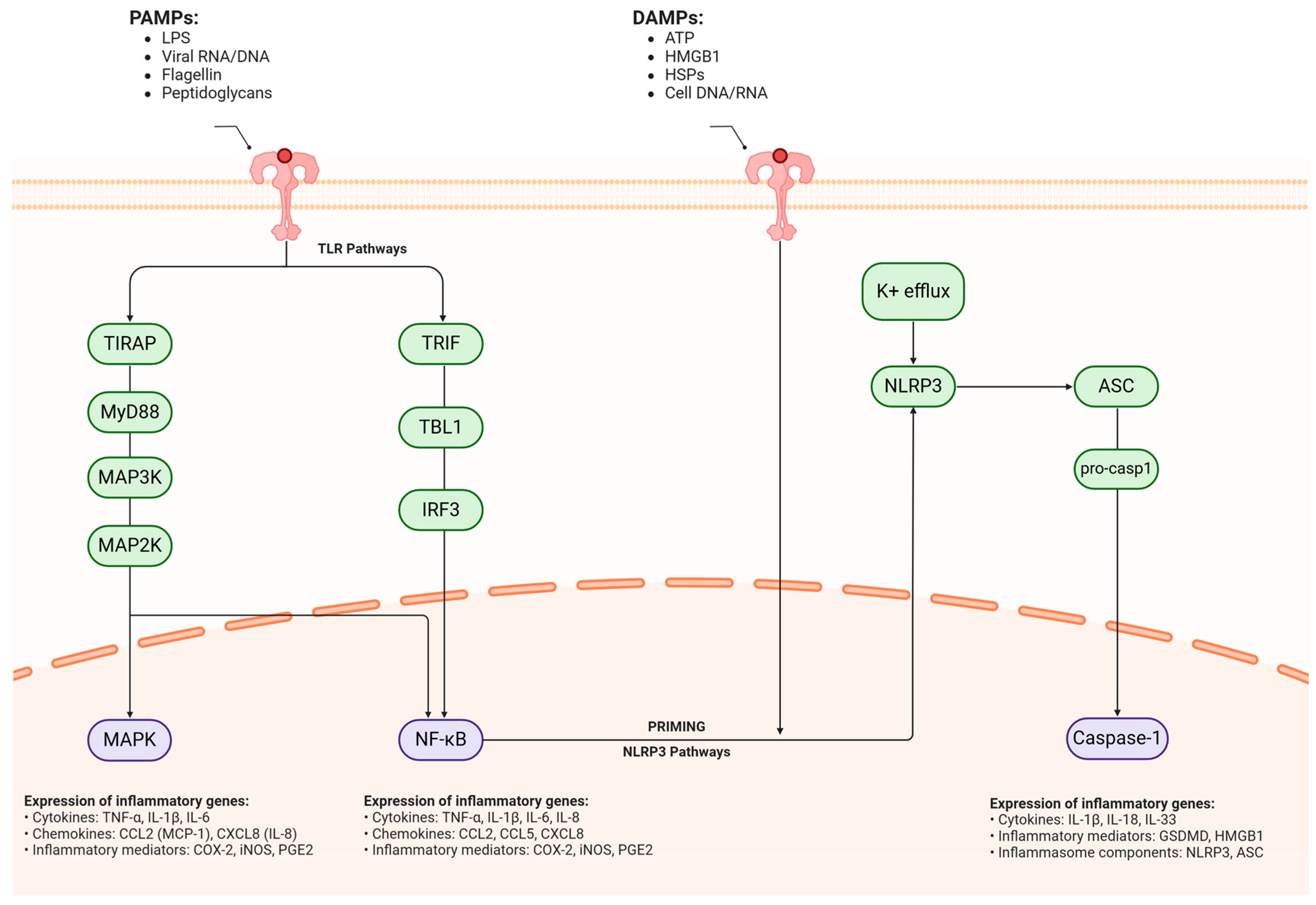
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).