
Metabolomic Profiling of Leptadenia reticulata: Unveiling Therapeutic Potential for Inflammatory Diseases through Network Pharmacology and Docking Studies

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Identification of Phytochemicals Using HR-LCMS/MS(Q-TOF)
2.2. ADMET Profiling
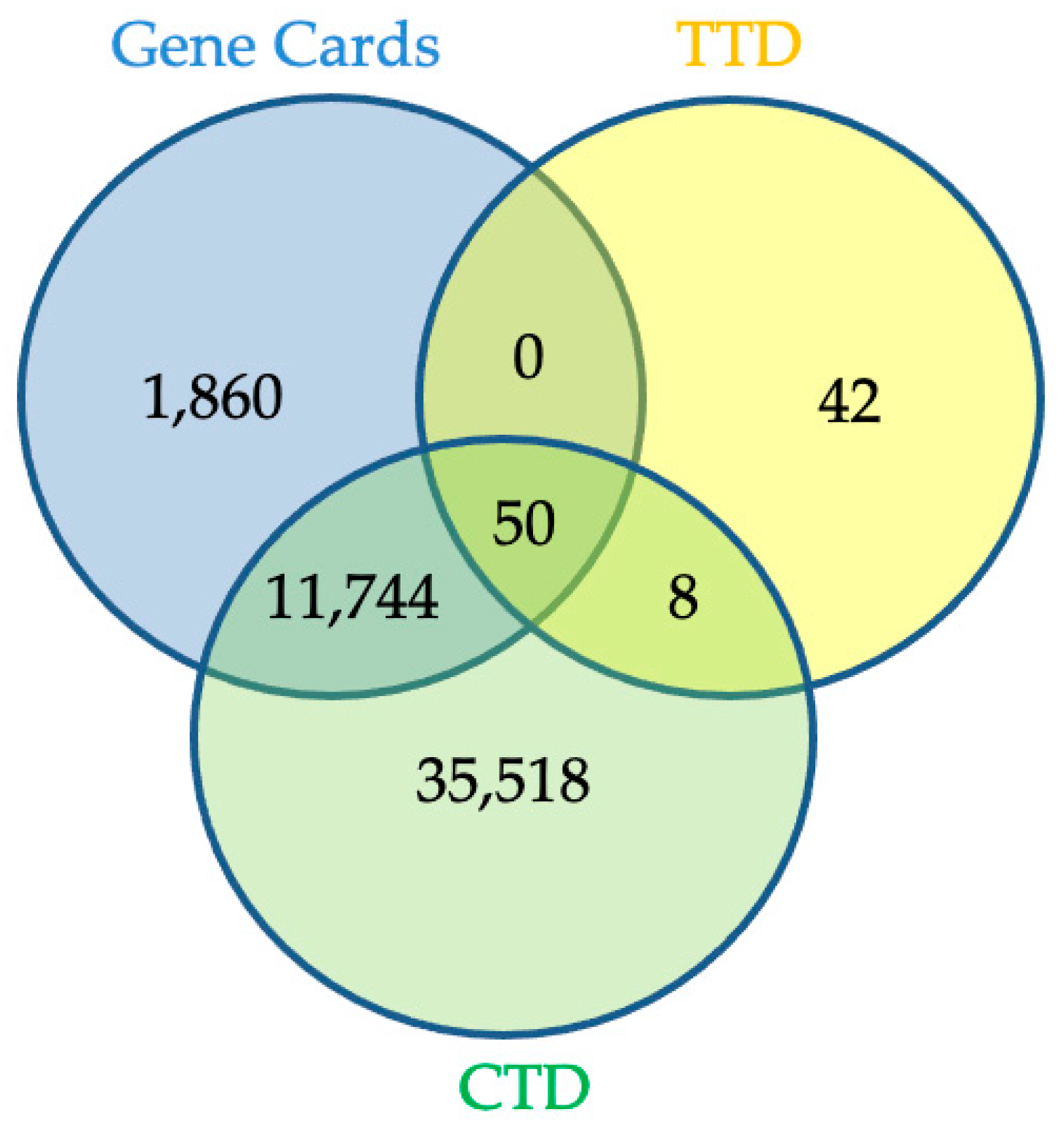
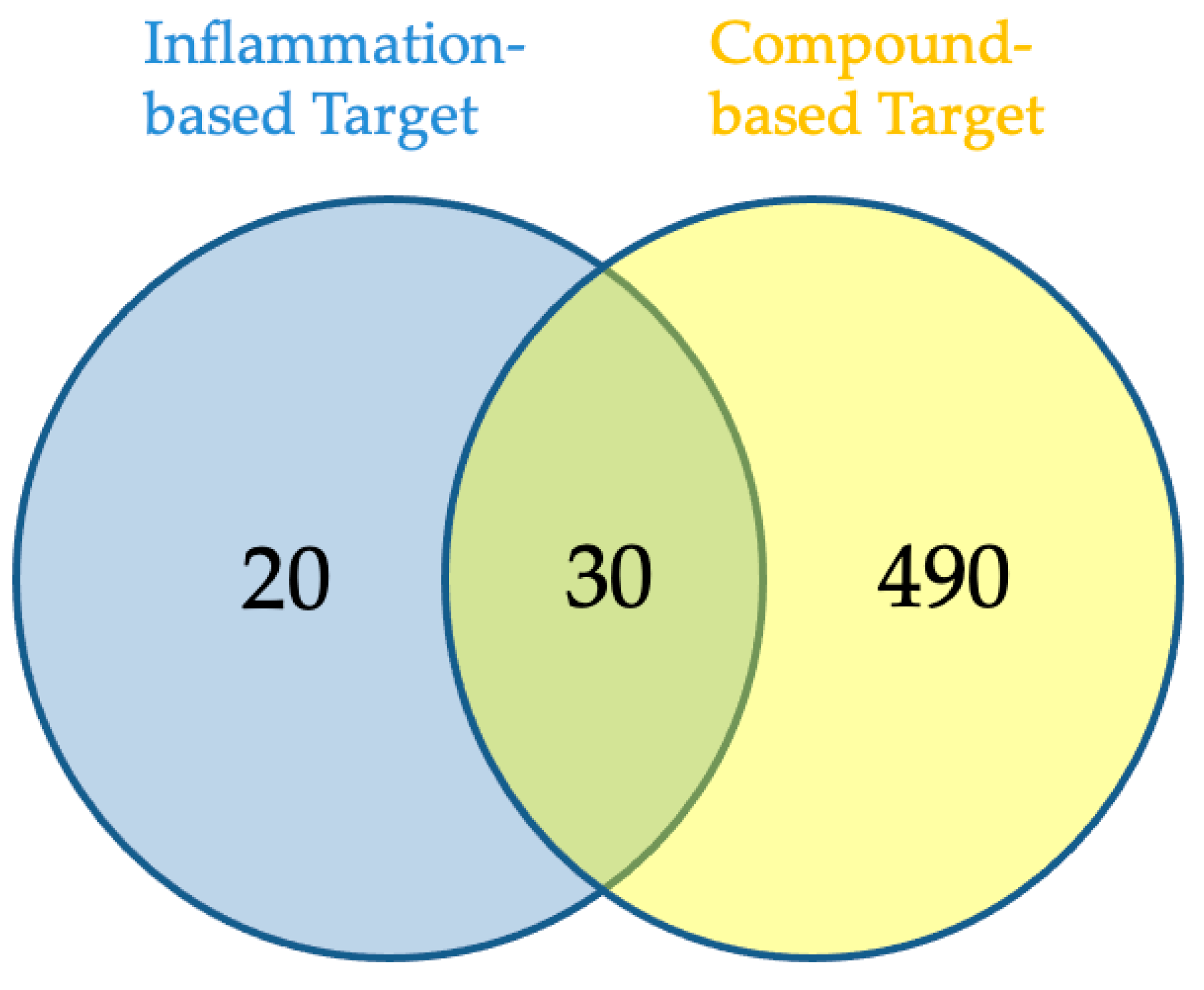
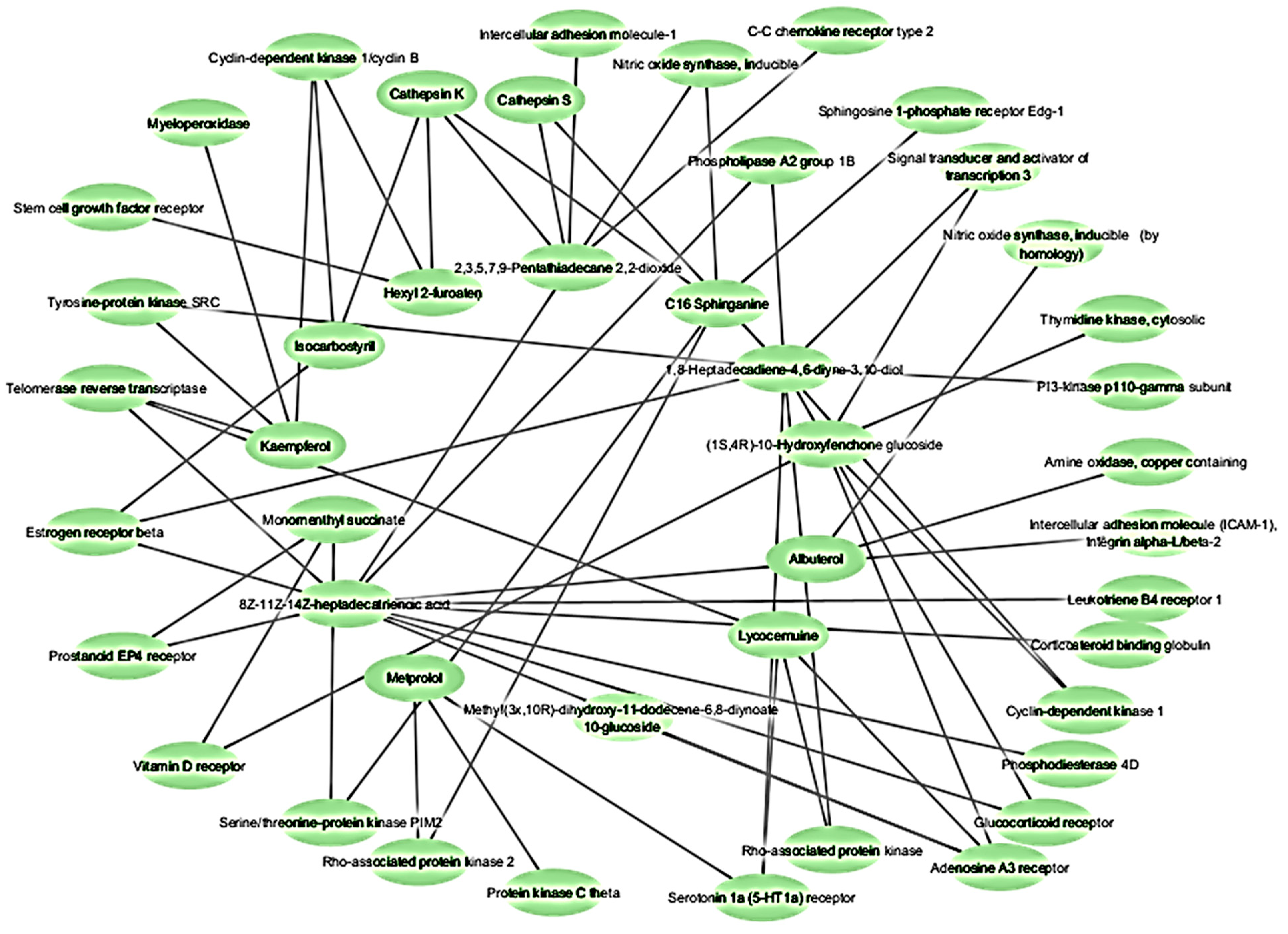
2.3. Network Pharmacology Analysis for Potential Active Compound Targets and Anti-Inflammatory Targets
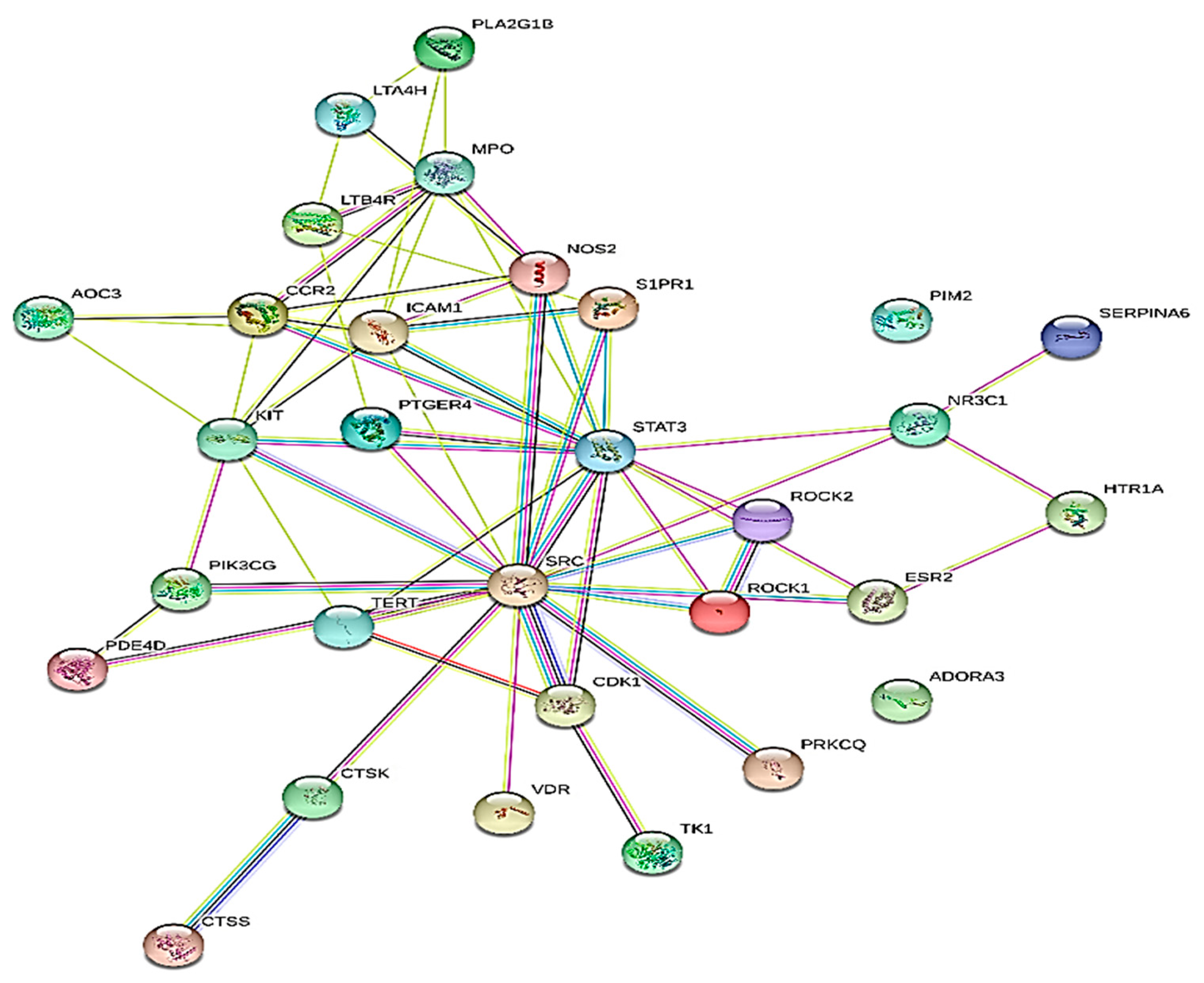
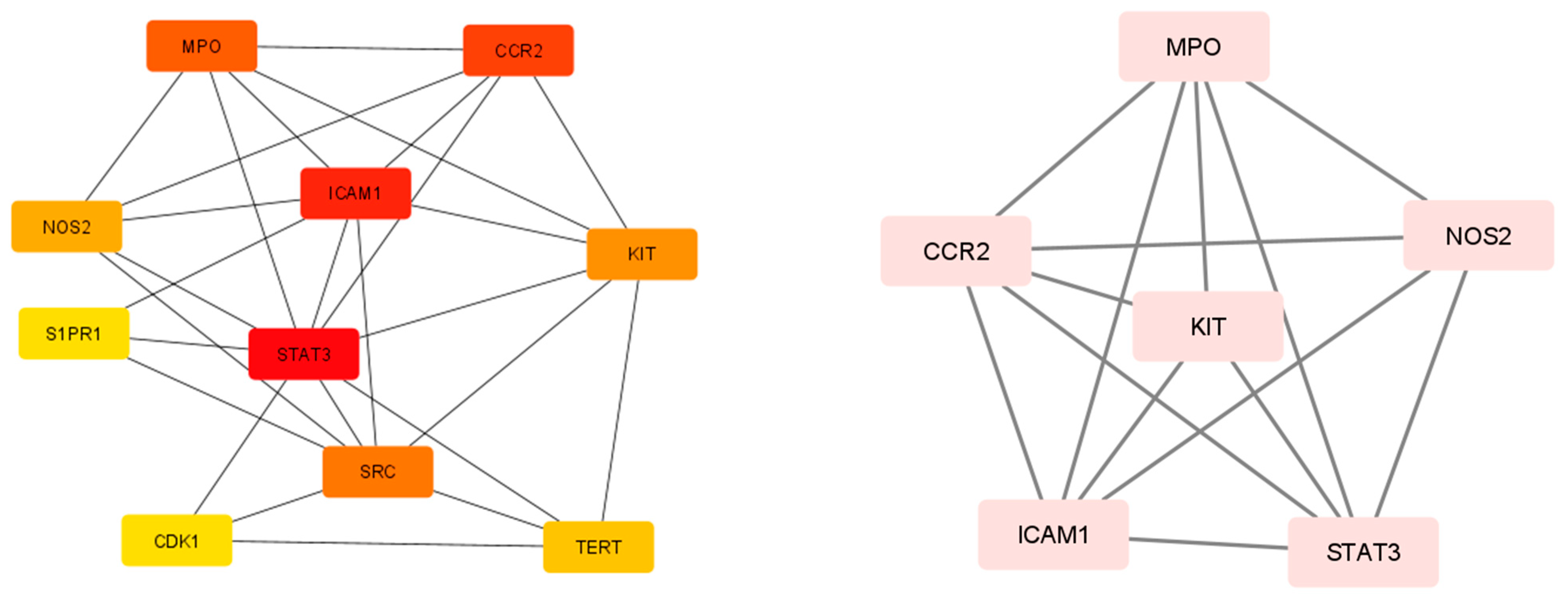
2.4. Protein–Protein Interaction
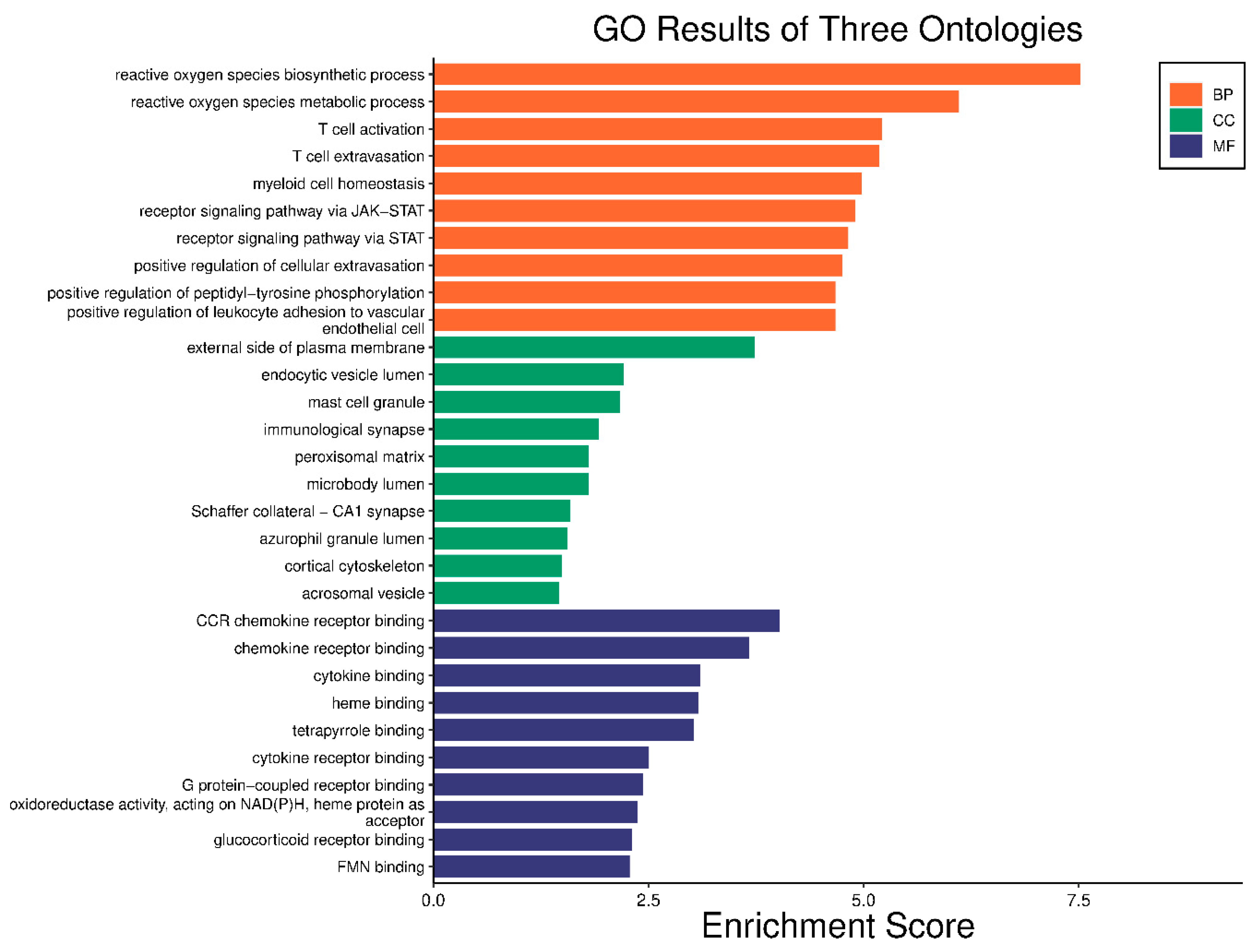
2.5. GO Enrichment and KEGG Analysis
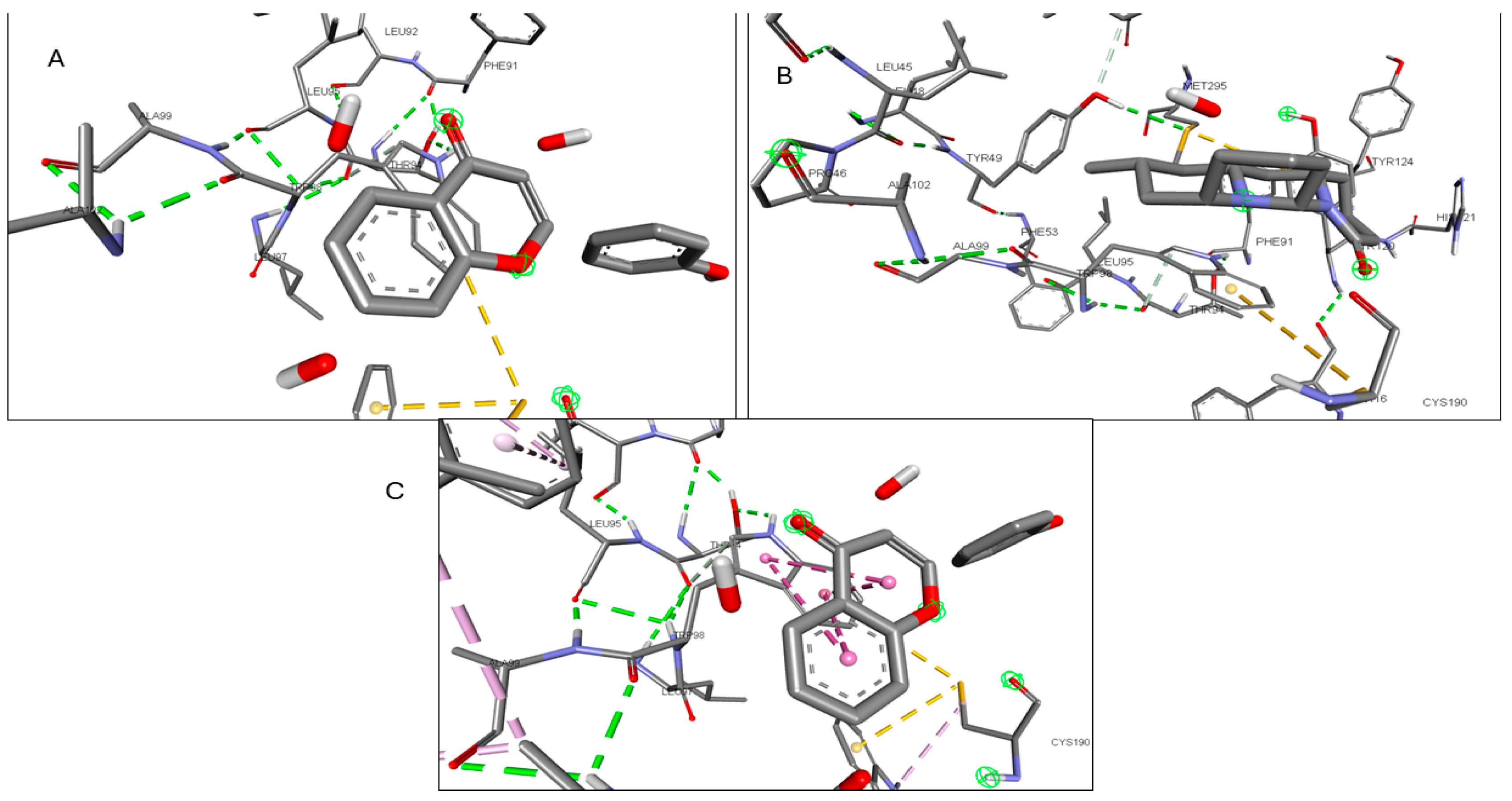
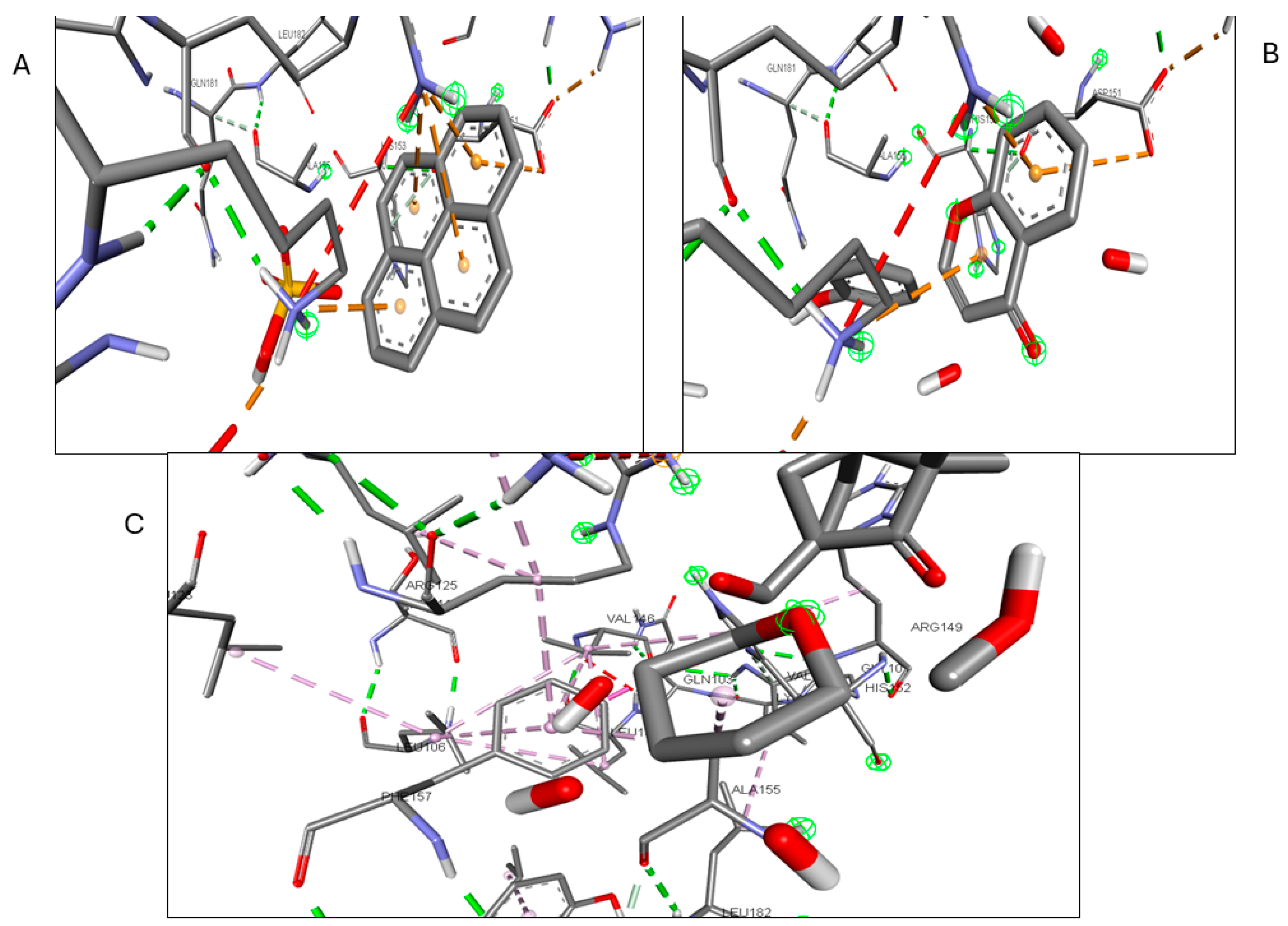
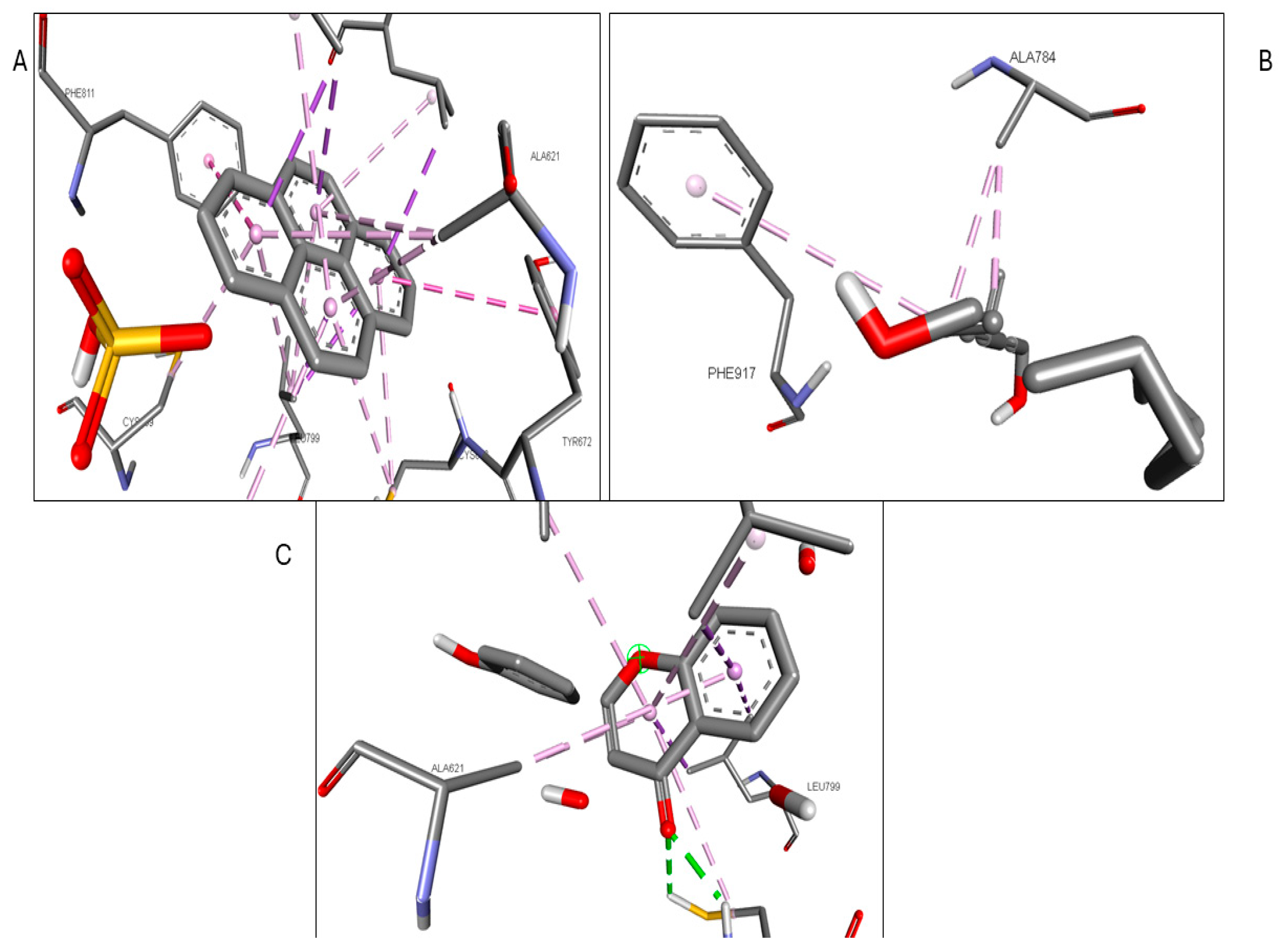
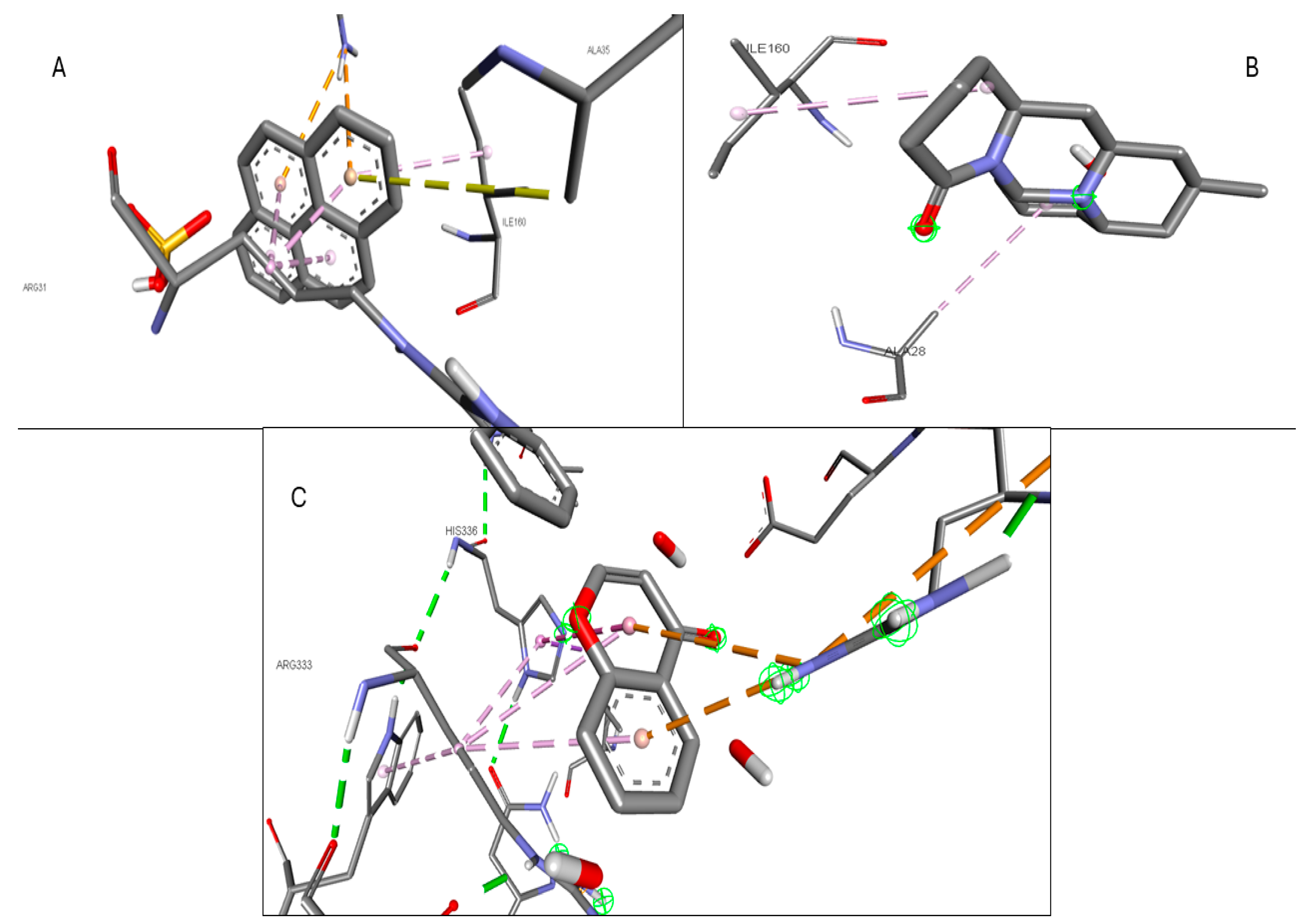
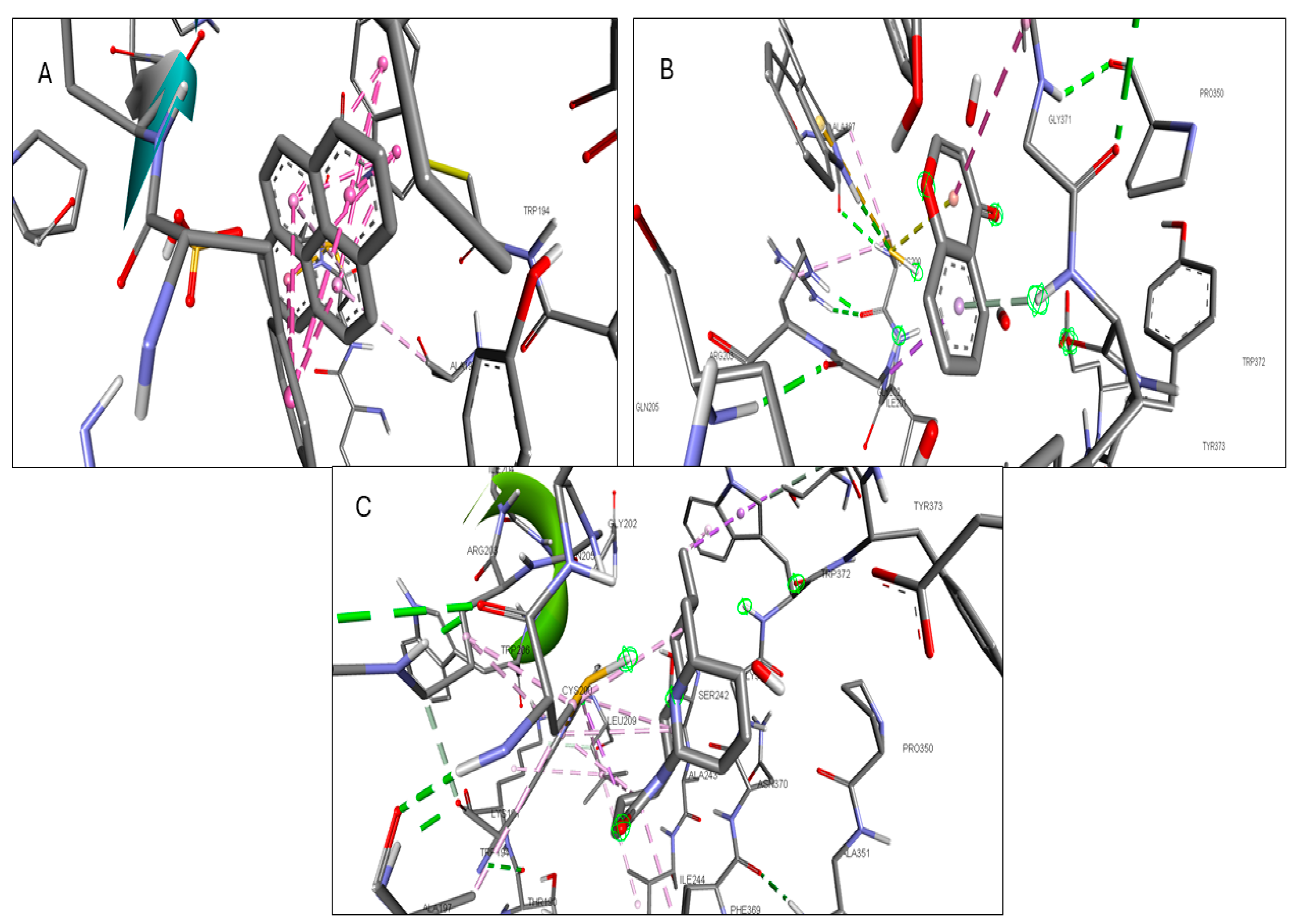
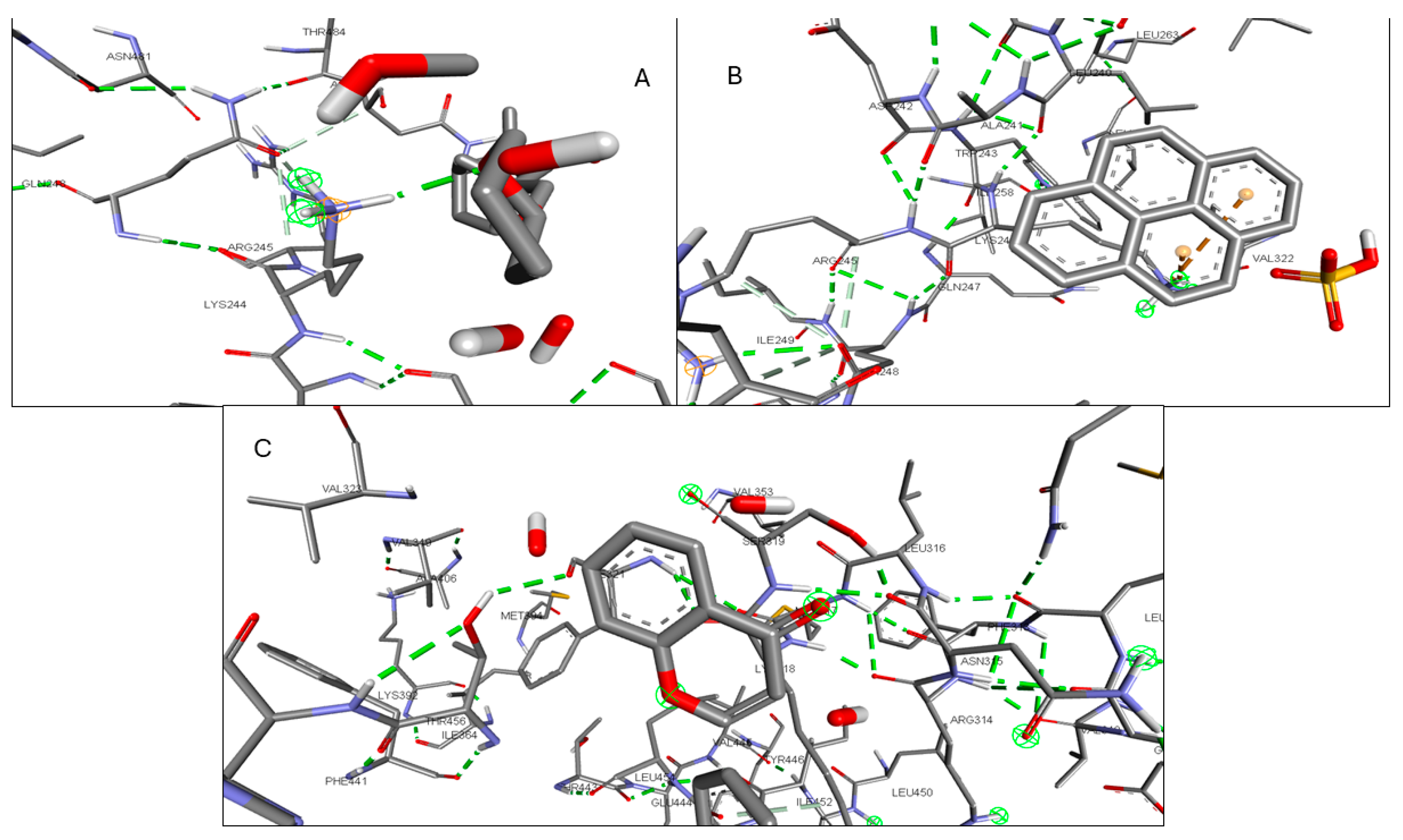
2.6. Molecular Docking of Active Compounds and Key Targets
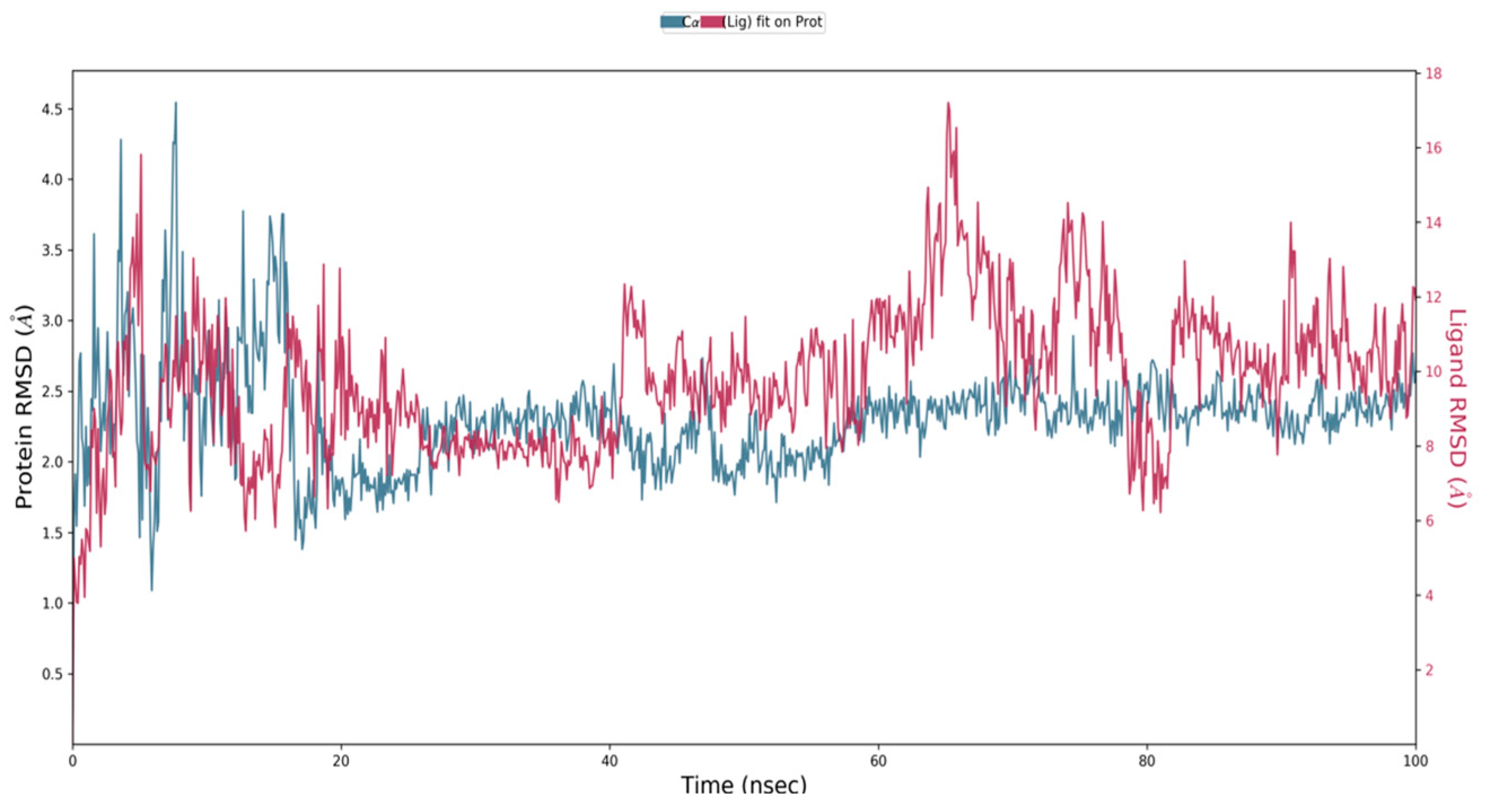
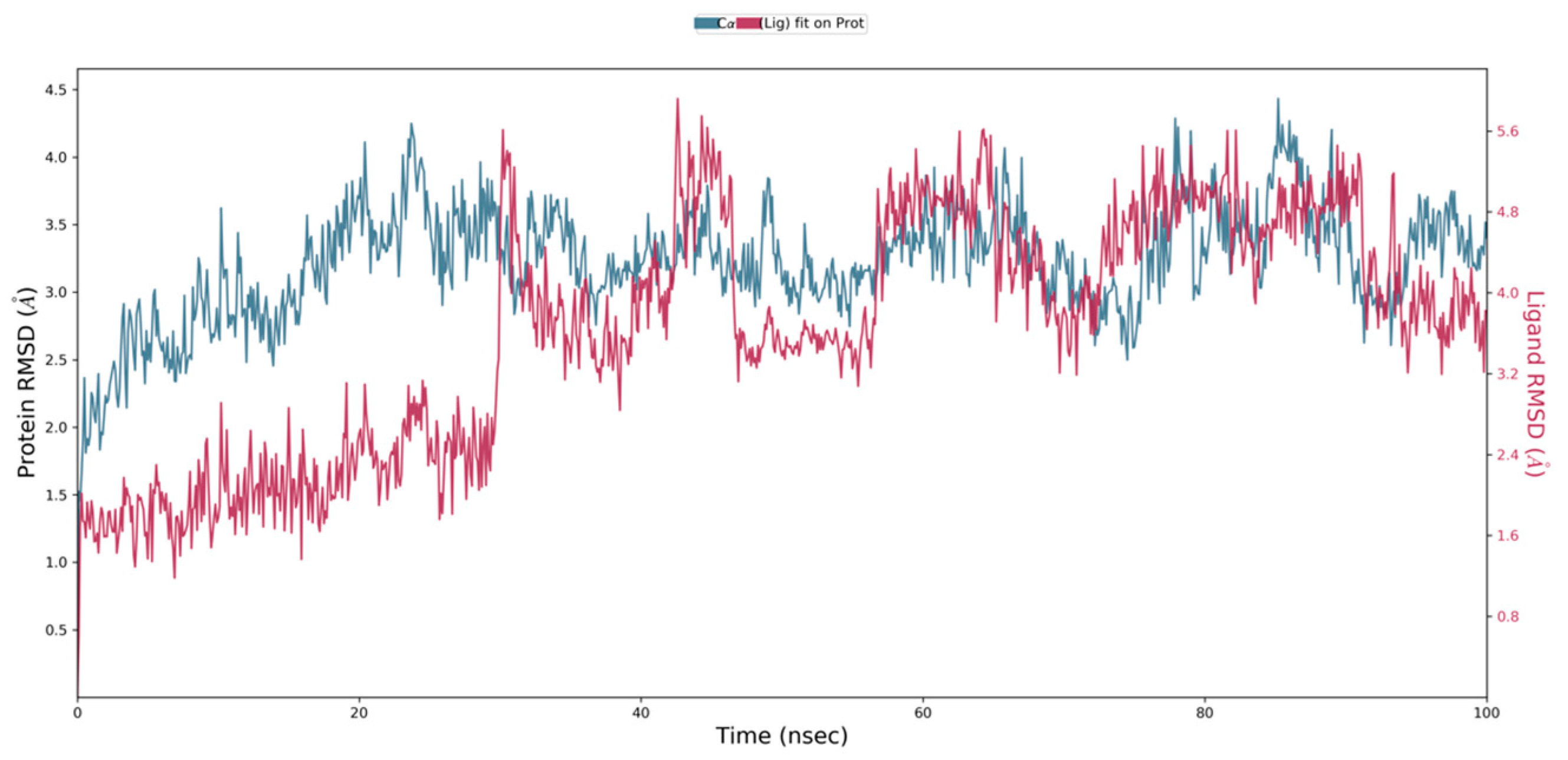
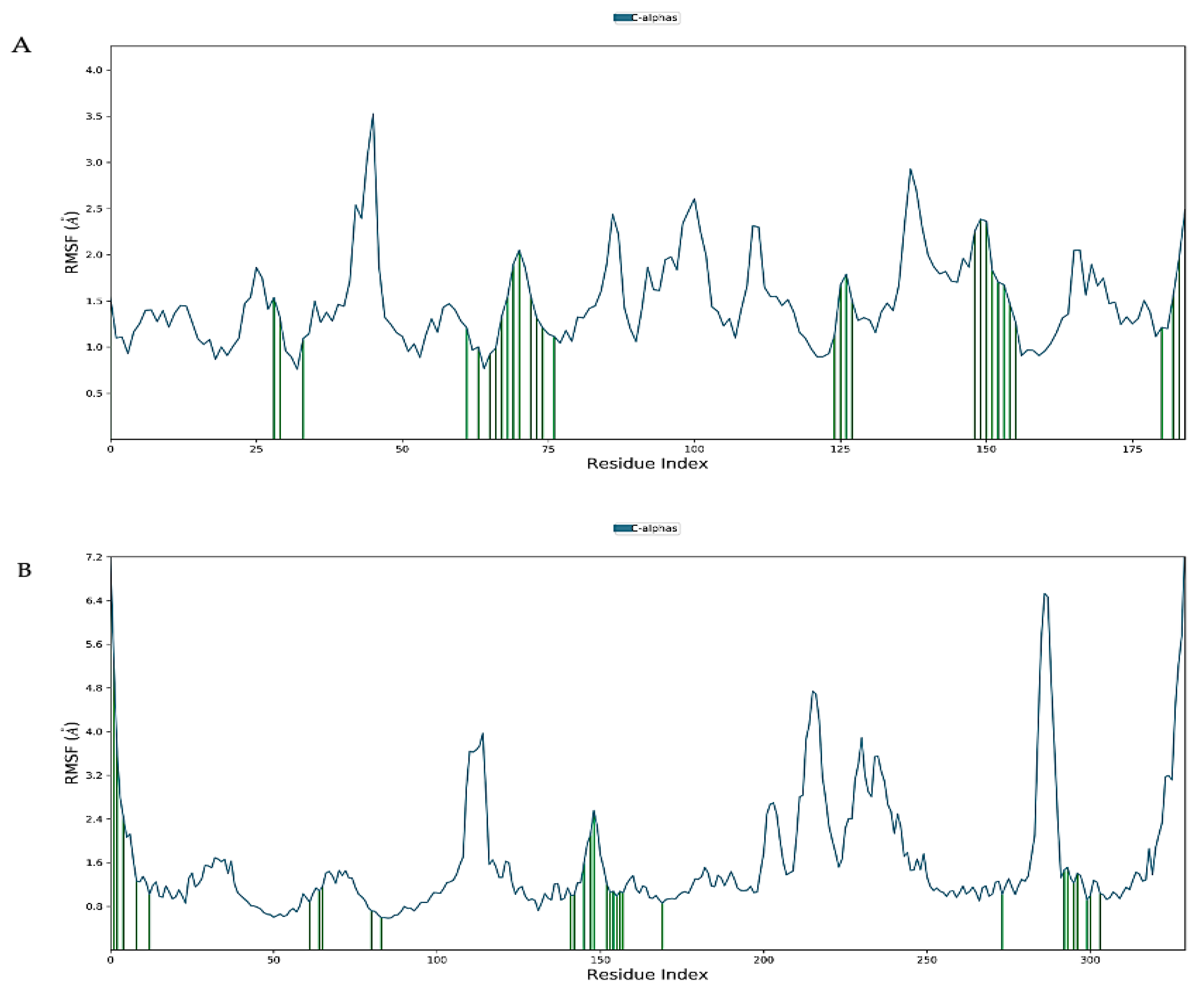
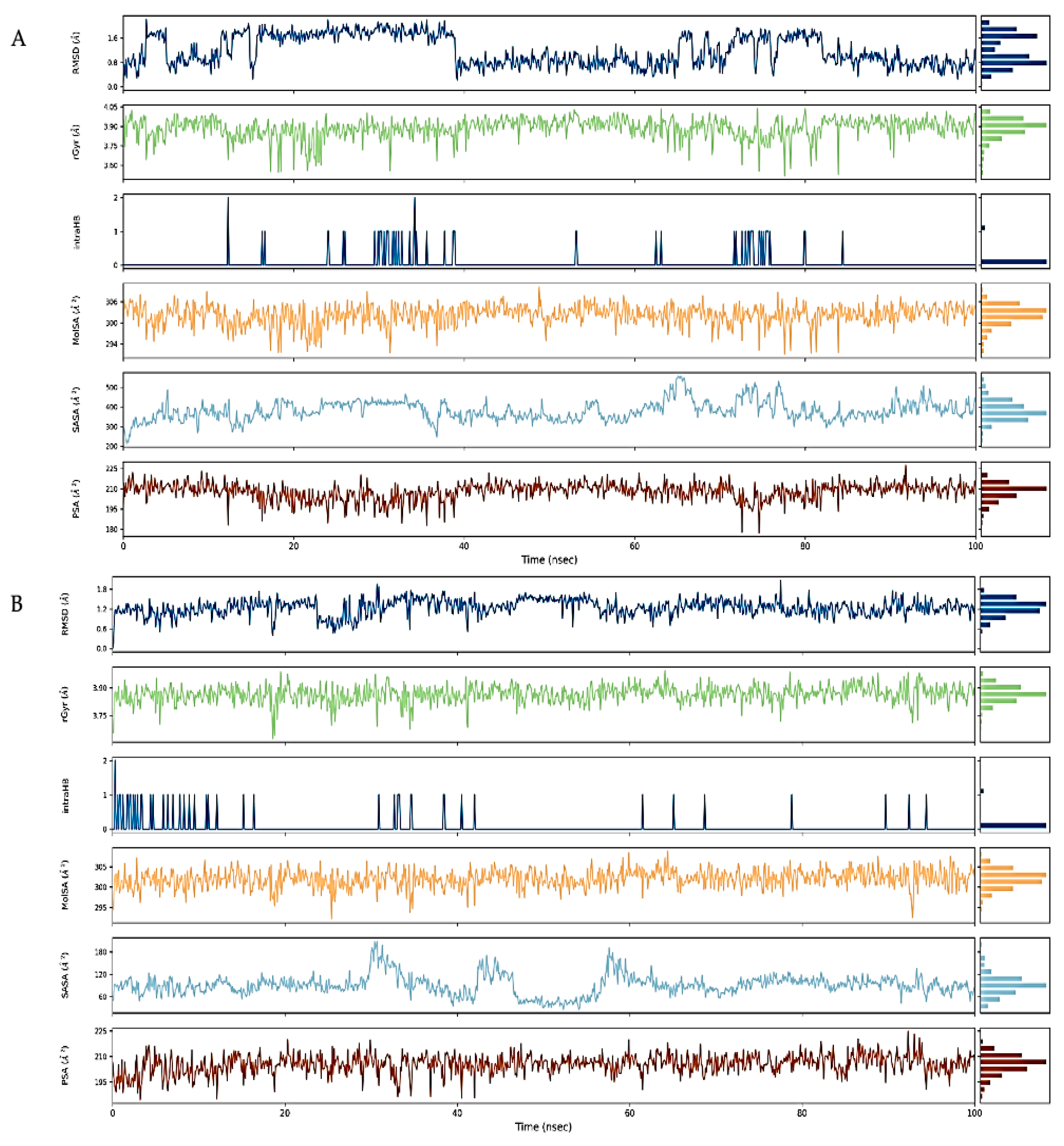
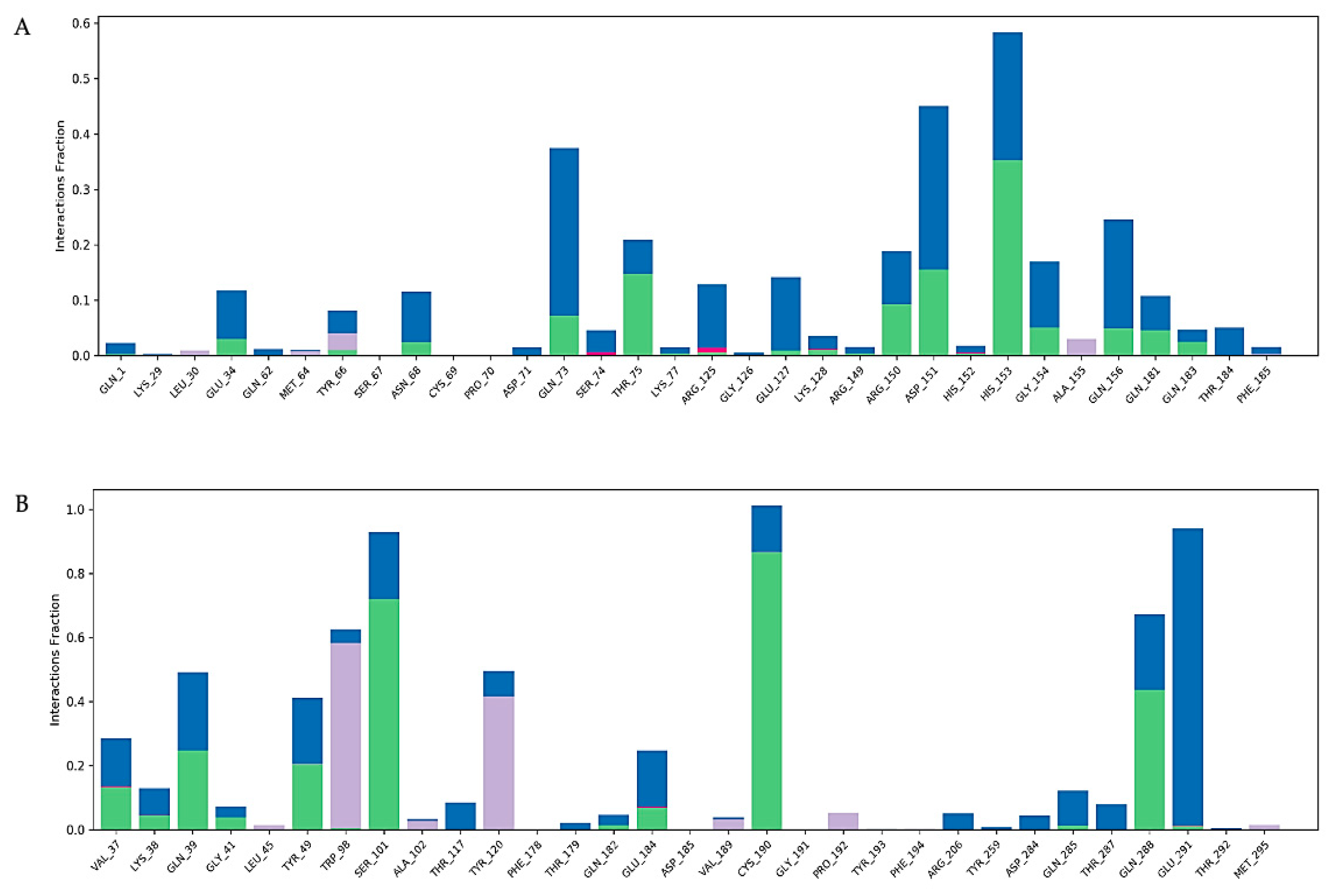
2.7. Molecular Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Extraction of Phytochemicals from L. reticulata
4.3. Identification of Phytochemicals Using HR-LCMS/MS(Q-TOF)
4.4. Active Ingredient Screening
4.5. Inflammation-Related Target and Associated Drug Target Screening
4.6. Protein–Protein Interaction
4.7. GO Enrichment and KEGG Analysis
4.8. Molecular Docking
4.9. Molecular Docking Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashley, N.T.; Weil, Z.M.; Nelson, R.J. Inflammation: Mechanisms, Costs, and Natural Variation. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 385–406. [Google Scholar] [CrossRef]
- Pfitzner, E.; Kliem, S.; Baus, D.; Litterst, C.M. The Role of STATs in Inflammation and Inflammatory Diseases. Curr. Pharm. Des. 2004, 10, 2839–2850. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Points of Control in Inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic Inflammation in the Etiology of Disease across the Life Span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Pahwa, R.; Goyal, A.; Jialal, I. Chronic Inflammation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Germolec, D.R.; Shipkowski, K.A.; Frawley, R.P.; Evans, E. Markers of Inflammation. Methods Mol. Biol. 2018, 1803, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and Cancer. Ann. Afr. Med. 2019, 18, 121. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From Inflammation to Cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef]
- AAbdulla, A.; Adams, N.; Bone, M.; Elliott, A.M.; Gaffin, J.; Jones, D.; Knaggs, R.; Martin, D.; Sampson, L.; Schofield, P. Guidance on the Management of Pain in Older People. Age Ageing 2013, 42 (Suppl. S1), i1–i57. [Google Scholar] [CrossRef]
- Onder, G.; Pellicciotti, F.; Gambassi, G.; Bernabei, R. NSAID-Related Psychiatric Adverse Events: Who Is at Risk? Drugs 2004, 64, 2619–2627. [Google Scholar] [CrossRef]
- Wongrakpanich, S.; Wongrakpanich, A.; Melhado, K.; Rangaswami, J. A Comprehensive Review of Non-Steroidal Anti-Inflammatory Drug Use in The Elderly. Aging Dis. 2018, 9, 143–150. [Google Scholar] [CrossRef]
- Rajčević, N.; Bukvički, D.; Dodoš, T.; Marin, P.D. Interactions between Natural Products—A Review. Metabolites 2022, 12, 1256. [Google Scholar] [CrossRef]
- Harish, K.; Sivasankaran, S.M.; Manoharan, S. Indian Medicinal Plants with Multiple Pharmacological Efficacies: A Comprehensive Review. J. Drug Deliv. Ther. 2024, 14, 143–154. [Google Scholar] [CrossRef]
- Bamola, N.; Verma, P.; Negi, C. A Review on Some Traditional Medicinal Plants. Int. J. Life-Sci. Sci. Res. 2018, 4, 1550–1556. [Google Scholar] [CrossRef]
- Sharma, G.; Agarwal, S.; Verma, K.; Bhardwaj, R.; Mathur, V. Therapeutic Compounds from Medicinal Plant Endophytes: Molecular and Metabolic Adaptations. J. Appl. Microbiol. 2023, 134, lxad074. [Google Scholar] [CrossRef]
- Padmanabhan, D.; Siddiqui, M.H.; Natarajan, P.; Palanisamy, S. Hecogenin a Plant Derived Small Molecule as an Antagonist to BACE-1: A Potential Target for Neurodegenerative Disorders. Metabolites 2023, 13, 758. [Google Scholar] [CrossRef]
- Bawra, B.; Dixit, M.; Chauhan, N.; Dixit, V.; Saraf, D.K. Leptadenia Reticulata a Rasayana Herbs: A Review. Asian J. Plant Sci. 2010, 9, 314–319. [Google Scholar] [CrossRef]
- Mohanty, S.K.; Swamy, M.K.; Sinniah, U.R.; Anuradha, M. Leptadenia reticulata (Retz.) Wight & Arn. (Jivanti): Botanical, Agronomical, Phytochemical, Pharmacological, and Biotechnological Aspects. Molecules 2017, 22, 1019. [Google Scholar] [CrossRef]
- Pullaiah, T. (Ed.) Phytochemical Composition and Pharmacy of Medicinal Plants: 2-Volume Set; Apple Academic Press: New York, NY, USA, 2023; ISBN 978-1-00-339049-7. [Google Scholar]
- Božunović, J.; Ivanov, M.; Petrović, J.; Gašić, U.; Nakarada, Đ.; Milutinović, M.; Aničić, N.; Giba, Z.; Mišić, D.; Stojković, D. Solvent System-Guided Extraction of Centaurium spicatum (L.) Fritch Provides Optimized Conditions for the Biological and Chemical Characteristics of the Herbal Extracts. Pharmaceuticals 2023, 16, 245. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Nie, S.; Wang, S.; Wang, H.; Gong, J.; Yan, W.; Tian, D.; Liu, M. Therapeutic Effect of Costunolide in Autoimmune Hepatitis: Network Pharmacology and Experimental Validation. Pharmaceuticals 2023, 16, 316. [Google Scholar] [CrossRef]
- Ramírez, G.L.S.; López Rincón, A.; García, J.M.; Correa, J.B.; Martínez, M.A. In Silico Screening of Drugs that Target Different Forms of E Protein for Potential Treatment of COVID-19. Pharmaceuticals 2023, 16, 296. [Google Scholar] [CrossRef]
- Periferakis, A.; Periferakis, K.; Badarau, I.A.; Petran, E.M.; Popa, D.C.; Caruntu, A.; Costache, R.S.; Scheau, C.; Caruntu, C.; Costache, D.O. Kaempferol: Antimicrobial Properties, Sources, Clinical, and Traditional Applications. Int. J. Mol. Sci. 2022, 23, 15054. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Malar, D.S.; Nabavi, S.F.; Sureda, A.; Xiao, J.; Nabavi, S.M.; Daglia, M. Kaempferol and Inflammation: From Chemistry to Medicine. Pharmacol. Res. 2015, 99, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Taheri, Y.; Sharifi-Rad, J.; Antika, G.; Yılmaz, Y.B.; Tumer, T.B.; Abuhamdah, S.; Chandra, S.; Saklani, S.; Kılıç, C.S.; Sestito, S.; et al. Paving Luteolin Therapeutic Potentialities and Agro-Food-Pharma Applications: Emphasis on In Vivo Pharmacological Effects and Bioavailability Traits. Oxidative Med. Cell. Longev. 2021, 2021, 1987588. [Google Scholar] [CrossRef]
- Li, D.; Rui, Y.-X.; Guo, S.-D.; Luan, F.; Liu, R.; Zeng, N. Ferulic Acid: A Review of Its Pharmacology, Pharmacokinetics and Derivatives. Life Sci. 2021, 284, 119921. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, G.; Sun, C.; Peng, F.; Yu, L.; Chen, Y.; Tan, Y.; Cao, X.; Tang, Y.; Xie, X.; et al. Chemistry, Pharmacokinetics, Pharmacological Activities, and Toxicity of Quercitrin. Phytother. Res. 2022, 36, 1545–1575. [Google Scholar] [CrossRef] [PubMed]
- Ganeshpurkar, A.; Saluja, A. The Pharmacological Potential of Catechin. Indian J. Biochem. Biophys. 2020, 57, 285–299. [Google Scholar]
- Kumari, S.; Saini, R.; Bhatnagar, A.; Mishra, A. A Comprehensive Review on Ayurvedic Herb Leptadenia reticulata (Jeevanti): A Phytochemistry and Pharmacological Perspective. Nat. Prod. Res. 2023, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Fang, Y.; Wang, H.; Tan, X.; Zhu, X.; Xu, Z.; Jiang, H.; Wu, X.; Hong, W.; Wang, X.; et al. Role of Chemokine Receptor 2 in Rheumatoid Arthritis: A Research Update. Int. Immunopharmacol. 2023, 116, 109755. [Google Scholar] [CrossRef]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 Signaling in Cancer Pathogenesis. Cell Commun. Signal. 2020, 18, 82. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A Master Regulator of Cellular Responses in Inflammation, Injury Resolution, and Tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Kargapolova, Y.; Geißen, S.; Zheng, R.; Baldus, S.; Winkels, H.; Adam, M. The Enzymatic and Non-Enzymatic Function of Myeloperoxidase (MPO) in Inflammatory Communication. Antioxidants 2021, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.M.; Ridnour, L.A.; Cheng, R.Y.S.; Wink, D.A.; Thomas, D.D. The Chemical Biology of NO That Regulates Oncogenic Signaling and Metabolism: NOS2 and Its Role in Inflammatory Disease. Crit. Rev. Oncog. 2023, 28, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, S.; Chen, Y.; Saravanan, D.; Sundram, K.M.; Yoga Latha, L. Extraction, Isolation and Characterization of Bioactive Compounds from Plants’ Extracts. Afr. J. Tradit. Complement. Altern. Med. 2010, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Taamalli, A.; Arráez-Román, D.; Abaza, L.; Iswaldi, I.; Fernández-Gutiérrez, A.; Zarrouk, M.; Segura-Carretero, A. LC-MS-Based Metabolite Profiling of Methanolic Extracts from the Medicinal and Aromatic Species Mentha Pulegium and Origanum Majorana. Phytochem. Anal. 2015, 26, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, J.; Zhao, C.; Xiao, H.; Fang, X.; Zheng, J. LC-Q-TOF-MS/MS Detection of Food Flavonoids: Principle, Methodology, and Applications. Crit. Rev. Food Sci. Nutr. 2023, 63, 3750–3770. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P. Proton NMR and HR-LC/MS Based Phytochemical Analysis of Methanolic Fraction of Alectra Parasitica A. Rich. Rhizomes. Heliyon 2020, 6, e03171. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Zhao, D.; Yu, X.; Shen, X.; Zhou, Y.; Wang, S.; Qiu, Y.; Chen, Y.; Zhu, F. TTD: Therapeutic Target Database Describing Target Druggability Information. Nucleic Acids Res. 2024, 52, D1465–D1477. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Wiegers, T.C.; Wiegers, J.; Wyatt, B.; Johnson, R.J.; Sciaky, D.; Barkalow, F.; Strong, M.; Planchart, A.; Mattingly, C.J. CTD Tetramers: A New Online Tool that Computationally Links Curated Chemicals, Genes, Phenotypes, and Diseases to Inform Molecular Mechanisms for Environmental Health. Toxicol. Sci. 2023, 195, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Mehryary, F.; Nastou, K.; Ohta, T.; Jensen, L.J.; Pyysalo, S. STRING-Ing Together Protein Complexes: Extracting Physical Protein Interactions from the Literature. bioRxiv 2023. bioRxiv:2023.12.10.570999. [Google Scholar]
- Liao, Y.; Ding, Y. Exploring the Mechanism of Alisma Orientale for the Treatment of Pregnancy Induced Hypertension and Potential Hepato-Nephrotoxicity by Using Network Pharmacology, Network Toxicology, Molecular Docking and Molecular Dynamics Simulation. Front. Pharmacol. 2022, 13, 1027112. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium, T. Expansion of the Gene Ontology Knowledgebase and Resources. Nucleic Acids Res. 2017, 45, D331. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.Org): Delivery of Experimentally-Determined PDB Structures alongside One Million Computed Structure Models of Proteins from Artificial Intelligence/Machine Learning. Nucleic Acids Res. 2023, 51, D488–D508. [Google Scholar] [CrossRef]
- Gökşen Tosun, N. Enhancing Therapeutic Efficacy in Breast Cancer: A Study on the Combined Cytotoxic Effects of Doxorubicin and MPC-3100. Naunyn Schmiedebergs Arch. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Houshmand, F.; Houshmand, S. Potentially Highly Effective Drugs for COVID-19: Virtual Screening and Molecular Docking Study through PyRx-Vina Approach. Front. Health Inform. 2023, 12, 150. [Google Scholar] [CrossRef]
- Umi Baroroh, S.S.; Muscifa, Z.S.; Destiarani, W.; Rohmatullah, F.G.; Yusuf, M. Molecular Interaction Analysis and Visualization of Protein-Ligand Docking Using Biovia Discovery Studio Visualizer. Indones. J. Comput. Biol. 2023, 2, 22–30. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 Update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial No | Compound Name | m/z | Molecular Formula | Expressed in Root (R), Leaf (L), Stem (S) |
|---|---|---|---|---|
| 1 | Brassilexin | 175.03 | C9 H6 N2 S | S |
| 2 | 1-Pyrenylsulfate | 299.04 | C16 H10 O4 S | R |
| 3 | 12-Tridecene-4,6,8,10tetraynal | 203.048 | C13 H8 O | S |
| 4 | Methyl N-methylanthranilate | 188.066 | C9 H11 N O₂ | S, L, R |
| 5 | Fenapanil | 254.169 | C16 H19 N3 | S |
| 6 | 2,4,6-Triethyl-1,3,5-trioxane | 197.113 | C9 H18 O3 | S, L |
| 7 | 11-Methoxy-vinorine | 387.172 | C22 H24 N2 O3 | S |
| 8 | Naltrindole | 415.203 | C26 H26 N2 O3 | S |
| 9 | Hydroxyprolyl-Alanine | 203.102 | C8 H14 N2 O4 | S |
| 10 | 5-Acetoxydihydrotheaespirane | 277.174 | C15 H26 O3 | S, R |
| 11 | Monomenthyl succinate | 279.153 | C14 H24 O4 | S |
| 12 | Grossamide | 625.254 | C36 H36 N2 O8 | S |
| 13 | Somniferine | 609.259 | C36 H36 N2 O7 | S |
| 14 | Campestanol | 425.369 | C28 H50 O | S |
| 15 | Octadecyl fumarate | 391.277 | C22 H40 O4 | S |
| 16 | Archaeol | 675.663 | C43 H88 O3 | S |
| 17 | 1-Eicosanol | 321.308 | C20 H42 O | S |
| 18 | Chinomethionat | 256.979 | C10 H6 N2 O S2 | L |
| 19 | Neotussilagine | 222.111 | C10 H17 N O3 | L, R |
| 20 | Neuraminic acid | 268.102 | C9 H17 N O8 | L, R |
| 21 | Gabapentin | 172.132 | C9 H17 N O2 | L, R |
| 22 | 1-Phenylbiguanide | 200.09 | C8 H11 N5 | L |
| 23 | L-Tryptophan | 205.095 | C11 H12 N2 O2 | L, R |
| 24 | 6-Methylquinoline | 144.08 | C10 H9 N | L, R |
| 25 | Isocarbostyril | 146.059 | C9 H7 N O | L, R |
| 26 | Methyprylon | 206.116 | C10 H17 N O2 | L |
| 27 | Pirbuterol | 263.137 | C12 H20 N2 O3 | L |
| 28 | 2-Ethyl-5-methylpyridine | 144.079 | C8 H11 N | L |
| 29 | Citrinin | 273.073 | C13 H14 O5 | L |
| 30 | [2,2-bis (2-methylpropoxy) ethyl]benzene | 273.178 | C16 H26 O2 | L |
| 31 | Hexyl 2-furoate | 197.116 | C11 H16 O3 | L |
| 32 | Maritimetin | 287.053 | C15 H10 O6 | L |
| 33 | Ismine | 258.113 | C15 H15 N O3 | L |
| 34 | Lenacil | 257.126 | C13 H18 N2 O2 | L |
| 35 | 3-Hydroxynonyl acetate | 225.147 | C11 H22 O3 | L, R |
| 36 | C16 Sphinganine | 274.273 | C16 H35 N O2 | L, R |
| 37 | Symlandine | 404.203 | C20 H31 N O6 | L |
| 38 | Lauroyl diethanolamide | 288.251 | C16 H33 N O3 | L, R |
| 39 | Gibberellin A74 | 387.177 | C20 H28 O6 | L, R |
| 40 | Thiamylal | 255.12 | C12 H18 N2 O2 S | L |
| 41 | 2,6-Di-tert-butyl-4-ethylphenol | 257.188 | C16 H26 O | L |
| 42 | Nigakilactone B | 415.208 | C22 H32 O6 | L, R |
| 43 | Sphinganine | 302.303 | C18 H39 N O2 | L, R |
| 44 | 18-Nor-4(19),8,11,13abietatetraene | 277.195 | C19 H26 | L |
| 45 | Oxidized dinoflagellate luciferin | 625.261 | C33 H38 N4 O7 | L |
| 46 | Irinotecan | 609.266 | C33 H38 N4 O6 | L |
| 47 | 3-[(3-Methylbutyl) nitrosoamino]-2butanone | 187.142 | C9 H18 N2 O2 | R |
| 48 | Metoprolol | 268.188 | C15 H25 N O3 | R |
| 49 | (Z)-3-(1-Formyl-1propenyl) pentanedioic acid | 223.058 | C9 H12 O5 | R |
| 50 | Luciduline | 208.168 | C13 H21 N O | R |
| 51 | methyl (2E,6E,10R,11S)-10,11epoxy-3,7,11-trimethyltrideca-2,6-dienoate | 303.193 | C17 H28 O3 | R |
| 52 | 3-Hydroxy-4-deoxypaxilline | 444.246 | C27 H35 N O3 | R |
| 53 | Licoagrodione | 357.131 | C20 H20 O6 | R |
| 54 | Gibberellin A91 | 387.14 | C19 H24 O7 | R |
| 55 | Riesling acetal | 249.147 | C13 H22 O3 | R |
| 56 | 11-Hydroxy-9-tridecenoic acid | 251.162 | C13 H24 O3 | R |
| 57 | Ginsenoyne D | 285.183 | C17 H26 O2 | R |
| 58 | Sulfadimidine | 279.091 | C12 H14 N4 O2 S | R |
| 59 | 8Z,11Z,14Z-heptadecatrienoic acid | 287.198 | C17 H28 O2 | R |
| 60 | 1,8-Heptadecadiene-4,6-diyne-3,10-diol | 283.167 | C17 H24 O2 | R |
| 61 | Albuterol | 262.142 | C13 H21 N O3 | R |
| 62 | Caffeic aldehyde | 163.042 | C9 H8 O3 | L |
| 63 | Quercitrin | 447.096 | C21 H20 O11 | L |
| 64 | Kaempferol | 285.042 | C15 H10 O6 | L |
| 65 | Luteolin | 285.042 | C15 H10 O6 | L |
| 66 | Colnelenic acid | 291.199 | C18 H28 O3 | L |
| 67 | 9-HOTE | 293.214 | C18 H30 O3 | L, R |
| 68 | Ferulic acid | 193.052 | C10 H10 O4 | L |
| 69 | Ellagic acid | 301.001 | C14 H6 O8 | L |
| 70 | Malic acid | 133.015 | C₄ H₆ O₅ | L |
| 71 | Ribose-1-arsenate | 272.961 | C5 H11 As O8 | L, S |
| 72 | 2,3,5,7,9-Pentathiadecane 2,2-dioxide | 262.932 | C5 H12 O2 S5 | L |
| 73 | Apigenin 7-[rhamnosyl-(1->2)-galacturonide] | 591.139 | C27 H28 O15 | L |
| 74 | CMP-N-glycoloylneuraminate | 629.132 | C20 H31 N4 O17 P | L |
| 75 | Nicotiflorin | 593.156 | C27 H30 O15 | L |
| 76 | Genistein 8-C-glucoside | 431.102 | C21 H20 O10 | L |
| 77 | Biorobin | 593.156 | C27 H30 O15 | L |
| 78 | Glafenine | 431.102 | C19 H17 Cl N2 O4 | L |
| 79 | Tetradecyl sulfate | 293.178 | C14 H30 O4 S | L, R |
| 80 | Hexazinone | 297.156 | C12 H20 N4 O2 | L, S |
| 81 | Magnesium protoporphyrin monomethyl ester | C35 H34 Mg N4 O4 | L | |
| 82 | Lamprolobine | 309.177 | C15 H24 N2 O2 | L, R |
| 83 | Kanokoside D | 623.257 | C27 H44 O16 | L |
| 84 | 19-Hydroxycinnzeylanol 19-glucoside | 607.261 | C26 H42 O13 | L |
| 85 | Muricatalin | 671.471 | C35 H64 O8 | L, S |
| 86 | 14,19-Dihydroaspidospermatine | 339.203 | C21 H28 N2 O2 | L, R |
| 87 | Lycocernuine | 337.209 | C16 H26 N2 O2 | L |
| 88 | Malvalic acid | 339.256 | C18 H32 O2 | R |
| 89 | 6-Feruloylglucose 2,3,4-trihydroxy-3-methylbutylglycoside | 473.168 | C21 H30 O12 | R |
| 90 | Lusitanicoside | 487.184 | C21 H30 O10 | R |
| 91 | Thiazopyr | 455.106 | C16 H17 F5 N2 O2 S | R |
| 92 | beta-D-3-[5-Deoxy-5-(dimethylarsinyl)ribofuranosyl oxy]-2-hydroxy-1-propanesulfonic acid | 451.023 | C10 H21 As O9 S | R |
| 93 | Tosyllysine chloromethyl ketone | 377.09 | C14 H21 Cl N2 O3 S | R |
| 94 | Dictyoquinazol C | 341.113 | C18 H18 N2 O5 | R |
| 95 | Methyl (3×,10R)-dihydroxy-11-dodecene-6,8-diynoate 10-glucoside | 443.16 | C19 H26 O9 | R |
| 96 | Haemocorin | 687.199 | C32 H34 O14 | R |
| 97 | Sudachiin A | 521.136 | C24 H26 O13 | R |
| 98 | 4-(4-Hydroxyphenyl)-2-butanone O-[2-galloyl-6-p-coumaroylglucoside] | 669.189 | C32 H32 O13 | R |
| 99 | Phytolaccoside E | 825.437 | C42 H66 O16 | R |
| 100 | (1S,4R)-10-Hydroxyfenchone glucoside | 329.157 | C16 H26 O7 | R |
| 101 | Madasiatic acid | 487.348 | C30 H48 O5 | R, S |
| 102 | Provincialin | 517.207 | C27 H34 O10 | R |
| 103 | 2-Hexaprenyl-3-methyl-6-methoxy-1,4 benzoquinone | 605.411 | C38 H56 O3 | R |
| 104 | Omega-hydroxy behenic acid | 335.326 | C22 H44 O3 | R |
| 105 | Catechin | 349.094 | C15 H14 O6 | S |
| 106 | Cauleprin | 457.141 | C24 H18 N2 O4 | S |
| 107 | Dracorubin | 487.152 | C32 H24 O5 | S |
| 108 | 1,4-beta-D-Glucan | 595.174 | C18 H32 O18 | S |
| 109 | Daidzin 4′-O-glucuronide | 591.143 | C27 H28 O15 | S |
| 110 | Aurasperone C | 591.144 | C31 H28 O12 | S |
| 111 | Nb-Stearoyltryptamine | 471.355 | C28 H46 N2 O | S |
| 112 | Tetrahexosylceramide (d18:1/24:0) | 668.442 | C68 H126 N2 O23 | S |
| 113 | Hydroquinidine | 325.19 | C20 H26 N2 O2 | S |
| Molecule | Molecular Formula | MW g/mol | No. of H-Bond Acceptors | No. of H-Bond Donors | TPSA | GI Absorption | BBB Permeant | No. of Lipinski Violations |
|---|---|---|---|---|---|---|---|---|
| Neotussilagine | C10 H17 NO3 | 199.25 | 4 | 1 | 49.77 | High | No | 0 |
| Isocarbostyril | C9 H7 N O | 145.16 | 1 | 1 | 32.86 | High | Yes | 0 |
| Hexyl 2-furoateṇ | C11 H16 O3 | 196.24 | 3 | 0 | 39.44 | High | Yes | 0 |
| 1-Pyrenylsulfate | C16 H10 O4 S | 298.31 | 4 | 1 | 71.98 | High | No | 0 |
| C16 Sphinganine | C16 H35 N O2 | 273.45 | 3 | 3 | 66.48 | High | Yes | 0 |
| 2,3,5,7,9-Pentathiadecane 2,2-dioxide | C5 H12 O2 S5 | 264.47 | 2 | 0 | 143.72 | Low | No | 0 |
| Lycocernuine | C16 H26 N2 O2 | 278.39 | 3 | 1 | 43.78 | High | Yes | 0 |
| Kaempferol | C15 H10 O6 | 286.24 | 6 | 4 | 111.13 | High | No | 0 |
| Malic acid | C4 H6 O5 | 134.09 | 5 | 3 | 94.83 | High | No | 0 |
| Metoprolol | C15 H25 N O3 | 267.36 | 4 | 2 | 50.72 | High | Yes | 0 |
| (Z)-3-(1-Formyl-1-propenyl)pentanedioic acid | C9 H12 O5 | 200.19 | 5 | 2 | 91.67 | High | No | 0 |
| (1S,4R)-10-Hydroxyfenchone glucoside | C16 H26 O7 | 330.37 | 7 | 4 | 116.45 | High | No | 0 |
| beta-D-3[5-Deoxy-5-(dimethylarsinyl)ribofuranosyloxy]-2-hydroxy-1-propanesulfonic acid | C10 H21 As O9 S | 392.25 | 9 | 4 | 161.96 | Low | No | 0 |
| Albuterol | C13 H21 N O3 | 239.31 | 4 | 4 | 72.72 | High | No | 0 |
| 8Z-11Z-14Z-heptadecatrienoic acid | C17 H28 O2 | 264.4 | 2 | 1 | 37.3 | High | Yes | 0 |
| Methyl(3×,10R)-dihydroxy-11-dodecene-6,8-diynoate 10-glucoside | C19 H26 O9 | 398.4 | 9 | 5 | 145.91 | Low | No | 0 |
| Monomenthyl succinate | C14 H24 O4 | 256.34 | 4 | 1 | 63.6 | High | Yes | 0 |
| 1,8-Heptadecadiene-4,6-diyne-3,10-diol | C17 H24 O2 | 260.37 | 2 | 2 | 40.46 | High | Yes | 0 |
| Compound Name | Oral LD50 Value (mg/kg) | Predicted Toxicity Class | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity |
|---|---|---|---|---|---|---|---|
| Neotussilagine | 1240 | 4 | Inactive (−0.92) | Active (−0.59) | Inactive (−0.98) | Inactive (−0.75) | Inactive (−0.72) |
| Isocarbostyril | 360 | 4 | Inactive (−0.51) | Inactive (−0.58) | Inactive (−0.99) | Inactive (−0.66) | Inactive (−0.85) |
| Hexyl 2-furoateṇ | 1500 | 4 | Inactive (−0.8) | Active (−0.51) | Inactive (−0.9) | Inactive (−0.84) | Inactive (−0.74) |
| 1-Pyrenylsulfate | 2793 | 5 | Inactive (−0.73) | Inactive (−0.73) | Inactive (−0.83) | Inactive (−0.79) | Inactive (−0.83) |
| C16 Sphinganine | 3500 | 5 | Inactive (−0.76) | Inactive (−0.54) | Inactive (−0.99) | Inactive (−0.9) | Inactive (−0.71) |
| 2,3,5,7,9-Pentathiadecane 2,2-dioxide | 3200 | 5 | Inactive (−0.69) | Inactive (−0.64) | Inactive (−0.99) | Inactive (−0.62) | Inactive (−0.78) |
| Lycocernuine | 4000 | 5 | Inactive (−0.73) | Inactive (−0.61) | Inactive (−0.84) | Inactive (−0.74) | Inactive (−0.74) |
| Kaempferol | 3919 | 5 | Inactive (−0.68) | Inactive (−0.72) | Inactive (−0.96) | Inactive (−0.52) | Inactive (−0.98) |
| Malic acid | 2497 | 5 | Inactive (−0.9) | Inactive (−0.71) | Inactive (−0.99) | Inactive (−0.97) | Inactive (−0.74) |
| Metprolol | 1050 | 4 | Inactive (−0.94) | Inactive (−0.82) | Inactive (−0.88) | Inactive (−0.93) | Inactive (−0.73) |
| (Z)-3-(1-Formyl-1-propenyl)pentanedioic acid | 2140 | 5 | Inactive (−0.73) | Inactive (−0.73) | Inactive (−0.99) | Inactive (−0.9) | Inactive (−0.69) |
| 1,8-Heptadecadiene-4,6-diyne-3,10-diol | 5600 | 6 | Inactive (−0.7) | Inactive (−0.65) | Inactive (−0.95) | Inactive (−0.95) | Inactive (−0.79) |
| 8Z-11Z-14Z-heptadecatrienoic acid | 10,000 | 6 | Inactive (−0.54) | Inactive (−0.63) | Inactive (−0.99) | Inactive (−0.95) | Inactive (−0.71) |
| Albuterol | 660 | 4 | Inactive (−0.98) | Inactive (−0.86) | Inactive (−0.88) | Inactive (−0.75) | Inactive (−0.66) |
| beta-D-3[5-Deoxy-5-(dimethylarsinyl)ribofuranosyloxy]-2-hydroxy-1-propanesulfonic acid | 8000 | 6 | Inactive (−0.78) | Inactive (−0.68) | Inactive (−0.84) | Inactive (−0.54) | Inactive (−0.72) |
| (1S,4R)-10-Hydroxyfenchone glucoside | 190 | 3 | Inactive (−0.9) | Inactive (−0.83) | Inactive (−0.96) | Inactive (−0.7) | Inactive (−0.63) |
| Methyl(3×,10R)-dihydroxy-11-dodecene-6,8-diynoate 10-glucoside | 10,000 | 6 | Inactive (−0.87) | Inactive (−0.83) | Inactive (−0.99) | Inactive (−0.69) | Inactive (−0.76) |
| Monomenthyl succinate | 930 | 4 | Inactive (−0.63) | Inactive (−0.66) | Inactive (−0.99) | Inactive (−0.86) | Inactive (−0.81) |
| Serial No. | Target | Common Name | Uniport ID |
|---|---|---|---|
| 1 | Intercellular adhesion molecule 1 | ICAM1 | P05362 |
| 2 | Signal transducer and activator of transcription 3 | STAT3 | P40763 |
| 3 | Myeloperoxidase | MPO | P05164 |
| 4 | Nitric oxide synthase, inducible | NOS2 | P35228 |
| 5 | PI3-kinase p110-gamma subunit | PIK3CG | P48736 |
| 6 | Tyrosine-protein kinase SRC | SRC | P12931 |
| 7 | Vitamin D receptor | VDR | P11473 |
| 8 | Glucocorticoid receptor | NR3C1 | P04150 |
| 9 | Leukotriene B4 receptor 1 | LTB4R | Q15722 |
| 10 | C-C chemokine receptor type 2 | CCR2 | P41597 |
| 11 | Telomerase reverse transcriptase | TERT | O14746 |
| 12 | Protein kinase C theta | PRKCQ | Q04759 |
| 13 | Leukotriene A4 hydrolase | LTA4H | |
| 14 | Prostanoid EP4 receptor | PTGER4 | P35408 |
| 15 | Amine oxidase, copper containing | AOC3 | Q16853 |
| 16 | Estrogen receptor beta | ESR2 | Q92731 |
| 17 | Corticosteroid binding globulin | SERPINA6 | P08185 |
| 18 | Serotonin 1a (5-HT1a) receptor | HTR1A | P08908 |
| 19 | Adenosine A3 receptor | ADORA3 | P0DMS8 |
| 20 | Phospholipase A2 group 1B | PLA2G1B | P04054 |
| 21 | Sphingosine 1-phosphate receptor Edg-1 | S1PR1 | P21453 |
| 22 | Stem cell growth factor receptor | KIT | P10721 |
| 23 | Cathepsin S | CTSS | P25774 |
| 24 | Phosphodiesterase 4D | PDE4D | Q08499 |
| 25 | Rho-associated protein kinase | ROCK1 | Q13464 |
| 26 | Cathepsin K | CTSK | P43235 |
| 27 | Rho-associated protein kinase 2 | ROCK2 | O75116 |
| 28 | Serine/threonine-protein kinase PIM2 | PIM2 | Q9P1W9 |
| 29 | Cyclin-dependent kinase 1/cyclin B | CDK1 | P06493 |
| 30 | Thymidine kinase, cytosolic | TK1 | P04183 |
| Gene | Phytocompounds | Binding Affinity kcal/mol | Interactions |
|---|---|---|---|
| CCR2 | Lycocernuine | −8 | |
| (1S,4R)-10-Hydroxyfenchone glucoside | −7.8 | TRP A:98, SER A:101 | |
| Kaempferol | −7.7 | THR A:179, SER A:101 | |
| ICAM1 | 1-Pyrenylsulfate | −5.9 | LYS A:128, GLN A:156, HIS A:153 |
| Kaempferol | −5.5 | GLN A:156, HIS A:152 | |
| (1S,4R)-10-Hydroxyfenchone glucoside | −5.2 | GLN A:156, HIS A:153, LYS A:128, GLY A:154, HIS A:152 | |
| KIT | Kaempferol | −9.9 | CYSA:673 |
| 1-Pyrenylsulfate | −9.4 | LYS A:623 | |
| 1,8-Heptadecadiene-4,6-diyne-3,10-diol | −7.4 | ||
| MPO | Kaempferol | −8.5 | ARG C:323, ARG D:161 |
| 1-Pyrenylsulfate | −8.3 | ARG C:323 | |
| Lycocernuine | −8 | ARG C:323 | |
| NOS2 | 1-Pyrenylsulfate | −10.3 | GLY D:371, GLU D:377 |
| Kaempferol | −9.5 | TRP D:372 | |
| Lycocernuine | −8.3 | ||
| STAT3 | 1-Pyrenylsulfate | −7 | THR A:456, LYS A:318, LYS A:244 |
| Kaempferol | −6.7 | PHE A:321, THR A:456, LYS A:318 | |
| (1S,4R)-10-Hydroxyfenchone glucoside | −6.6 | SER A:319, PHE A:321, GLU A:455, THR A:456, LYS A:244 |
| S. No | Compound Name | PubChem ID | Molecular Weight (g/mol) | Molecular Formula |
|---|---|---|---|---|
| 1 | (1S,4R)-10-Hydroxyfenchone glucoside | 85257992 | 330.37 | C16 H26 O7 |
| 2 | (Z)-3-(1-Formyl-1-propenyl)pentanedioic acid | 22394751 | 200.19 | C9 H12 O5 |
| 3 | 1,8-Heptadecadiene-4,6-diyne-3,10-diol | 5318010 | 260.399 | C17 H24 O2 |
| 4 | 1-Pyrenylsulfate | 9543290 | 298.3 | C16 H10 O4 S |
| 5 | 2,3,5,7,9-Pentathiadecane 2,2-dioxide | 11777600 | 264.5 | C5 H12 O2 S2 |
| 6 | 8Z-11Z-14Z-Heptadecatrienoic acid | 16061034 | 264.4 | C17 H28 O2 |
| 7 | Albuterol | 2083 | 239.31 | C13 H21 N O3 |
| 8 | C16 Sphinganine | 656816 | 273.45 | C16 H35 N O2 |
| 9 | Isocarbostyril | 10284 | 145.16 | C9 H7 N O |
| 10 | Kaempferol | 5280863 | 286.24 | C15 H10 O6 |
| 11 | Lycocernuine | 442481 | 278.39 | C16 H26 N2 O2 |
| 12 | Malic acid | 525 | 134.09 | C4 H6 O5 |
| 13 | Methyl(3×,10R)-dihydroxy-11-dodecene-6,8-diynoate 10-glucoside | 131752977 | 398.4 | C19 H26 O9 |
| 14 | Metprolol | 4171 | 267.36 | C15 H25 N O3 |
| 15 | Monomenthyl succinate | 10199004 | 256.339 | C14 H24 O4 |
| 16 | Neotussilagine | 4484216 | 199.25 | C10 H7 N O3 |
| 17 | Hexyl 2-furoate | 61984 | 196.24 | C11 H16 O3 |
| 18 | beta-D-3[5-Deoxy-5-(dimethylarsinyl)ribofuranosyloxy]-2-hydroxy-1-propanesulfonic acid | 131751282 | 392.26 | C10 H21 As O9 S |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallepura Adinarayanaswamy, Y.; Padmanabhan, D.; Natarajan, P.; Palanisamy, S. Metabolomic Profiling of Leptadenia reticulata: Unveiling Therapeutic Potential for Inflammatory Diseases through Network Pharmacology and Docking Studies. Pharmaceuticals 2024, 17, 423. https://doi.org/10.3390/ph17040423
Mallepura Adinarayanaswamy Y, Padmanabhan D, Natarajan P, Palanisamy S. Metabolomic Profiling of Leptadenia reticulata: Unveiling Therapeutic Potential for Inflammatory Diseases through Network Pharmacology and Docking Studies. Pharmaceuticals. 2024; 17(4):423. https://doi.org/10.3390/ph17040423
Chicago/Turabian StyleMallepura Adinarayanaswamy, Yashaswini, Deepthi Padmanabhan, Purushothaman Natarajan, and Senthilkumar Palanisamy. 2024. "Metabolomic Profiling of Leptadenia reticulata: Unveiling Therapeutic Potential for Inflammatory Diseases through Network Pharmacology and Docking Studies" Pharmaceuticals 17, no. 4: 423. https://doi.org/10.3390/ph17040423
APA StyleMallepura Adinarayanaswamy, Y., Padmanabhan, D., Natarajan, P., & Palanisamy, S. (2024). Metabolomic Profiling of Leptadenia reticulata: Unveiling Therapeutic Potential for Inflammatory Diseases through Network Pharmacology and Docking Studies. Pharmaceuticals, 17(4), 423. https://doi.org/10.3390/ph17040423