
Discovery of Small Molecule Glycolytic Stimulants for Enhanced ApoE Lipidation in Alzheimer’s Disease Cell Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
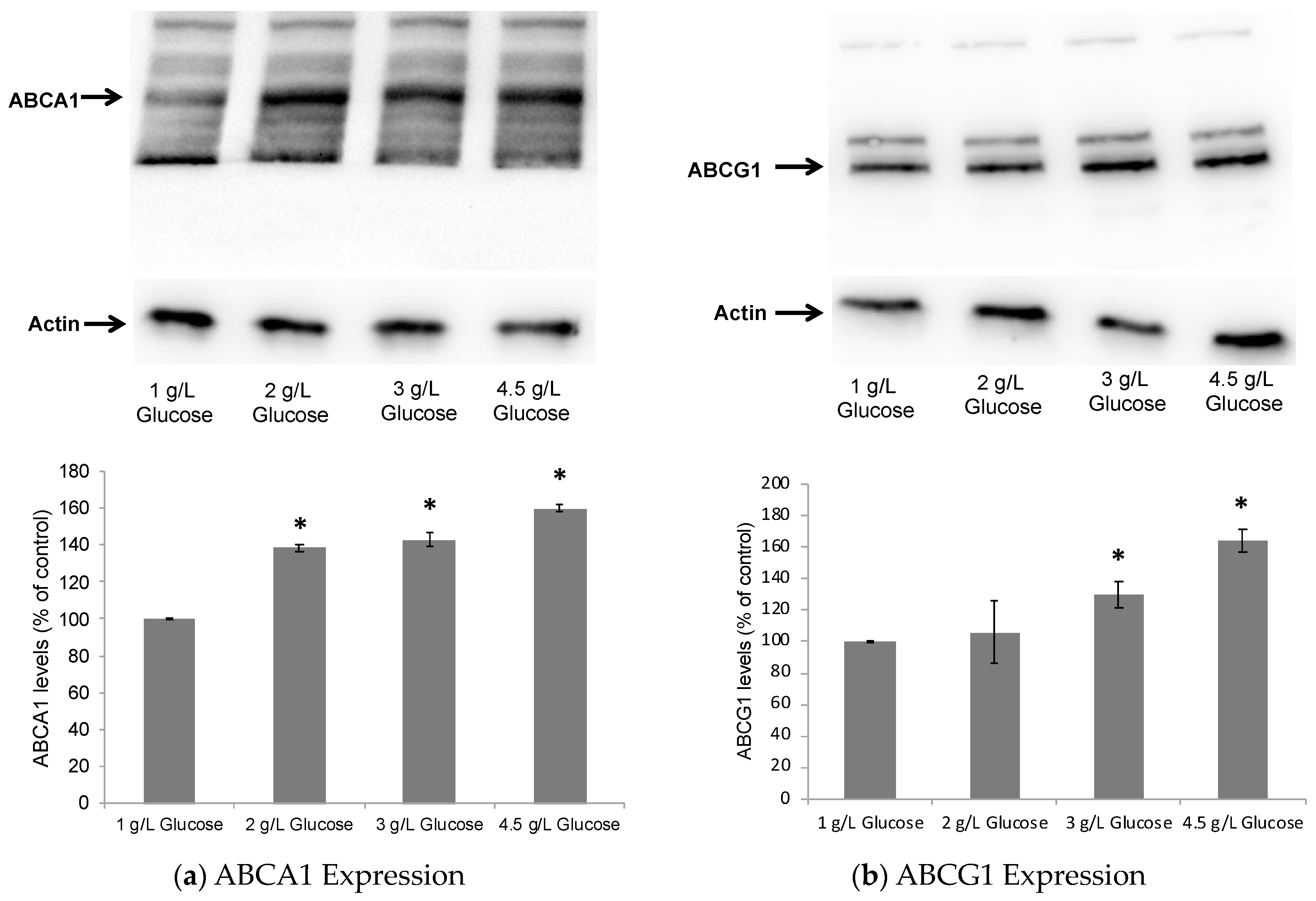
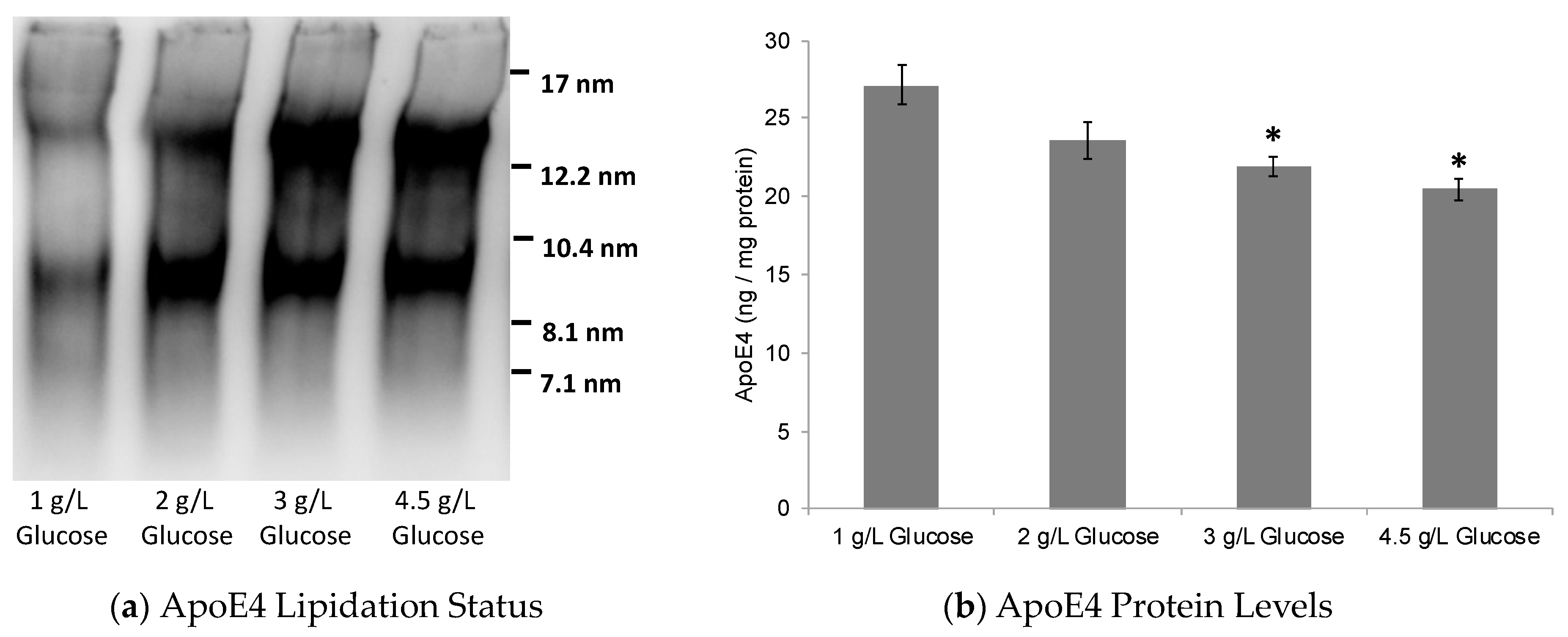
2.1. Dose-Dependent Effect of Glucose on ABCA1/G1 Expression and ApoE Lipidation
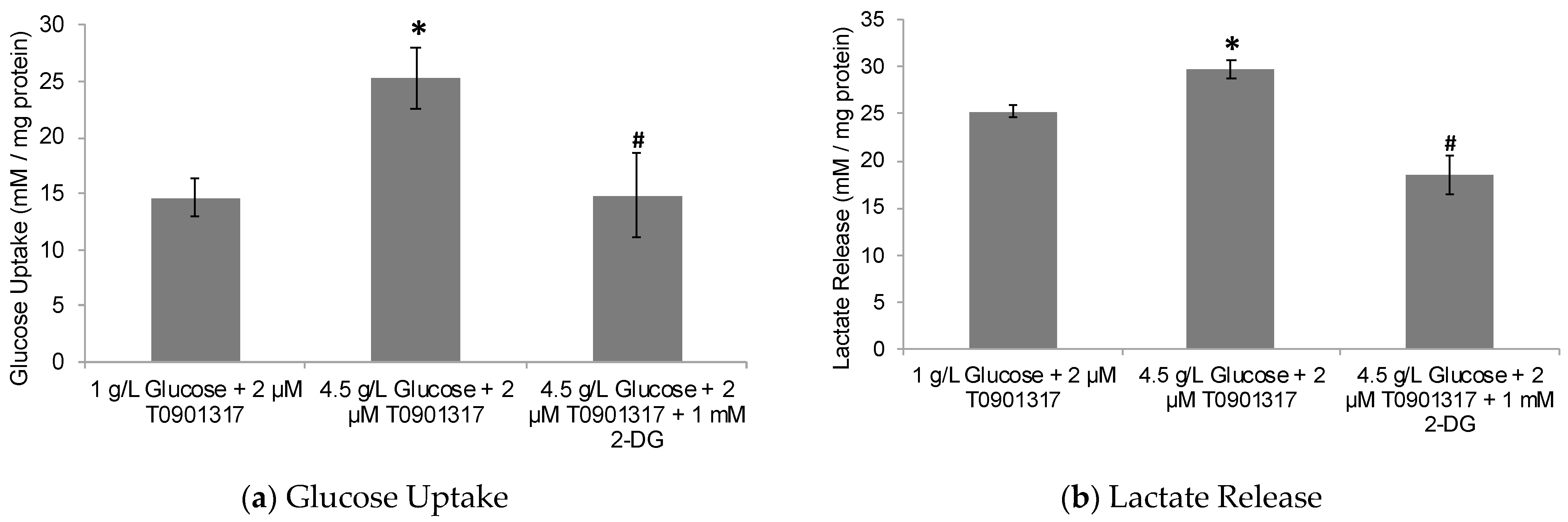
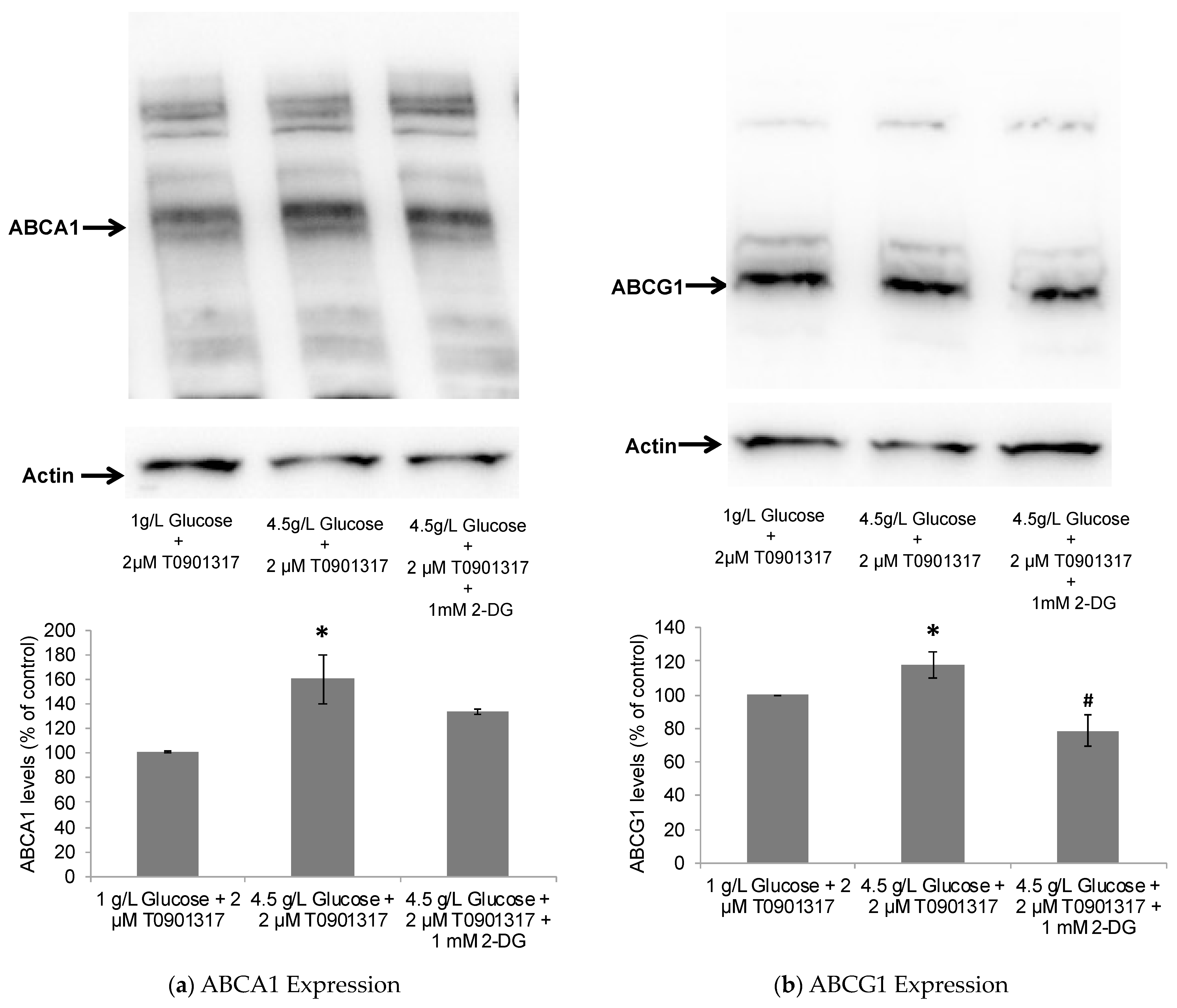
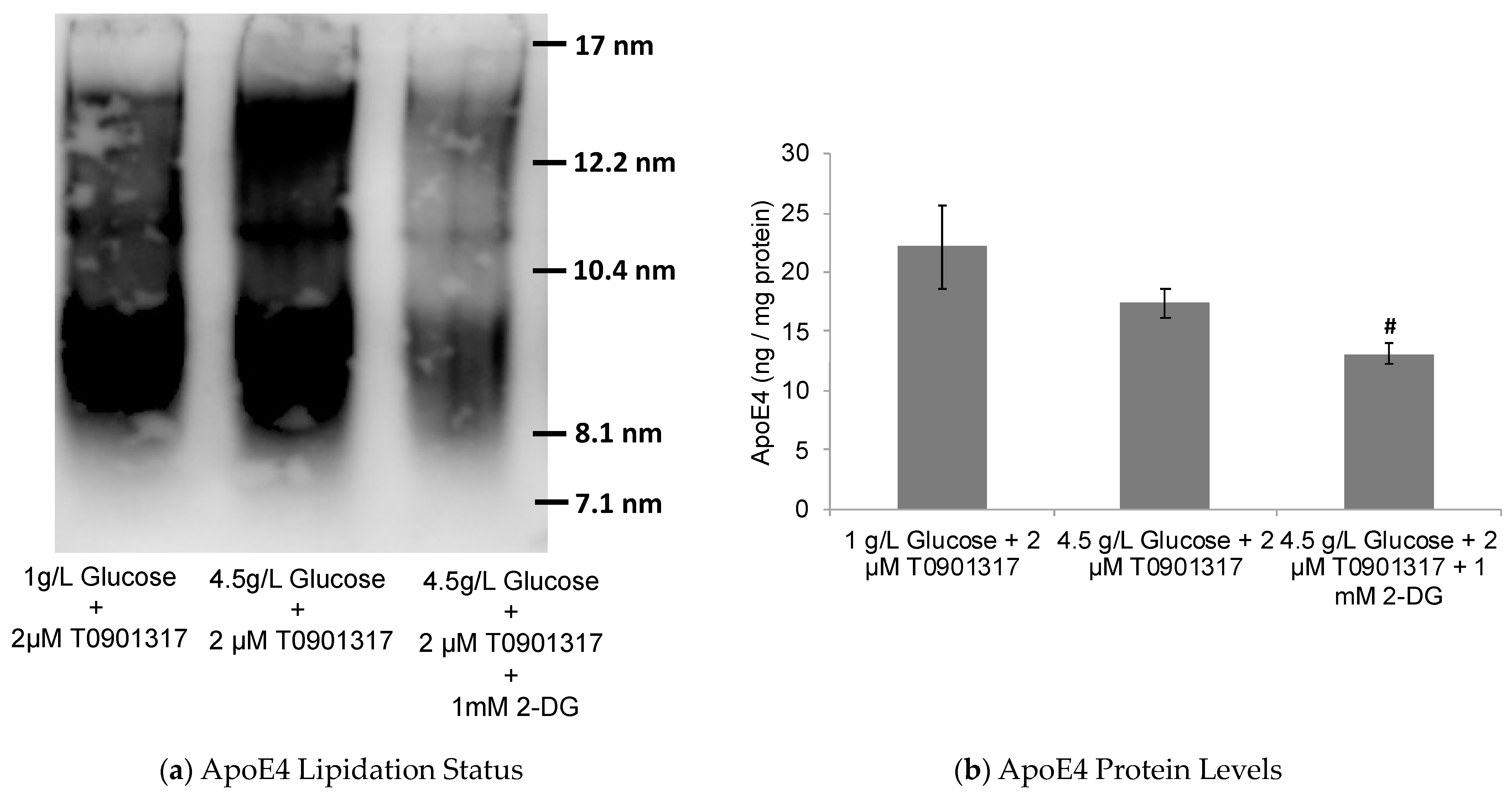
2.2. Glycolytic Control of LXR-Induced ABCA1/G1 Expression and ApoE Lipidation
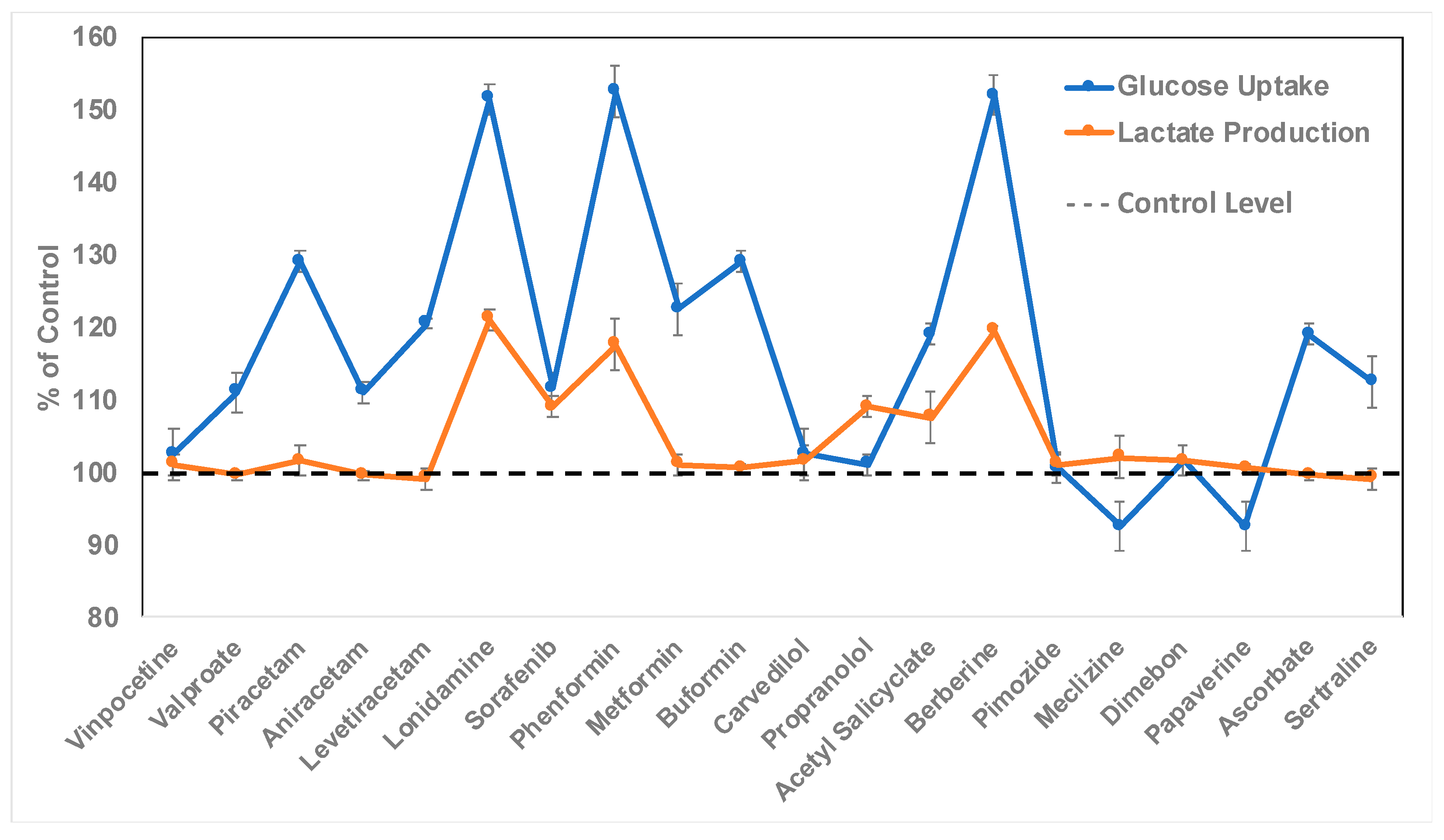
2.3. Discovery of Bioactive Compounds for Enhanced Astrocytic Glycolysis
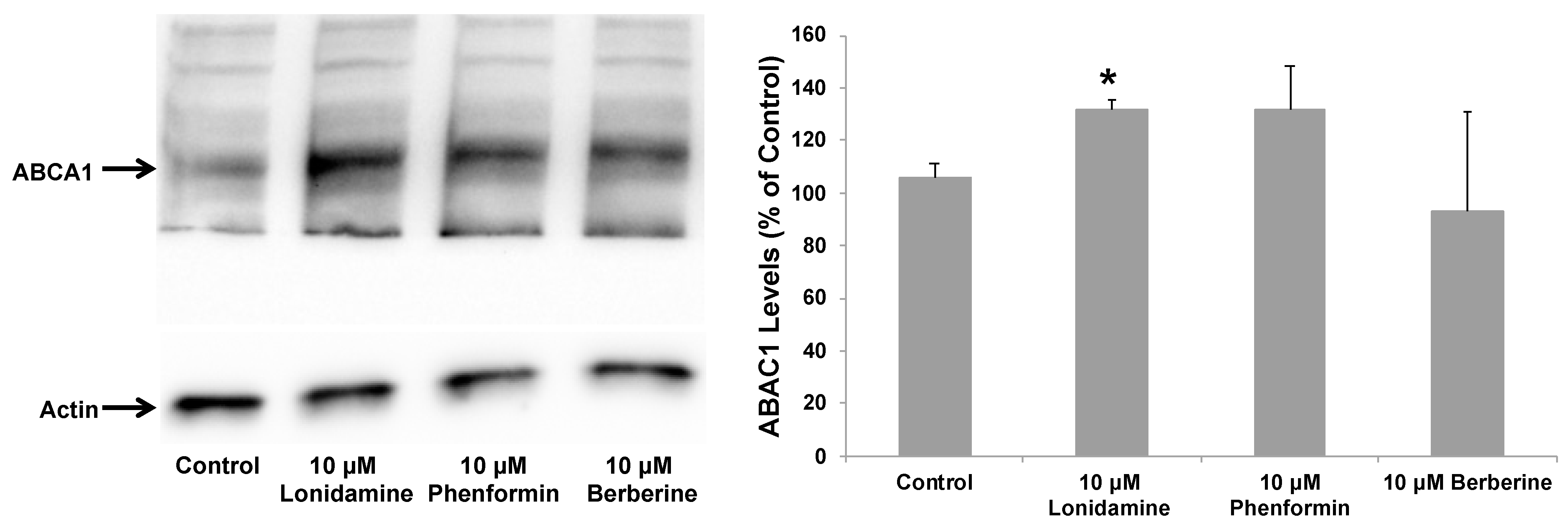
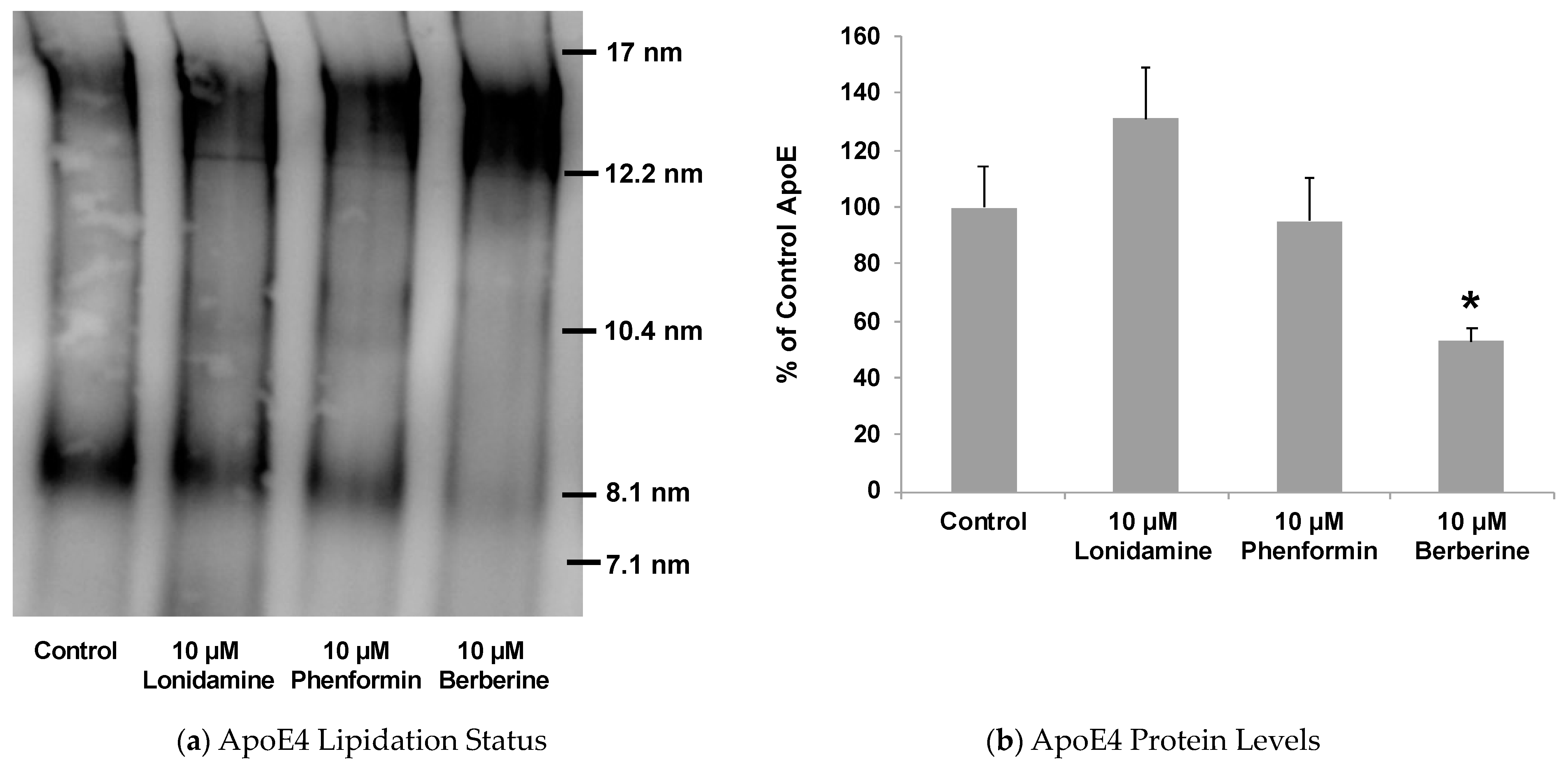
2.4. Effect of Glycolytic Stimulants on ApoE Lipidation and Secretion by Astrocytes
3. Materials and Methods
3.1. Cell Culture and Treatment
3.2. Glucose-Metabolism Studies
3.3. Western Blotting Analysis
3.4. ApoE Native Gel Electrophoresis and ELISA
3.5. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Chapter 13—Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Lee, H.; Hsieh, Y.C.; Chou, V.; Selkoe, D.J. Moving beyond amyloid and tau to capture the biological heterogeneity of Alzheimer’s disease. Trends Neurosci. 2023, 46, 426–444. [Google Scholar] [CrossRef]
- Patil, S.; Chan, C. Palmitic and stearic fatty acids induce Alzheimer-like hyperphosphorylation of tau in primary rat cortical neurons. Neurosci. Lett. 2005, 384, 288–293. [Google Scholar] [CrossRef]
- Patil, S.; Melrose, J.; Chan, C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur. J. Neurosci. 2007, 26, 2131–2141. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Amyloid β-protein and beyond: The path forward in Alzheimer’s disease. Curr. Opin. Neurobiol. 2020, 61, 116–124. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves second anti-amyloid antibody for Alzheimer disease. Nat. Rev. Drug Discov. 2023, 22, 89. [Google Scholar] [CrossRef]
- Cummings, J. Anti-Amyloid Monoclonal Antibodies are Transformative Treatments that Redefine Alzheimer’s Disease Therapeutics. Drugs 2023, 83, 569–576. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef]
- Bateman, R.J.; Munsell, L.Y.; Morris, J.C.; Swarm, R.; Yarasheski, K.E.; Holtzman, D.M. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 2006, 12, 856–861. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Goedert, M.; Weisgraber, K.H.; Dong, L.M.; Jakes, R.; Huang, D.Y.; Pericak-Vance, M.; Schmechel, D.; Roses, A.D. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 11183–11186. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Ou, Y.N.; Xu, W.; Li, J.Q.; Guo, Y.; Cui, M.; Chen, K.L.; Huang, Y.Y.; Dong, Q.; Tan, L.; Yu, J.T. Alzheimer’s Disease Neuroimaging Initiative. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alzheimers Res. Ther. 2019, 11, 57. [Google Scholar] [CrossRef]
- Szablewski, L. Brain Glucose Transporters: Role in Pathogenesis and Potential Targets for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8142. [Google Scholar] [CrossRef]
- Hunt, A.; Schönknecht, P.; Henze, M.; Seidl, U.; Haberkorn, U.; Schröder, J. Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease. Psychiatry Res. 2007, 155, 147–154. [Google Scholar] [CrossRef]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1, 217–233. [Google Scholar] [CrossRef]
- Patil, S.P.; Ballard, R.; Sanchez, S.; Osborn, J.; Santangelo, D., Jr. ApoE: The link between Alzheimer’s-related glucose hypometabolism and Aβ deposition? Med. Hypotheses 2012, 78, 494–496. [Google Scholar] [CrossRef]
- Kim, W.S.; Rahmanto, A.S.; Kamili, A.; Rye, K.A.; Guillemin, G.J.; Gelissen, I.C.; Jessup, W.; Hill, A.F.; Garner, B. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J. Biol. Chem. 2007, 282, 2851–2861. [Google Scholar] [CrossRef]
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779. [Google Scholar] [CrossRef]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef]
- Tokuda, T.; Calero, M.; Matsubara, E.; Vidal, R.; Kumar, A.; Permanne, B.; Zlokovic, B.; Smith, J.D.; Ladu, M.J.; Rostagno, A.; et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem. J. 2000, 348 Pt 2, 359–365. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017, 168, 427–441.e21. [Google Scholar] [CrossRef]
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef]
- Lee, C.Y.; Tse, W.; Smith, J.D.; Landreth, G.E. Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 2012, 287, 2032–2044. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef]
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef]
- Heinsinger, N.M.; Gachechiladze, M.A.; Rebeck, G.W. Apolipoprotein E Genotype Affects Size of ApoE Complexes in Cerebrospinal Fluid. J. Neuropathol. Exp. Neurol. 2016, 75, 918–924. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef]
- Michikawa, M.; Fan, Q.W.; Isobe, I.; Yanagisawa, K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J. Neurochem. 2000, 74, 1008–1016. [Google Scholar] [CrossRef]
- Hara, M.; Matsushima, T.; Satoh, H.; Iso-o, N.; Noto, H.; Togo, M.; Kimura, S.; Hashimoto, Y.; Tsukamoto, K. Isoform-dependent cholesterol efflux from macrophages by apolipoprotein E is modulated by cell surface proteoglycans. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 269–274. [Google Scholar] [CrossRef]
- Donkin, J.J.; Stukas, S.; Hirsch-Reinshagen, V.; Namjoshi, D.; Wilkinson, A.; May, S.; Chan, J.; Fan, J.; Collins, J.; Wellington, C.L. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J. Biol. Chem. 2010, 285, 34144–34154. [Google Scholar] [CrossRef]
- Terwel, D.; Steffensen, K.R.; Verghese, P.B.; Kummer, M.P.; Gustafsson, J.Å.; Holtzman, D.M.; Heneka, M.T. Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J. Neurosci. 2011, 31, 7049–7059. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef]
- Dumanis, S.B.; Tesoriero, J.A.; Babus, L.W.; Nguyen, M.T.; Trotter, J.H.; Ladu, M.J.; Weeber, E.J.; Turner, R.S.; Xu, B.; Rebeck, G.W.; et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 2009, 29, 15317–15322. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Jiang, X.C.; Beyer, T.P.; Li, Z.; Liu, J.; Quan, W.; Schmidt, R.J.; Zhang, Y.; Bensch, W.R.; Eacho, P.I.; Cao, G. Enlargement of high density lipoprotein in mice via liver X receptor activation requires apolipoprotein E and is abolished by cholesteryl ester transfer protein expression. J. Biol. Chem. 2003, 278, 49072–49078. [Google Scholar] [CrossRef]
- Li, G.; Bien-Ly, N.; Andrews-Zwilling, Y.; Xu, Q.; Bernardo, A.; Ring, K.; Halabisky, B.; Deng, C.; Mahley, R.W.; Huang, Y. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 2009, 5, 634–645. [Google Scholar] [CrossRef]
- Chang, S.; Ma, T.R.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef]
- Gibson, G.E.; Haroutunian, V.; Zhang, H.; Park, L.C.; Shi, Q.; Lesser, M.; Mohs, R.C.; Sheu, R.K.; Blass, J.P. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann. Neurol. 2000, 48, 297–303. [Google Scholar] [CrossRef]
- Cirrito, J.R.; May, P.C.; O’Dell, M.A.; Taylor, J.W.; Parsadanian, M.; Cramer, J.W.; Audia, J.E.; Nissen, J.S.; Bales, K.R.; Paul, S.M.; et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J. Neurosci. 2003, 23, 8844–8853. [Google Scholar] [CrossRef] [PubMed]
- Mitro, N.; Mak, P.A.; Vargas, L.; Godio, C.; Hampton, E.; Molteni, V.; Kreusch, A.; Saez, E. The nuclear receptor LXR is a glucose sensor. Nature 2007, 445, 219–223. [Google Scholar] [CrossRef]
- Patil, S.; Sheng, L.; Masserang, A.; Chan, C. Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci. Lett. 2006, 406, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Salvadó, G.; Milà-Alomà, M.; Shekari, M.; Ashton, N.J.; Operto, G.; Falcon, C.; Cacciaglia, R.; Minguillon, C.; Fauria, K.; Niñerola-Baizán, A.; et al. Reactive astrogliosis is associated with higher cerebral glucose consumption in the early Alzheimer’s continuum. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4567–4579. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Bian, S.; Haque, R.U.; Carter, E.K.; Watson, C.M.; Gordon, B.A.; Ping, L.; Duong, D.M.; Epstein, M.P.; McDade, E.; et al. Cerebrospinal fluid proteomics define the natural history of autosomal dominant Alzheimer’s disease. Nat. Med. 2023, 29, 1979–1988. [Google Scholar] [CrossRef]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.; Huang, S.C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L.; et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 6, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ushio, Y.; Morino, Y.; Ohta, T.; Matsukado, Y. Immunohistochemical localization of apolipoprotein E in human glial neoplasms. J. Clin. Investig. 1988, 82, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Uchihara, T.; Duyckaerts, C.; He, Y.; Kobayashi, K.; Seilhean, D.; Amouyel, P.; Hauw, J.J. ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain. Neurosci. Lett. 1995, 195, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.T.; Gilbert, J.R.; Qiu, H.L.; Ervin, J.; Rothrock-Christian, T.R.; Hulette, C.; Schmechel, D.E. Specific regional transcription of apolipoprotein E in human brain neurons. Am. J. Pathol. 1999, 154, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Zalocusky, K.A.; Najm, R.; Taubes, A.L.; Hao, Y.; Yoon, S.Y.; Koutsodendris, N.; Nelson, M.R.; Rao, A.; Bennett, D.A.; Bant, J.; et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Jasmin, S.B.; Pearson, V.; Lalonde, D.; Domenger, D.; Théroux, L.; Poirier, J. Differential regulation of ABCA1 and ABCG1 gene expressions in the remodeling mouse hippocampus after entorhinal cortex lesion and liver-X receptor agonist treatment. Brain Res. 2014, 1562, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jiang, H.; Park, S.; Eltorai, A.E.; Stewart, F.R.; Yoon, H.; Basak, J.M.; Finn, M.B.; Holtzman, D.M. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J. Neurosci. 2011, 31, 18007–18012. [Google Scholar] [CrossRef] [PubMed]
- Chemparathy, A.; Guen, Y.L.; Chen, S.; Lee, E.-G.; Leong, L.; Gorzynski, J.; Xu, G.; Belloy, M.; Kasireddy, N.; Tauber, A.P.; et al. APOE loss-of-function variants: Compatible with longevity and associated with resistance to Alzheimer’s Disease pathology. Neuron 2024, 112, 1110–1116. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc. Natl. Acad. Sci. USA 2005, 102, 8299–8302. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Vaishnavi, S.N.; Couture, L.; Sacco, D.; Shannon, B.J.; Mach, R.H.; Morris, J.C.; Raichle, M.E.; Mintun, M.A. Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc. Natl. Acad. Sci. USA 2010, 107, 17763–17767. [Google Scholar] [CrossRef] [PubMed]
- Gohil, V.M.; Sheth, S.A.; Nilsson, R.; Wojtovich, A.P.; Lee, J.H.; Perocchi, F.; Chen, W.; Clish, C.B.; Ayata, C.; Brookes, P.S.; et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol. 2010, 28, 249–255. [Google Scholar] [CrossRef]
- Lublin, A.; Isoda, F.; Patel, H.; Yen, K.; Nguyen, L.; Hajje, D. FDA-Approved Drugs that Protect Mammalian Neurons from Glucose Toxicity Slow Aging Dependent on Cbp and Protect Against Proteotoxicity. PLoS ONE 2011, 6, e27762. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef]
- Floridi, A.; Paggi, M.G.; D’Atri, S.; De Martino, C.; Marcante, M.L.; Silvestrini, B.; Caputo, A. Effect of lonidamine on the energy metabolism of Ehrlich ascites tumor cells. Cancer Res. 1981, 41 Pt 1, 4661–4666. [Google Scholar]
- Floridi, A.; Paggi, M.G.; Marcante, M.L.; Silvestrini, B.; Caputo, A.; De Martino, C. Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J. Natl. Cancer Inst. 1981, 66, 497–499. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.S.; Choi, J.Y. Synergistic anti-cancer effect of phenformin and oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef]
- Yin, J.; Xing, H.; Ye, J. Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism 2008, 57, 712–717. [Google Scholar] [CrossRef]
- Dresselhaus, E.; Duerr, J.M.; Vincent, F.; Sylvain, E.K.; Beyna, M.; Lanyon, L.F.; LaChapelle, E.; Pettersson, M.; Bales, K.R.; Ramaswamy, G. Class I HDAC inhibition is a novel pathway for regulating astrocytic apoE secretion. PLoS ONE 2018, 13, e0194661. [Google Scholar] [CrossRef]
- Fan, J.; Zareyan, S.; Zhao, W.; Shimizu, Y.; Pfeifer, T.A.; Tak, J.H.; Isman, M.B.; Van den Hoven, B.; Duggan, M.E.; Wood, M.W.; et al. Identification of a Chrysanthemic Ester as an Apolipoprotein E Inducer in Astrocytes. PLoS ONE 2016, 11, e0162384. [Google Scholar] [CrossRef] [PubMed]
- Boehm-Cagan, A.; Bar, R.; Liraz, O.; Bielicki, J.K.; Johansson, J.O.; Michaelson, D.M. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J. Alzheimers Dis. 2016, 54, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Litvinchuk, A.; Suh, J.H.; Guo, J.L.; Lin, K.; Davis, S.S.; Bien-Ly, N.; Tycksen, E.; Tabor, G.T.; Remolina Serrano, J.; Manis, M.; et al. Amelioration of Tau and ApoE4-linked glial lipid accumulation and neurodegeneration with an LXR agonist. Neuron 2023, 112, 384–403. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Yuan, Z.; Pan, R.; Su, X.; Wang, H.; Xue, J.; Zhuang, K.; Gao, J.; Chen, Z.; Lin, H.; et al. Microglial hexokinase 2 deficiency increases ATP generation through lipid metabolism leading to β-amyloid clearance. Nat. Metab. 2022, 4, 1287–1305. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, Y.; Ning, X.; Li, S.; Xue, D.; Wei, C.; Zhu, Z.; Sheng, L.; Lu, B.; Li, Y.; et al. Directly targeting ASC by lonidamine alleviates inflammasome-driven diseases. J. Neuroinflamm. 2022, 19, 315. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Fryer, J.D.; Sullivan, P.M.; Christopher, E.A.; Wahrle, S.E.; DeMattos, R.B.; O’Dell, M.A.; Fagan, A.M.; Lashuel, H.A.; Walz, T.; et al. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol. Dis. 2005, 19, 66–76. [Google Scholar] [CrossRef]
- Liang, W.S.; Chen, K.; Lee, W.; Sidhar, K.; Corneveaux, J.J.; Allen, A.N.; Myers, A.; Villa, S.; Meechoovet, B.; Pruzin, J.; et al. Association between GAB2 haplotype and higher glucose metabolism in Alzheimer’s disease-affected brain regions in cognitively normal APOEε4 carriers. Neuroimage 2011, 54, 1896–1902. [Google Scholar] [CrossRef][Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, S.P.; Kuehn, B.R. Discovery of Small Molecule Glycolytic Stimulants for Enhanced ApoE Lipidation in Alzheimer’s Disease Cell Model. Pharmaceuticals 2024, 17, 491. https://doi.org/10.3390/ph17040491
Patil SP, Kuehn BR. Discovery of Small Molecule Glycolytic Stimulants for Enhanced ApoE Lipidation in Alzheimer’s Disease Cell Model. Pharmaceuticals. 2024; 17(4):491. https://doi.org/10.3390/ph17040491
Chicago/Turabian StylePatil, Sachin P., and Bella R. Kuehn. 2024. "Discovery of Small Molecule Glycolytic Stimulants for Enhanced ApoE Lipidation in Alzheimer’s Disease Cell Model" Pharmaceuticals 17, no. 4: 491. https://doi.org/10.3390/ph17040491
APA StylePatil, S. P., & Kuehn, B. R. (2024). Discovery of Small Molecule Glycolytic Stimulants for Enhanced ApoE Lipidation in Alzheimer’s Disease Cell Model. Pharmaceuticals, 17(4), 491. https://doi.org/10.3390/ph17040491