
Orally Bioavailable Proteolysis-Targeting Chimeras: An Innovative Approach in the Golden Era of Discovering Small-Molecule Cancer Drugs

 ,
,  , , ,
, , ,
Abstract
1. Introduction
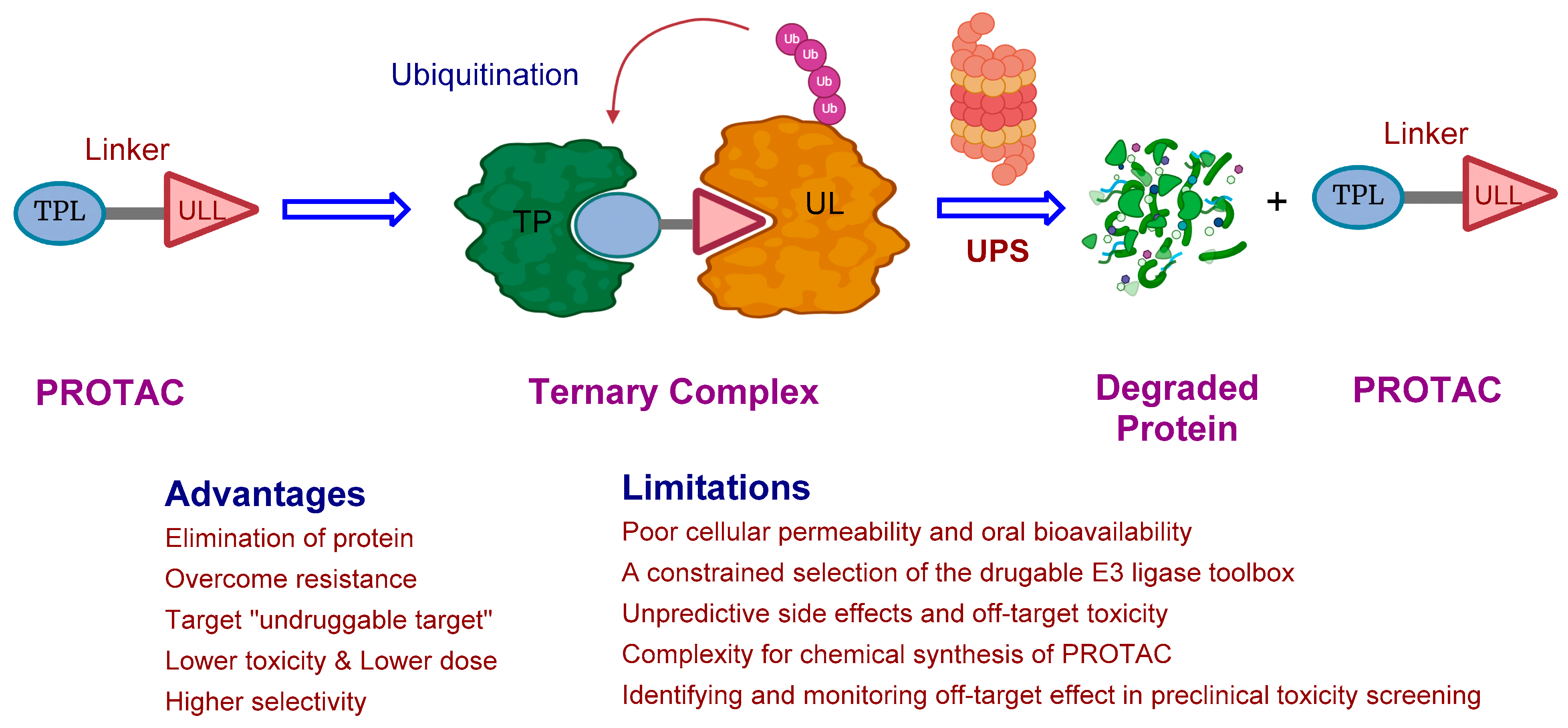
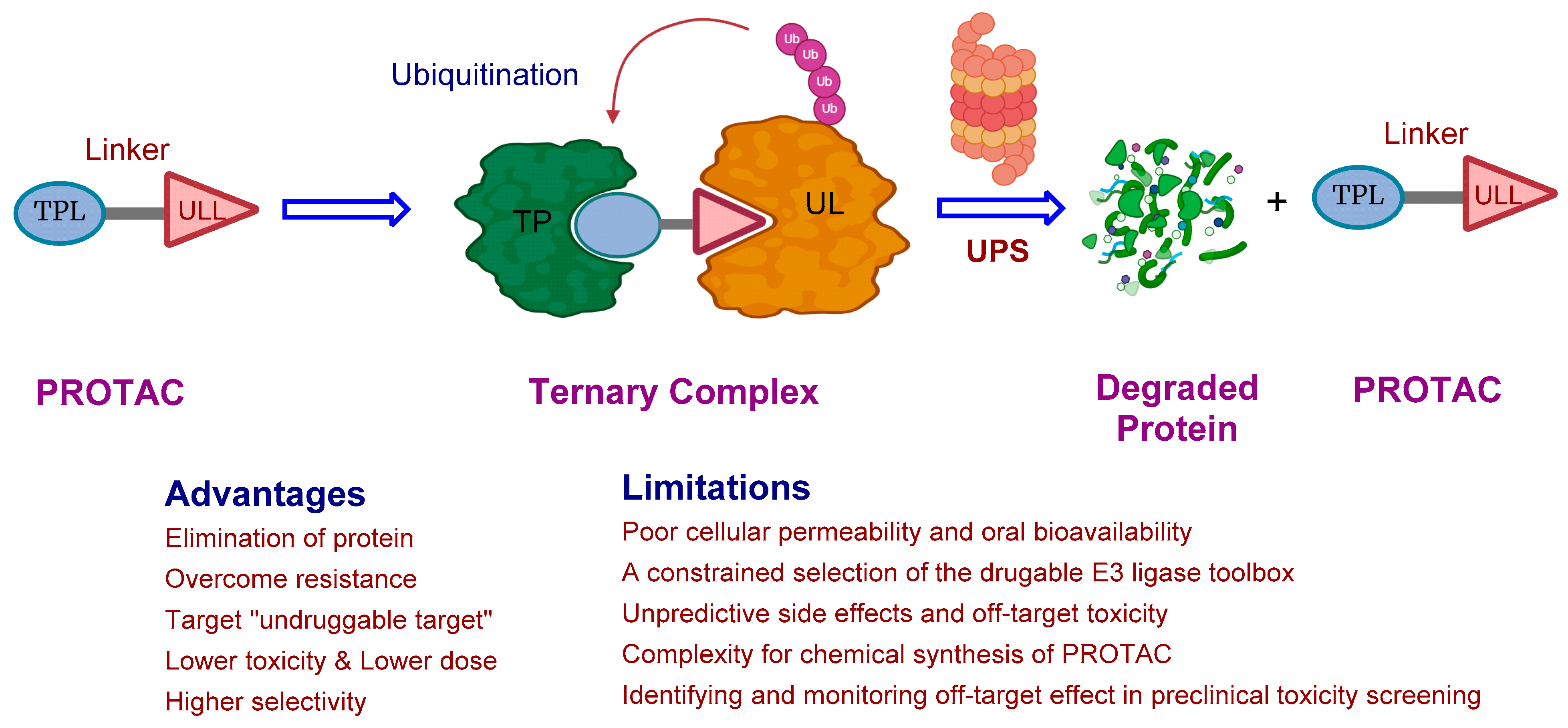
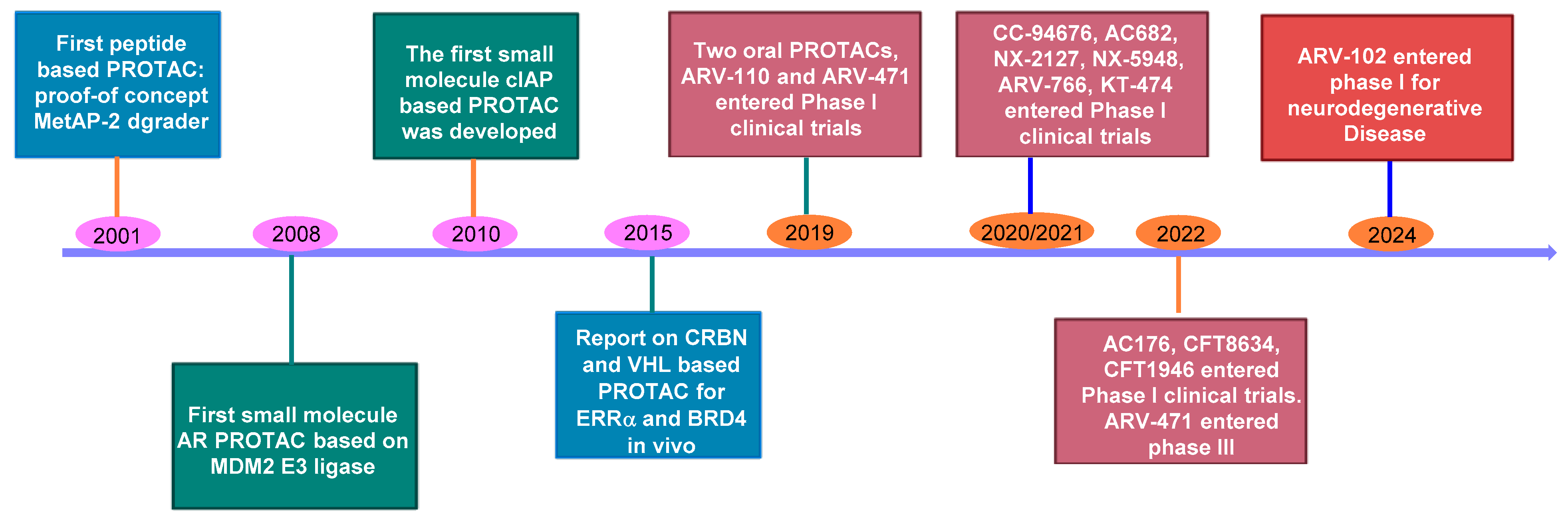
2. The Rise of PROTAC Technology: A Versatile Game-Changer
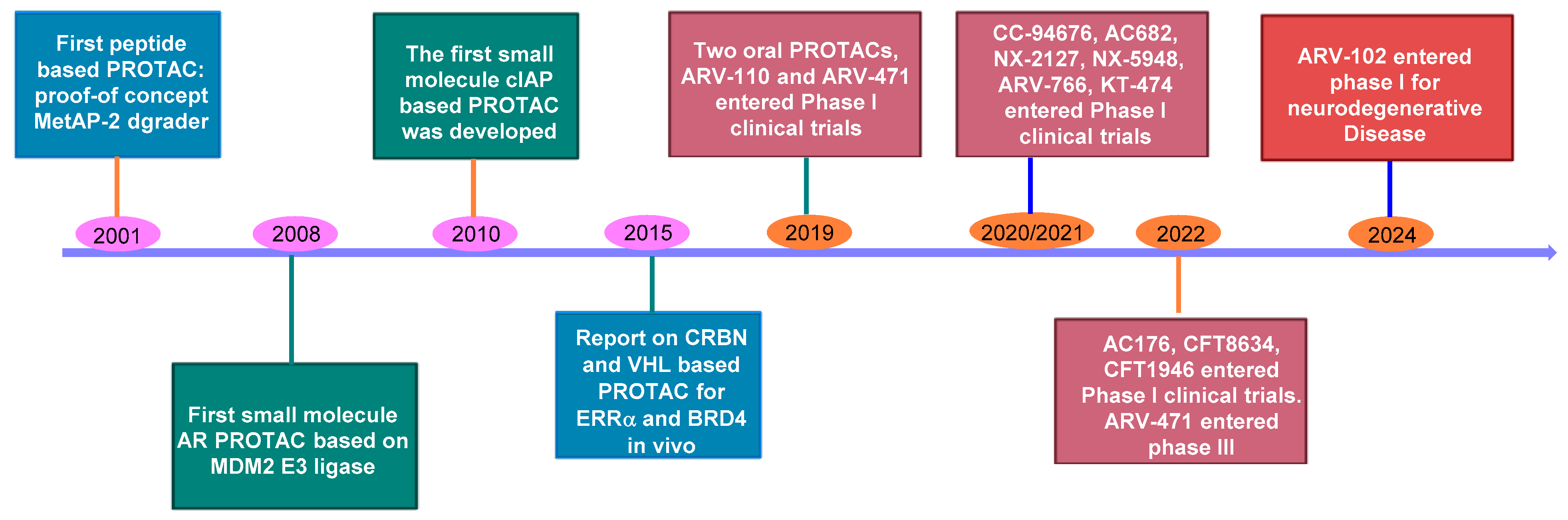
3. Oral Protein Degraders: Progress and Limitations
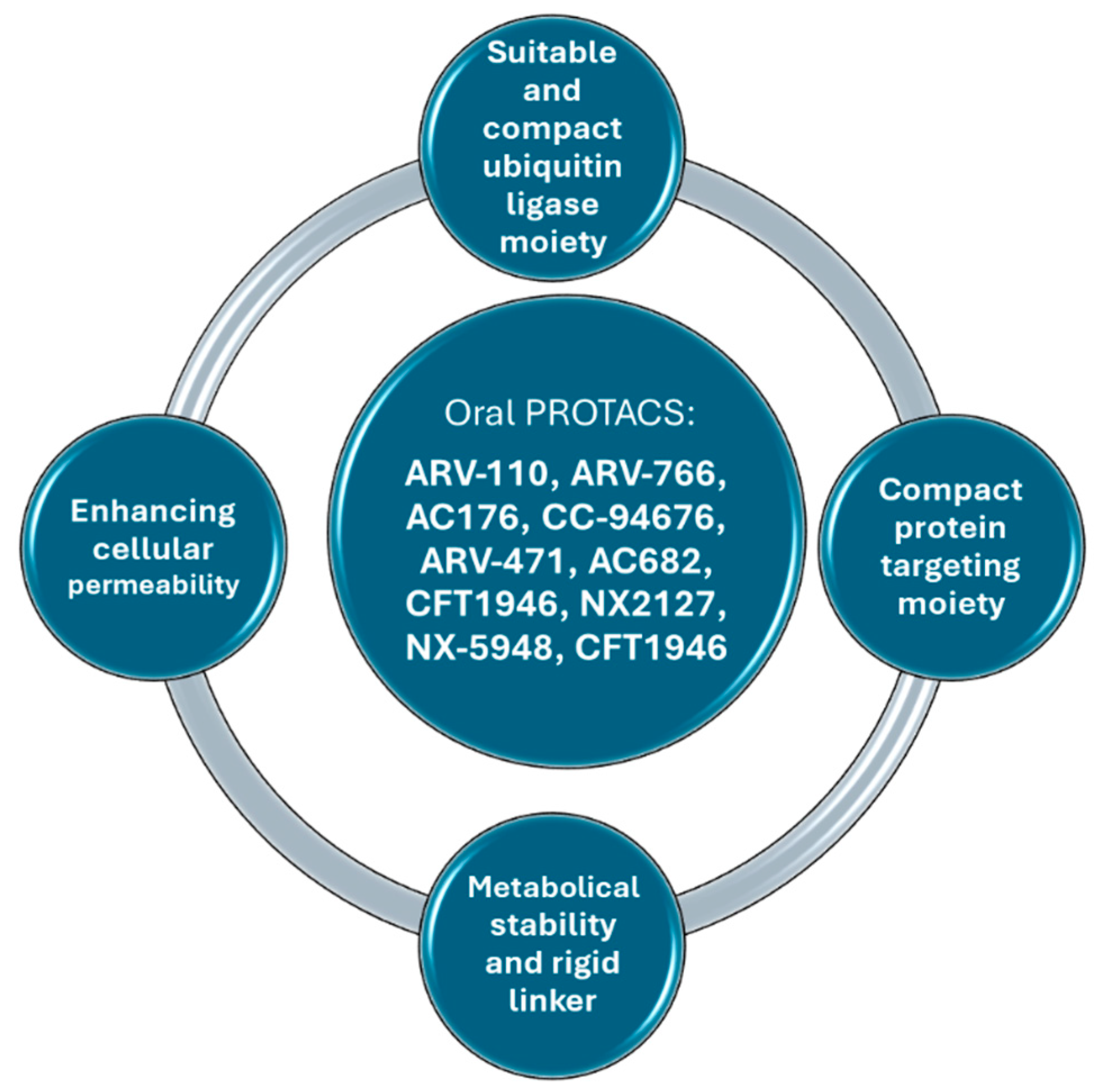
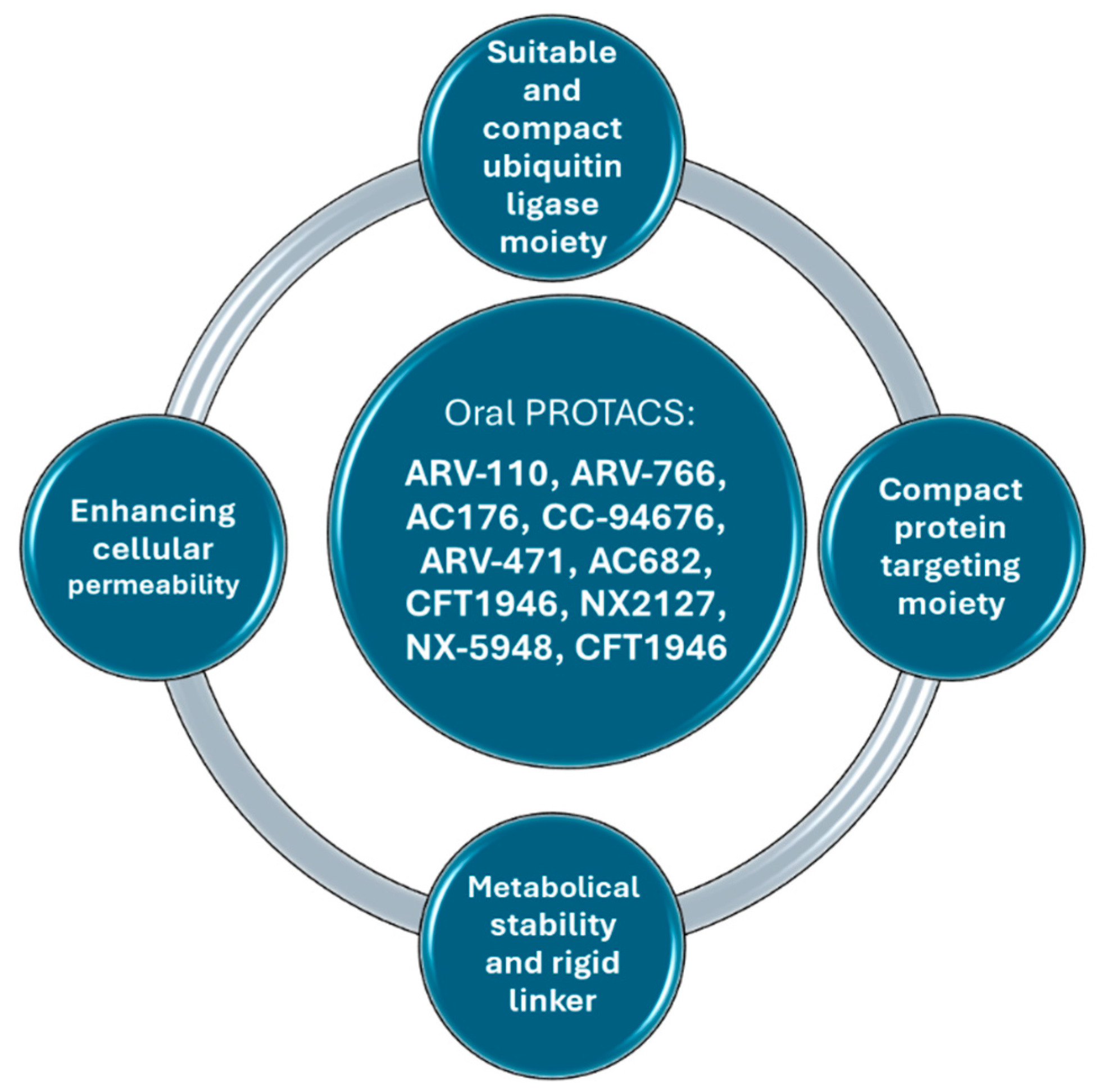
4. Several Ways to Improve PROTACs’ Oral Bioavailability
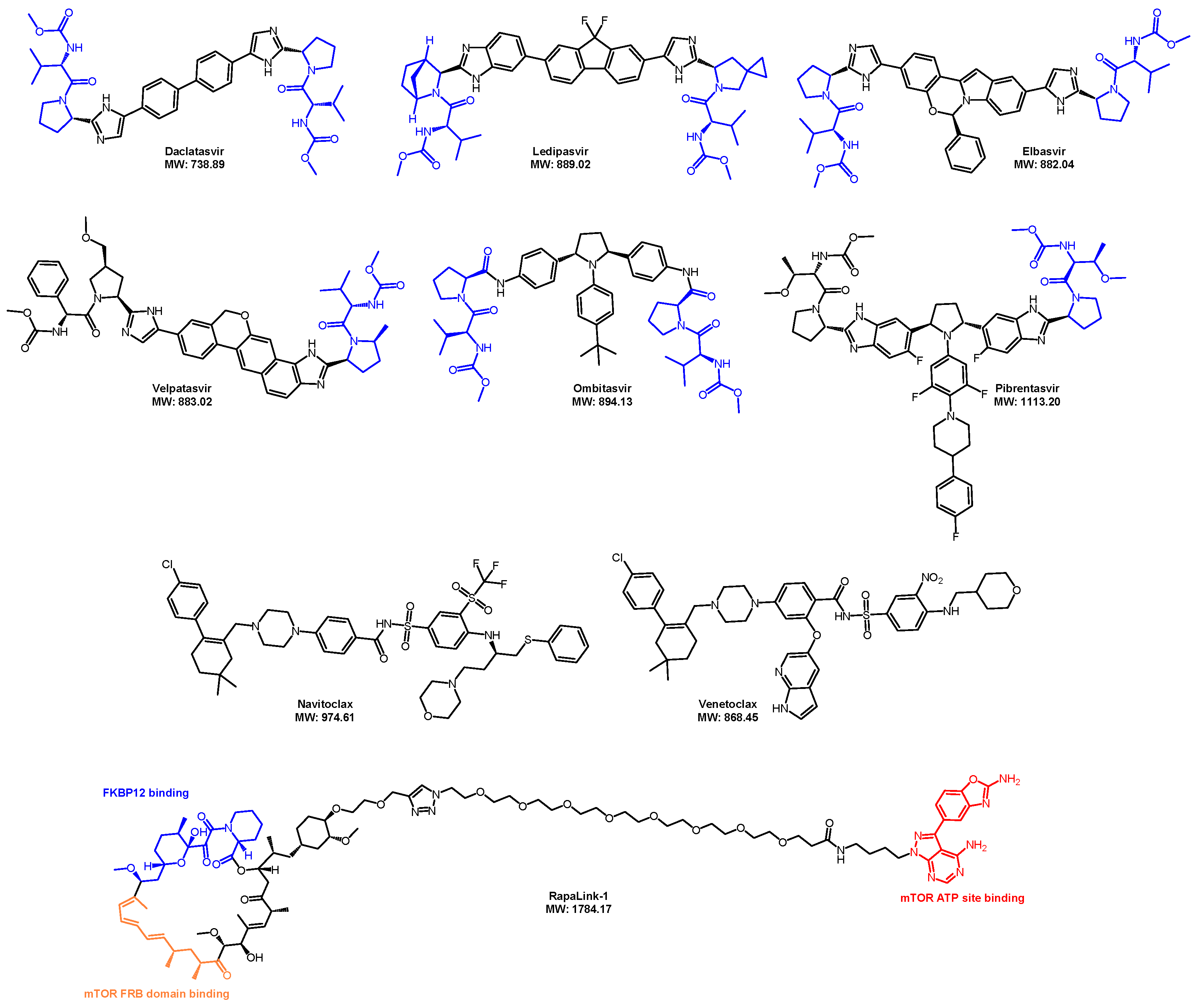
5. Recent Advancements in Orally Bioavailable PROTACs
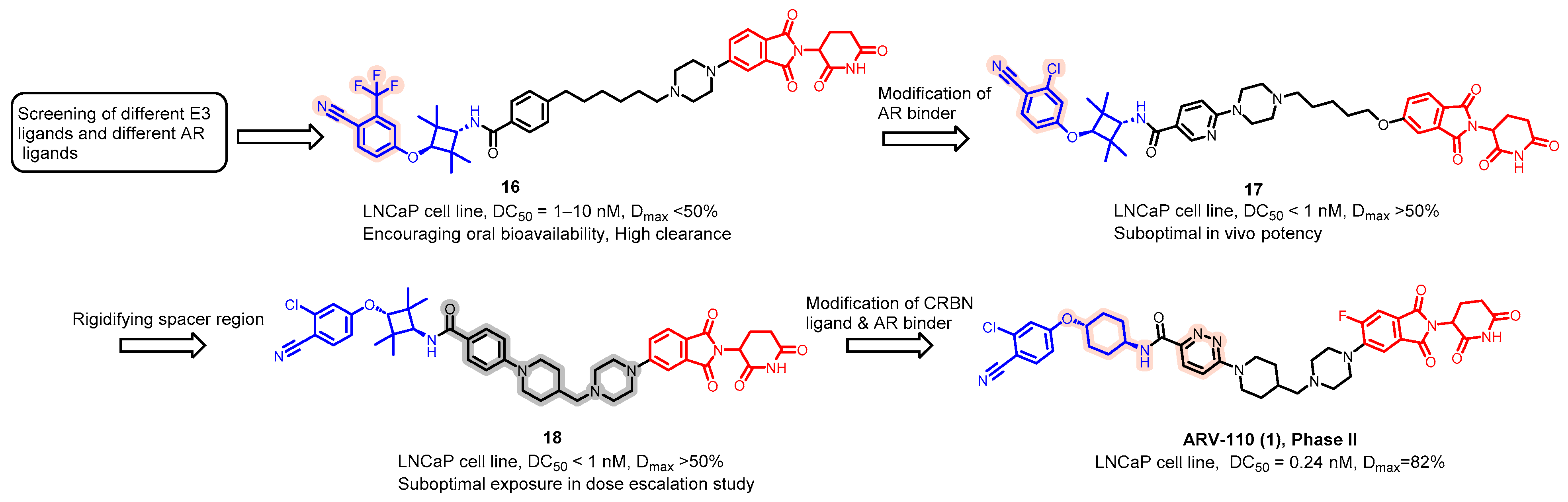
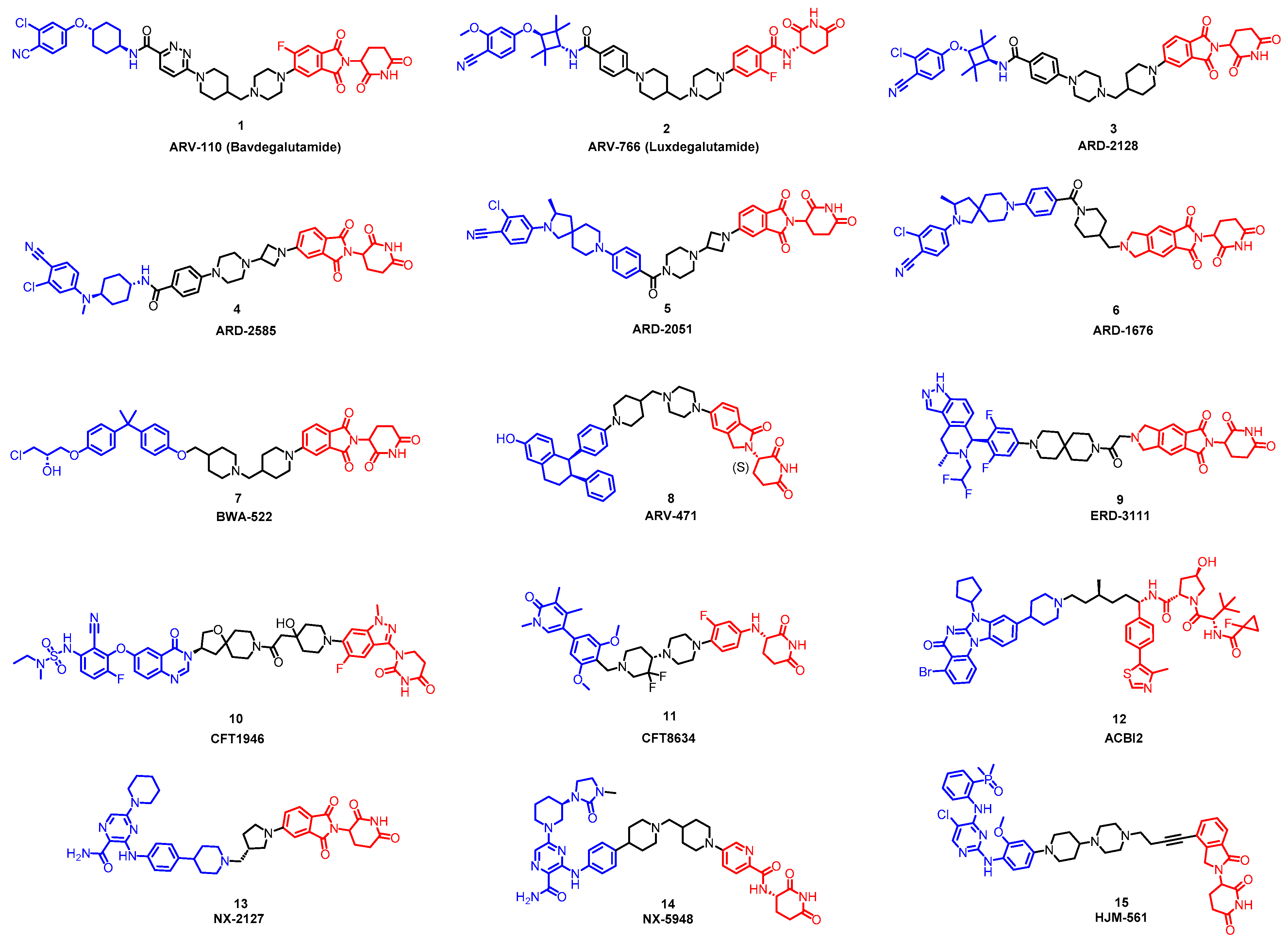
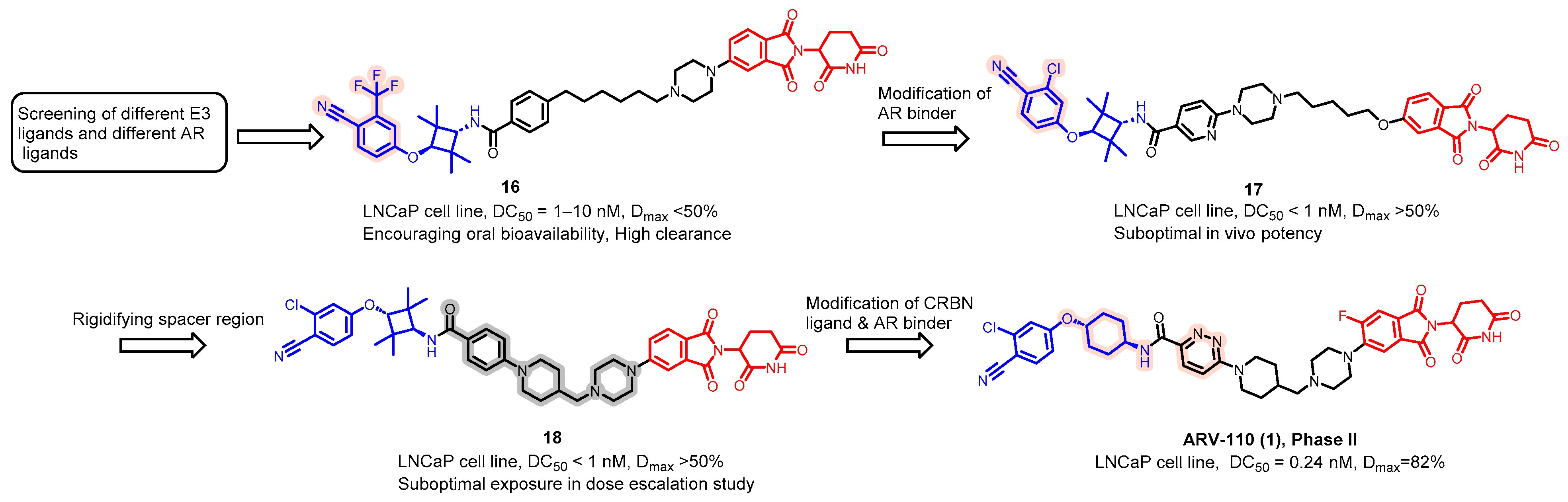
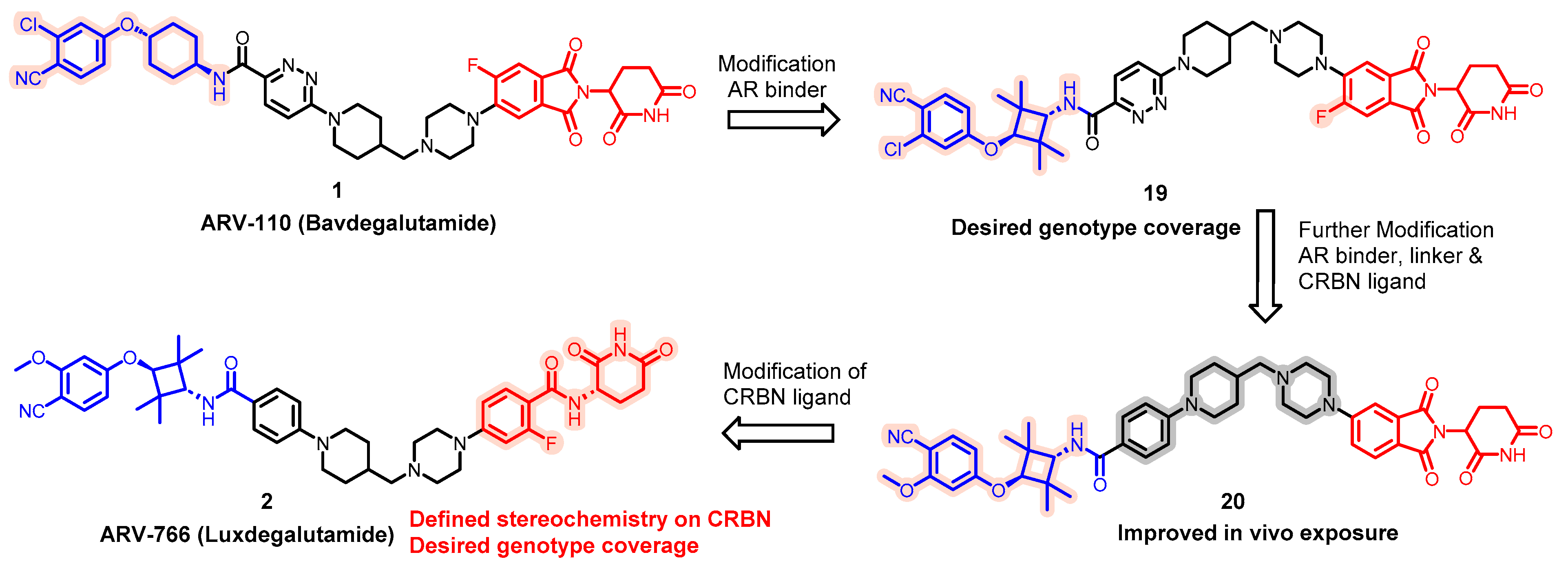
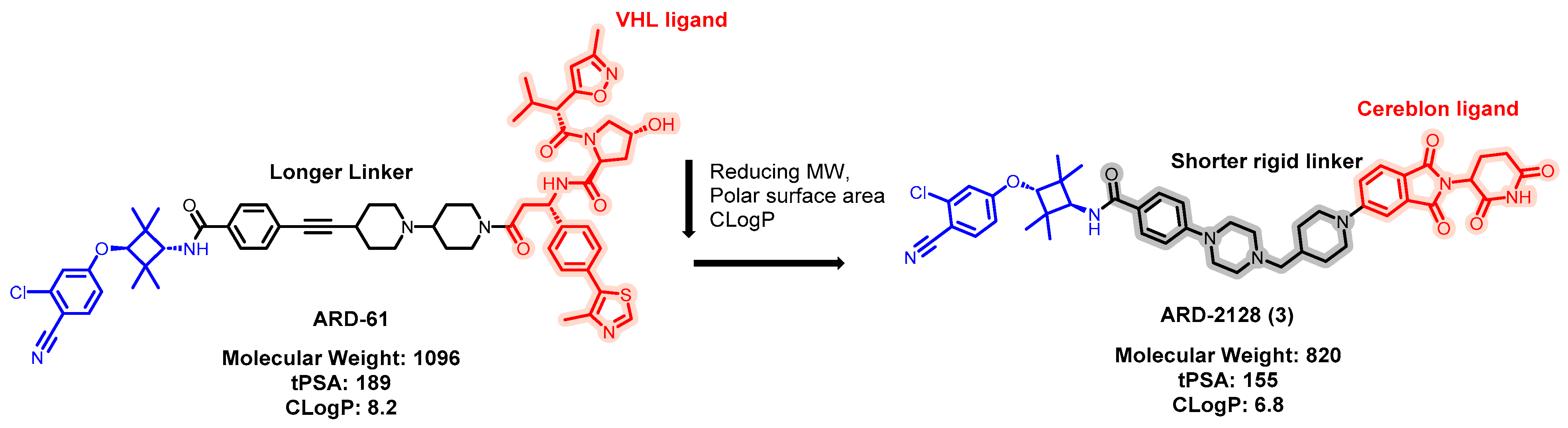
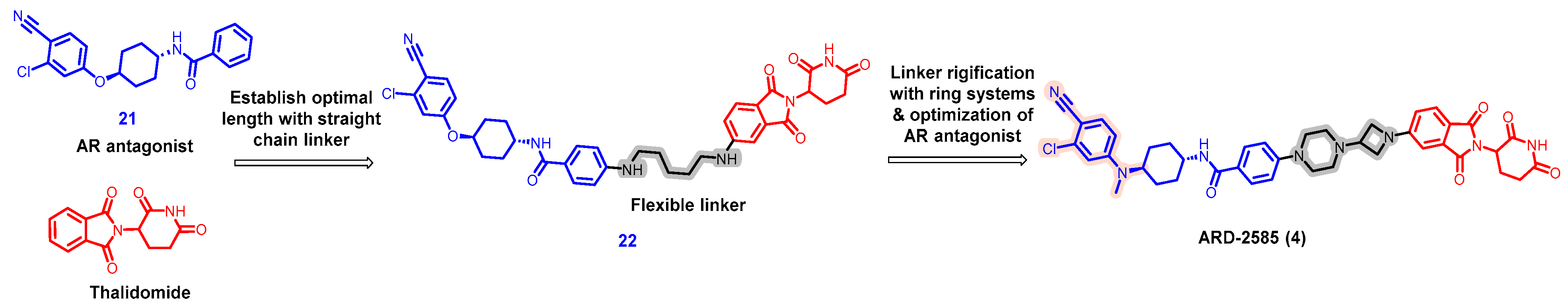
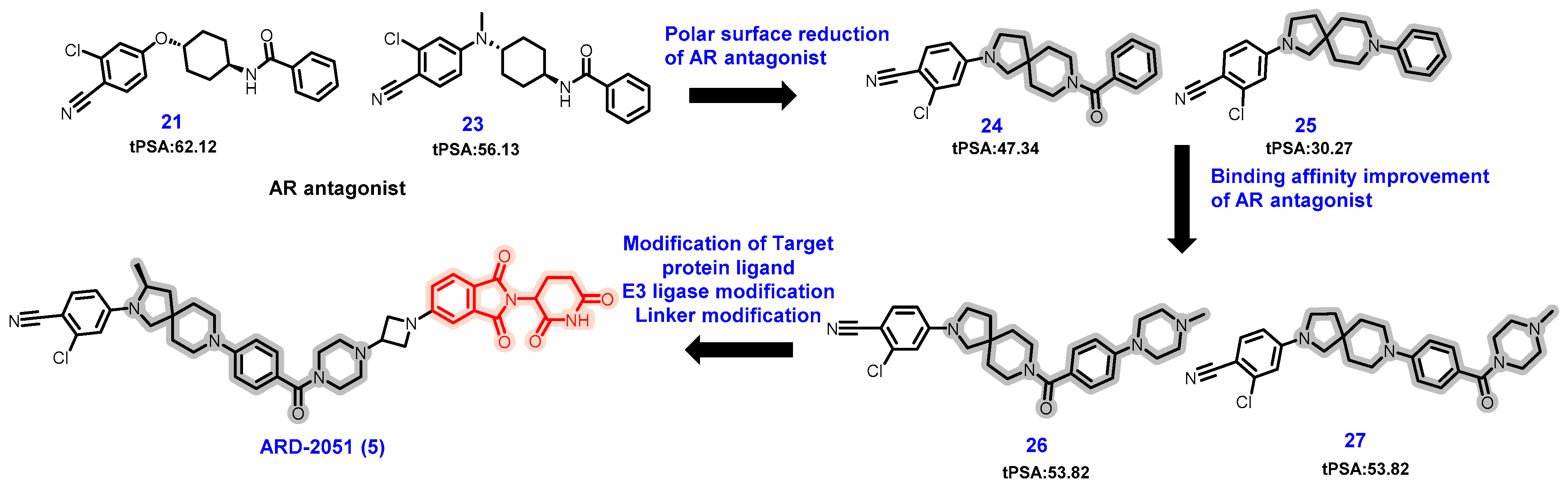
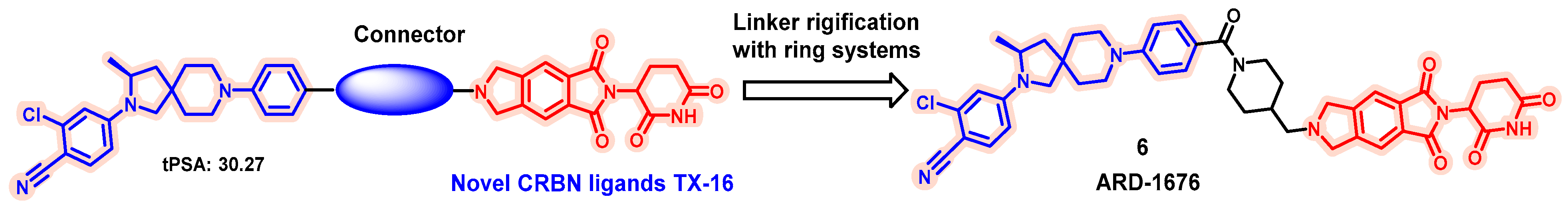
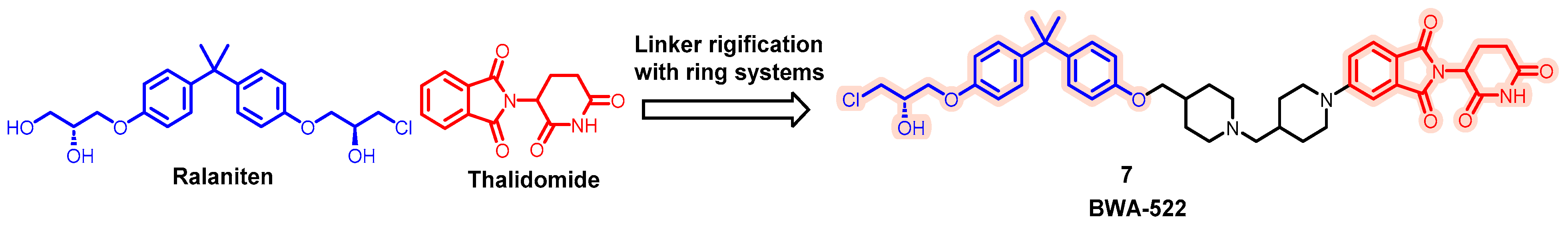
5.1. Discovery of Orally Bioavailable AR PROTAC Degraders for the Treatment of Metastatic Castration-Resistant Prostate Cancer (mCRPC)
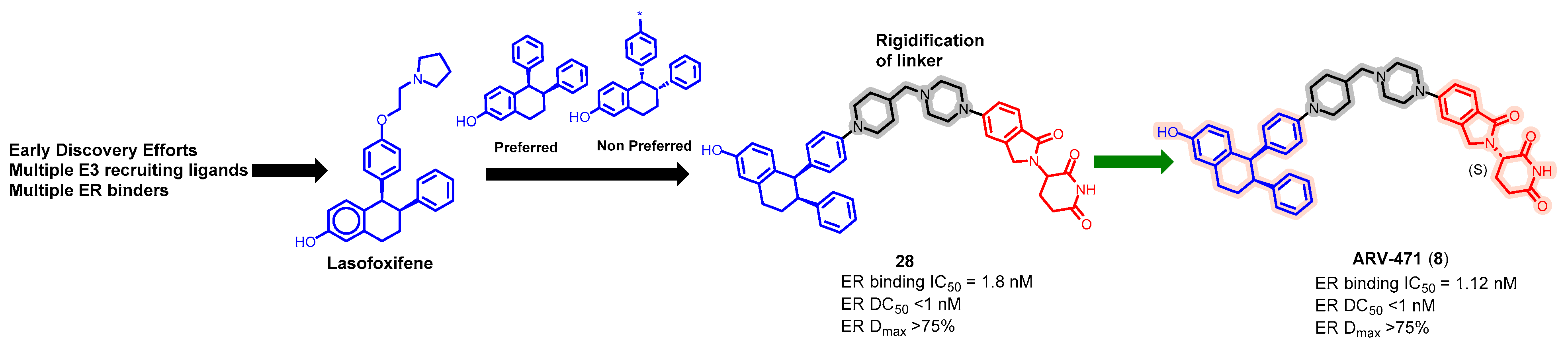
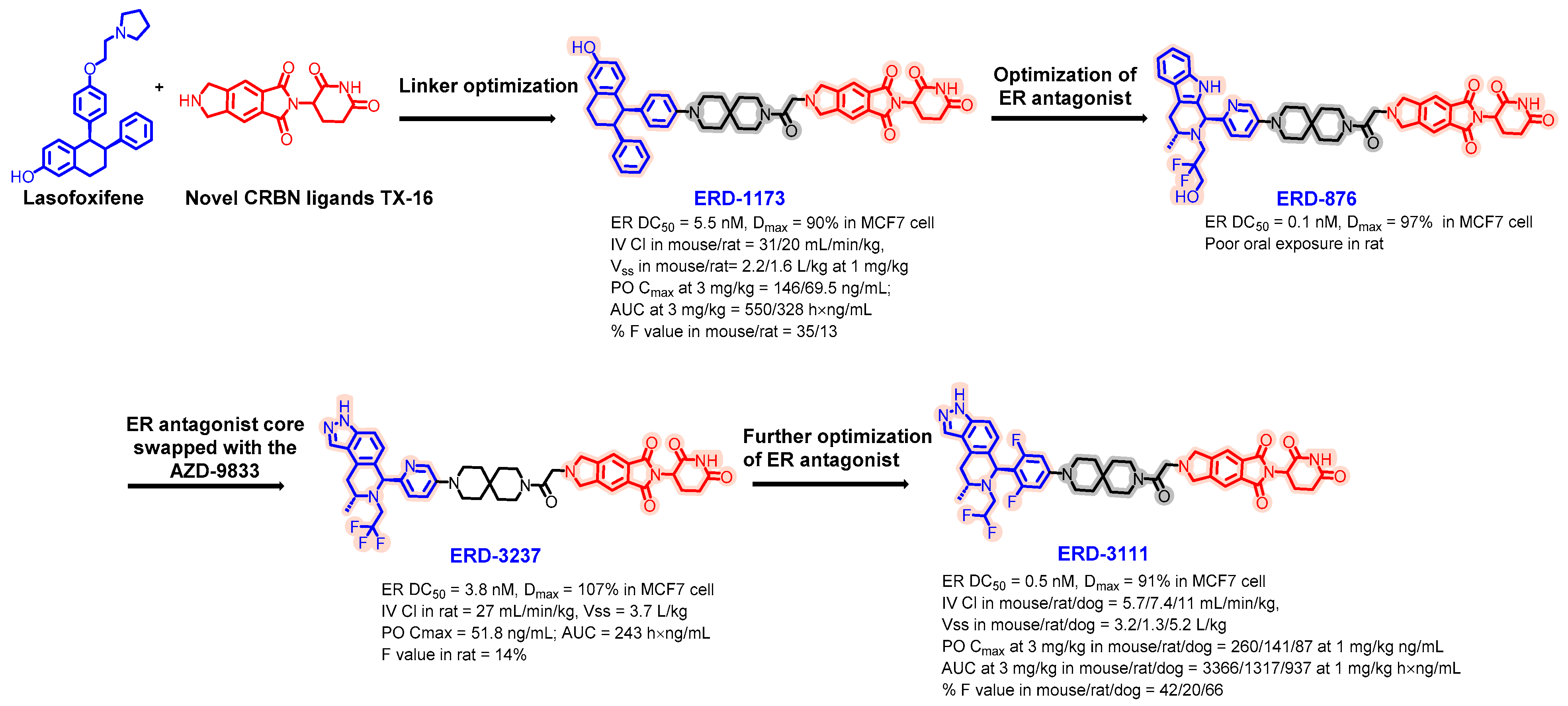
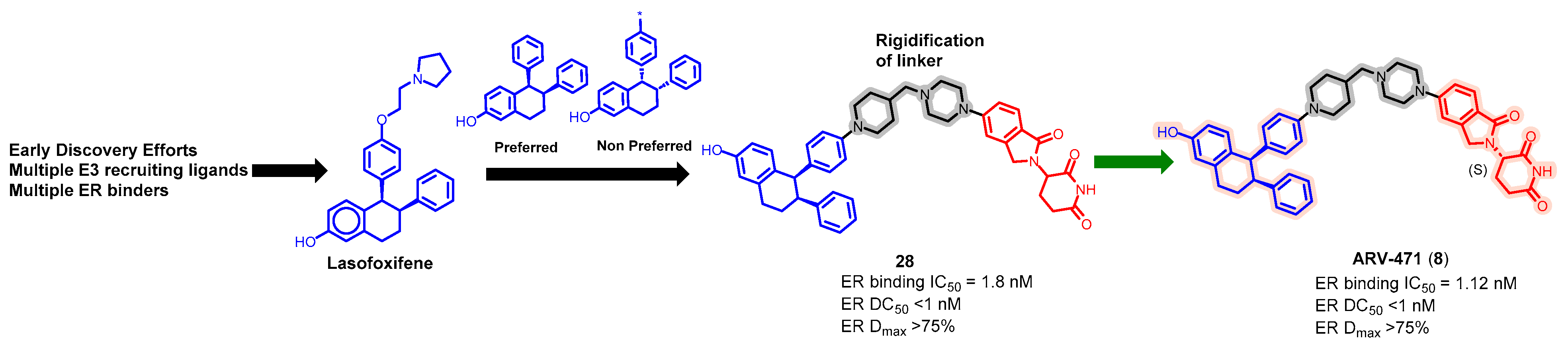
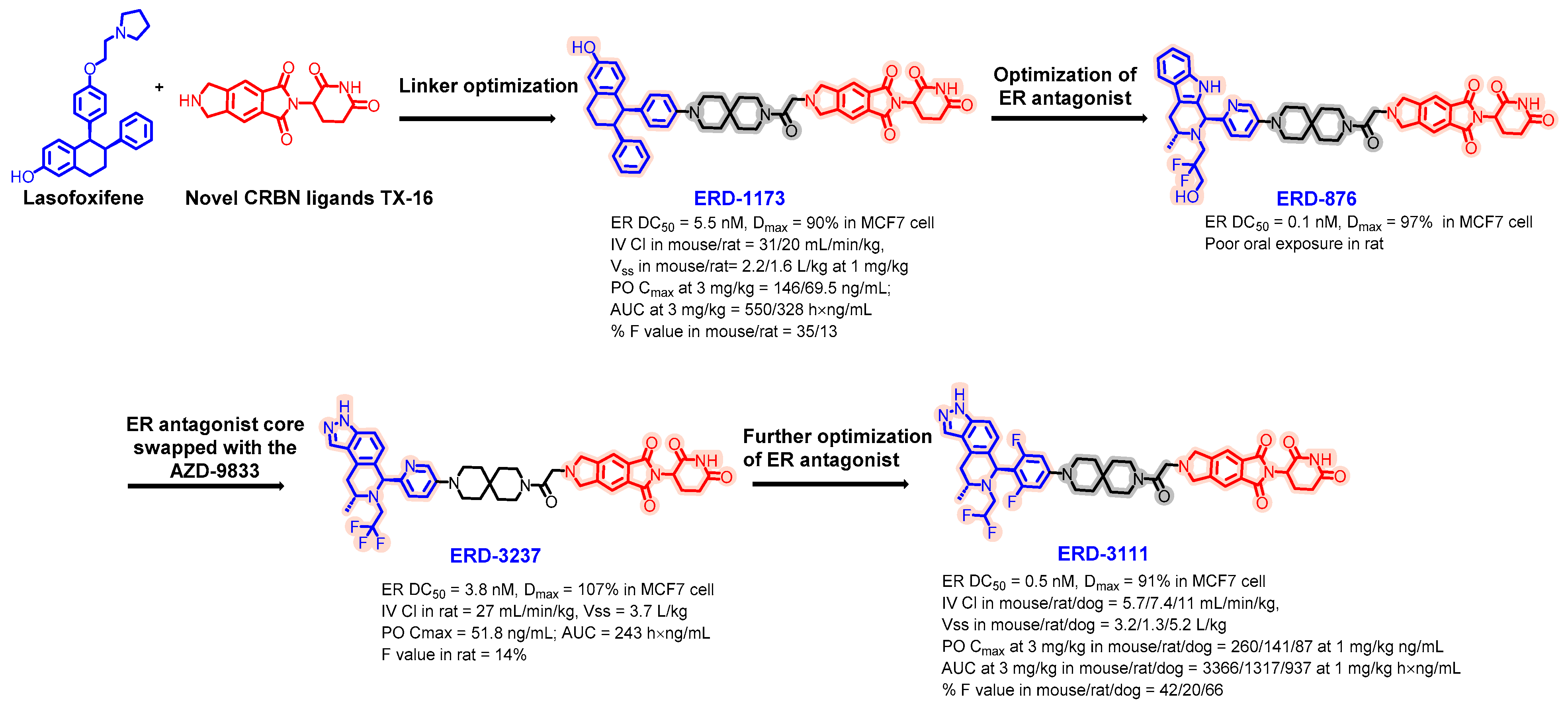
5.2. Discovery of Orally Bioavailable ER PROTAC Degraders for the Treatment of ER+/HER2− Advanced Breast Cancer
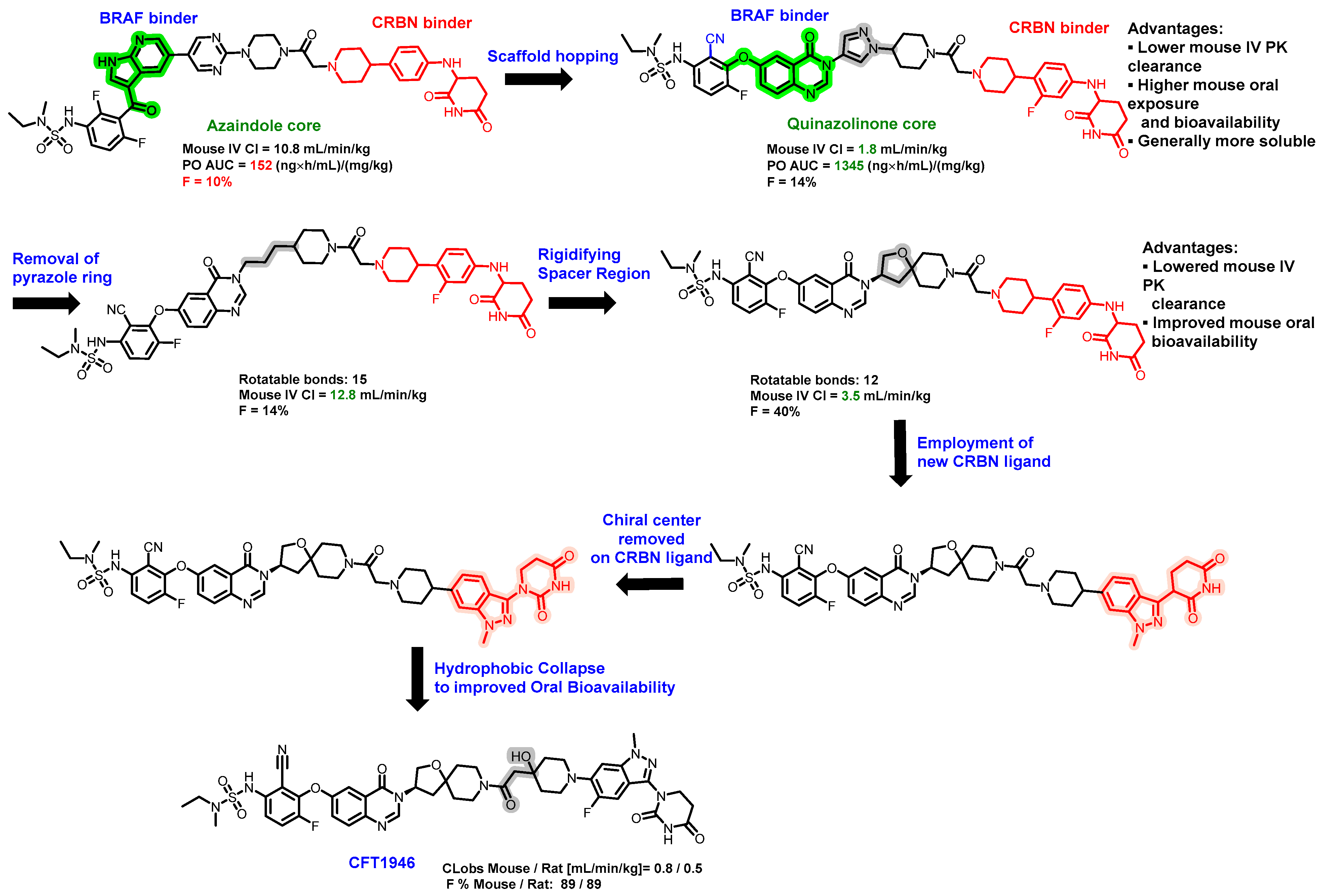
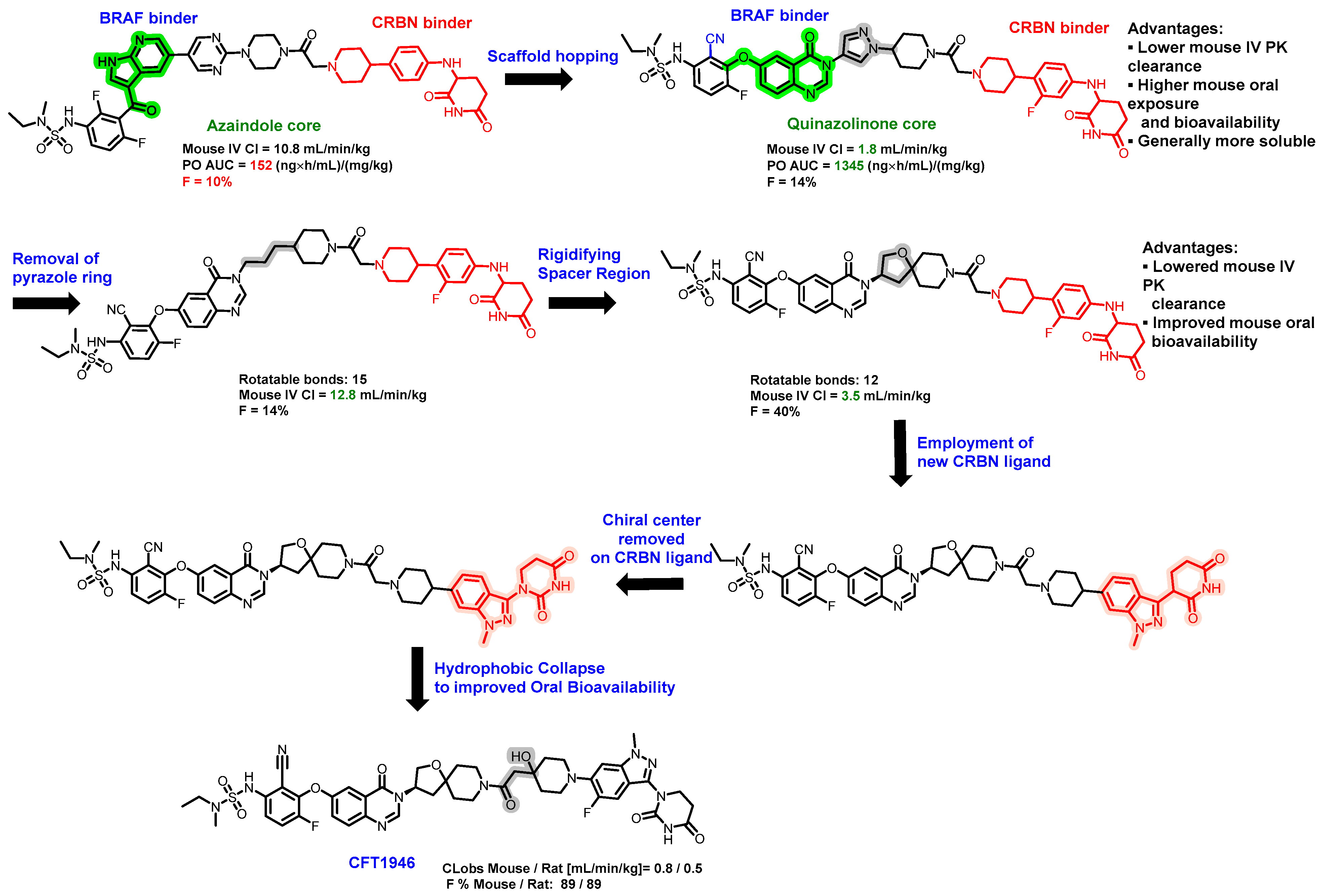
5.3. Discovery of Orally Bioavailable BRAF Degraders
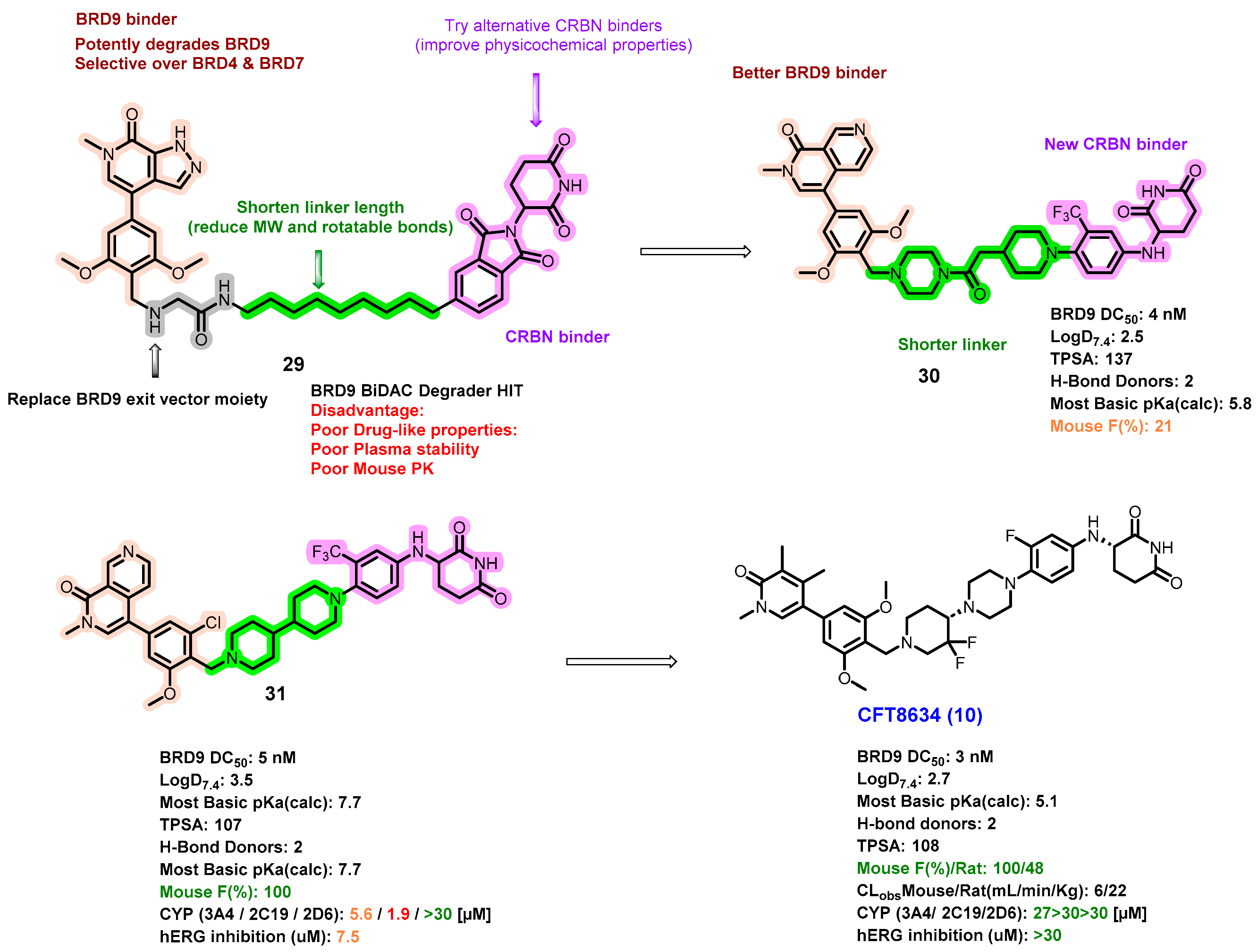
5.4. Discovery of Orally Bioavailable BRD9 Degraders
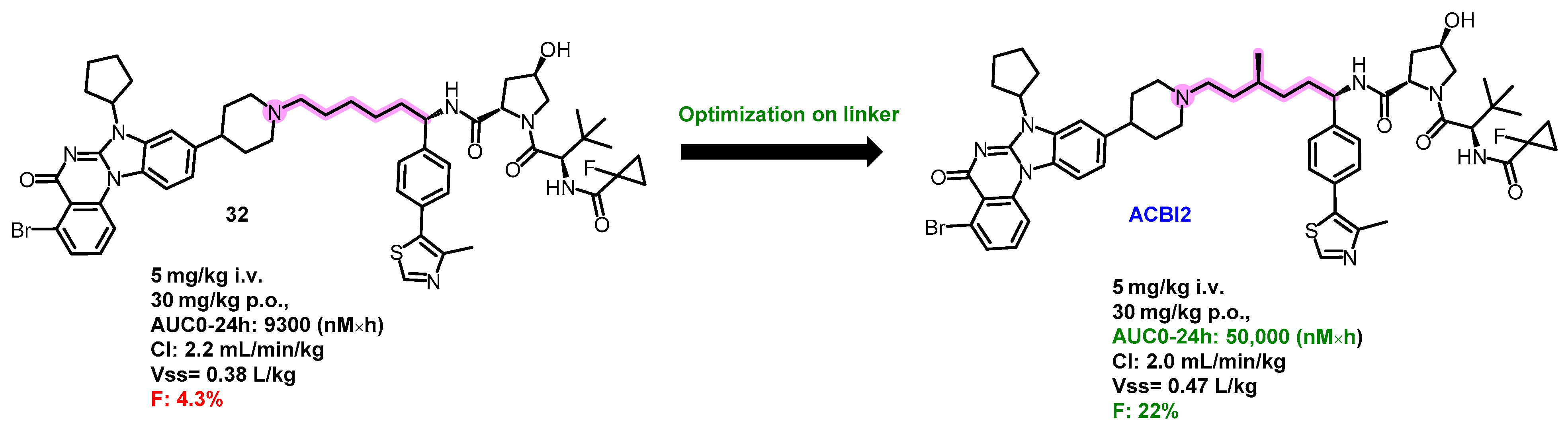
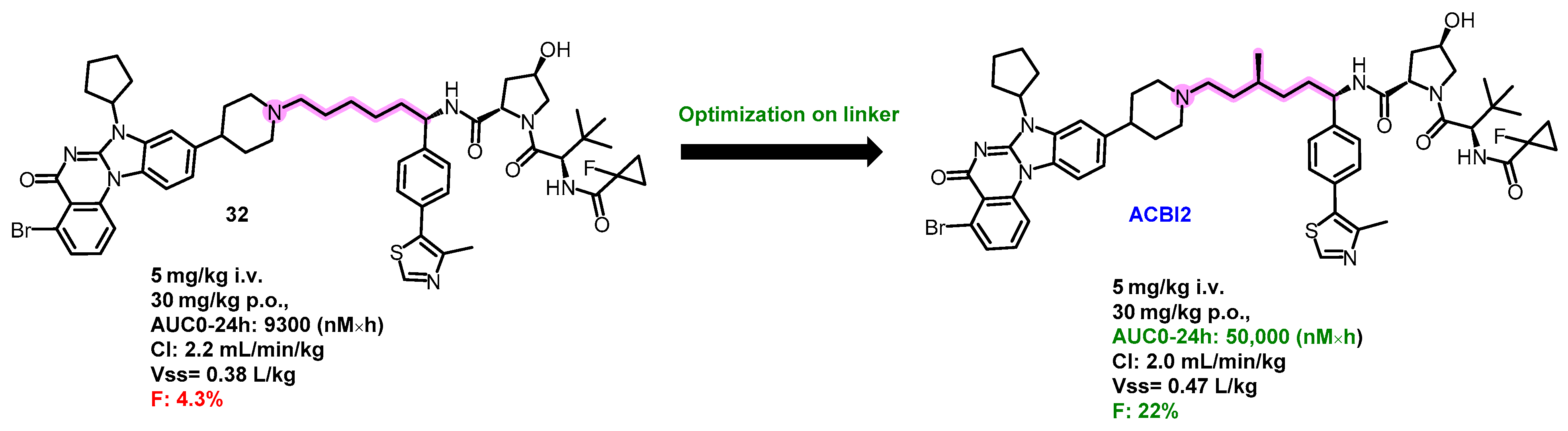
5.5. Oral PROTAC SMARCA2 Degraders
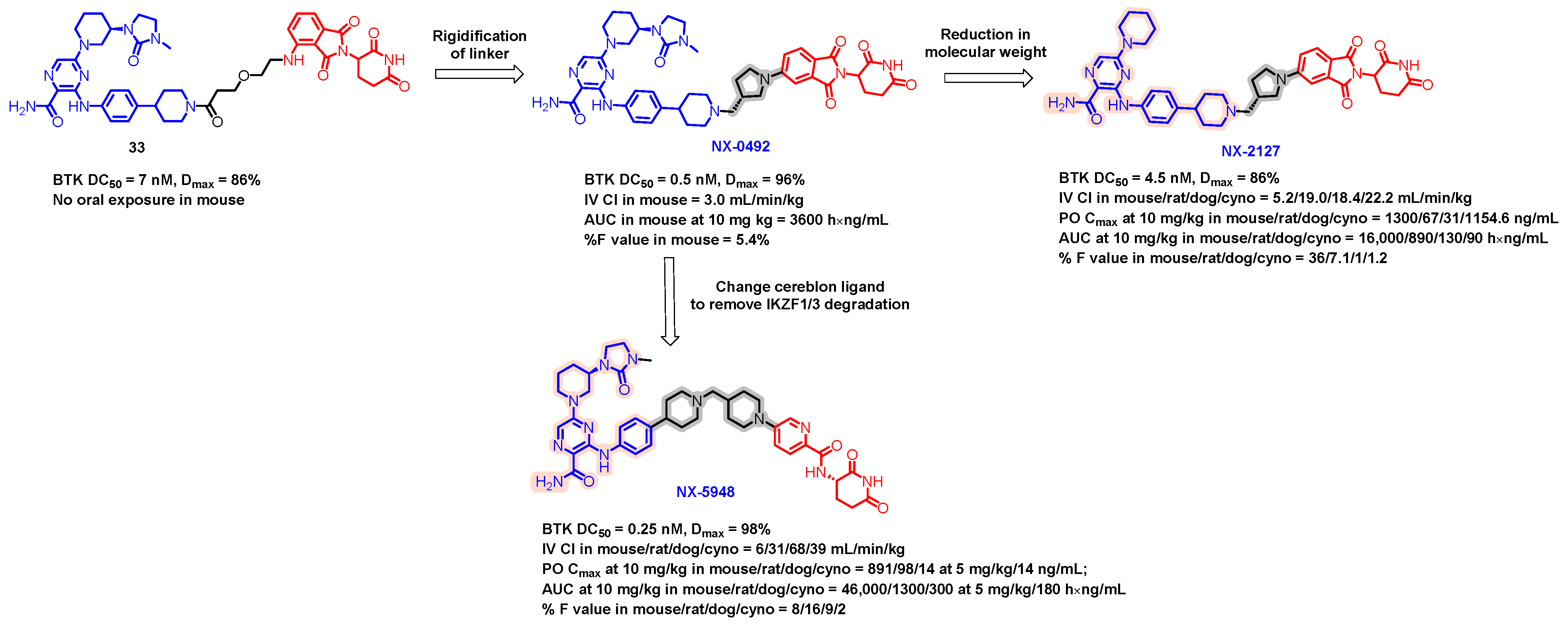
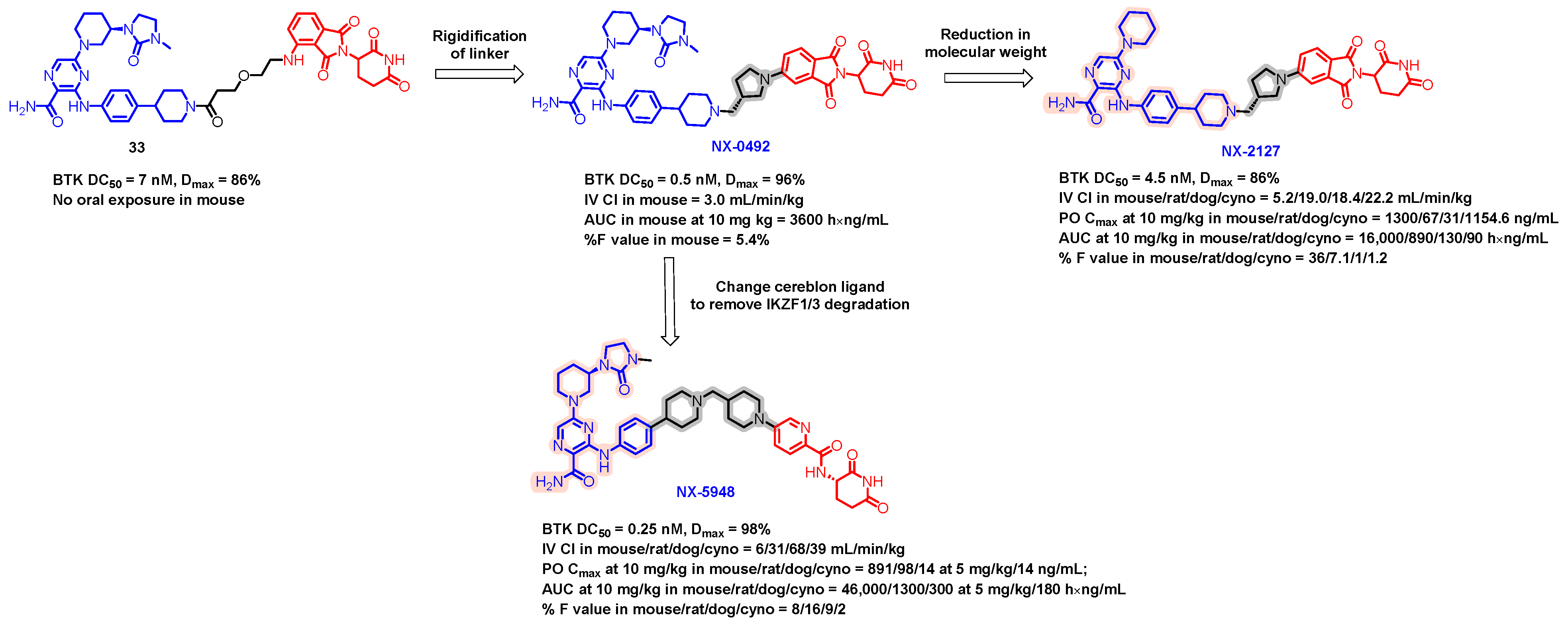
5.6. Discovery of Orally Bioavailable BTK Degraders
5.7. Discovery of Orally Bioavailable EGFR Degraders
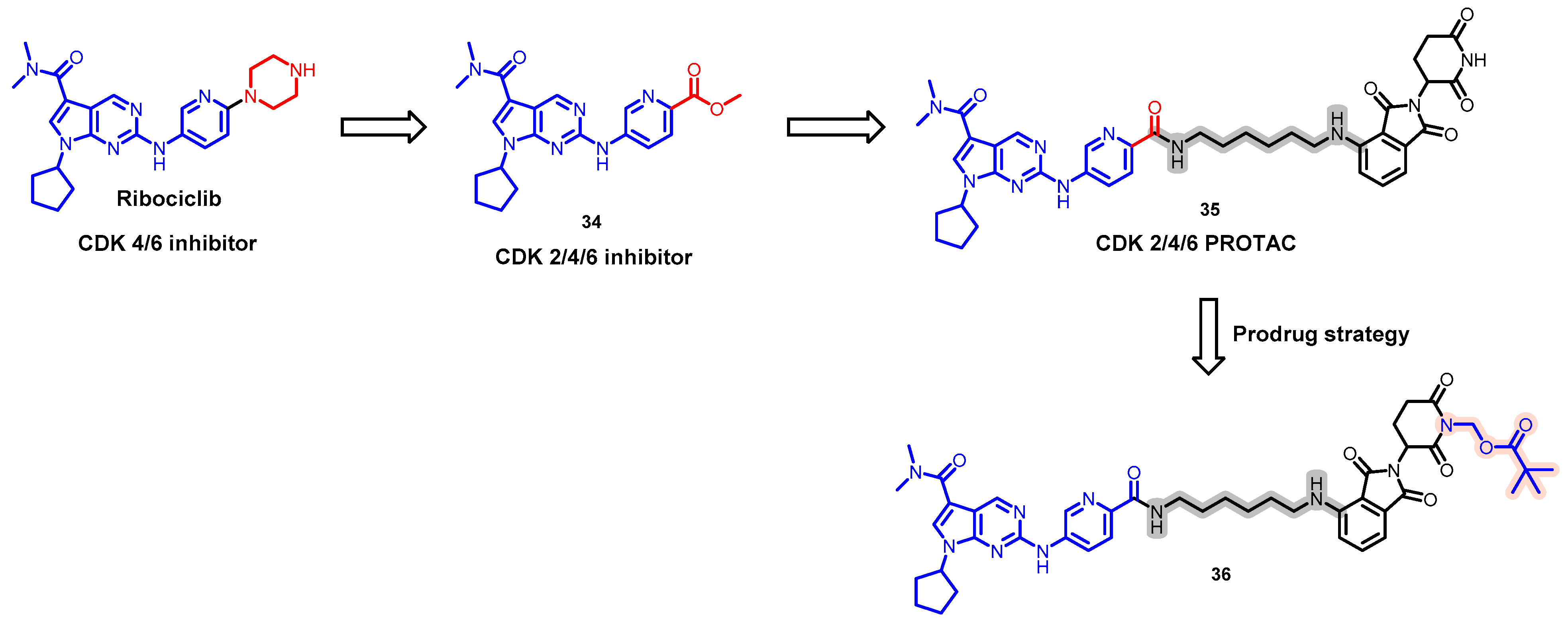
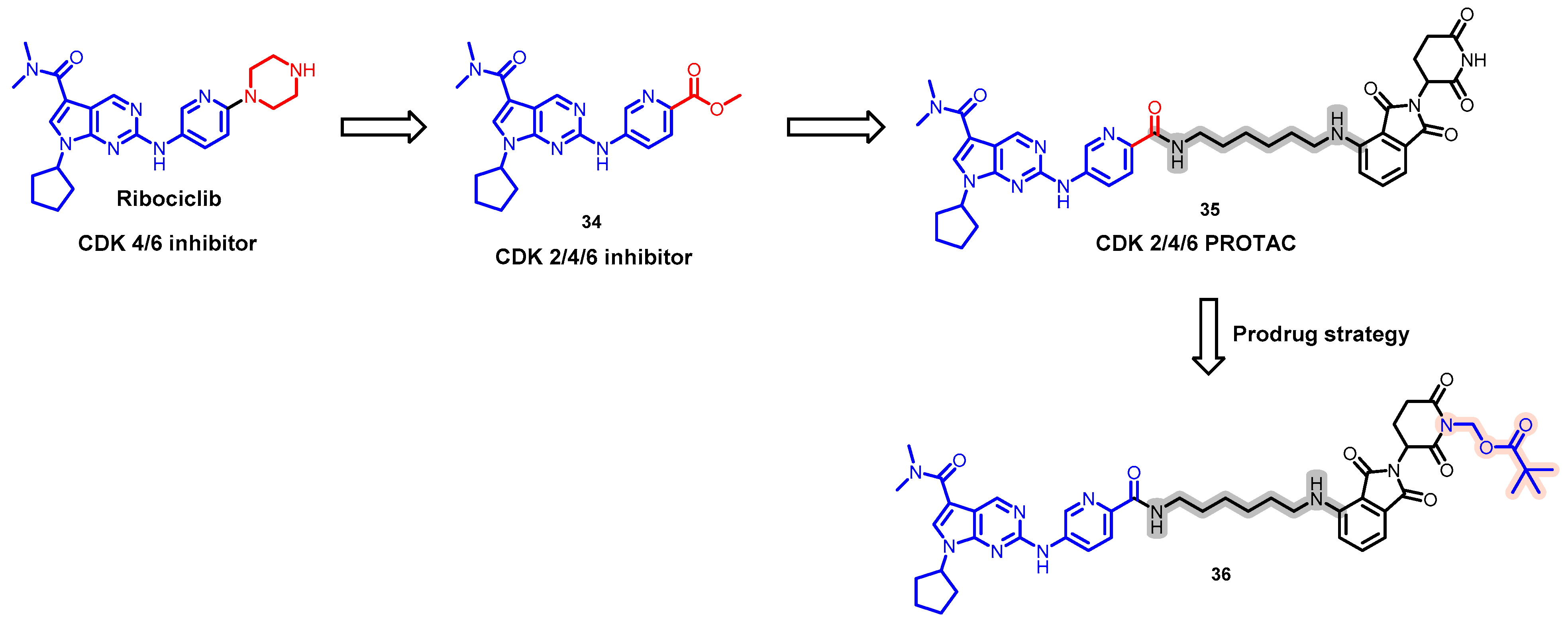
5.8. Discovery of an Orally Bioavailable CDK 2/4/6 Degrader by Employing a Prodrug Strategy
6. Overcoming the Limitations of Lipinski’s Rule of Five: Utilizing Uptake Transporters
7. New Technology Based on PROTACs and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef]
- Nguyen, K.M.; Busino, L. Targeting the E3 ubiquitin ligases DCAF15 and cereblon for cancer therapy. Semin. Cancer Biol. 2020, 67, 53–60. [Google Scholar] [CrossRef]
- Churcher, I. Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? J. Med. Chem. 2018, 61, 444–452. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.M.; Altieri, M.; Dong, H.Q.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Edmondson, S.D.; Yang, B.; Fallan, C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Maple, H.J.; Clayden, N.; Baron, A.; Stacey, C.; Felix, R. Developing degraders: Principles and perspectives on design and chemical space. MedChemComm 2019, 10, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Samarasinghe, K.T.G.; Crews, C.M. Targeted protein degradation: A promise for undruggable proteins. Cell Chem. Biol. 2021, 28, 934–951. [Google Scholar] [CrossRef] [PubMed]
- Dale, B.; Cheng, M.; Park, K.S.; Kaniskan, H.Ü.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef]
- Kannan, M.P.; Sreeraman, S.; Somala, C.S.; Kushwah, R.B.; Mani, S.K.; Sundaram, V.; Thirunavukarasou, A. Advancement of targeted protein degradation strategies as therapeutics for undruggable disease targets. Future Med. Chem. 2023, 15, 867–883. [Google Scholar] [CrossRef]
- Li, R.; Liu, M.; Yang, Z.; Li, J.; Gao, Y.; Tan, R. Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future. Molecules 2022, 27, 8828. [Google Scholar] [CrossRef] [PubMed]
- Cantrill, C.; Chaturvedi, P.; Rynn, C.; Petrig Schaffland, J.; Walter, I.; Wittwer, M.B. Fundamental aspects of DMPK optimization of targeted protein degraders. Drug Discov. Today 2020, 25, 969–982. [Google Scholar] [CrossRef]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I.; Roth, A.G.; Tae, H.S.; Gareiss, P.C.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1a. Angew. Chem. Int. Edit 2012, 51, 11463–11467. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Foley, C.A.; Potjewyd, F.; Lamb, K.N.; James, L.I.; Frye, S.V. Assessing the Cell Permeability of Bivalent Chemical Degraders Using the Chloroalkane Penetration Assay. ACS Chem. Biol. 2020, 15, 290–295. [Google Scholar] [CrossRef]
- Liu, T.T.; Chang, L.J.; Uss, A.; Chu, I.; Morrison, R.A.; Wang, L.; Prelusky, D.; Cheng, K.C.; Li, C. The impact of protein on Caco-2 permeability of low mass balance compounds for absorption projection and efflux substrate identification. J. Pharm. Biomed. 2010, 51, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical high throughput screening: Parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Rooney, T.P.C.; Bayle, E.D.; Mirza, T.; Willems, H.M.G.; Clarke, J.H.; Andrews, S.P.; Skidmore, J. Systematic Investigation of the Permeability of Androgen Receptor PROTACs. ACS Med. Chem. Lett. 2020, 11, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Waters, N.J.; Jones, R.; Williams, G.; Sohal, B. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J. Pharm. Sci. 2008, 97, 4586–4595. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, K.; Cawley, S.; Yates, P.D.; Chang, C.; Funk, C.; Niosi, M.; Lin, J.; Di, L. Plasma Protein Binding of Challenging Compounds. J. Pharm. Sci. 2015, 104, 2627–2636. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.V.; Lake, B.G.; Dickins, M. Quantitative structure-activity relationships (QSARs) in inhibitors of various cytochromes P450: The importance of compound lipophilicity. J. Enzym. Inhib. Med. Chem. 2007, 22, 1–6. [Google Scholar] [CrossRef]
- Pike, A.; Williamson, B.; Harlfinger, S.; Martin, S.; McGinnity, D.F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: A drug metabolism and pharmacokinetics perspective. Drug Discov. Today 2020, 25, 1793–1800. [Google Scholar] [CrossRef]
- Saraswat, A.L.; Vartak, R.; Hegazy, R.; Patel, A.; Patel, K. Drug delivery challenges and formulation aspects of proteolysis targeting chimera (PROTACs). Drug Discov. Today 2023, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pöstges, F.; Kayser, K.; Appelhaus, J.; Monschke, M.; Gütschow, M.; Steinebach, C.; Wagner, K.G. Solubility Enhanced Formulation Approaches to Overcome Oral Delivery Obstacles of PROTACs. Pharmaceutics 2023, 15, 156. [Google Scholar] [CrossRef]
- Alex, A.; Millan, D.S.; Perez, M.; Wakenhut, F.; Whitlock, G.A. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. MedChemComm 2011, 2, 669–674. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.M.; Wu, Y.; Chen, J.J.; Shen, Y.W.; Zhang, L.J.; Zhang, H.; Chen, L.L.; Yuan, H.B.; Chen, H.Z.; Zhang, W.D.; et al. The peptide PROTAC modality: A novel strategy for targeted protein ubiquitination. Theranostics 2020, 10, 10141–10153. [Google Scholar] [CrossRef] [PubMed]
- Rathod, D.; Fu, Y.; Patel, K. BRD4 PROTAC as a novel therapeutic approach for the treatment of vemurafenib resistant melanoma: Preformulation studies, formulation development and evaluation. Eur. J. Pharm. Sci. 2019, 138, 105039. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Qiu, M.; Ma, F.H.; Yang, L.; Glass, Z.; Xu, Q.B. Enhanced protein degradation by intracellular delivery of pre-fused PROTACs using lipid-like nanoparticles. J. Control Release 2021, 330, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Klein, S. The Use of Biorelevant Dissolution Media to Forecast the Performance of a Drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef]
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653. [Google Scholar] [CrossRef]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [CrossRef]
- Anestis, A.; Zoi, I.; Papavassiliou, A.G.; Karamouzis, M.V. Androgen Receptor in Breast Cancer-Clinical and Preclinical Research Insights. Molecules 2020, 25, 358. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.; Collazo, J.; Kyprianou, N. Androgen Receptor as a Driver of Therapeutic Resistance in Advanced Prostate Cancer. Int. J. Biol. Sci. 2014, 10, 588–595. [Google Scholar] [CrossRef]
- Beltran, H.; Beer, T.M.; Carducci, M.A.; de Bono, J.; Gleave, M.; Hussain, M.; Kelly, W.K.; Saad, F.; Sternberg, C.; Tagawa, S.T.; et al. New Therapies for Castration-Resistant Prostate Cancer: Efficacy and Safety. Eur. Urol. 2011, 60, 279–290. [Google Scholar] [CrossRef]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef] [PubMed]
- Mohler, M.L.; Sikdar, A.; Ponnusamy, S.; Hwang, D.J.; He, Y.L.; Miller, D.D.; Narayanan, R. An Overview of Next-Generation Androgen Receptor-Targeted Therapeutics in Development for the Treatment of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 2124. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.; Blair, I.A.; Van Duyne, G.; Hilser, V.J.; Moiseenkova-Bell, V.; Plymate, S.; Sprenger, C.; Wand, A.J.; Penning, T.M. Using biochemistry and biophysics to extinguish androgen receptor signaling in prostate cancer. J. Biol. Chem. 2021, 296, 100240. [Google Scholar] [CrossRef] [PubMed]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.Q.; Jin, M.Z.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
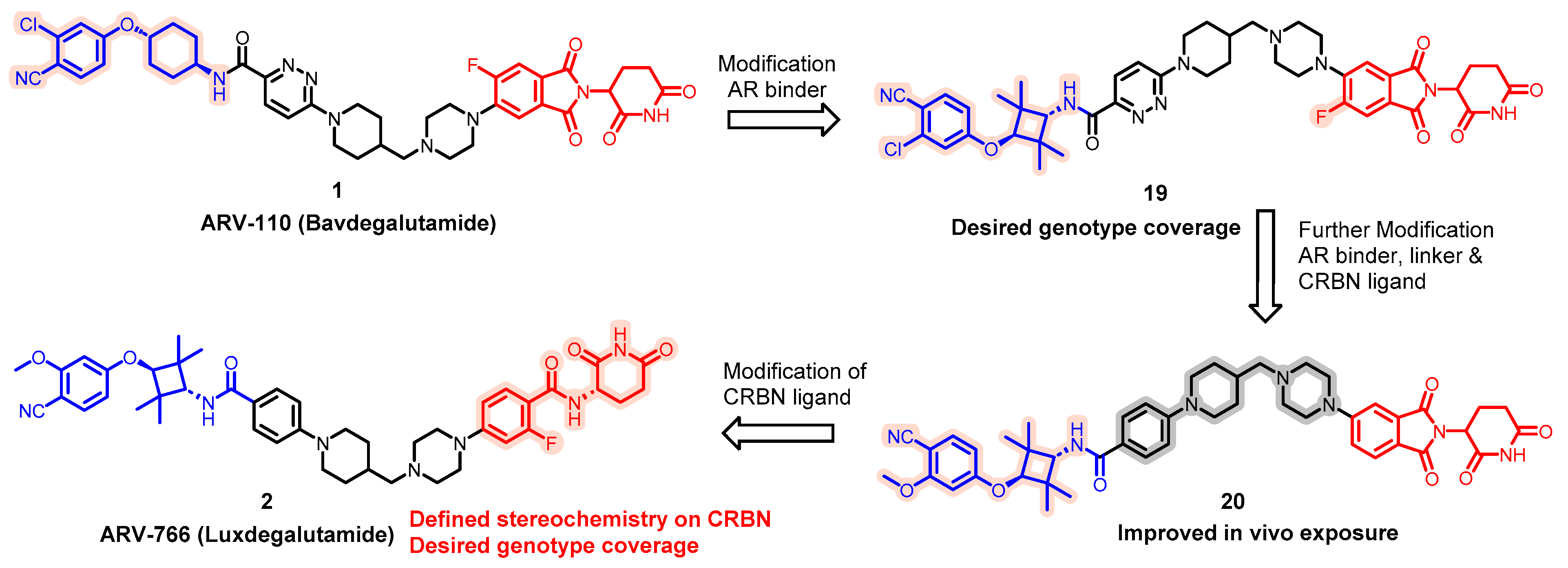
- Snyder, L.B.; Neklesa, T.K.; Chen, X.; Dong, H.; Ferraro, C.; Gordon, D.A.; Macaluso, J.; Pizzano, J.; Wang, J.; Willard, R.R.; et al. Discovery of ARV-110, a first in class androgen receptor degrading PROTAC for the treatment of men with metastatic castration resistant prostate cancer. Cancer Res. 2021, 81, 43. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.Q.; Ferraro, C.; et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef]
- Guo, C.X.; Linton, A.; Kephart, S.; Ornelas, M.; Pairish, M.; Gonzalez, J.; Greasley, S.; Nagata, A.; Burke, B.J.; Edwards, M.; et al. Discovery of Aryloxy Tetramethylcyclobutanes as Novel Androgen Receptor Antagonists. J. Med. Chem. 2011, 54, 7693–7704. [Google Scholar] [CrossRef]
- Han, X.; Sun, Y. Strategies for the discovery of oral PROTAC degraders aimed at cancer therapy. Cell Rep. Phys. Sci. 2022, 3, 101062. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Raina, K.; Pizzano, J.; Gordon, D.; Bookbinder, M.; Macaluso, J.; Dong, H.Q.; et al. ARV-110: An androgen receptor PROTAC degrader for prostate cancer. Cancer Res. 2018, 78, 5236. [Google Scholar] [CrossRef]
- Mullard, A. First targeted protein degrader hits the clinic. Nat. Rev. Drug Discov. 2019, 18, 237–239. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Garmezy, B.; Shen, J.; Kalebasty, A.R.; Sartor, O.; Dreicer, R.; Agarwal, N.; Hussain, M.; Percent, I.; Heath, E.; et al. Phase I/II study of bavdegalutamide, a PROTAC androgen receptor (AR) degrader in metastatic castration-resistant prostate cancer (mCRPC): Radiographic progression-free survival (rPFS) in patients (pts) with AR ligand-binding domain (LBD) mutations. Ann. Oncol. 2023, 34, S973–S974. [Google Scholar] [CrossRef]
- Shore, N.D.; Shen, J.; Devitt, M.E.; Lu, H.; Alicea, J.; Parameswaran, J.; Chirnomas, D.; Gao, X.; McKean, M. Phase 1b study of bavdegalutamide, an androgen receptor PROTAC degrader, combined with abiraterone in patients with metastatic prostate cancer. J. Clin. Oncol. 2022, 40, TPS5106. [Google Scholar] [CrossRef]
- Branch, J.R.; Bush, T.L.; Pande, V.; Connolly, P.J.; Zhang, Z.M.; Hickson, I.; Ondrus, J.; Jaensch, S.; Bischoff, J.R.; Habineza, G.; et al. Discovery of JNJ-63576253, a Next-Generation Androgen Receptor Antagonist Active Against Wild-Type and Clinically Relevant Ligand Binding Domain Mutations in Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2021, 20, 763–774. [Google Scholar] [CrossRef]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted Next-generation Sequencing of Advanced Prostate Cancer Identifies Potential Therapeutic Targets and Disease Heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Zhang, N.; Saha, J.; Nevalaita, L.; Shell, S.A.; Garratt, C.; Ikonen, T.; Fizazi, K. Real-world assessment of AR-LBD mutations in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2023, 41, 204. [Google Scholar] [CrossRef]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; McEachern, D.; Wang, Y.; Metwally, H.; Wang, L.; Matvekas, A.; et al. Strategies toward Discovery of Potent and Orally Bioavailable Proteolysis Targeting Chimera Degraders of Androgen Receptor for the Treatment of Prostate Cancer. J. Med. Chem. 2021, 64, 12831–12854. [Google Scholar] [CrossRef]
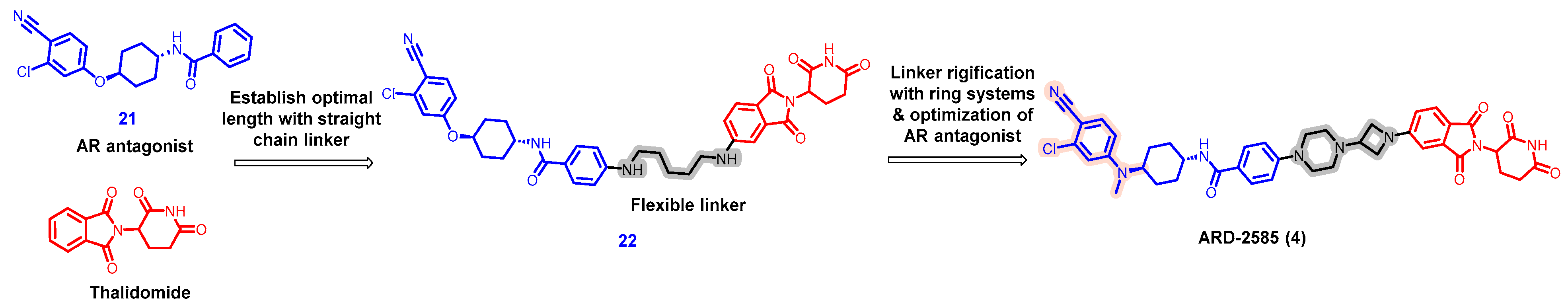
- Xiang, W.; Zhao, L.; Han, X.; Qin, C.; Miao, B.; McEachern, D.; Wang, Y.; Metwally, H.; Kirchhoff, P.D.; Wang, L.; et al. Discovery of ARD-2585 as an Exceptionally Potent and Orally Active PROTAC Degrader of Androgen Receptor for the Treatment of Advanced Prostate Cancer. J. Med. Chem. 2021, 64, 13487–13509. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhao, L.; Xiang, W.; Miao, B.; Qin, C.; Wang, M.; Xu, T.; McEachern, D.; Lu, J.; Wang, Y.; et al. Discovery of ARD-2051 as a Potent and Orally Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor for the Treatment of Advanced Prostate Cancer. J. Med. Chem. 2023, 66, 8822–8843. [Google Scholar] [CrossRef]
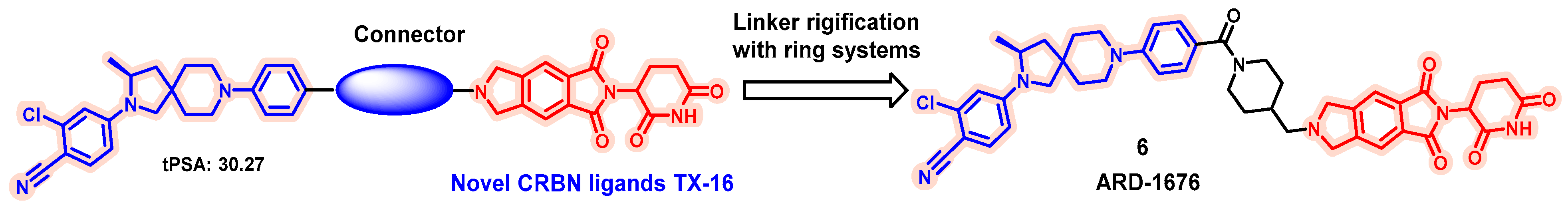
- Xiang, W.; Zhao, L.; Han, X.; Xu, T.; Kregel, S.; Wang, M.; Miao, B.; Qin, C.; Wang, M.; McEachern, D.; et al. Discovery of ARD-1676 as a Highly Potent and Orally Efficacious AR PROTAC Degrader with a Broad Activity against AR Mutants for the Treatment of AR + Human Prostate Cancer. J. Med. Chem. 2023, 66, 13280–13303. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; Xu, T.; Wang, M.; Yang, C.Y.; Chinnaswamy, K.; Stuckey, J.; et al. Discovery of Highly Potent and Efficient PROTAC Degraders of Androgen Receptor (AR) by Employing Weak Binding Affinity VHL E3 Ligase Ligands. J. Med. Chem. 2019, 62, 11218–11231. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Han, X.; Lu, J.; McEachern, D.; Wang, S. A highly potent PROTAC androgen receptor (AR) degrader ARD-61 effectively inhibits AR-positive breast cancer cell growth in vitro and tumor growth in vivo. Neoplasia 2020, 22, 522–532. [Google Scholar] [CrossRef]
- Hornberger, K.R.; Araujo, E.M.V. Physicochemical Property Determinants of Oral Absorption for PROTAC Protein Degraders. J. Med. Chem. 2023, 66, 8281–8287. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Ye, Y.; Xia, H.; Min, J.; Xu, J.; Wang, Z.; Pan, Y.; Zhou, X.; Huang, W. Current advances and development strategies of orally bioavailable PROTACs. Eur. J. Med. Chem. 2023, 261, 115793. [Google Scholar] [CrossRef] [PubMed]
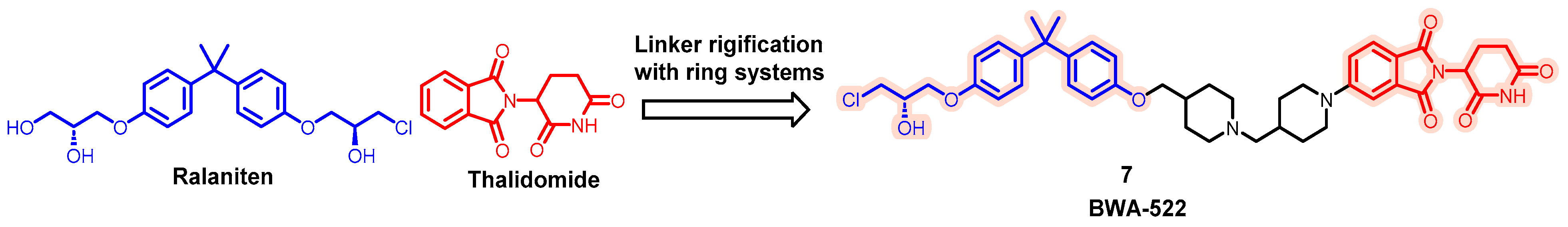
- Zhang, B.; Liu, C.; Yang, Z.; Zhang, S.; Hu, X.; Li, B.; Mao, M.; Wang, X.; Li, Z.; Ma, S.; et al. Discovery of BWA-522, a First-in-Class and Orally Bioavailable PROTAC Degrader of the Androgen Receptor Targeting N-Terminal Domain for the Treatment of Prostate Cancer. J. Med. Chem. 2023, 66, 11158–11186. [Google Scholar] [CrossRef] [PubMed]
- Huppert, L.A.; Gumusay, O.; Idossa, D.; Rugo, H.S. Systemic therapy for hormone receptor-positive/human epidermal growth factor receptor 2-negative early stage and metastatic breast cancer. CA Cancer J. Clin. 2023, 73, 480–515. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Vahdat, L.; Han, H.S.; Ranciato, J.; Gedrich, R.; Keung, C.F.; Chirnomas, D.; Hurvitz, S. First-in-human safety and activity of ARV-471, a novel PROTAC® estrogen receptor degrader, in ER+/HER2-locally advanced or metastatic breast cancer. Cancer Res. 2022, 82, PD13-08. [Google Scholar] [CrossRef]
- Campone, M.; Ma, C.X.; De Laurentiis, M.; Iwata, H.; Hurvitz, S.A.; Wander, S.A.; Danso, M.A.; Lu, D.R.; Smith, J.P.; Liu, Y.; et al. VERITAC-2: A global, randomized phase 3 study of ARV-471, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, vs fulvestrant in ER plus /human epidermal growth factor receptor 2 (HER2)- advanced breast cancer. J. Clin. Oncol. 2023, 41, TPS1122. [Google Scholar] [CrossRef]
- Teh, J.; Bortolon, E.; Pizzano, J.; Pannone, M.; Landrette, S.; Gedrich, R.; Taylor, I. Enhanced efficacy of ARV-471, a novel PROTAC® estrogen receptor degrader, in combination with targeted agents in estrogen receptor-positive (ER plus) breast cancer models. Cancer Res. 2023, 83, 3075. [Google Scholar] [CrossRef]
- Rej, R.K.; Roy, J.; Allu, S.R. Therapies for the Treatment of Advanced/Metastatic Estrogen Receptor-Positive Breast Cancer: Current Situation and Future Directions. Cancers 2024, 16, 552. [Google Scholar] [CrossRef] [PubMed]
- Layman, R.M.; Jerzak, K.J.; Hilton, J.F.; Clifton, K.; Alkuzweny, B.; Mo, G.; Valota, O.; Smith, J.P.; Anderson, S.; Ma, C.X. TACTIVE-U: Phase 1b/2 umbrella study of ARV-471, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, combined with other anticancer treatments in ER plus advanced or metastatic breast cancer. J. Clin. Oncol. 2023, 41, TPS1121. [Google Scholar] [CrossRef]
- Rej, R.K.; Thomas, J.I.I.; Acharyya, R.K.; Rae, J.M.; Wang, S.M. Targeting the Estrogen Receptor for the Treatment of Breast Cancer: Recent Advances and Challenges. J. Med. Chem. 2023, 66, 8339–8381. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, B.; Rej, R.K.; Wu, D.; Acharyya, R.K.; Wang, M.; Xu, T.; Lu, J.; Metwally, H.; Wang, Y.; et al. Discovery of ERD-3111 as a Potent and Orally Efficacious Estrogen Receptor PROTAC Degrader with Strong Antitumor Activity. J. Med. Chem. 2023, 66, 12559–12585. [Google Scholar] [CrossRef]
- Alabi, S.; Jaime-Figueroa, S.; Yao, Z.; Gao, Y.; Hines, J.; Samarasinghe, K.T.G.; Vogt, L.; Rosen, N.; Crews, C.M. Mutant-selective degradation by BRAF-targeting PROTACs. Nat. Commun. 2021, 12, 920. [Google Scholar] [CrossRef] [PubMed]
- Sowa, M.E.; Kreger, B.; Baddour, J.; Liang, Y.K.; Simard, J.R.; Poling, L.; Li, P.; Yu, R.; Hart, A.; Agafonov, R.V.; et al. Preclinical evaluation of CFT1946 as a selective degrader of mutant BRAF for the treatment of BRAF driven cancers. Cancer Res. 2022, 82, 2158. [Google Scholar] [CrossRef]
- Liang, Y.K.; Sowa, M.E.; Jackson, K.L.; Simard, J.R.; Kreger, B.; Li, P.; Poling, L.; Baddour, J.; Good, A.; Huang, H.W.; et al. The discovery and characterization of CFT1946: A potent, selective, and orally bioavailable degrader of mutant BRAF for the treatment of BRAF-driven cancers. Cancer Res. 2023, 83, 3425. [Google Scholar] [CrossRef]
- McKean, M.; Spira, A.I.; Rosen, E.; Subbiah, V.; Moreno, V.; Gambardella, V.; Vieito, M.; Saavedra, O.; Cousin, S.; Cassier, P.A.; et al. A phase 1/2 study of CFT1946, a novel, bifunctional degradation activating compound (BIDAC) degrader, of mutant BRAF V600 as monotherapy and in combination with trametinib, in mutant BRAF V600 solid tumors. J. Clin. Oncol. 2023, 41, TPS3163. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Dou, C.; Sun, L.; Wang, L.; Cheng, J.; Wu, W.; Zhang, C.; Xu, Q.; Tu, K.; Liu, J. Bromodomain-containing protein 9 promotes the growth and metastasis of human hepatocellular carcinoma by activating the TUFT1/AKT pathway. Cell Death Dis. 2020, 11, 730. [Google Scholar] [CrossRef]
- Alpsoy, A.; Utturkar, S.M.; Carter, B.C.; Dhiman, A.; Torregrosa-Allen, S.E.; Currie, M.P.; Elzey, B.D.; Dykhuizen, E.C. BRD9 Is a Critical Regulator of Androgen Receptor Signaling and Prostate Cancer Progression. Cancer Res. 2021, 81, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, X.; Lu, A.; Liang, C. Targeted protein degradation in cancers: Orthodox PROTACs and beyond. Innovation 2023, 4, 100413. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.L.; Agafonov, R.V.; Carlson, M.W.; Chaturvedi, P.; Cocozziello, D.; Cole, K.; Deibler, R.; Eron, S.J.; Good, A.; Hart, A.A.; et al. The discovery and characterization of CFT8634: A potent and selective degrader of BRD9 for the treatment of SMARCB1-perturbed cancers. Cancer Res. 2022, 82, ND09. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Pollack, J.R. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PLoS ONE 2013, 8, e55119. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.R.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E.; Bagdasarian, L.; Huber, J.; Lindeman, A.; Chen, D.S.; et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 3128–3133. [Google Scholar] [CrossRef] [PubMed]
- Oike, T.; Ogiwara, H.; Tominaga, Y.; Ito, K.; Ando, O.; Tsuta, K.; Mizukami, T.; Shimada, Y.; Isomura, H.; Komachi, M.; et al. A Synthetic Lethality-Based Strategy to Treat Cancers Harboring a Genetic Deficiency in the Chromatin Remodeling Factor BRG1. Cancer Res. 2013, 73, 5508–5518. [Google Scholar] [CrossRef]
- Kofink, C.; Trainor, N.; Mair, B.; Wohrle, S.; Wurm, M.; Mischerikow, N.; Roy, M.J.; Bader, G.; Greb, P.; Garavel, G.; et al. A selective and orally bioavailable VHL-recruiting PROTAC achieves SMARCA2 degradation in vivo. Nat. Commun. 2022, 13, 5969. [Google Scholar] [CrossRef]
- Robbins, D.W.; Noviski, M.A.; Tan, Y.S.; Konst, Z.A.; Kelly, A.; Auger, P.; Brathaban, N.; Cass, R.; Chan, M.L.; Cherala, G.; et al. Discovery and Preclinical Pharmacology of NX-2127, an Orally Bioavailable Degrader of Bruton’s Tyrosine Kinase with Immunomodulatory Activity for the Treatment of Patients with B Cell Malignancies. J. Med. Chem. 2024, 67, 2321–2336. [Google Scholar] [CrossRef]
- Danilov, A.; Tees, M.T.; Patel, K.; Wierda, W.G.; Patel, M.; Flinn, I.W.; Latif, T.; Ai, W.Y.; Thompson, M.C.; Wang, M.L.; et al. A First-in-Human Phase 1 Trial of NX-2127, a First-in-Class Bruton’s Tyrosine Kinase (BTK) Dual-Targeted Protein Degrader with Immunomodulatory Activity, in Patients with Relapsed/Refractory B Cell Malignancies. Blood 2023, 142, 4463. [Google Scholar] [CrossRef]
- Montoya, S.; Bourcier, J.; Noviski, M.; Lu, H.; Thompson, M.C.; Chirino, A.; Jahn, J.; Sondhi, A.K.; Gajewski, S.; Tan, Y.S.M.; et al. Kinase-impaired BTK mutations are susceptible to clinical-stage BTK and IKZF1/3 degrader NX-2127. Science 2024, 383, eadi5798. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.R.; Wierda, W.G.; Ai, W.Z.; Flinn, I.W.; Tees, M.; Patel, M.R.; Patel, K.; O’Brien, S.; Bond, D.A.; Roeker, L.E.; et al. NX-2127-001, a First-in-Human Trial of NX-2127, a Bruton’s Tyrosine Kinase-Targeted Protein Degrader, in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia and B-Cell Malignancies. Blood 2022, 140, 2329–2332. [Google Scholar] [CrossRef]
- Huynh, T.; Rodriguez-Rodriguez, S.; Roleder, C.; Powers, J.; Danilov, A. NX-2127 and NX-5948, two clinical stage cereblon-recruiting BTK degraders, facilitate T-cell functionality in CLL. Leuk. Lymphoma 2023, 64, S181–S182. [Google Scholar]
- Linton, K.; Forconi, F.; Lewis, D.; Riches, J.; El-Sharkawi, D.; Gleeson, M.; Injac, S.G.; Nandakumar, S.; Tan, M.; Cherala, G.; et al. Robust Bruton’s Tyrosine Kinase (BTK) Degradation with NX-5948, an Oral BTK Degrader, in a First-in-Human Phase 1a Trial in Relapsed/Refractory B cell Malignancies. Hematol. Oncol. 2023, 41, 573–574. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Du, Y.; Chen, Y.F.; Wang, Y.X.; Chen, J.J.; Lu, X.R.; Zhang, L.; Li, Y.; Wang, Z.F.; Ye, G.Z.; Zhang, G.R. HJM-561, a Potent, Selective, and Orally Bioavailable EGFR PROTAC that Overcomes Osimertinib-Resistant EGFR Triple Mutations. Mol. Cancer Ther. 2022, 21, 1060–1066. [Google Scholar] [CrossRef]
- Johnson, D.G.; Walker, C.L. Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 295–312. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Zhao, R.; Cao, Y.; Wei, Y.; Li, M.; Dong, Z.; Liu, Y.; Ruan, H.; Li, Y.; Cao, S.; et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo. Eur. J. Med. Chem. 2021, 209, 112903. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Gray, N.S. The rise of degrader drugs. Cell Chem. Biol. 2023, 30, 864–878. [Google Scholar] [CrossRef]
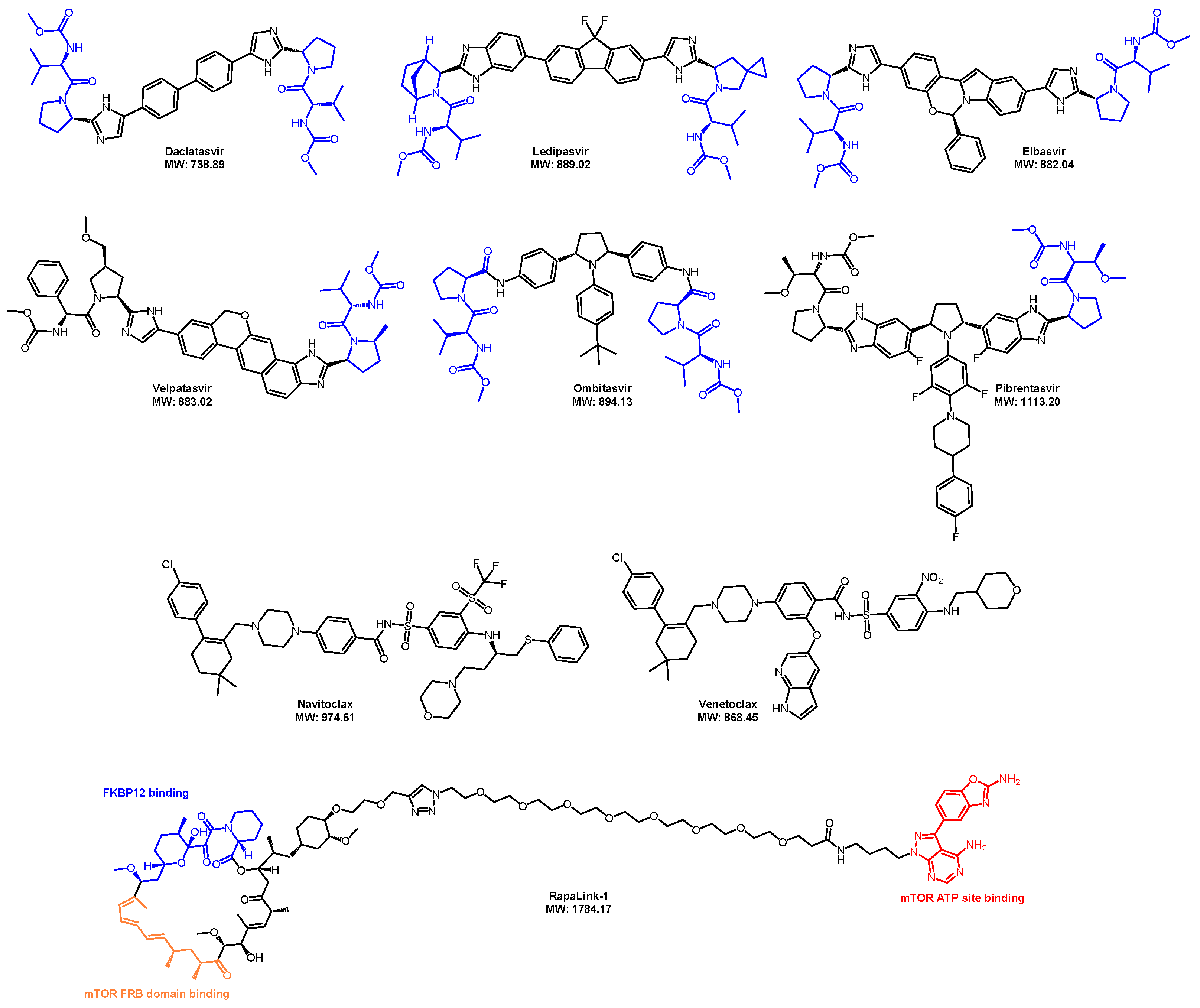
- Belema, M.; Nguyen, V.N.; Bachand, C.; Deon, D.H.; Goodrich, J.T.; James, C.A.; Lavoie, R.; Lopez, O.D.; Martel, A.; Romine, J.L.; et al. Hepatitis C virus NS5A replication complex inhibitors: The discovery of daclatasvir. J. Med. Chem. 2014, 57, 2013–2032. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R., 2nd; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Park, C.M.; Bruncko, M.; Adickes, J.; Bauch, J.; Ding, H.; Kunzer, A.; Marsh, K.C.; Nimmer, P.; Shoemaker, A.R.; Song, X.; et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J. Med. Chem. 2008, 51, 6902–6915. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, J.A.; Wang, S.W.J.; Knemeyer, I.W.; Wirth, M.A.; Alton, K.B. Intestinal lymphatic transport for drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 923–942. [Google Scholar] [CrossRef]
- Lou, K.; Wassarman, D.R.; Yang, T.P.; Paung, Y.; Zhang, Z.Y.; O’Loughlin, T.A.; Moore, M.K.; Egan, R.K.; Greninger, P.; Benes, C.H.; et al. IFITM proteins assist cellular uptake of diverse linked chemotypes. Science 2022, 378, 1097–1104. [Google Scholar] [CrossRef]
- Burnett, G.L.; Yang, Y.C.; Aggen, J.B.; Pitzen, J.; Gliedt, M.K.; Semko, C.M.; Marquez, A.; Evans, J.W.; Wang, G.; Won, W.S.; et al. Discovery of RMC-5552, a Selective Bi-Steric Inhibitor of mTORC1, for the Treatment of mTORC1-Activated Tumors. J. Med. Chem. 2023, 66, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.Y.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed]
- Poongavanam, V.; Doak, B.C.; Kihlberg, J. Opportunities and guidelines for discovery of orally absorbed drugs in beyond rule of 5 space. Curr. Opin. Chem. Biol. 2018, 44, 23–29. [Google Scholar] [CrossRef]
- He, M.; Cao, C.; Ni, Z.; Liu, Y.; Song, P.; Hao, S.; He, Y.; Sun, X.; Rao, Y. PROTACs: Great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target. Ther. 2022, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Gao, F.; Ma, J.; Ma, H.; Dong, G.; Sheng, C. Aptamer-PROTAC Conjugates (APCs) for Tumor-Specific Targeting in Breast Cancer. Angew. Chem. Int. Ed. Engl. 2021, 60, 23299–23305. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Huo, J.; Gu, X.; Wang, Y.; Wu, C.; Zhang, Q.; Wang, W.; Liu, Y.; Liu, Y.; Zhou, X.; et al. Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP. J. Med. Chem. 2021, 64, 7839–7852. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Liu, Y.; Shen, Y.; Meng, F.; Kaniskan, H.U.; Jin, J.; Wei, W. Cancer Selective Target Degradation by Folate-Caged PROTACs. J. Am. Chem. Soc. 2021, 143, 7380–7387. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Kaniskan, H.U.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Degrader | Target | Title | Conditions | Clinical Trial Number (Status) |
|---|---|---|---|---|
| ARV-110 | AR | Trial of ARV-110 and Abiraterone in Patients With Metastatic Castration-Resistant Prostate Cancer (mCRPC) | Prostate Cancer Metastatic | NCT05177042 (Phase I) |
| Trial of ARV-110 in Patients With Metastatic Castration-Resistant Prostate Cancer | Prostate Cancer Metastatic | NCT03888612 (Phase II) | ||
| ARV-766 | AR | A Study of ARV-766 Given by Mouth in Men With Metastatic Prostate Cancer | Prostate Cancer Metastatic | NCT05067140 (Phase II) |
| AC176 | AR | A Study of AC176 for the Treatment of Metastatic Castration-Resistant Prostate Cancer | Metastatic Castration-Resistant Prostate Cancer | NCT05241613 (Phase I) |
| CC-94676 | AR | Study to Evaluate the Safety and Tolerability of CC-94676 in Participants With Metastatic Castration-Resistant Prostate Cancer | Prostatic Neoplasms | NCT04428788 (Phase I) |
| ARV-471 | ER | A Phase 1/2 Trial of ARV-471 Alone and in Combination With Palbociclib (IBRANCE®) in Patients With ER+/HER2− Locally Advanced or Metastatic Breast Cancer | Breast Cancer | NCT04072952 (Phase I/II) |
| ARV-471 in Combination With Everolimus for the Treatment of Advanced or Metastatic ER+, HER2− Breast Cancer | Breast Cancer | NCT05501769 (Phase 1) | ||
| A Study of ARV-471 (PF-07850327) Plus Palbociclib Versus Letrozole Plus Palbociclib in Participants With Estrogen Receptor Positive, Human Epidermal Growth Factor Negative Advanced Breast Cancer | Breast Cancer | NCT05909397 (Phase 3) | ||
| A Study to Learn About a New Medicine Called ARV-471 (PF-07850327) in People Who Have Advanced Metastatic Breast Cancer. | Advanced Breast Cancer | NCT05654623 (Phase 3) | ||
| AC682 | ER | A Study of AC682 for the Treatment of Locally Advanced or Metastatic ER+ Breast Cancer | Breast Cancer | NCT05080842 (Phase 1) |
| NX-2127 | BTK | A Study of NX-2127 in Adults With Relapsed/Refractory B-cell Malignancies | B cell malignancies | NCT04830137 (Phase I) |
| NX-5948 | BTK | A Study of NX-5948 in Adults With Relapsed/Refractory B-cell Malignancies | B cell malignancies | NCT05131022 (Phase I) |
| CFT8634 | BRD9 | A Study to Assess the Safety and Tolerability of CFT8634 in Locally Advanced or Metastatic SMARCB1-Perturbed Cancers, Including Synovial Sarcoma and SMARCB1-Null Tumors | Synovial Sarcoma, Soft Tissue Sarcoma | NCT05355753 (Phase I) |
| CFT1946 | BRAFV600E | A Study to Characterize the Safety, Tolerability, and Preliminary Efficacy of CFT1946 as Monotherapy and in Combination With Trametinib in Subjects With BRAFV600 Mutant Solid Tumors | Solid Tumors, Melanoma, NSCLC | NCT05668585 (Phase 1/II) |
| Estimates of AR LBD Mutation Prevalence | |||
|---|---|---|---|
| AR LBD Mutation | 2016 | 2020 | 2023 |
| L702H | ~2% | ~9% | ~11% |
| T878X | ~6% | ~6% | ~8% |
| H875Y | ~4% | ~4% | ~5% |
| Potential to Improve Outcomes in Patients with Prostate Cancer | Bavdegalutamide | ARV-766 |
|---|---|---|
| Degrades wild type and amplified AR |  | |
| Targets all AR LBD mutations | No L702H | |
| Tolerability suitable for mCRPC and mCSPC | | |
| PSA50 in patients with tumors harboring the L702H mutation | 7% (2 of 24) | 50% (4 of 8) |
| Addressable mCRPC patient population | ~11,000 (6–9%) | ~35,000 (~25%) |
| ID | Oral Degraders | MW | HBD | HBA | RB | TPSA | NAr | CLogP | Fraction Csp3 |
|---|---|---|---|---|---|---|---|---|---|
| ARV-110 |  | 812 | 2 | 11 | 10 | 181 | 3 | 4.3 | 0.46 |
| ARV-766 | 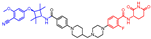 | 807 | 3 | 9 | 13 | 156 | 3 | 5.9 | 0.49 |
| ARD-2128 |  | 820 | 2 | 8 | 10 | 155 | 3 | 6.8 | 0.47 |
| ARD-2585 |  | 763 | 2 | 7 | 9 | 149 | 3 | 4.7 | 0.41 |
| ARD-2051 |  | 789 | 1 | 7 | 7 | 140 | 3 | 4.7 | 0.44 |
| ARD-1676 | 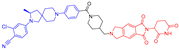 | 788 | 1 | 7 | 7 | 137 | 3 | 4.1 | 0.45 |
| BWA-522 | 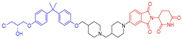 | 770 | 2 | 8 | 13 | 128 | 3 | 5.9 | 0.49 |
| ARV-471 | 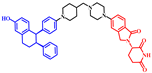 | 724 | 2 | 5 | 7 | 96 | 4 | 6.7 | 0.40 |
| ERD-3111 |  | 854 | 2 | 12 | 8 | 142 | 4 | 4.8 | 0.47 |
| CFT1946 | 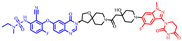 | 957 | 3 | 15 | 12 | 245 | 4 | -0.55 | 0.44 |
| CFT8634 | 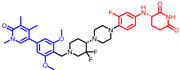 | 710 | 2 | 10 | 9 | 108 | 3 | 4.5 | 0.49 |
| ACBI2 | 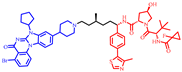 | 1064 | 3 | 9 | 19 | 182 | 5 | 10.3 | 0.54 |
| NX-2127 |  | 719 | 3 | 8 | 9 | 174 | 3 | 4.4 | 0.46 |
| NX-5918 | 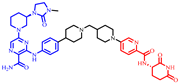 | 807 | 4 | 9 | 12 | 202 | 3 | 3.6 | 0.52 |
| HJM-561 | 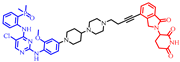 | 864 | 3 | 9 | 11 | 151 | 4 | 2.1 | 0.40 |
| Ro5/Veber | 500 | 5 | 10 | 10 | 140 | ND | 5 | ND | |
| bRo5 | 1000 | 6 | 15 | 20 | 250 | ND | 7.5 | ND | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rej, R.K.; Allu, S.R.; Roy, J.; Acharyya, R.K.; Kiran, I.N.C.; Addepalli, Y.; Dhamodharan, V. Orally Bioavailable Proteolysis-Targeting Chimeras: An Innovative Approach in the Golden Era of Discovering Small-Molecule Cancer Drugs. Pharmaceuticals 2024, 17, 494. https://doi.org/10.3390/ph17040494
Rej RK, Allu SR, Roy J, Acharyya RK, Kiran INC, Addepalli Y, Dhamodharan V. Orally Bioavailable Proteolysis-Targeting Chimeras: An Innovative Approach in the Golden Era of Discovering Small-Molecule Cancer Drugs. Pharmaceuticals. 2024; 17(4):494. https://doi.org/10.3390/ph17040494
Chicago/Turabian StyleRej, Rohan Kalyan, Srinivasa Rao Allu, Joyeeta Roy, Ranjan Kumar Acharyya, I. N. Chaithanya Kiran, Yesu Addepalli, and V. Dhamodharan. 2024. "Orally Bioavailable Proteolysis-Targeting Chimeras: An Innovative Approach in the Golden Era of Discovering Small-Molecule Cancer Drugs" Pharmaceuticals 17, no. 4: 494. https://doi.org/10.3390/ph17040494
APA StyleRej, R. K., Allu, S. R., Roy, J., Acharyya, R. K., Kiran, I. N. C., Addepalli, Y., & Dhamodharan, V. (2024). Orally Bioavailable Proteolysis-Targeting Chimeras: An Innovative Approach in the Golden Era of Discovering Small-Molecule Cancer Drugs. Pharmaceuticals, 17(4), 494. https://doi.org/10.3390/ph17040494