

Structural Investigations on 2-Amidobenzimidazole Derivatives as New Inhibitors of Protein Kinase CK1 Delta
 , ,
, ,  , , , , ,
, , , , ,  and
and
Abstract
:
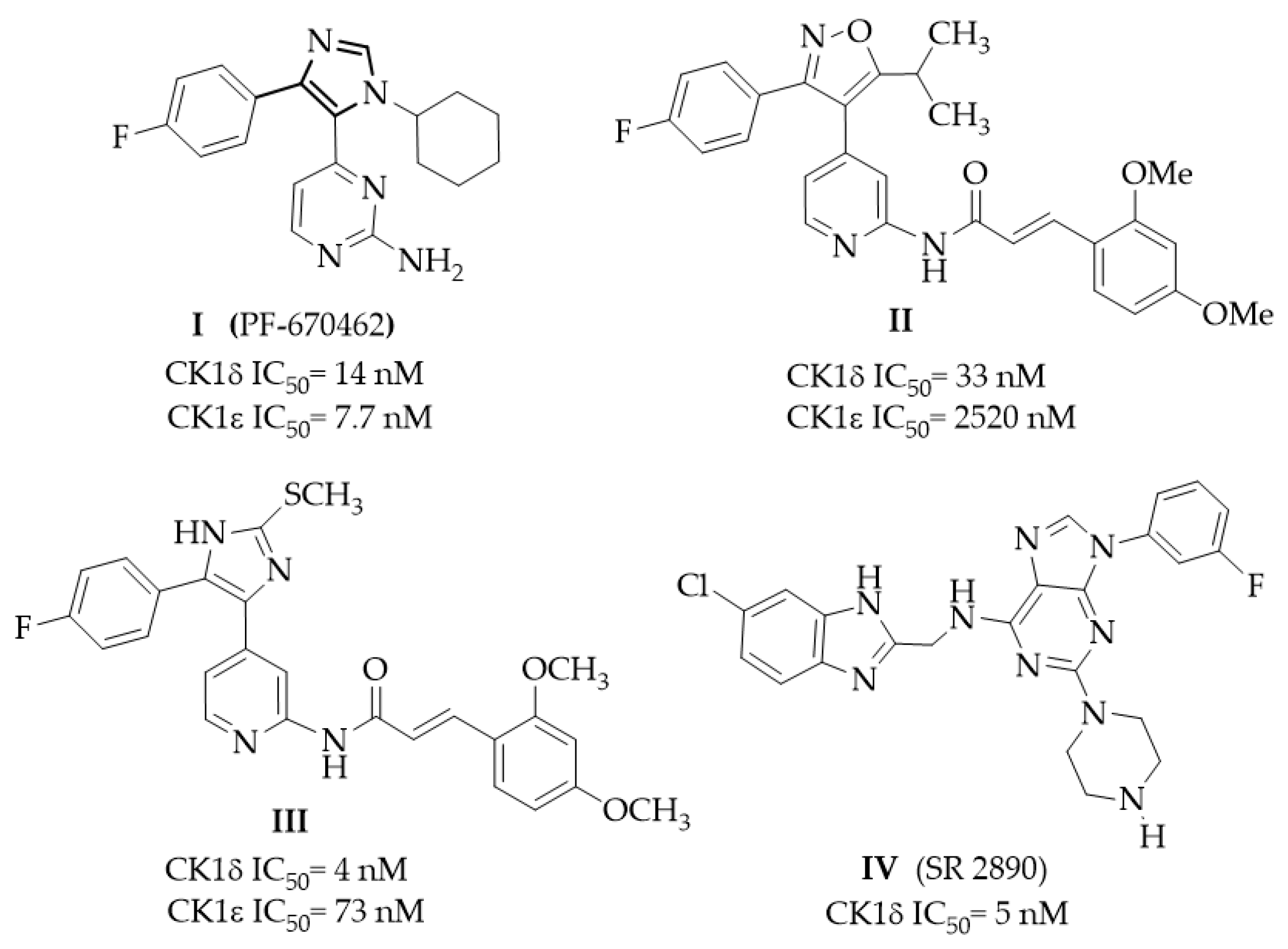
1. Introduction
2. Results and Discussion
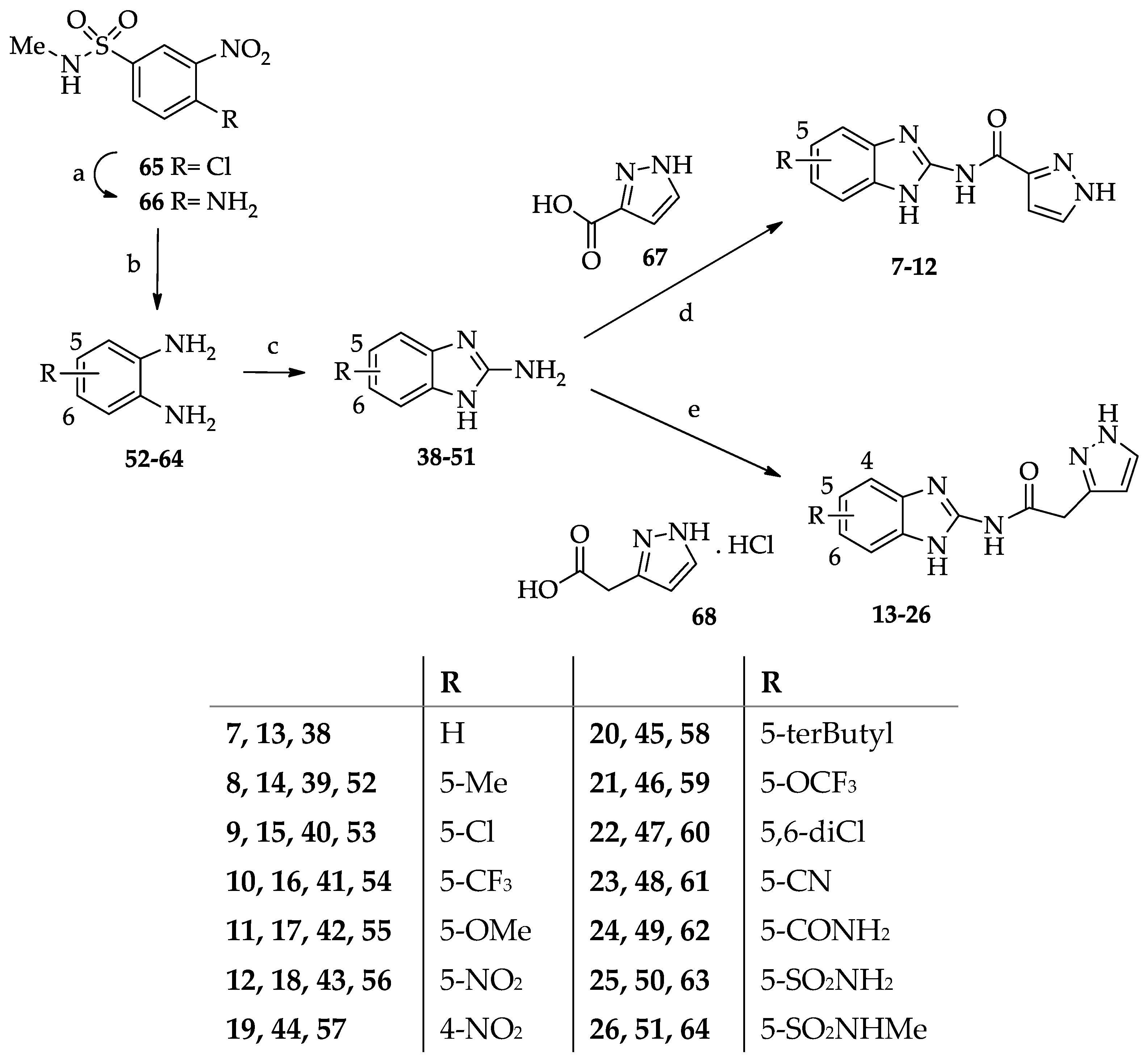
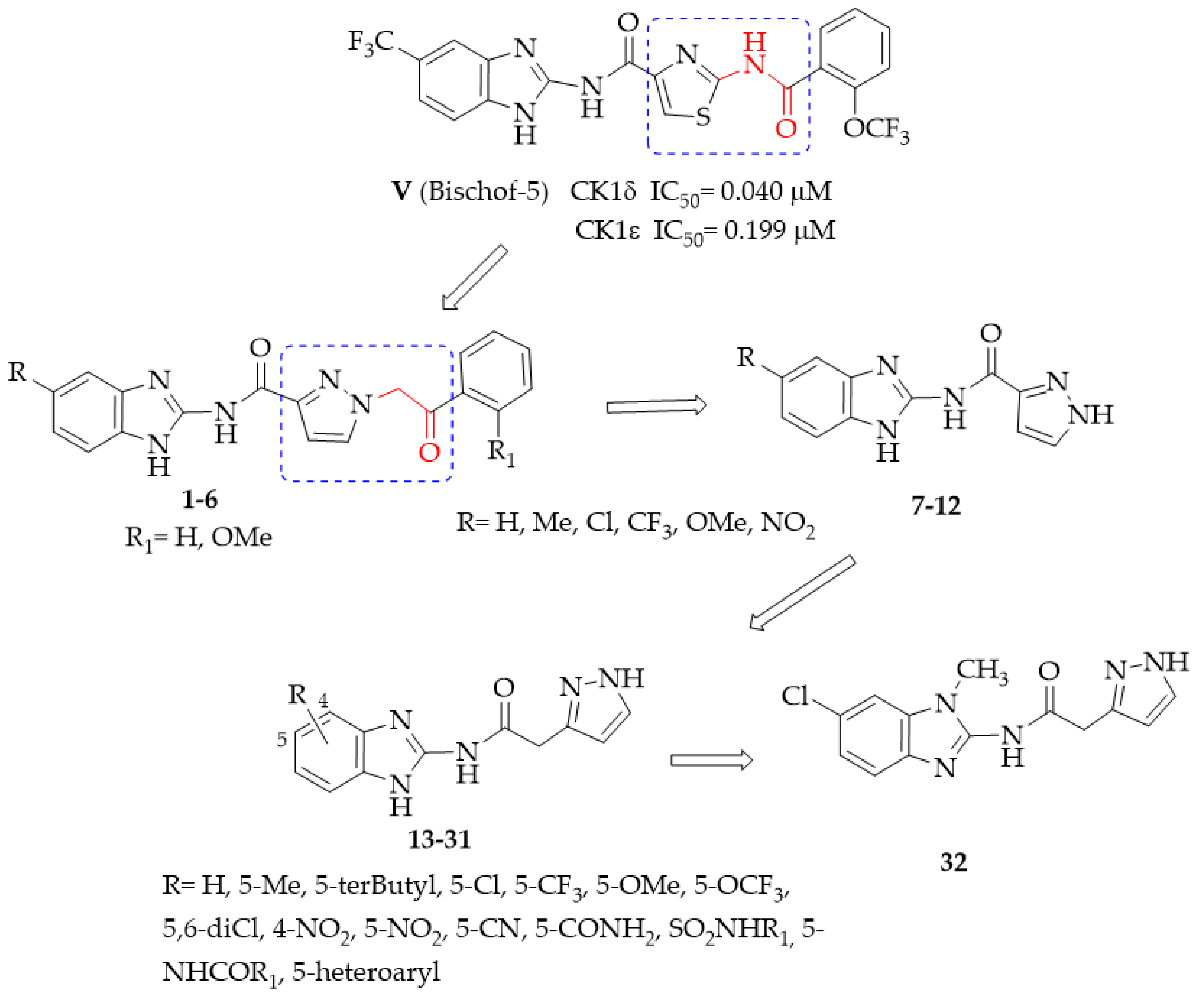
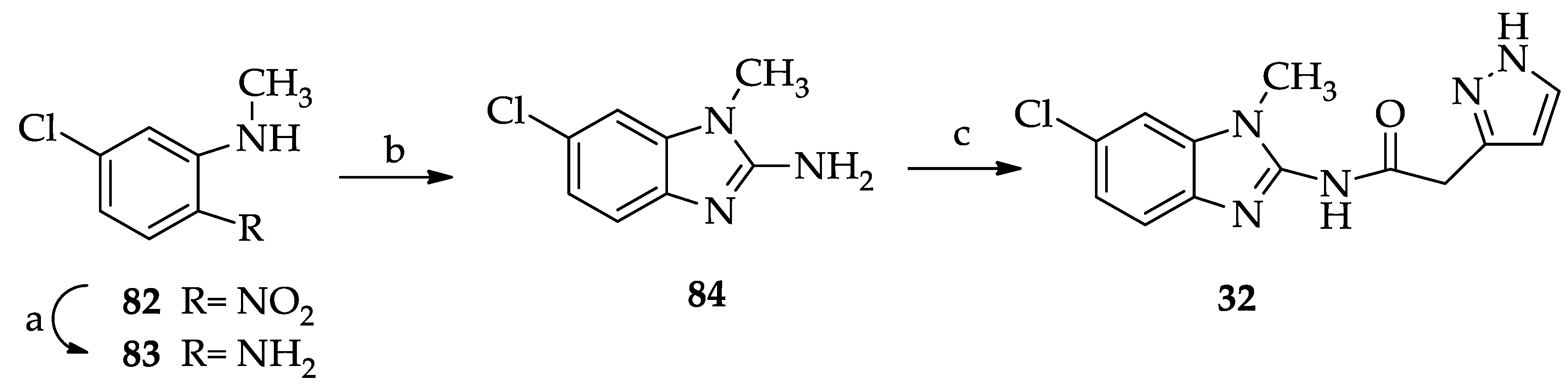
2.1. Chemistry
2.2. Structure–Activity Relationship (SAR) Studies
2.3. Physicochemical and Pharmacokinetic Parameters
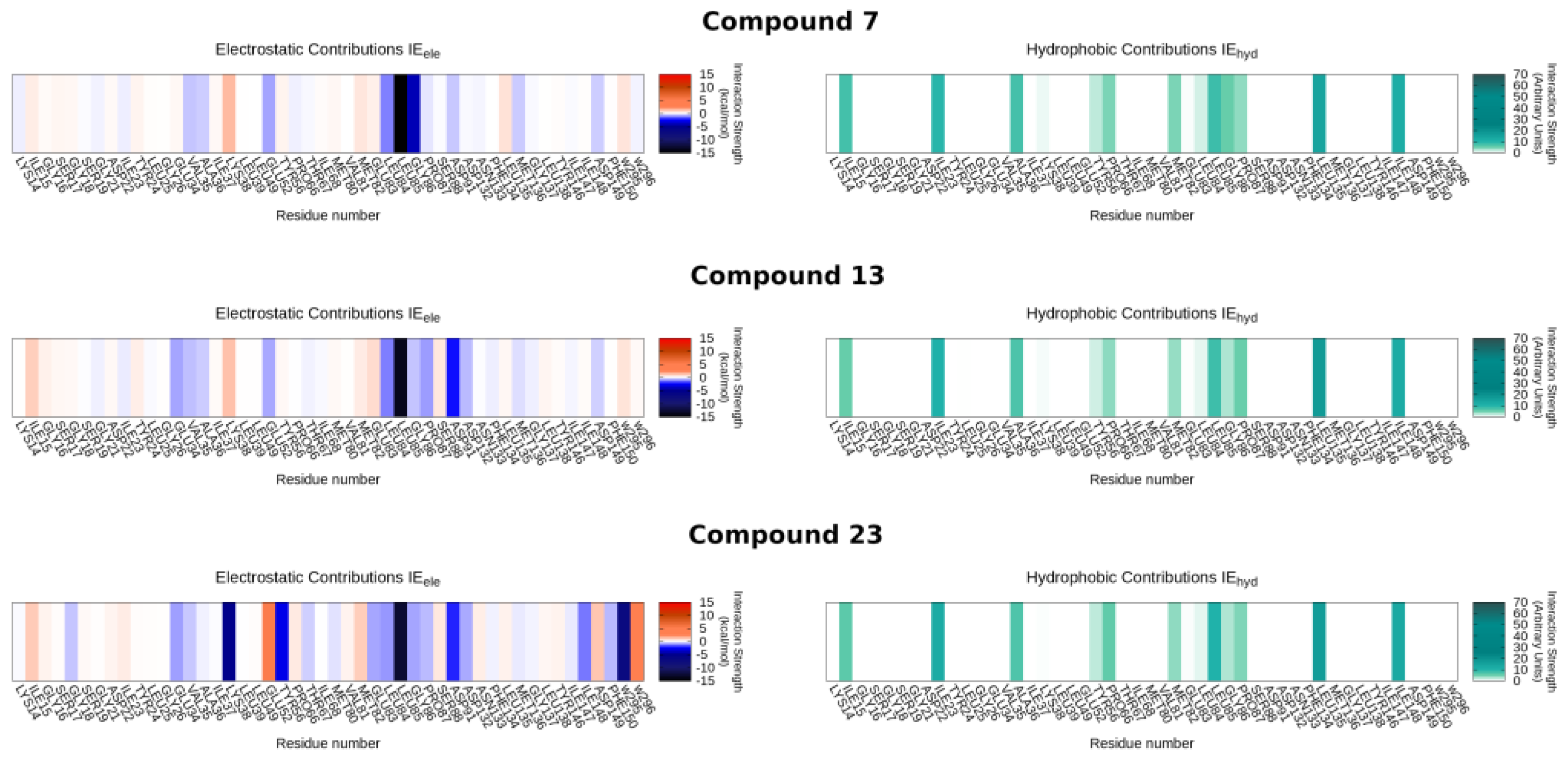
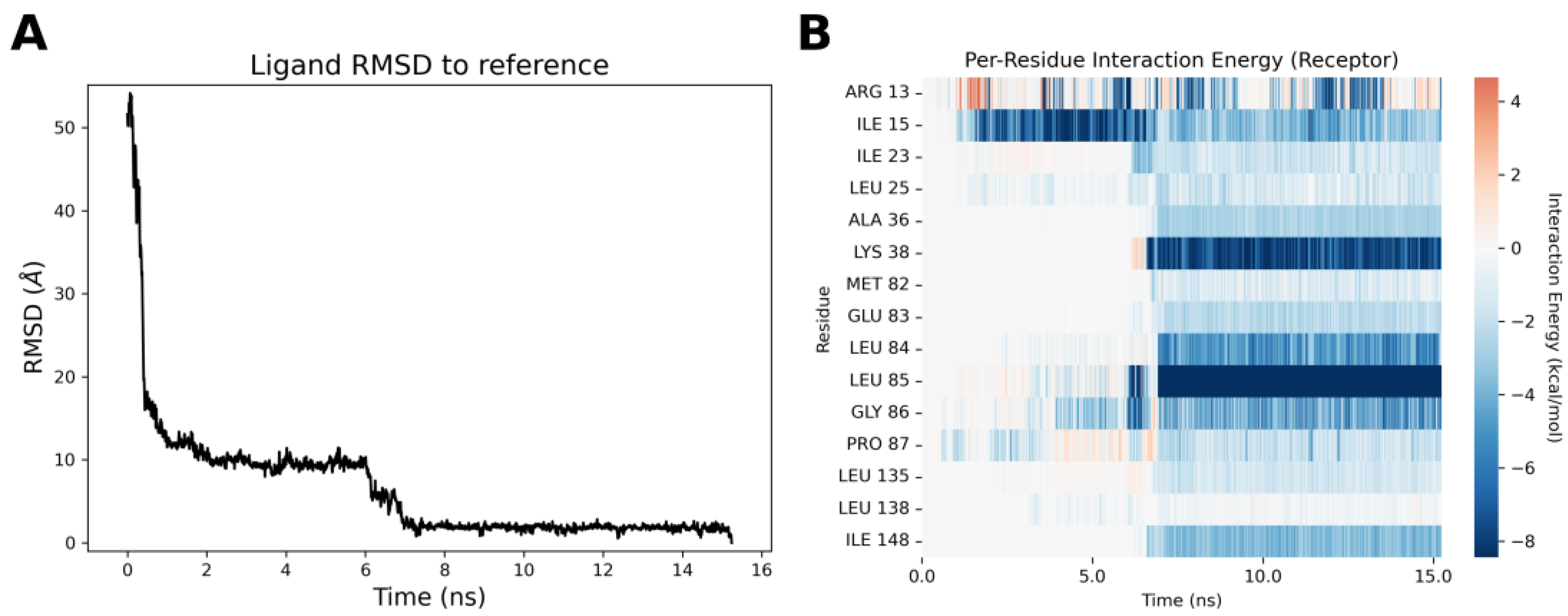
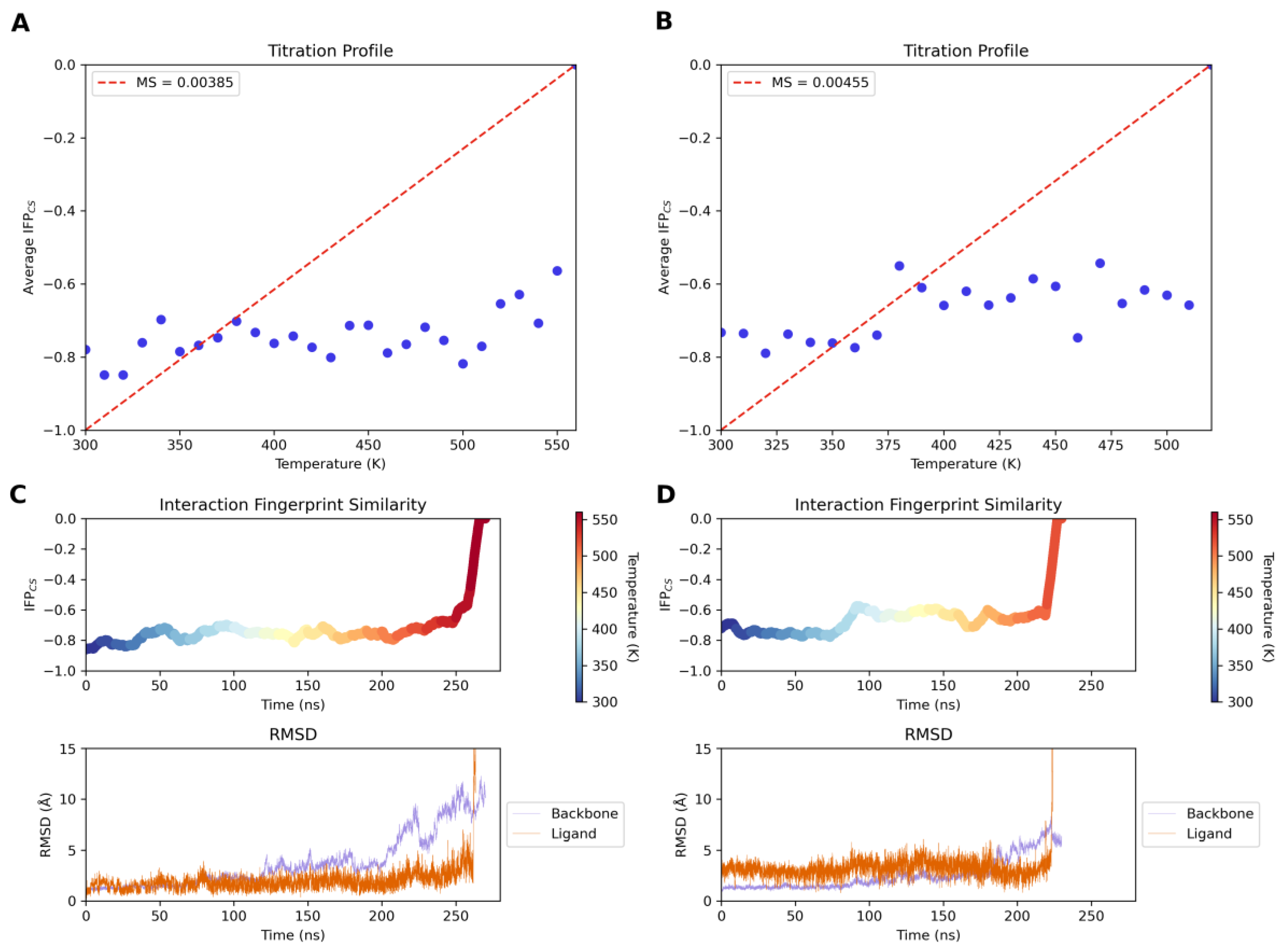
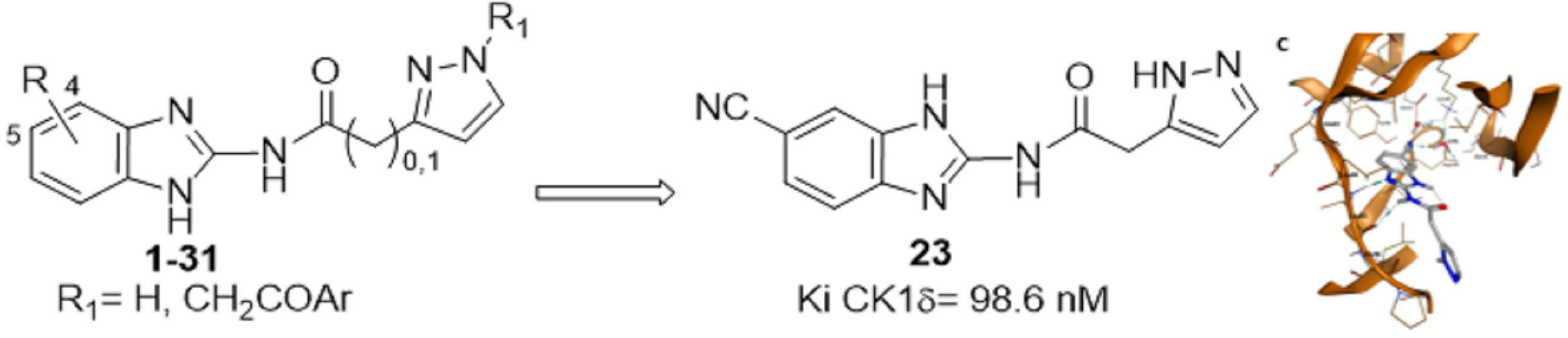
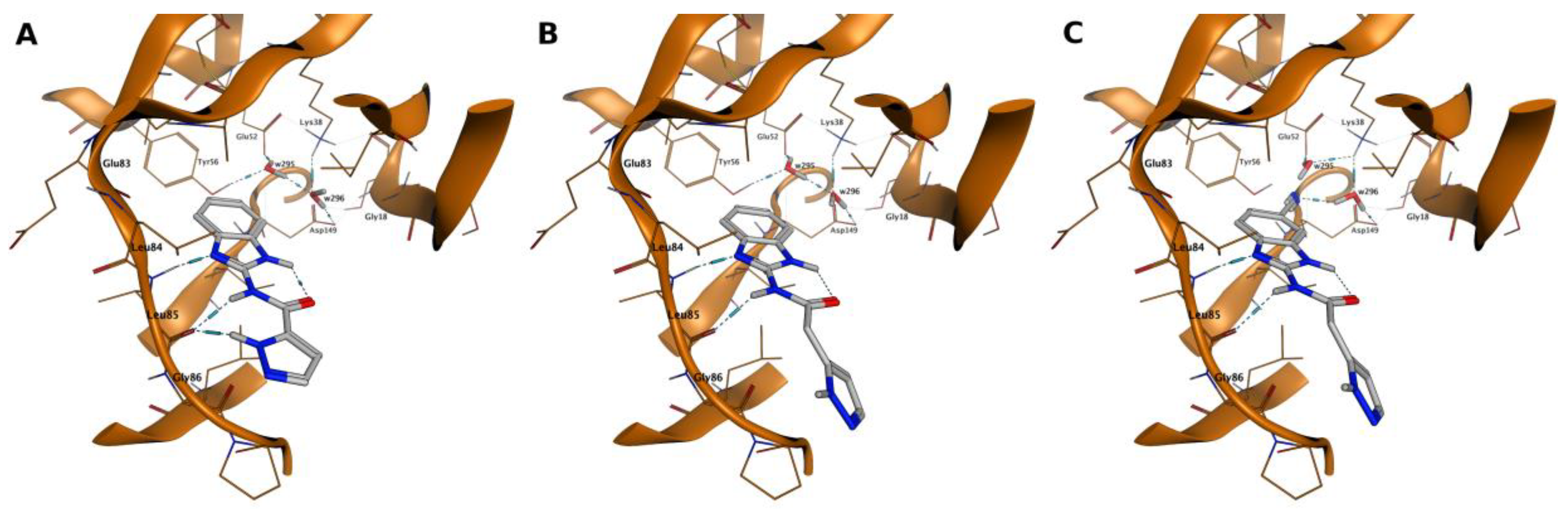
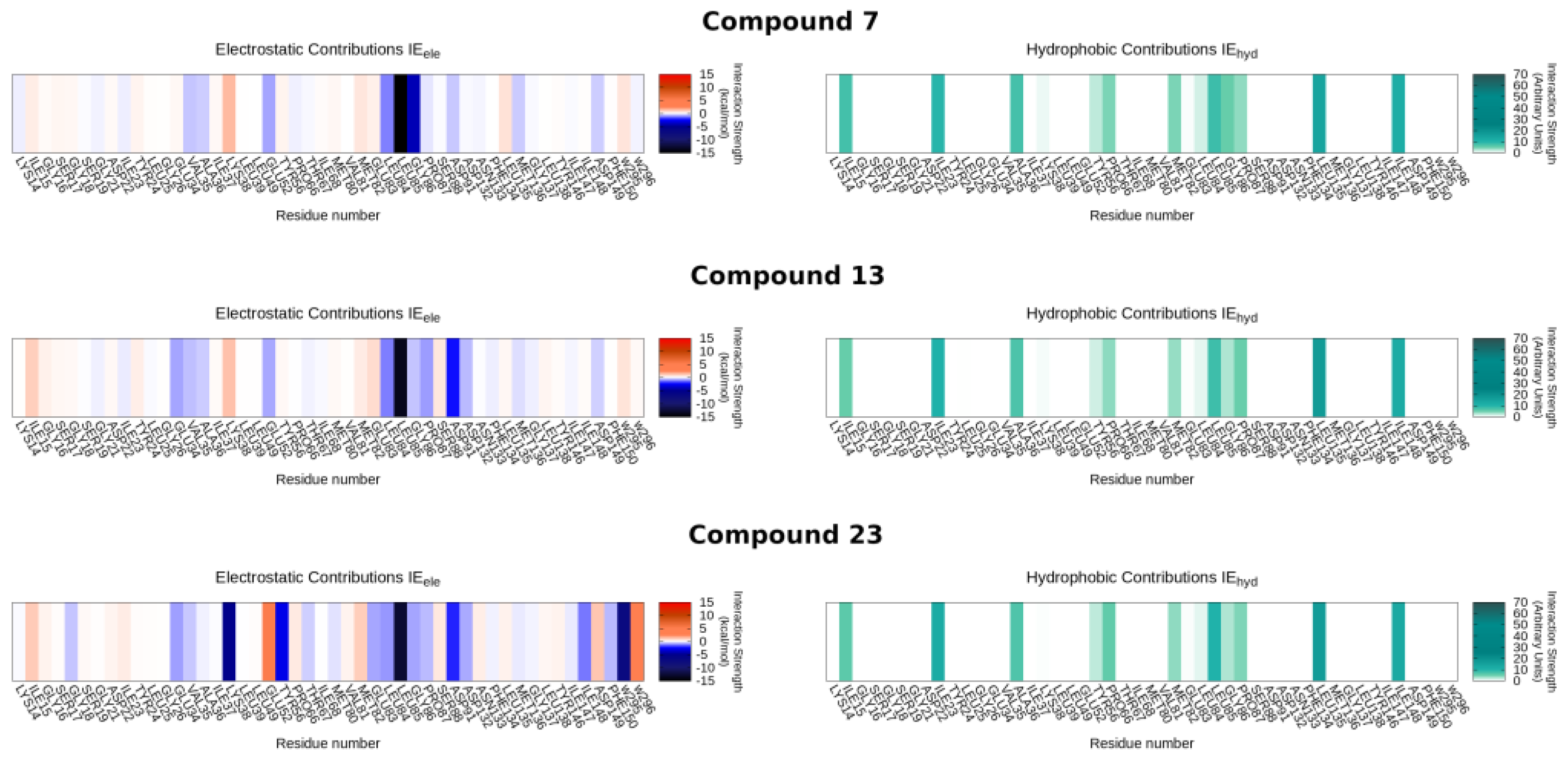
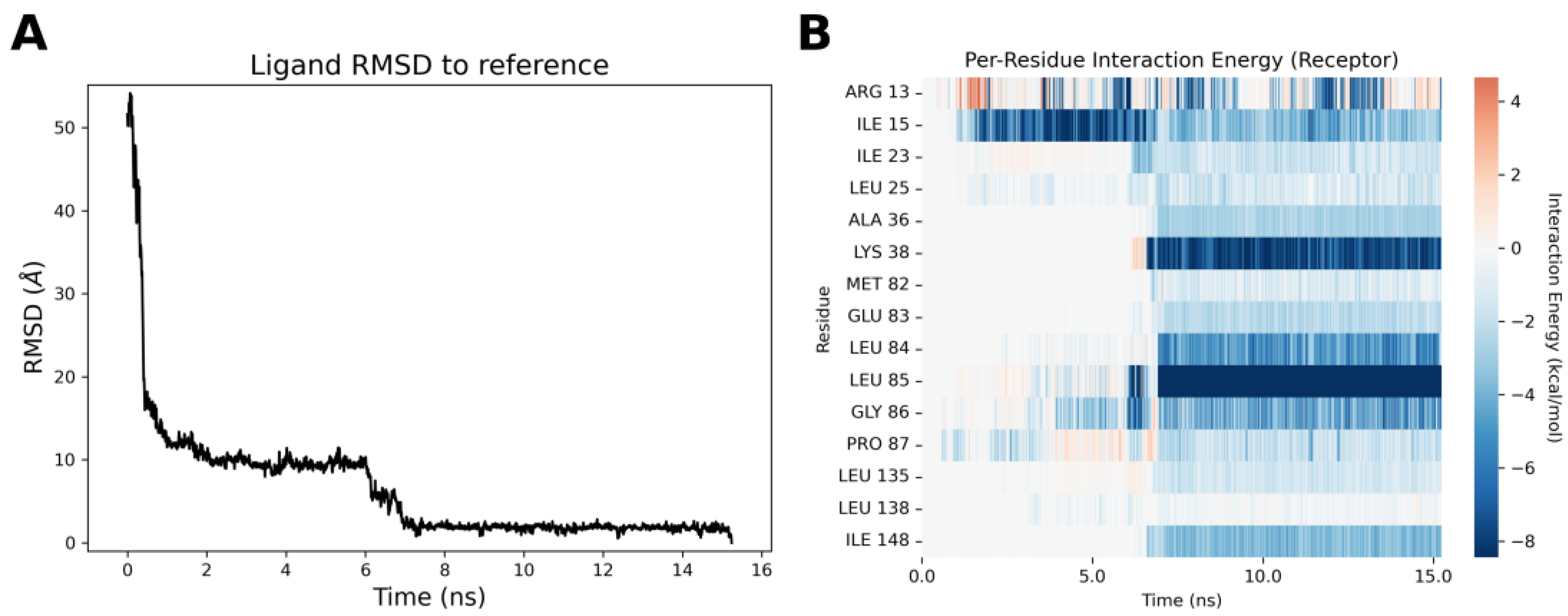
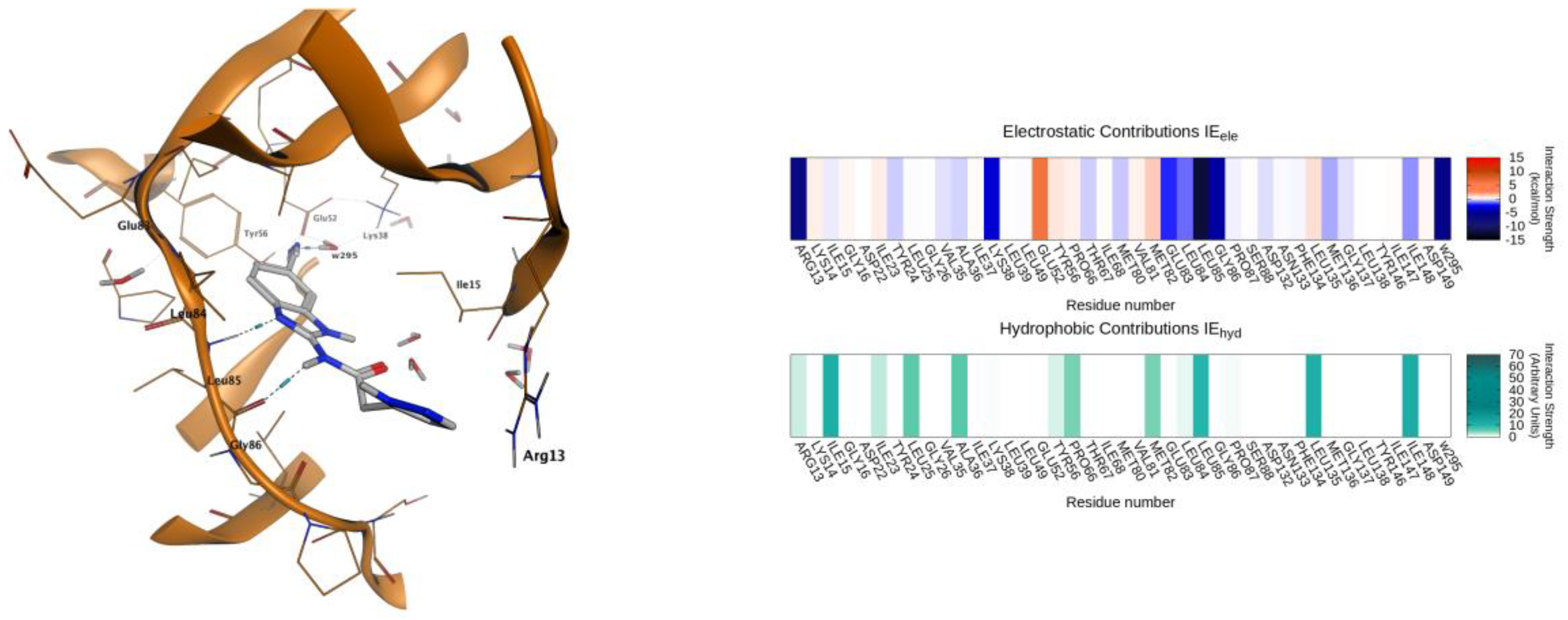
2.4. Molecular Modeling Studies
3. Materials and Methods
3.1. Chemistry
General Methods
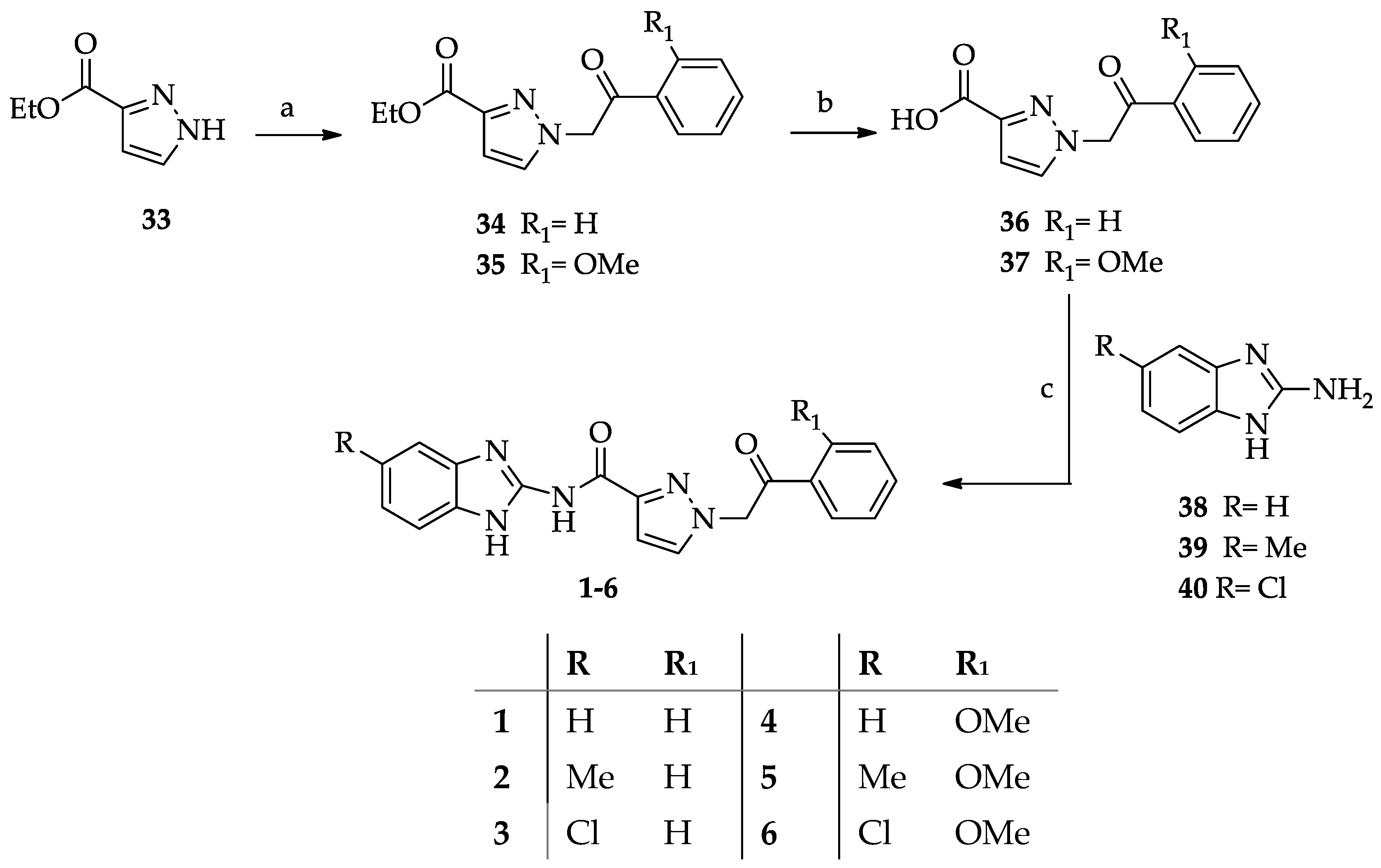
- General procedure for the synthesis of N-(1H-benzimidazol-2yl)-1-(2-arylethyl-2-oxo)-1H-pyrazole-3-carboxamides 1–6.
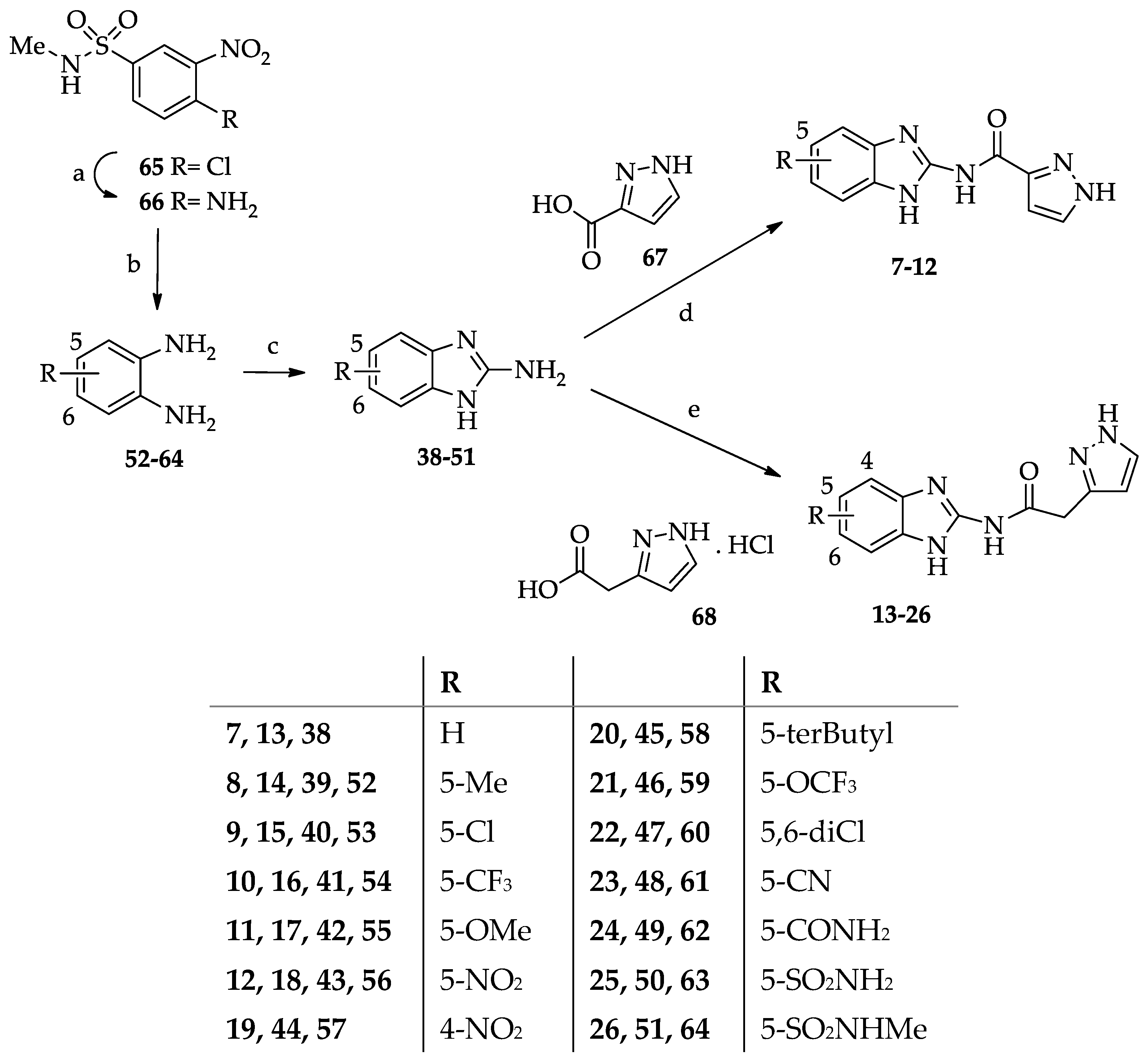
- General procedure for the synthesis of N-(benzimidazol-2-yl)pyrazole-3(5)-carboxamide derivatives 7–12.
- General procedure for the synthesis of N-(Benzimidazol-2-yl)(pyrazol-3(5)-yl)acetamide derivatives 13–26.
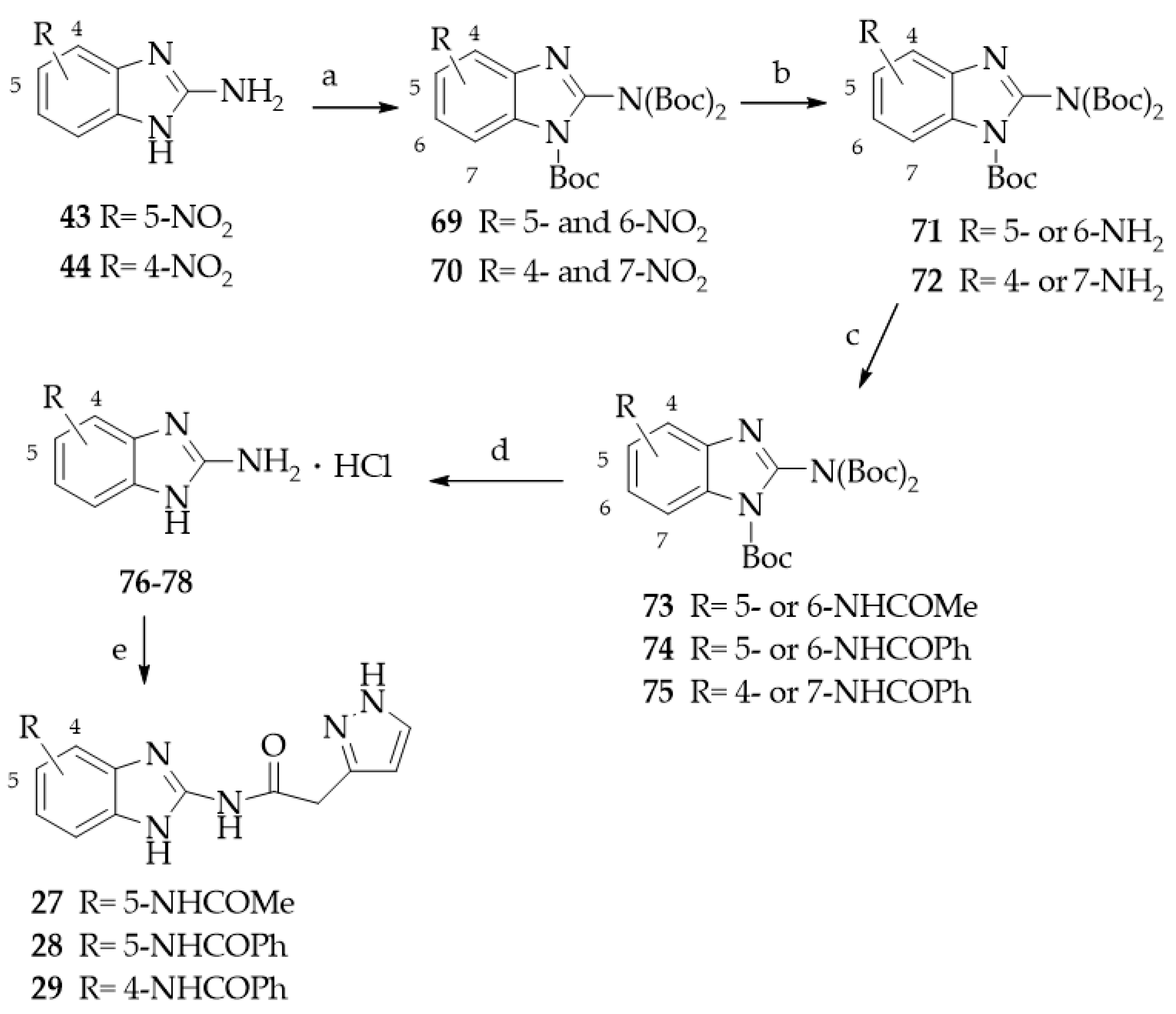
- General procedure for the synthesis of N-(Benzimidazol-2-yl)(pyrazol-3(5)-yl)acetamide derivatives 27–29.
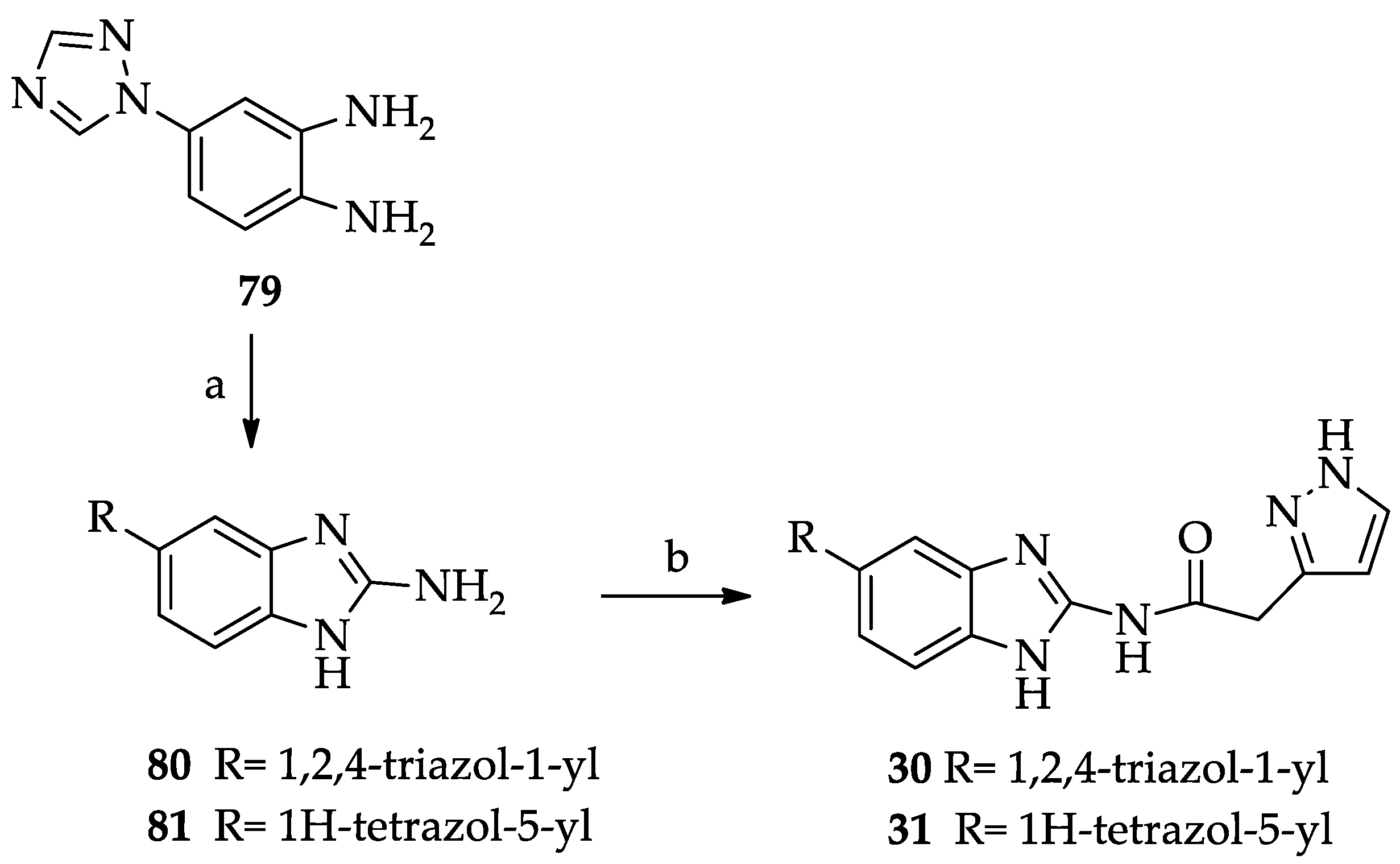
- General procedure for the synthesis of the 5(6)-heterocyclic-substituted N-(1H-benzimidazol-2-yl)(1H-pyrazol-3(5)-yl)acetamides 30 and 31.
- General procedure for the synthesis of ethyl 1-(2-aryl-2-oxoethyl)-1H-pyrazole-3-carboxylates (34–35).
- General procedure for the synthesis of 1-(2-aryl-2-oxoethyl)-1H-pyrazole-3-carboxylic acids 36–37.
- General procedure for the synthesis of 2-amino-1H-benzimidazole-5(7)-sulfonamide (50) and 2-amino-N-methyl-1H-benzimidazole-5(7)-sulfonamide (51).
- General procedure for the synthesis of nitro-substituted 1-tert-butoxycarbonyl-2-(N,N-bis-tert-butoxycarbonylamino)-benzimidazole derivatives 69 and 70.
- General procedure for the synthesis of amino-substituted 1-tert-Butoxycarbonyl-2-(N,N-bis-tert-butoxycarbonylamino)-benzimidazole derivatives 71 and 72.
- General procedure for the synthesis of 4- or 5-amido substituted 1-tert-Butoxycarbonyl-2-(N,N-bis-tert-butoxycarbonylamino)-benzimidazole derivatives 73–75.
- General procedure for the synthesis of 4- or 5-amido substituted 2-aminobenzimidazole hydrochlorides 76–78.
3.2. CK1δ Activity Assays
3.3. Molecular Modeling Studies
3.3.1. Hardware and Software Overview
3.3.2. Structure Preparation
3.3.3. Molecular Docking
3.3.4. Pharmacophoric Filter
3.3.5. Interaction Energy Fingerprints (IEF)
3.3.6. Docking Video Maker
3.3.7. System Setup for MD Simulations and Equilibration Protocol
3.3.8. Supervised Molecular Dynamics (SuMD) Simulations
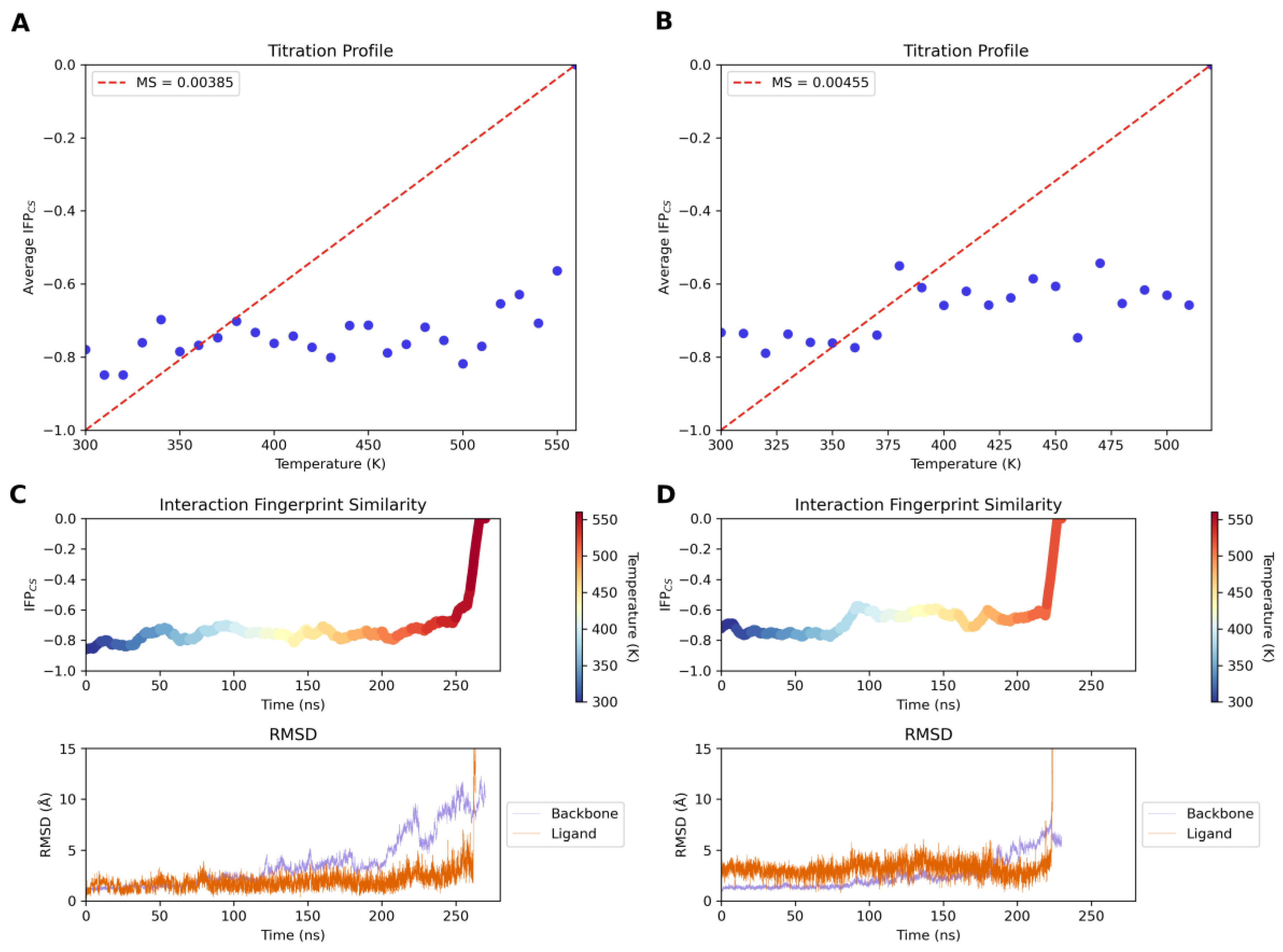
3.3.9. Thermal Titration Molecular Dynamics (TTMD) Simulations
3.3.10. Physicochemical and Pharmacokinetic Parameters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, P.; Ianes, C.; Gärtner, F.; Liu, C.; Burster, T.; Bakulev, V.; Rachidi, N.; Knippschild, U.; Bischof, J. Structure, regulation, and (patho-)physiological functions of the stress-induced protein kinase CK1 delta (CSNK1D). Gene 2019, 715, 144005. [Google Scholar] [CrossRef]
- Lohler, J.; Hirner, H.; Schmidt, B.; Kramer, K.; Fischer, D.; Thal, D.R.; Leithauser, F.; Knippschild, U. Immunohistochemical characterization of cell-type specific expression of CK1δelta in various tissues of young adult BALB/c mice. PLoS ONE 2009, 4, e4174. [Google Scholar] [CrossRef] [PubMed]
- Behrend, L.; Stoter, M.; Kurth, M.; Rutter, G.; Heukeshoven, J.; Deppert, W.; Knippschild, U. Interaction of casein kinase 1 delta (CK1δelta) with post-Golgi structures, microtubules, and the spindle apparatus. Eur. J. Cell. Biol. 2000, 79, 240–251. [Google Scholar] [CrossRef]
- Cunningham, P.S.; Ahern, S.A.; Smith, L.C.; da Silva Santos, C.S.; Wager, T.T.; Bechtold, D.A. Targeting of the circadian clock via CK1δ/ε to improve glucose homeostasis in obesity. Sci. Rep. 2016, 6, 29983. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.-M. Casein kinase 1 and Wnt/-catenin signaling. Curr. Opin. Cell. Biol. 2014, 31, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B.; Sinnberg, T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol. Cancer 2014, 13, 231. [Google Scholar] [CrossRef]
- Knippschild, U.; Krüger, M.; Richter, J.; Xu, P.; García-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. The CK1 family: Contribution to cellular stress response and its role in carcinogenesis. Front. Oncol. 2014, 4, 96. [Google Scholar] [CrossRef]
- Fulcher, L.J.; Sapkota, G.P. Functions and regulation of the serine/threonine protein kinase CK1 family: Moving beyond promiscuity. Biochem. J. 2020, 477, 4603–4621. [Google Scholar] [CrossRef]
- Richter, J.; Rudeck, S.; Kretz, A.L.; Kramer, K.; Just, S.; Henne-Bruns, D.; Hillenbrand, A.; Leithauser, F.; Lemke, J.; Knippschild, U. Decreased CK1δ expression predicts prolonged survival in colorectal cancer patients. Tumor Biol. 2016, 37, 8731–8739. [Google Scholar] [CrossRef]
- Eng, G.W.L.; Edison; Virshup, D.M. Site-specific phosphorylation of casein kinase 1 δ (CK1δ) regulates its activity towards the circadian regulator PER2. PLoS ONE 2017, 12, e0177834. [Google Scholar] [CrossRef]
- Yasojima, K.; Kuret, J.; DeMaggio, A.J.; McGeer, E.; McGeer, P.L. Casein kinase 1 delta mRNA is upregulated in Alzheimer disease brain. Brain Res. 2000, 865, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, H.; Kuret, J. Casein kinase 1 delta phosphorylates tau and disrupts its binding to microtubules. J. Biol. Chem. 2004, 279, 15938–15945. [Google Scholar] [CrossRef] [PubMed]
- Martínez-González, L.; Rodríguez-Cueto, C.; Cabezudo, D.; Bartolomé, F.; Andrés-Benito, P.; Ferrer, I.; Gil, C.; Martín-Requero, Á.; Fernández-Ruiz, J.; Martínez, A.; et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1 kinase inhibitor treatment. Sci. Rep. 2020, 10, 4449. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Huntley, M.L.; Perry, G.; Wang, X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018, 146, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, L.; Cuevas, E.P.; Tosat-Bitrián, C.; Nozal, V.; Gil, C.; Palomo, V.; Martín-Requero, Á.; Martinez, A. TTBK1 and CK1 inhibitors restore TDP-43 pathology and avoid disease propagation in lymphoblast from Alzheimer’s disease patients. Front. Mol. Neurosci. 2023, 16, 1243277. [Google Scholar] [CrossRef]
- Alquezar, C.; Salado, I.G.; de la Encarnacion, A.; Perez, D.I.; Moreno, F.; Gil, C.; de Munain, A.L.; Martinez, A.; Martin-Requero, A. Targeting TDP-43 phosphorylation by casein kinase-1δ inhibitors: A novel strategy for the treatment of frontotemporal dementia. Mol. Neurodegener. 2016, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, F.; Yung, K.K.L.; Zhang, S.; Qu, S. Effects of α-synuclein-associated post-translational modifications in Parkinson’s disease. ACS Chem. Neurosci. 2021, 12, 1061–1071. [Google Scholar] [CrossRef]
- Okochi, M.; Walter, J.; Koyama, A.; Nakajo, S.; Baba, M.; Iwatsubo, T.; Meijer, L.; Kahle, P.J.; Haass, C. Constitutive phosphorylation of the Parkinson’s disease-associated alpha-synuclein. J. Biol. Chem. 2000, 275, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Li, F.; Sonoustoun, B.; Kondru, N.C.; Yuka, A.; Martens, Y.A.; Qiao, W.; Heckman, M.G.; Ikezu, T.C.; Li, Z.; et al. APOE4 exacerbates α-synuclein seeding activity and contributes to neurotoxicity in Alzheimer’s disease with Lewy body pathology. Acta Neuropathol. 2022, 143, 641–662. [Google Scholar] [CrossRef]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef]
- Madsen, D.A.; Schmidt, S.I.; Blaabjerg, M.; Meyer, M. Interaction between parkin and α-synuclein in PARK2-mediated Parkinson’s disease. Cells 2021, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Catarzi, D.; Varano, F.; Vigiani, E.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Colotta, V. Casein kinase 1 delta inhibitors as promising therapeutic agents for neurodegenerative disorders. Curr. Med. Chem. 2022, 29, 4698–4737. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Calenda, S.; Vigiani, E.; Colotta, V. CK1 delta inhibition: An emerging strategy to combat neurodegenerative diseases. Future Med. Chem. 2022, 14, 1111–1113. [Google Scholar] [CrossRef]
- Badura, L.; Swanson, T.; Adamowicz, W.; Adams, J.; Cianfrogna, J.; Fisher, K.; Holland, J.; Kleiman, R.; Nelson, F.; Reynolds, L.; et al. An inhibitor of casein kinase Iε induces phase delays in circadian rhythms under free-running and entrained conditions. J. Pharmacol. Exp. Ther. 2007, 322, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Adler, P.; Mayne, J.; Wlaker, K.; Ning, Z.; Figeys, D.J. Therapeutic targeting of casein kinase 1δ/ε in an Alzheimer’s disease mouse model. J. Proteome Res. 2019, 18, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Long, A.; Zhao, H.; Huang, X. Structural basis for the interaction between casein kinase 1 delta and a potent and selective inhibitor. J. Med. Chem. 2012, 55, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Peifer, C.; Abadleh, M.; Bischof, J.; Hauser, D.; Schattel, V.; Hirner, H.; Knippschild, U.; Laufer, S. 3,4-Diaryl-isoxazoles and -imidazoles as potent dual inhibitors of p38alpha mitogen-activated protein kinase and casein kinase 1delta. J. Med. Chem. 2009, 52, 7618–7630. [Google Scholar] [CrossRef] [PubMed]
- Halekotte, J.; Witt, L.; Ianes, C.; Krüger, M.; Bührmann, M.; Rauh, D.; Pichlo, C.; Brunstein, E.; Luxenburger, A.; Baumann, U.; et al. Optimized 4,5-diarylimidazoles as potent/selective inhibitors of protein kinase CK1δ and their structural relation to p38α MAPK. Molecules 2017, 22, 522. [Google Scholar] [CrossRef] [PubMed]
- Bibian, M.; Rahaim, R.J.; Choi, J.Y.; Noguchi, Y.; Schürer, S.; Chen, W.; Nakanishi, S.; Licht, K.; Rosenberg, L.H.; Li, L.; et al. Development of highly selective casein kinase 1/1 (CK1) inhibitors with potent antiproliferative properties. Bioorg. Med. Chem. Lett. 2013, 23, 4374–4380. [Google Scholar] [CrossRef]
- Bischof, J.; Leban, J.; Zaja, M.; Grothey, A.; Radunsky, B.; Othersen, O.; Strobl, S.; Vitt, D.; Knippschild, U. 2-Benzamido-N-(1H-benzo[d]imidazol-2-yl)thiazole-4-carboxamide derivatives as potent inhibitors of CK1δelta/epsilon. Amino Acids 2012, 43, 1577–1591. [Google Scholar] [CrossRef]
- Richter, J.; Bischof, J.; Zaja, M.; Kohlhof, H.; Othersen, O.; Vitt, D.; Alscher, V.; Pospiech, I.; García-Reyes, B.; Berg, S.; et al. Difluoro-dioxolo-benzoimidazol-benzamides as potent inhibitors of CK1δ and ε with nanomolar inhibitory activity on cancer cell proliferation. J. Med. Chem. 2014, 57, 7933–7946. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.W.; Arnold, E.P.; Yang, X. Steric redirection of alkylation in 1H-pyrazole-3-carboxylate esters. Tetrahedron Lett. 2018, 59, 402–405. [Google Scholar] [CrossRef]
- Jones, R.G.; Mann, M.J. New methods of synthesis of S-aminoethylpyrazoles. J. Am. Chem. Soc. 1953, 75, 4048–4050. [Google Scholar] [CrossRef]
- Manuel, L.R.; Porter, C.C.; Kueh, F.A.; Jr Jensen, N.P.; Schmitt, S.M.; Windholz, T.B.; Beattie, T.R.; Carty, J.A.; Christensen, B.G.; Shen, T.Y. Inhibition of adrenal phenethanolamine /V-methyltransferase by substituted benzimidazoles. J. Med. Chem. 1970, 13, 1043–1047. [Google Scholar]
- Bella, M.; Milata, V.; Larina, L.I. 2-Amino-X-nitrobenzimidazoles as precursors of food-borne carcinogens: A new approach to IQ synthesis. J. Het. Chem. 2011, 49, 293–296. [Google Scholar] [CrossRef]
- Frei, R.; Breitbach, A.S.; Blackwell, H.E. 2-Aminobenzimidazole derivatives strongly inhibit and disperse pseudomonas aeruginosa biofilms. Angew. Chem. Int. Ed. 2012, 51, 5226–5229. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lau, F.; Liu, K.; Wood, H.B.; Zhou, G.; Chen, Y.; Li, Y.; Akiyama, T.E.; Castriota, G.; Einstein, M.; et al. Benzimidazolones: A new class of selective peroxisome proliferator-activated receptor γ (PPARγ) modulators. J. Med. Chem. 2011, 54, 8541–8554. [Google Scholar] [CrossRef] [PubMed]
- Arienti, K.L.; Brunmark, A.; Axe, F.U.; McClure, K.; Lee, A.; Blevitt, J.; Neff, D.K.; Huang, L.; Crawford, S.; Pandit, C.R.; et al. Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. J. Med. Chem. 2005, 48, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Püsküll, M.O.; Yıldız, S.; Göker, H. Synthesis and antistaphylococcal activity of N-substituted-1H-benzimidazole-sulphonamides. Arch. Pharm. Chem. Life Sci. 2010, 343, 31–39. [Google Scholar] [CrossRef]
- Hofmans, S.; Vanden Berghe, T.; Devisscher, L.; Hassannia, B.; Lyssens, S.; Joossens, J.; Van Der Veken, P.; Vandenabeele, P.; Augustyns, K. Novel ferroptosis inhibitors with improved potency and ADME properties. J. Med. Chem. 2016, 59, 2041–2053. [Google Scholar] [CrossRef]
- Kikuchi, K.; Hannak, R.B.; Guo, M.-J.; Kirbyd, A.J.; Hilvert, D. Toward bifunctional antibody catalysis. Bioorg. Med. Chem. 2006, 14, 6189–6196. [Google Scholar] [CrossRef] [PubMed]
- Huigens III, R.W.; Reyes, S.; Reed, C.S.; Bunders, C.; Rogers, S.A.; Steinhauer, A.T.; Melander, C. The chemical synthesis and antibiotic activity of a diverse library of 2-aminobenzimidazole small molecules against MRSA and multidrug-resistant A. baumannii. Bioorg. Med. Chem. 2010, 18, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Güngór, T.; Fouquet, A.; Teuton, J.-M.; Provost, D.; Gazes, M.; Cloarec, A. Cardiotonic agents. Synthesis and cardiovascular properties of novel 2-arylbenzimidazoles and azabenzimidazole. J. Med. Chem. 1992, 35, 4455–4463. [Google Scholar] [CrossRef] [PubMed]
- Duffin, G.R.; Lyddiatt, A.; Dolan, V.J.; Angus, K.L. Benzimidazole Compounds and Their Use as Chromatographic Ligands. EP1944311A1, 6 June 2012. [Google Scholar]
- Voskresensky, S.; Makosza, M. Selective one-pot N-monomethylation of 2-nitroanilines under Ptc conditions. Synth. Commun. 2000, 30, 3523–3526. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235. [Google Scholar] [CrossRef] [PubMed]
- Sciabola, S.; Benedetti, P.; D’Arrigo, G.; Torella, R.; Baroni, M.; Cruciani, G.; Spyrakis, F. Discovering new casein kinase 1δ inhibitors with an innovative molecular dynamics enabled virtual screening workflow. ACS Med. Chem. Lett. 2019, 10, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, C.; Jharia, P.; Waiker, D.K.; Nusbaum, A.C.; Amawi, H.; Kirwen, E.M.; Christman, R.; Arudra, S.K.C.; Meijer, L.; Tiwari, A.K.; et al. N-(1H-Pyrazol-3-Yl)quinazolin-4-amines as a novel class of casein kinase 1δ/ε inhibitors: Synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. Lett. 2017, 27, 2663–2667. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Gianoncelli, A.; Montopoli, M.; Caparrotta, L.; Venerando, A.; Meggio, F.; Pinna, L.A.; Zagotto, G.; Moro, S. Identification of novel protein kinase CK1 delta (CK1delta) inhibitors through structure-based virtual screening. Bioorg Med. Chem. Lett. 2008, 18, 5672–5675. [Google Scholar] [CrossRef] [PubMed]
- Grieco, I.; Bissaro, M.; Tiz, D.B.; Perez, D.I.; Perez, C.; Martinez, A.; Redenti, S.; Mariotto, E.; Bortolozzi, R.; Viola, G.; et al. Developing novel classes of protein kinase CK1δ inhibitors by fusing [1,2,4] triazole with different bicyclic heteroaromatic systems. Eur. J. Med. Chem. 2021, 216, 113331. [Google Scholar] [CrossRef]
- Sabbadin, D.; Salmaso, V.; Sturlese, M.; Moro, S. Supervised molecular dynamics (SuMD) approaches in drug design. Methods Mol. Biol. 2018, 1824, 287–298. [Google Scholar]
- Menin, S.; Pavan, M.; Salmaso, V.; Sturlese, M.; Moro, S. Thermal titration molecular dynamics (TTMD): Not your usual post-docking refinement. Int. J. Mol. Sci. 2023, 24, 3596. [Google Scholar] [CrossRef] [PubMed]
- Pavan, M.; Menin, S.; Bassani, D.; Sturlese, M.; Moro, S. Qualitative estimation of protein-ligand complex stability through thermal titration molecular dynamics simulations. J. Chem. Inf. Model. 2022, 62, 5715–5728. [Google Scholar] [CrossRef] [PubMed]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A.; et al. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755–2772. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE)|MOEsaic|PSILO. Available online: https://www.chemcomp.com/Products.htm (accessed on 7 August 2023).
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. Lect. Notes Comput. Sci. 2006, 4150, 247–258. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Molecular Modeling Software|OpenEye Scientific. Available online: https://www.eyesopen.com/ (accessed on 20 November 2023).
- Tautomers—Applications. Available online: https://docs.eyesopen.com/applications/quacpac/tautomers/tautomers.html (accessed on 7 August 2023).
- OMEGA 4.2.2.0—Applications. Available online: https://docs.eyesopen.com/applications/omega/index.html (accessed on 7 August 2023).
- FixpKa—Applications. Available online: https://docs.eyesopen.com/applications/quacpac/fixpka/fixpka.html (accessed on 7 August 2023).
- MolCharge—Applications. Available online: https://docs.eyesopen.com/applications/quacpac/molcharge/molcharge.html (accessed on 7 August 2023).
- Seidel, T.; Schuetz, D.A.; Garon, A.; Langer, T. The Pharmacophore Concept and Its Applications in Computer-Aided Drug Design. Prog. Chem. Org. Nat. Prod. 2019, 110, 99–141. [Google Scholar] [CrossRef] [PubMed]
- MPlayer—The Movie Player. Available online: http://www.mplayerhq.hu/design7/news.html (accessed on 14 November 2023).
- Gnuplot Homepage. Available online: http://www.gnuplot.info/index.html (accessed on 14 November 2023).
- RDKit. Available online: http://www.rdkit.org/ (accessed on 14 November 2023).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Case, D.A.; Walker, R.C.; Cheatham, T.E., III; Simmerling, C.; Roitberg, A.; Merz, K.M.; Luo, R.; Li, P.; Darden, T.; Sagui, C.; et al. Amber 2022 Reference Manual. Available online: https://ambermd.org/doc12/Amber22.pdf (accessed on 27 March 2024).
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the accuracy of protein side chain and backbone parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Kholmurodov, K.; Smith, W.; Yasuoka, K.; Darden, T.; Ebisuzaki, T.A. Smooth-particle mesh Ewald Method for DL_POLY Molecular Dynamics Simulation Package on the Fujitsu VPP700. J. Comput. Chem. 2000, 21, 1187–1191. [Google Scholar] [CrossRef]
- Faller, R.; De Pablo, J.J. Constant pressure hybrid molecular dynamics–Monte Carlo simulations. J. Chem. Phys. 2002, 116, 55–59. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Pavan, M.; Menin, S.; Bassani, D.; Sturlese, M.; Moro, S. Implementing a scoring function based on interaction fingerprint for Autogrow4: Protein Kinase CK1δ as a case study. Front. Mol. Biosci. 2022, 9, 909499. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A New Open-Source Player in the Drug Discovery Field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-1: QikProp; Schrödinger, LLC: New York, NY, USA, 2021.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| R | R | IC50 (μM) a | |
| 1 | H | CH2COC6H5 | >40 |
| 2 | Me | CH2COC6H5 | 14.6 ± 3.5 |
| 3 | Cl | CH2COC6H5 | 1.80 ± 0.50 |
| 4 | H | CH2COC6H4-2-OMe | >40 |
| 5 | Me | CH2COC6H4-2-OMe | >40 |
| 6 | Cl | CH2COC6H4-2-OMe | 13.2 ± 4.7 |
| 7 | H | H | >40 |
| 8 | Me | H | >40 |
| 9 | Cl | H | >40 |
| 10 | CF3 | H | >40 |
| 11 | OMe | H | >40 |
| 12 | NO2 | H | >40 |
 | ||
|---|---|---|
| R | IC50 (μM) a | |
| 13 | H | 4.21 ± 1.92 |
| 14 | 5-Me | 1.64 ± 0.31 |
| 15 | 5-Cl | 0.485 ± 0.310 |
| 16 | 5-CF3 | 1.74 ± 0.32 |
| 17 | 5-OMe | 6.27 ± 0.95 |
| 18 | 5-NO2 | 0.12 ± 0.083 |
| 19 | 4-NO2 | 1.22 ± 0.23 |
| 20 | 5-terBu | 1.00 ± 0.18 |
| 21 | 5-OCF3 | 5.99 ± 0.87 |
| 22 | 5,6-diCl | 0.98 ± 0.35 |
| 23 | 5-CN | 0.0986 ± 0.0394 |
| 24 | 5-CONH2 | 2.53 ± 0.38 |
| 25 | 5-SO2NH2 | >40 |
| 26 | 5-SO2NHMe | 10.7 ± 2.3 |
| 27 | 5-NHCOMe | >40 |
| 28 | 5-NHCOPh | >40 |
| 29 | 4-NHCOPh | >40 |
| 30 | 5-(1,2,4-triazol-1-yl) | 2.59 ± 0.5 |
| 31 | 5-(tetrazol-5-yl) | 1.54 ± 0.29 |
| 32 | - | 20.1 ± 7.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calenda, S.; Catarzi, D.; Varano, F.; Vigiani, E.; Volpini, R.; Lambertucci, C.; Spinaci, A.; Trevisan, L.; Grieco, I.; Federico, S.; et al. Structural Investigations on 2-Amidobenzimidazole Derivatives as New Inhibitors of Protein Kinase CK1 Delta. Pharmaceuticals 2024, 17, 468. https://doi.org/10.3390/ph17040468
Calenda S, Catarzi D, Varano F, Vigiani E, Volpini R, Lambertucci C, Spinaci A, Trevisan L, Grieco I, Federico S, et al. Structural Investigations on 2-Amidobenzimidazole Derivatives as New Inhibitors of Protein Kinase CK1 Delta. Pharmaceuticals. 2024; 17(4):468. https://doi.org/10.3390/ph17040468
Chicago/Turabian StyleCalenda, Sara, Daniela Catarzi, Flavia Varano, Erica Vigiani, Rosaria Volpini, Catia Lambertucci, Andrea Spinaci, Letizia Trevisan, Ilenia Grieco, Stephanie Federico, and et al. 2024. "Structural Investigations on 2-Amidobenzimidazole Derivatives as New Inhibitors of Protein Kinase CK1 Delta" Pharmaceuticals 17, no. 4: 468. https://doi.org/10.3390/ph17040468