
Dual Antitubercular and Antileishmanial Profiles of Quinoxaline Di-N-Oxides Containing an Amino Acidic Side Chain

 ,
,  , ,
, , 
Abstract
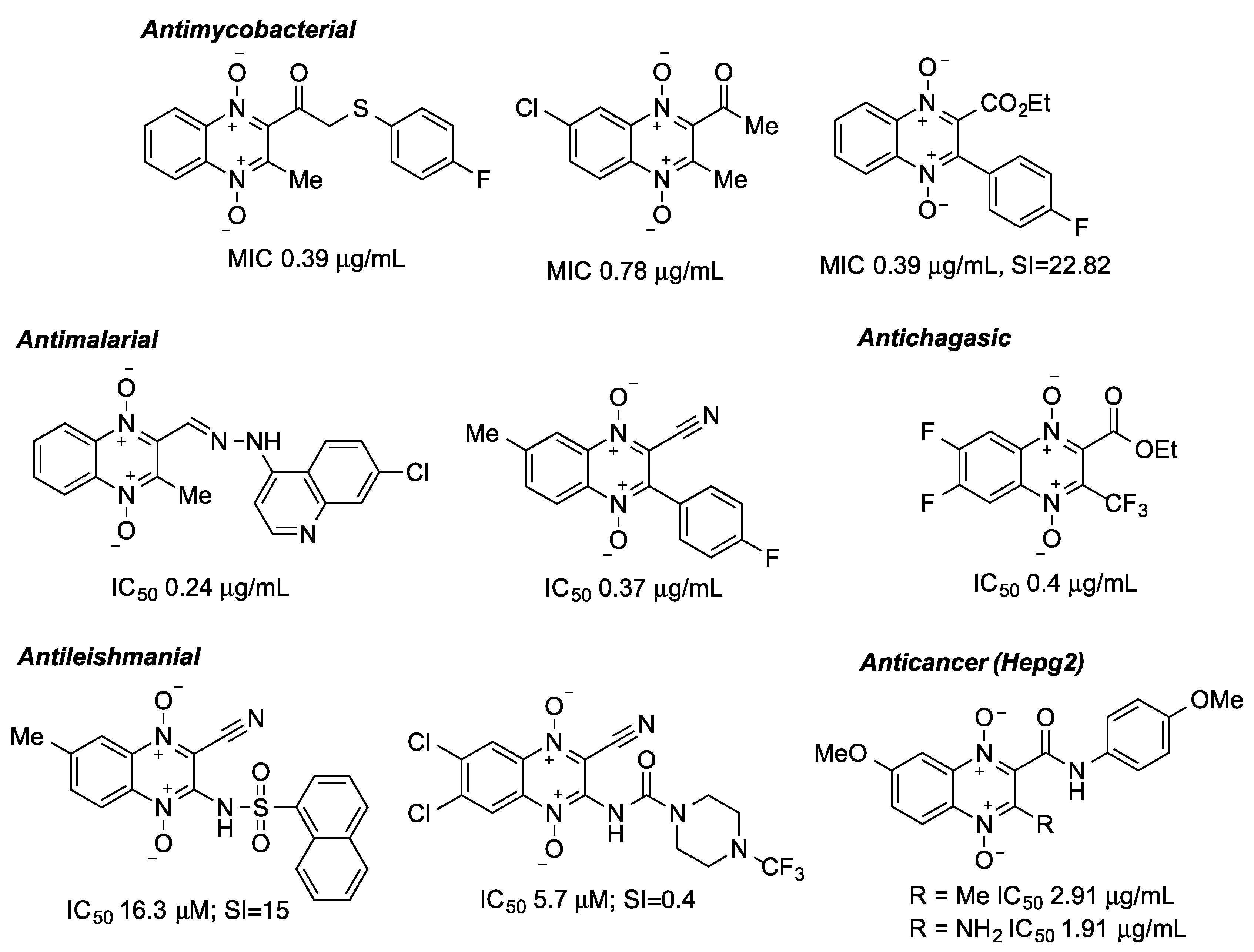
1. Introduction
2. Results
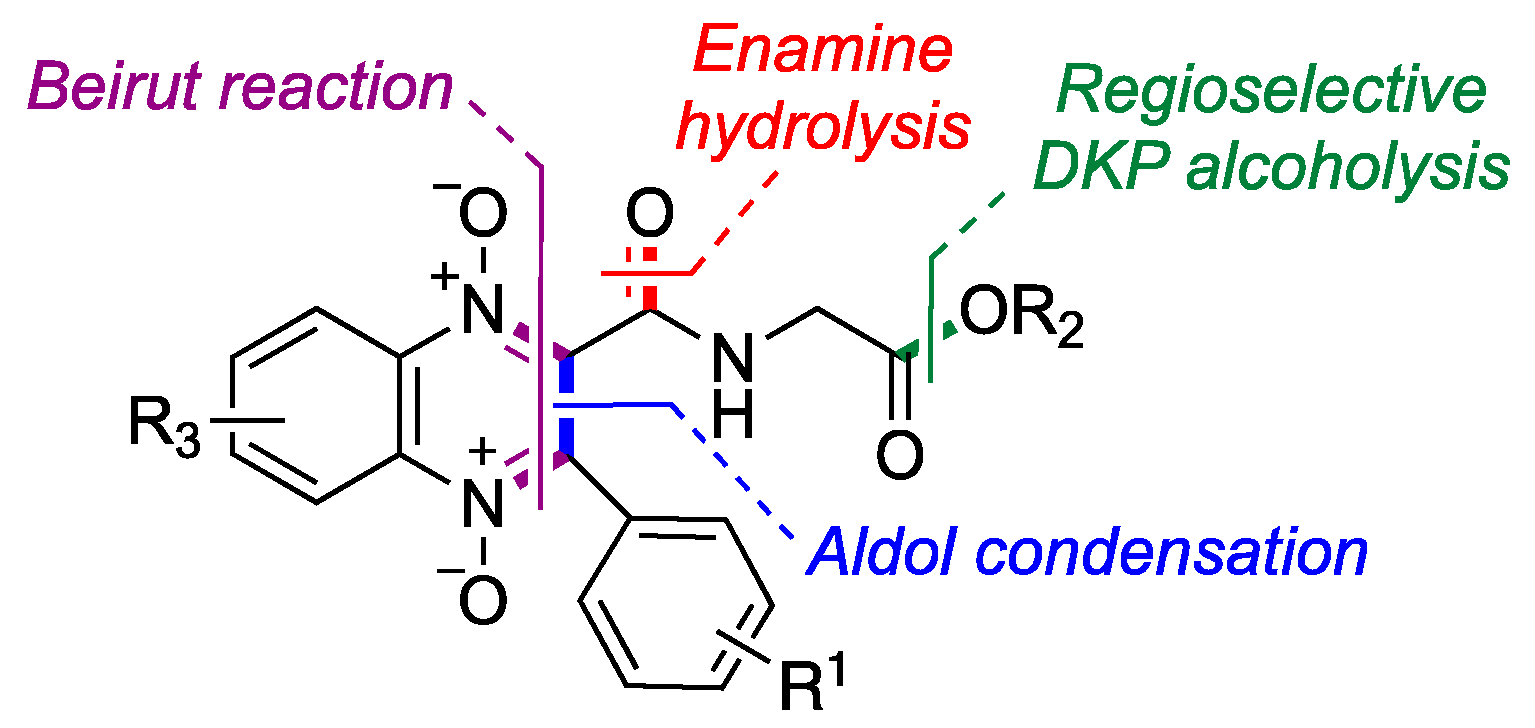
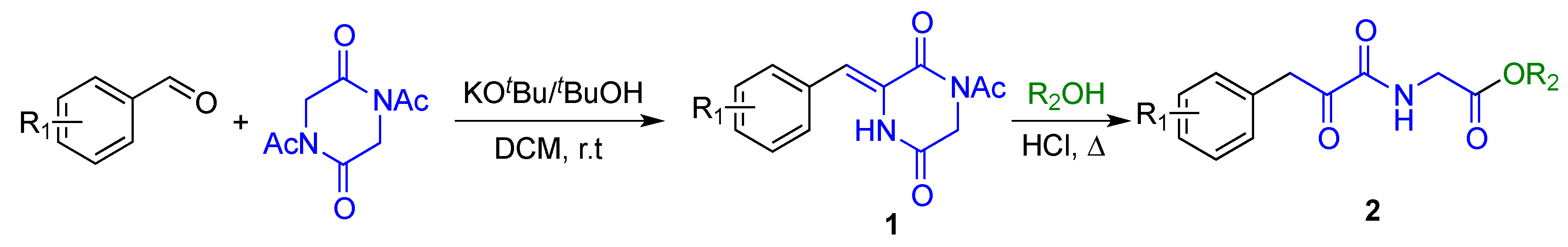
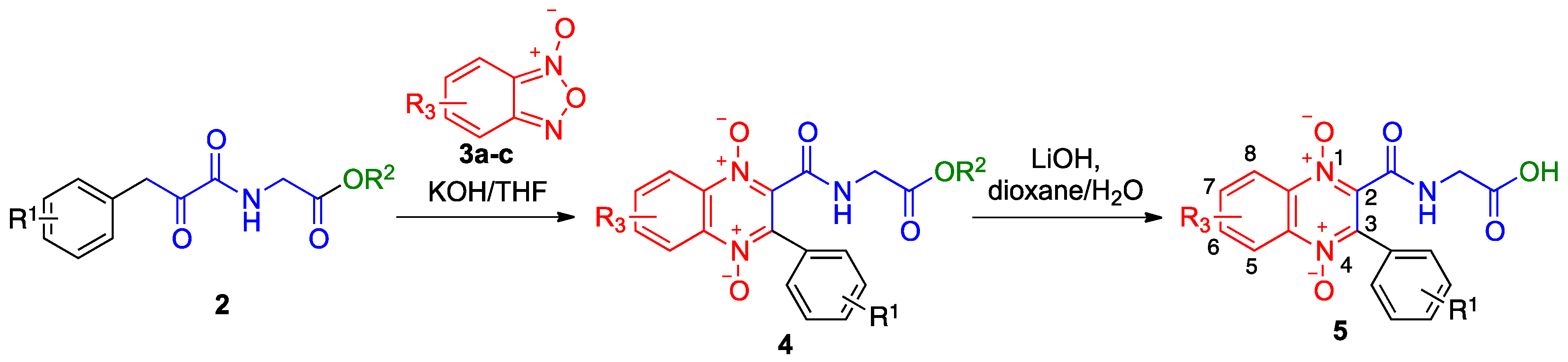
2.1. Synthesis
2.2. Biology
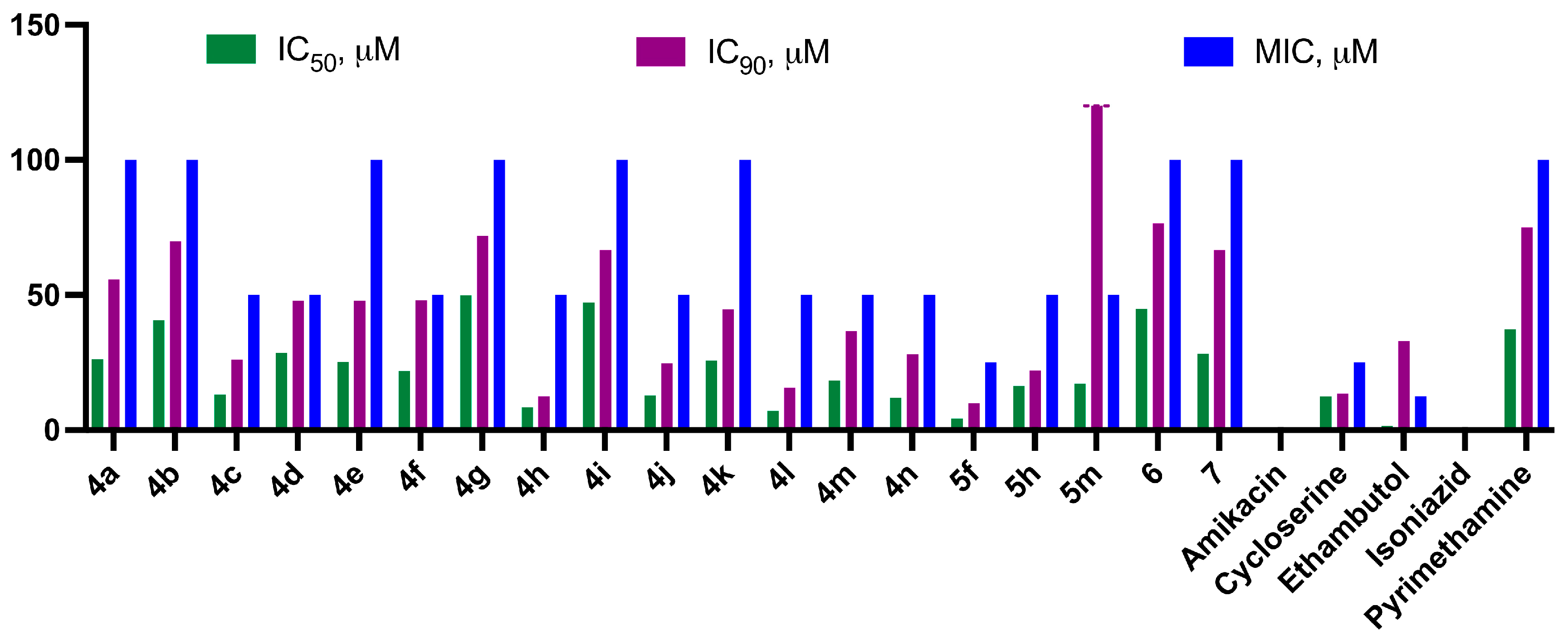
2.2.1. Antituberculosis Activity
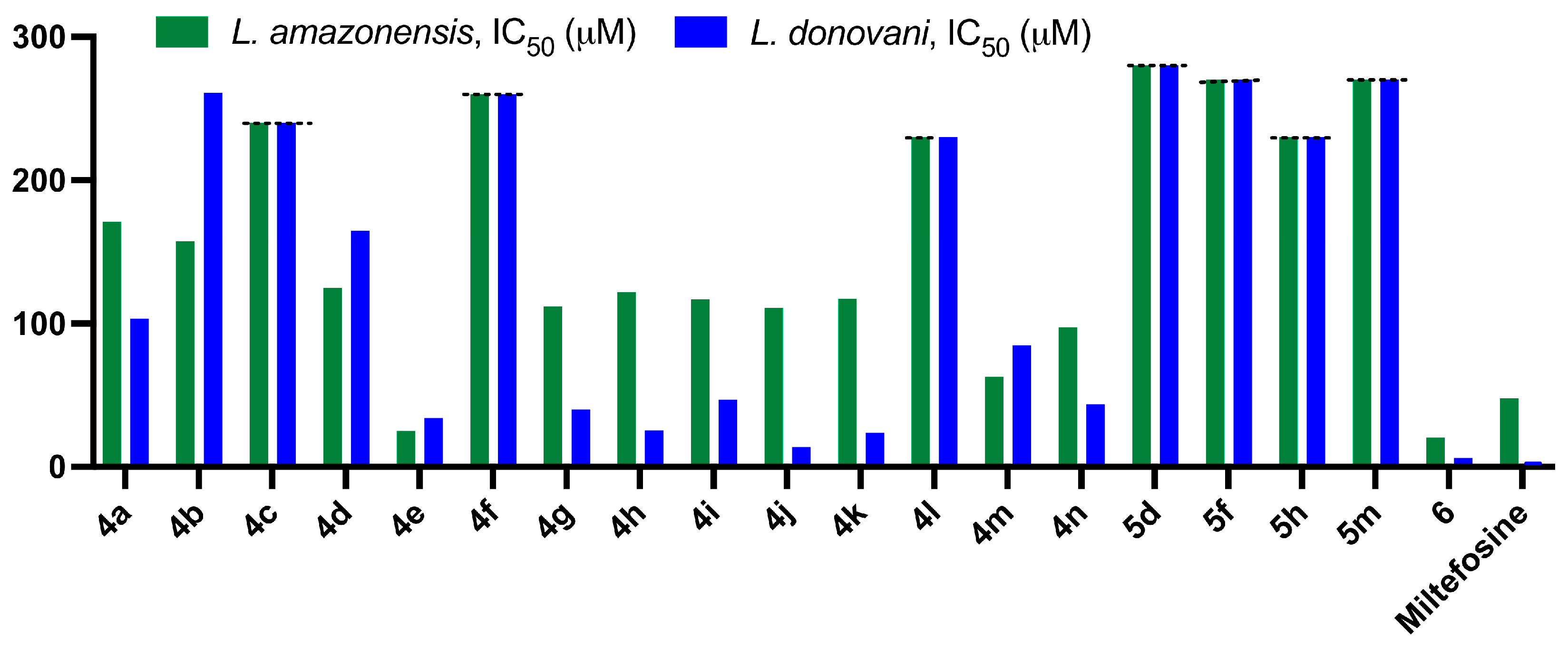
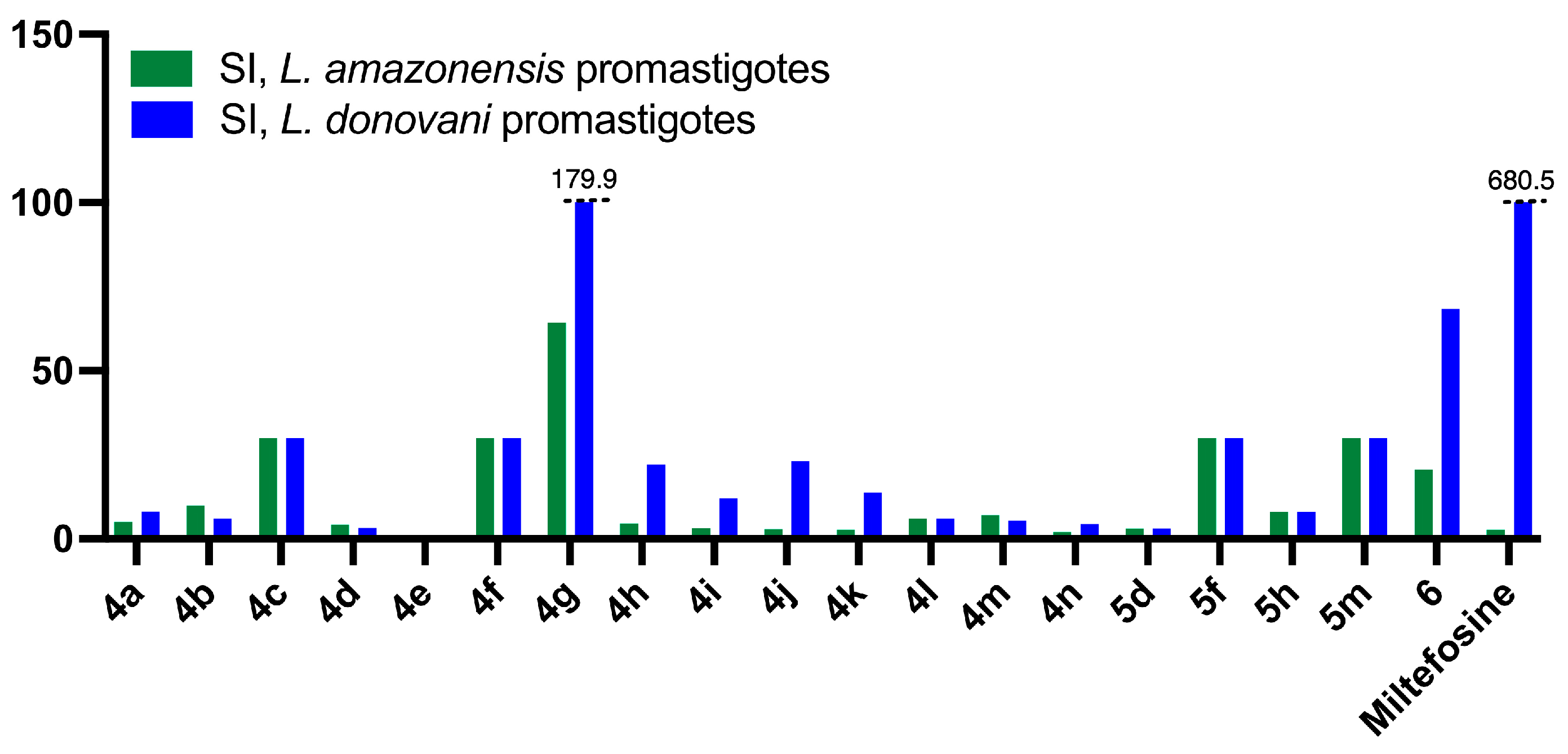
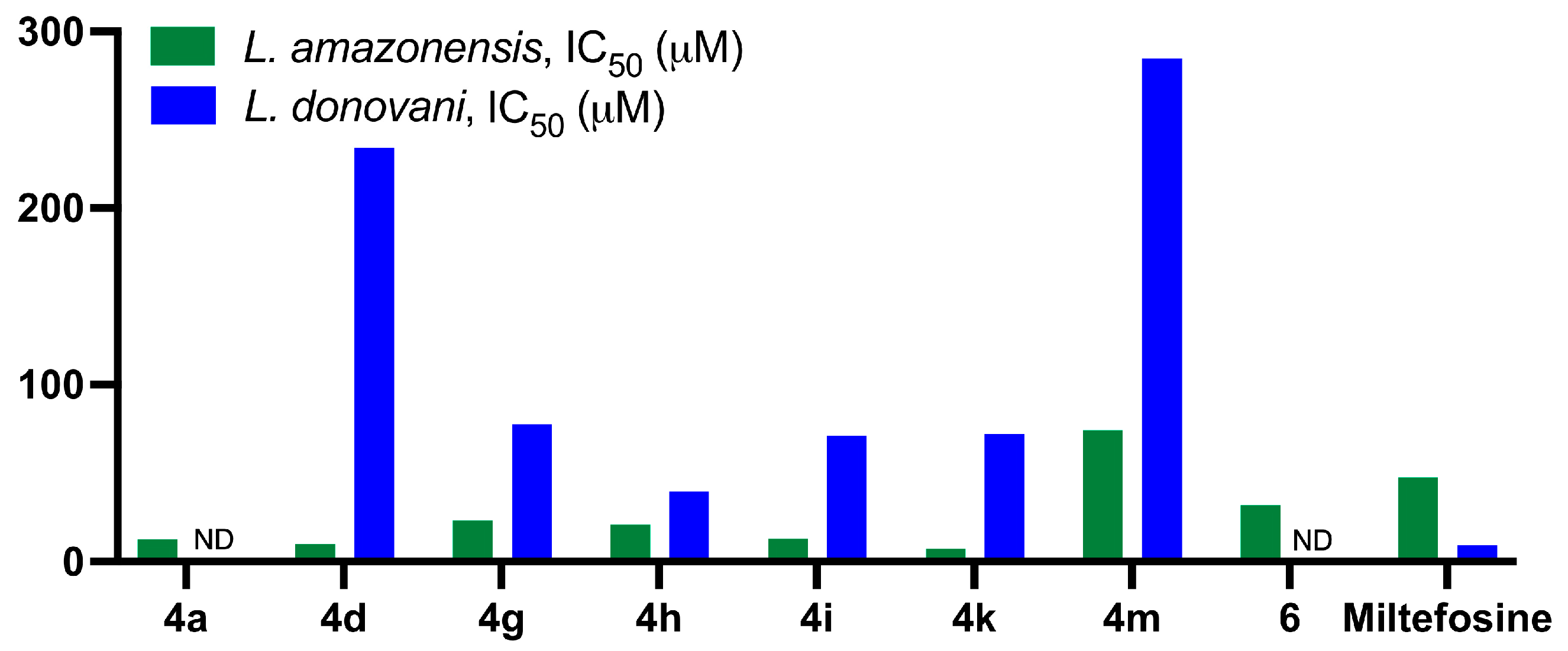
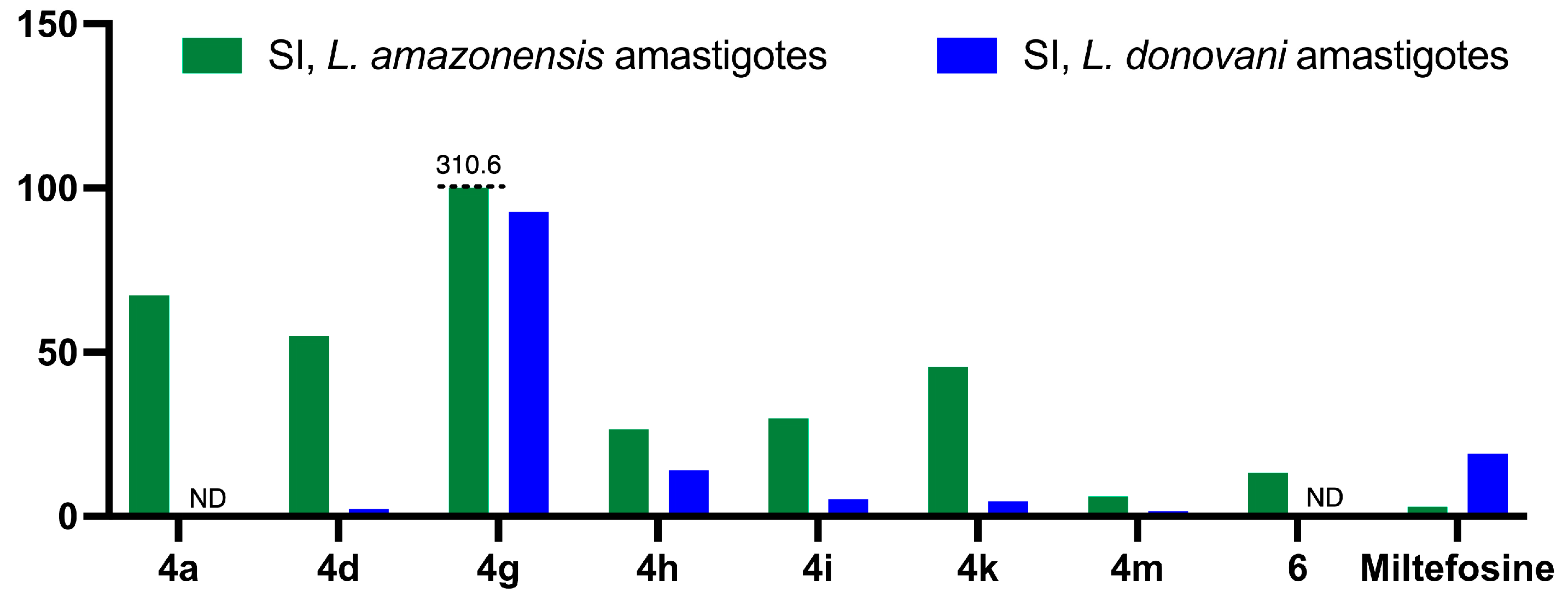
2.2.2. Antileishmanial Activity
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.1.1. General Synthetic Experimental Information
4.1.2. General Procedure for the Synthesis of 3-Arylmethylene-2,5-Piperazinediones (1)
4.1.3. General Procedure for the Synthesis of Alkyl 2-(2-Oxo-3-Arylpropanamido)Acetate Derivatives (2)
4.1.4. General Procedure for Preparation of Benzofurazane Oxides 3a–c
4.1.5. General Procedure for Preparation of 3-arylquinoxaline-1,4-di-N-oxides 4
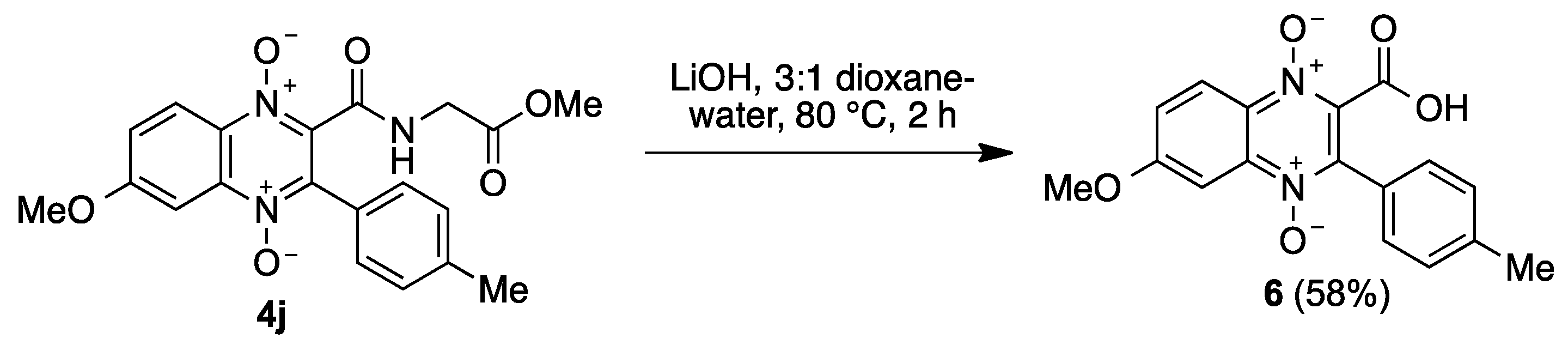
4.1.6. General Procedure for the Preparation of N-(1,4-Dioxide-3-Arylquinoxaline-2-Carbonyl)Glycine Derivatives 5
4.2. In Vitro Activity against Mycobacterium tuberculosis
4.2.1. First Protocol for Activity Measurement
4.2.2. Second Protocol for Activity Measurement
4.2.3. Autofluorescence
4.3. In Vitro Assays for Leishmanicidal Activity
4.3.1. Parasites and Culture Procedure
4.3.2. J774 Cell Cultivation
4.3.3. Promastigote Susceptibility Assay
4.3.4. Intracellular Amastigote Assay
4.3.5. Cytotoxicity Assay on Macrophages
4.3.6. Selectivity Index Calculations
5. Conclusions
- The presence of a peptide chain at C-2 improves the antitubercular activity compared to the presence of a carboxylic acid.
- A higher state of oxidation of the nitrogen atoms of the heterocyclic skeleton increases its activity.
- The presence of substituents in the benzene rings improves the activity of the compounds.
- Substituents at the para position of the aromatic side arm are more favorable than their ortho and meta analogues.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waldman, A.J.; Balskus, E.P. The human microbiota, infectious disease, and global health: Challenges and opportunities. ACS Infec. Dis. 2018, 4, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Aagaard-Hansen, J.; Nombela, N.; Alvar, J. Population movement: A key factor in the epidemiology of neglected tropical diseases. Trop. Med. Int. Health 2010, 15, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Georgiadou, S.P.; Makaritsis, K.P.; Dalekos, G.N. Leishmaniasis revisited: Current aspects on epidemiology, diagnosis and treatment. J. Transl. Intern. Med. 2015, 3, 43–50. [Google Scholar] [CrossRef]
- Mann, S.; Frasca, K.; Scherrer, S.; Henao-Martínez, A.F.; Newman, S.; Ramanan, P.; Suárez, J.Á. A Review of leishmaniasis: Current knowledge and future directions. Curr. Trop. Med. Rep. 2021, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Dinc, R. Leishmania vaccines: The current situation with its promising aspect for the future. Korean J. Parasitol. 2022, 60, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Rossi, M.; Fasel, N. The criminal association of Leishmania parasites and viruses. Curr. Opin. Microbiol. 2018, 46, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Abongomera, C.; Ritmeijer, K.; Vogt, F.; Buyze, J.; Mekonnen, Z.; Admassu, H.; Colebunders, R.; Mohammed, R.; Lynen, L.; Diro, E.; et al. Development and external validation of a clinical prognostic score for death in visceral leishmaniasis patients in a high HIV co-infection burden area in Ethiopia. PLoS ONE 2017, 12, e0178996. [Google Scholar] [CrossRef] [PubMed]
- Agusto, F.B.; Elmojtaba, I.M. Optimal control and cost-effective analysis of malaria/visceral leishmaniasis co-infection. PLoS ONE 2017, 12, e0171102. [Google Scholar] [CrossRef]
- Li, X.-X.; Zhou, X.-N. Co-infection of tuberculosis and parasitic diseases in humans: A systematic review. Parasit. Vectors 2013, 6, 79. [Google Scholar] [CrossRef]
- Desjeux, P. The increase in risk factors for leishmaniasis worldwide. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 239–243. [Google Scholar] [CrossRef]
- Karimi, A.; Amanati, A.; Mansour Ghanaie, R.; Mohajerzadeh, L. Co-infection of Leishmania and tuberculosis. Arch. Pediatr. Infect. Dis. 2013, 2, 183–187. [Google Scholar] [CrossRef]
- Shweta; Bhatnagar, S.; Gupta, A.K.; Murti, K.; Pandey, K. Co-infection of visceral leishmaniasis and pulmonary tuberculosis: A case study. Asian Pac. J. Trop. Dis. 2014, 4, 57–60. [Google Scholar] [CrossRef]
- Egbelowo, O.F.; Munyakazi, J.B.; Dlamini, P.G.; Osaye, F.J.; Simelane, S.M. Modeling visceral leishmaniasis and tuberculosis co-infection dynamics. Front. Appl. Math. Stat. 2023, 9, 1153666. [Google Scholar] [CrossRef]
- Makhoba, X.H.; Viegas, C.; Mosa, R.A.; Viegas, F.P.D.; Pooe, O.J. Potential impact of the multi-target drug approach in the treatment of some complex diseases. Drug Des. Devel. Ther. 2020, 14, 3235–3249. [Google Scholar] [CrossRef]
- Zhao, H.; Dietrich, J. Privileged scaffolds in lead generation. Expert Opin. Drug Discov. 2015, 10, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Schneider, G. Privileged structures revisited. Angew. Chem. Int. Ed. 2017, 56, 7971–7974. [Google Scholar] [CrossRef]
- Ancizu, S.; Moreno, E.; Torres, E.; Burguete, A.; Pérez-Silanes, S.; Benítez, D.; Villar, R.; Solano, B.; Marín, A.; Aldana, I.; et al. Heterocyclic-2-carboxylic acid (3-cyano-1,4-di-N-oxidequinoxalin-2-yl)amide derivatives as hits for the development of neglected disease drugs. Molecules 2009, 14, 2256–2272. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Sa, W.; Cao, C.; Guo, L.; Hao, H.; Liu, Z.; Wang, X.; Yuan, Z. Quinoxaline 1,4-di-N-oxides: Biological activities and mechanisms of actions. Front. Pharmacol. 2016, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Rivera, G. Quinoxaline 1,4-di-N-oxide derivatives: Are they unselective or selective inhibitors? Mini-Rev. Med. Chem. 2022, 22, 15–25. [Google Scholar] [CrossRef]
- Buravchenko, G.I.; Shchekotikhin, A.E. Quinoxaline 1,4-dioxides: Advances in chemistry and chemotherapeutic drug development. Pharmaceuticals 2023, 16, 1174. [Google Scholar] [CrossRef] [PubMed]
- Vicente, E.; Villar, R.; Pérez, S.; Aldana, I.; Goldman, R.; Monge, A. Quinoxaline 1,4-di-N-oxide and the potential for treating tuberculosis. Infect. Disord. Drug Targets 2011, 11, 196–204. [Google Scholar] [CrossRef]
- Zhao, Y.; Cheng, G.; Hao, H.; Pan, Y.; Liu, Z.; Dai, M.; Yuan, Z. In vitro antimicrobial activities of animal-used quinoxaline 1,4-di-N-oxides against mycobacteria, mycoplasma and fungi. BMC Vet. Res. 2016, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-Mycobacterium tuberculosis agents. J. Med. Chem. 2005, 48, 2019–2025. [Google Scholar] [CrossRef]
- Wilhelmsson, L.M.; Kingi, N.; Bergman, J. Interactions of antiviral indolo[2,3-b]quinoxaline derivatives with DNA. J. Med. Chem. 2008, 51, 7744–7750. [Google Scholar] [CrossRef]
- Benítez, D.; Cabrera, M.; Hernández, P.; Boiani, L.; Lavaggi, M.L.; Di Maio, R.; Yaluff, G.; Serna, E.; Torres, S.; Ferreira, M.E.; et al. 3-Trifluoromethylquinoxaline N,N’-dioxides as anti-trypanosomatid agents. Identification of optimal anti-T. cruzi agents and mechanism of action studies. J. Med. Chem. 2011, 54, 3624–3636. [Google Scholar] [CrossRef]
- Torres, E.; Moreno-Viguri, E.; Galiano, S.; Devarapally, G.; Crawford, P.W.; Azqueta, A.; Arbillaga, L.; Varela, J.; Birriel, E.; Di Maio, R.; et al. Novel quinoxaline 1,4-di-N-oxide derivatives as new potential antichagasic agents. Eur. J. Med. Chem. 2013, 66, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Barea, C.; Pabón, A.; Castillo, D.; Zimic, M.; Quiliano, M.; Galiano, S.; Pérez-Silanes, S.; Monge, A.; Deharo, E.; Aldana, I. New salicylamide and sulfonamide derivatives of quinoxaline 1,4-di-N-oxide with antileishmanial and antimalarial activities. Bioorg. Med. Chem. Lett. 2011, 21, 4498–4502. [Google Scholar] [CrossRef] [PubMed]
- Gali-Muhtasib, H.U.; Haddadin, M.J.; Rahhal, D.N.; Younes, I.H. Quinoxaline 1,4-dioxides as anticancer and hypoxia-selective drugs. Oncol. Rep. 2001, 8, 679–684. [Google Scholar] [CrossRef]
- Silva, L.; Coelho, P.; Soares, R.; Prudêncio, C.; Vieira, M. Quinoxaline-1,4-dioxide derivatives inhibitory action in melanoma and brain tumor cells. Future Med. Chem. 2019, 11, 645–657. [Google Scholar] [CrossRef]
- González, J.F.; Ortín, I.; de la Cuesta, E.; Menéndez, J.C. Privileged scaffolds in synthesis: 2,5-piperazinediones as templates for the preparation of structurally diverse heterocycles. Chem. Soc. Rev. 2012, 41, 6902–6915. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Ferreira, A.; Matos, J.; Fresco, P.; Gouveia, M.J. Amino acids in the development of prodrugs. Molecules 2018, 23, 2318. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Stevenazzi, A.; Marchini, M.; Sandrone, G.; Vergani, B.; Lattanzio, M. Amino acidic scaffolds bearing unnatural side chains: An old idea generates new and versatile tools for the life sciences. Bioorg. Med. Chem. Lett. 2014, 24, 5349–5356. [Google Scholar] [CrossRef] [PubMed]
- Niederweis, M. Nutrient acquisition by mycobacteria. Microbiology 2008, 154, 679–692. [Google Scholar] [CrossRef]
- Borthwick, A.D. 2,5-Diketopiperazines: Synthesis, reactions, medicinal chemistry, and bioactive natural products. Chem. Rev. 2012, 112, 3641–3716. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, J.D.; González, J.F.; Menéndez, J.C. Diketopiperazines as chiral building blocks. In Chiral Building Blocks in Modern Stereoselective Synthesis; Wojaczynska, E., Wojaczynski, J., Eds.; Wiley VCH: Hoboken, NJ, USA, 2022; Chapter 9; pp. 139–160. [Google Scholar]
- González, J.F.; de la Cuesta, E.; Avendaño, C. Atom-efficient synthesis of 2,6-diazacyclophane compounds through alcoholysis/reduction of 3-nitroarylmethylene-2,5-piperazinediones. Tetrahedron 2008, 64, 2762–2771. [Google Scholar] [CrossRef]
- Bull, S.D.; Davies, S.G.; Garner, A.C.; O’Shea, M.D. Conjugate additions of organocuprates to a 3-methylene-6-isopropyldiketopiperazine acceptor for the asymmetric synthesis of homochiral α-amino acids. J. Chem. Soc. Perkin Trans. 2001, 1, 3281–3287. [Google Scholar] [CrossRef]
- Farran, D.; Echalier, D.; Martinez, J.; Dewynter, G. Regioselective and sequential reactivity of activated 2,5-diketopiperazines. J. Pept. Sci. 2009, 15, 474–478. [Google Scholar] [CrossRef]
- González, J.F.; de la Cuesta, E.; Avendaño, C. Improvements in aldol reactions with diketopiperazines. Synth. Commun. 2004, 34, 1589–1597. [Google Scholar] [CrossRef]
- González, J.F.; de la Cuesta, E.; Avendaño, C. From cyclic dehydrodipeptides to uncommon acyclic peptide mimetics. Tetrahedron Lett. 2006, 47, 6711–6714. [Google Scholar] [CrossRef]
- Haddadin, M.; Issidorides, C.H. The Beirut reaction. Heterocycles 1993, 35, 1503–1525. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.; Amaral, D.N. Beirut reaction and its application in the synthesis of quinoxaline-N,N’-dioxides bioactive compounds. Rev. Virtual Quím. 2013, 5, 1075–1100. [Google Scholar] [CrossRef]
- Vicente, E.; Lima, L.M.; Bongard, E.; Charnaud, S.; Villar, R.; Solano, B.; Burguete, A.; Pérez-Silanes, S.; Aldana, I.; Vivas, L.; et al. Synthesis and structure-activity relationship of 3-phenylquinoxaline 1,4-di-N-oxide derivatives as antimalarial agents. Eur. J. Med. Chem. 2008, 43, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.; Franzblau, S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, M.; Inocêncio da Luz, R.; Toté, K.; Timmermans, J.-P.; Cos, P.; Maes, L. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: Practical relevance of stage-specific differences. Antimicrob. Agents Chemother. 2009, 53, 3855–3859. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Fan, W.Q.; Szajda, M.; Li, Q.-L.; Caster, K.C. Conjugated systems derived from piperazine-2,5-dione. J. Heterocycl. Chem. 1988, 25, 591–597. [Google Scholar] [CrossRef]
- Balducci, D.; Conway, P.A.; Sapuppo, G.; Muller-Bunz, H.; Paradisi, F. Novel approach to the synthesis of aliphatic and aromatic α-keto acids. Tetrahedron 2012, 68, 7374–7379. [Google Scholar] [CrossRef]
- Villemin, D.; Ben Alloum, A. Potassium fluoride on alumina: Condensation of 1,4-diacetylpiperazine-2,5-dione with aldehydes. Dry condensation under microwave irradiation. Synthesis of albonursin and analogs. Synth. Commun. 1990, 20, 3325–3331. [Google Scholar] [CrossRef]
- Mallory, F.B.; Smith, P.A.S.; Boyer, J.H. Benzofurazan oxide. Org. Synth. 1957, 37, 1. [Google Scholar]
- Dea-Ayuela, M.A.; Castillo, E.; González-Álvarez, M.; Vega, C.; Rolón, M.; Bolás-Fernández, F.; Borrás, J.; González-Rosende, M.E. In vivo and in vitro anti-leishmanial activities of 4-nitro-N-pyrimidin-2-ylbenzenesulfonamide and N2-(4-nitrophenyl)-N1-propylglycinamide. Bioorg. Med. Chem. 2009, 17, 7449–7456. [Google Scholar] [CrossRef]
- Bilbao-Ramos, P.; Sifontes-Rodríguez, S.; Dea-Ayuela, M.A.; Bolás-Fernández, F. A fluorometric method for evaluation of pharmacological activity against intracellular Leishmania amastigotes. J. Microbiol. Meth. 2012, 89, 8–11. [Google Scholar] [CrossRef]
- Ferreira, V.F.; Jorqueira, A.; Souza, A.M.T.; da Silva, M.N.; de Souza, M.C.; Gouvêa, R.M.; Rodrigues, C.R.; Pinto, A.V.; Castro, H.C.; Santos, D.O.; et al. Trypanocidal agents with low cytotoxicity to mammalian cell line: A comparison of the theoretical and biological features of lapachone derivatives. Bioorg. Med. Chem. 2006, 14, 5459–5466. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R1 | 1, Yield | R2 | 2, Yield |
|---|---|---|---|---|
| 1 | H | 1a, 82% | OMe | 2a, 71% |
| 2 | 4-CF3 | 1b, 73% | OMe | 2b, 68% |
| 3 | 4-F | 1c, 72% | OMe | 2c, 45% |
| 4 | 3-Cl | 1d, 97% | OMe | 2d, 98% |
| 5 | 4-Me | 1e, 65% | OMe | 2e, 54% |
| 6 | 2-Me | 1f, 50% | OMe | 2f, 55% |
| 7 | 3-MeO | 1g, 57% | OMe | 2g, 40% |
| 8 | 2-NO2-3,4-(MeO)2 | 1h, 80% | OMe | 2h, 68% |
| 9 | OBn | 2i, 63% |
| Entry | R1 | R2 | R3 | 4, Yield | 5, Yield |
|---|---|---|---|---|---|
| 1 | H | OMe | H | 4a, 82% | |
| 2 | 3-MeO | OMe | H | 4b, 98% | |
| 3 | 4-CF3 | OMe | H | 4c, 78% | |
| 4 | 4-F | OMe | H | 4d, 74% | 5d, 52% |
| 5 | 2-NO2, 3,4-(MeO)2 | OBn | H | 4e, 83% | |
| 6 | H | OMe | 6-OMe | 4f, 76% | 5f, 62% |
| 7 | 3-Cl | OMe | 6-OMe | 4g, 79% | |
| 8 | 4-CF3 | OMe | 6-OMe | 4h, 72% | 5h, 65% |
| 9 | 4-F | OMe | 6-OMe | 4i, 62% | |
| 10 | 4-Me | OMe | 6-OMe | 4j, 35% | |
| 11 | 2-Me | OMe | 6-OMe | 4k, 45% | |
| 12 | 4-CF3 | OMe | 6-Me | 4l, 28% | |
| 13 | 4-F | OMe | 6-Me | 4m, 60% | 5m, 58% |
| 14 | H | OMe | 6-CF3 | 4n, 40% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, J.F.; Dea-Ayuela, M.-A.; Huck, L.; Orduña, J.M.; Bolás-Fernández, F.; de la Cuesta, E.; Haseen, N.; Mohammed, A.A.; Menéndez, J.C. Dual Antitubercular and Antileishmanial Profiles of Quinoxaline Di-N-Oxides Containing an Amino Acidic Side Chain. Pharmaceuticals 2024, 17, 487. https://doi.org/10.3390/ph17040487
González JF, Dea-Ayuela M-A, Huck L, Orduña JM, Bolás-Fernández F, de la Cuesta E, Haseen N, Mohammed AA, Menéndez JC. Dual Antitubercular and Antileishmanial Profiles of Quinoxaline Di-N-Oxides Containing an Amino Acidic Side Chain. Pharmaceuticals. 2024; 17(4):487. https://doi.org/10.3390/ph17040487
Chicago/Turabian StyleGonzález, Juan F., María-Auxiliadora Dea-Ayuela, Lena Huck, José María Orduña, Francisco Bolás-Fernández, Elena de la Cuesta, Nazia Haseen, Ashraf Ali Mohammed, and J. Carlos Menéndez. 2024. "Dual Antitubercular and Antileishmanial Profiles of Quinoxaline Di-N-Oxides Containing an Amino Acidic Side Chain" Pharmaceuticals 17, no. 4: 487. https://doi.org/10.3390/ph17040487
APA StyleGonzález, J. F., Dea-Ayuela, M.-A., Huck, L., Orduña, J. M., Bolás-Fernández, F., de la Cuesta, E., Haseen, N., Mohammed, A. A., & Menéndez, J. C. (2024). Dual Antitubercular and Antileishmanial Profiles of Quinoxaline Di-N-Oxides Containing an Amino Acidic Side Chain. Pharmaceuticals, 17(4), 487. https://doi.org/10.3390/ph17040487