
Development and Validation of Novel HPLC Methods for Quantitative Determination of Vitamin D3 in Tablet Dosage Form


 , , and
, , and
Abstract
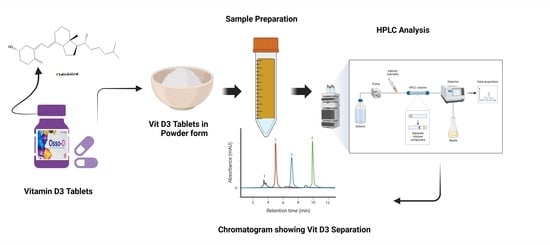
:
1. Introduction
2. Results and Discussion
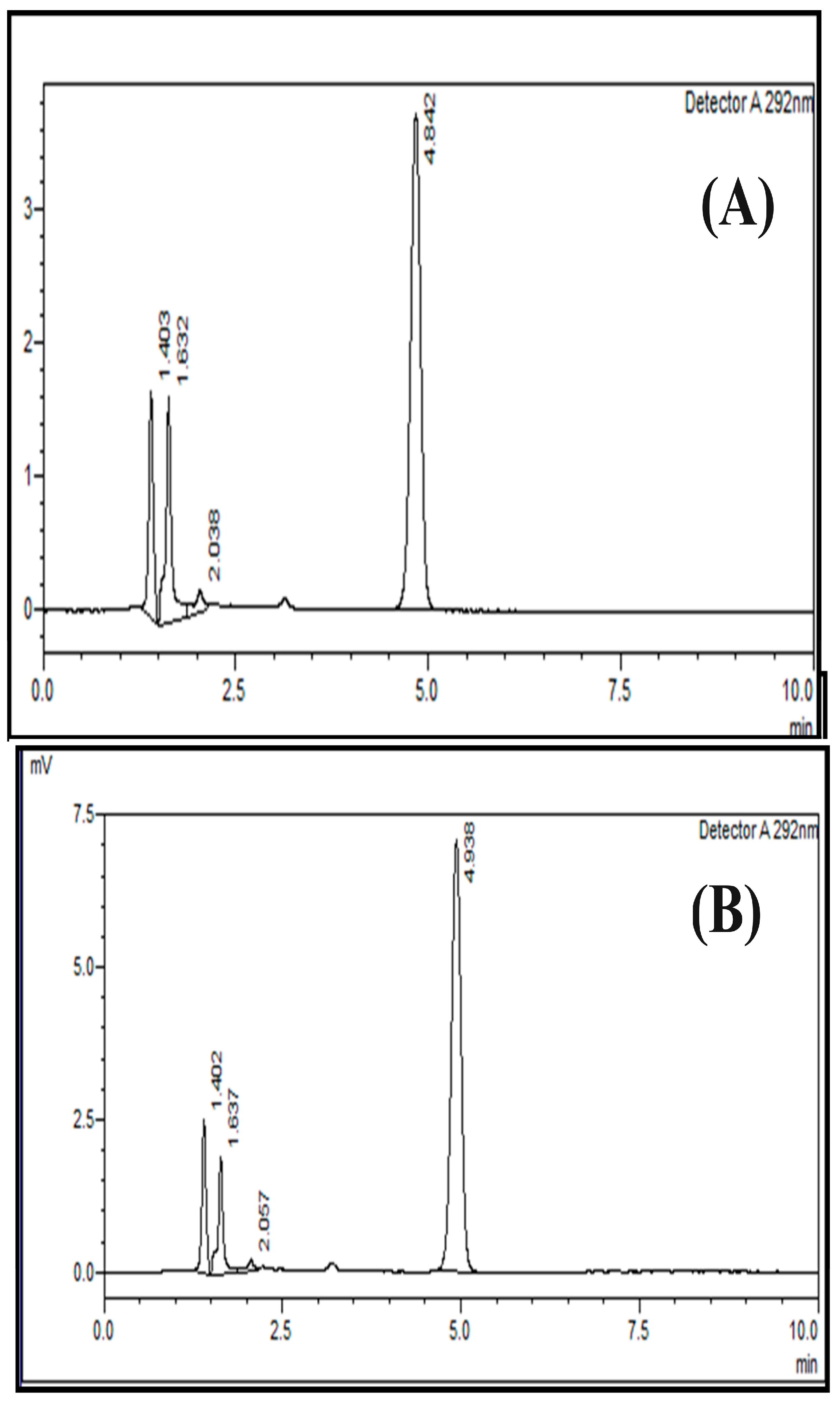
2.1. Method Development
2.2. Method Validation
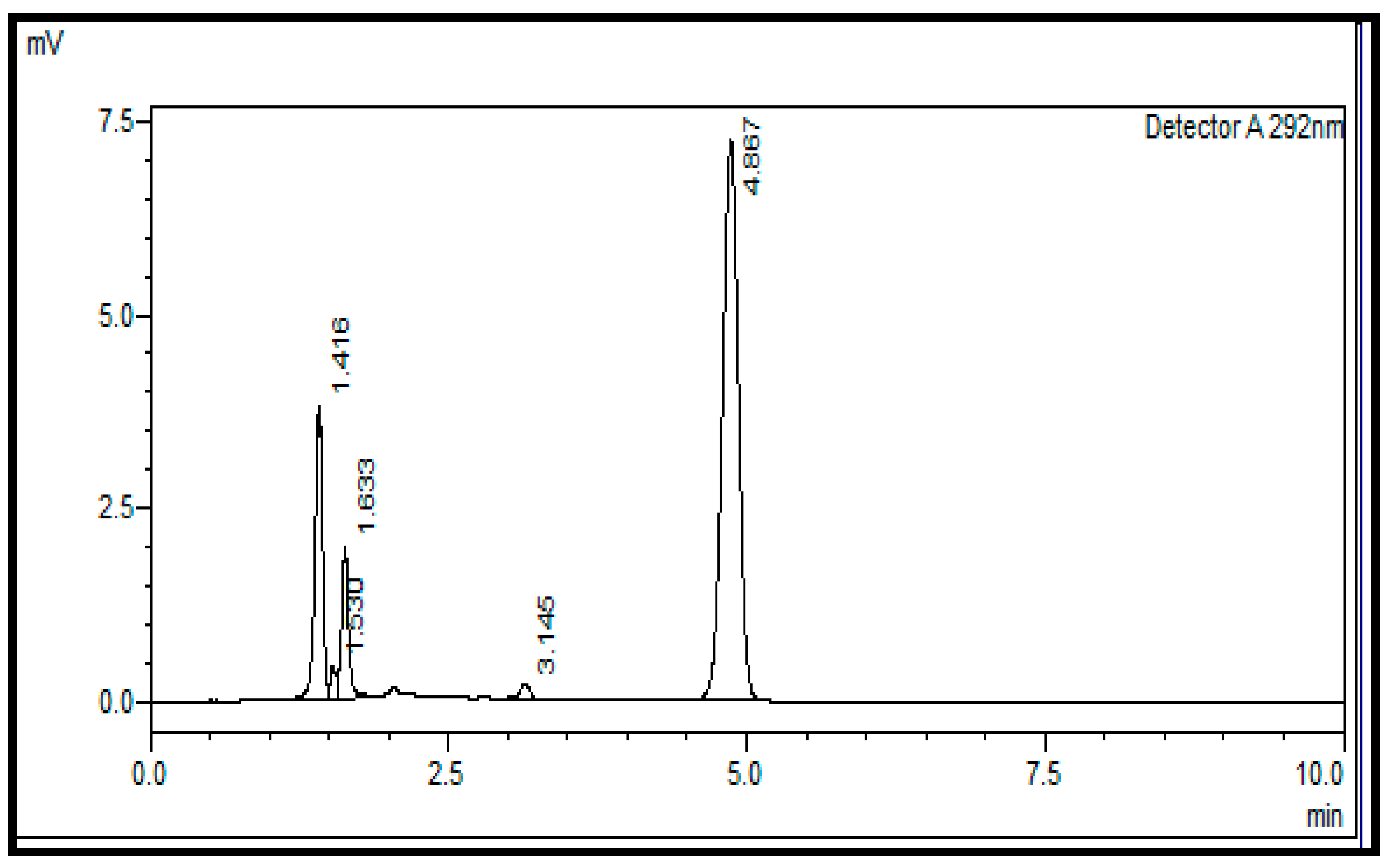
2.2.1. System Suitability
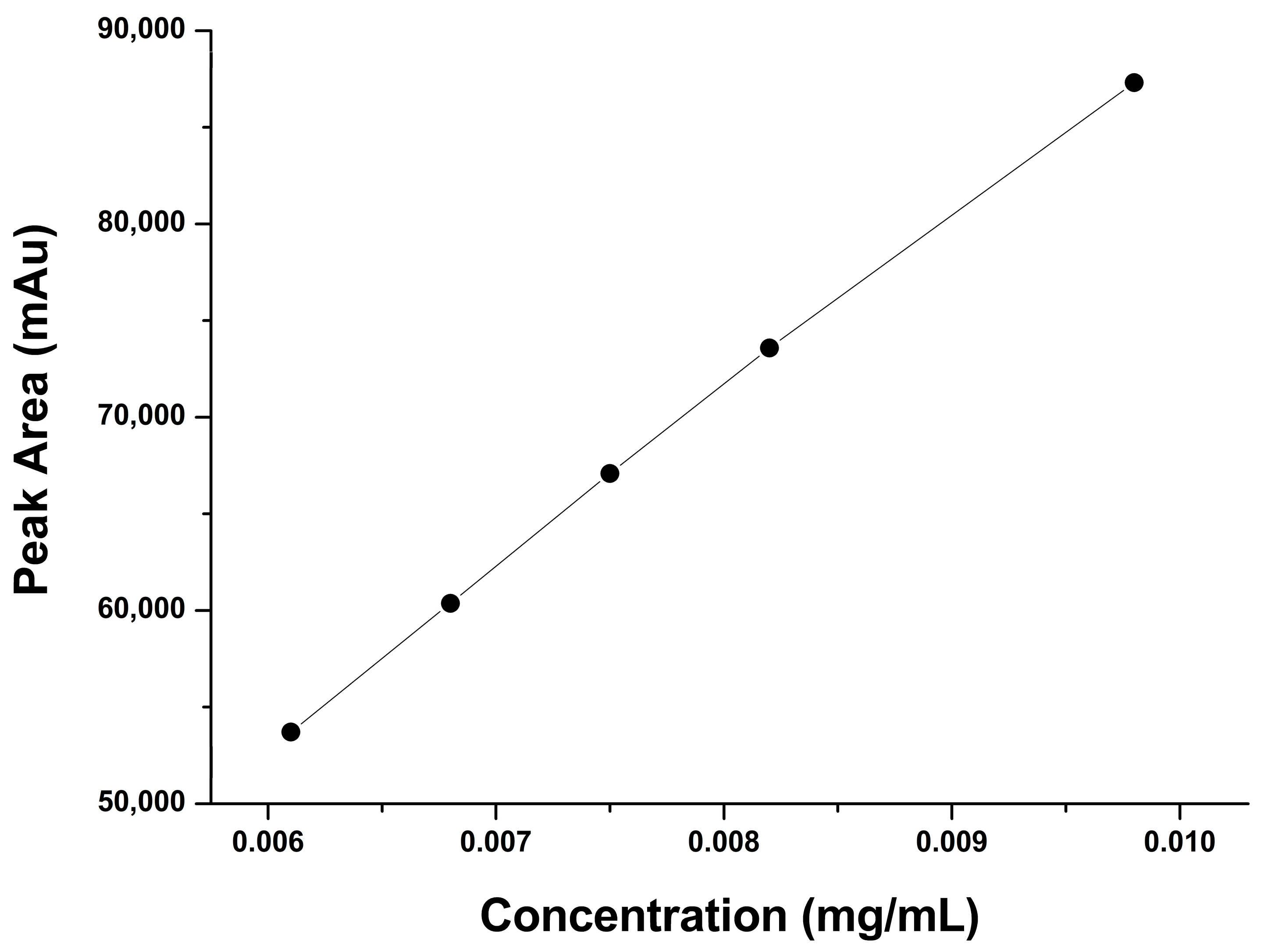
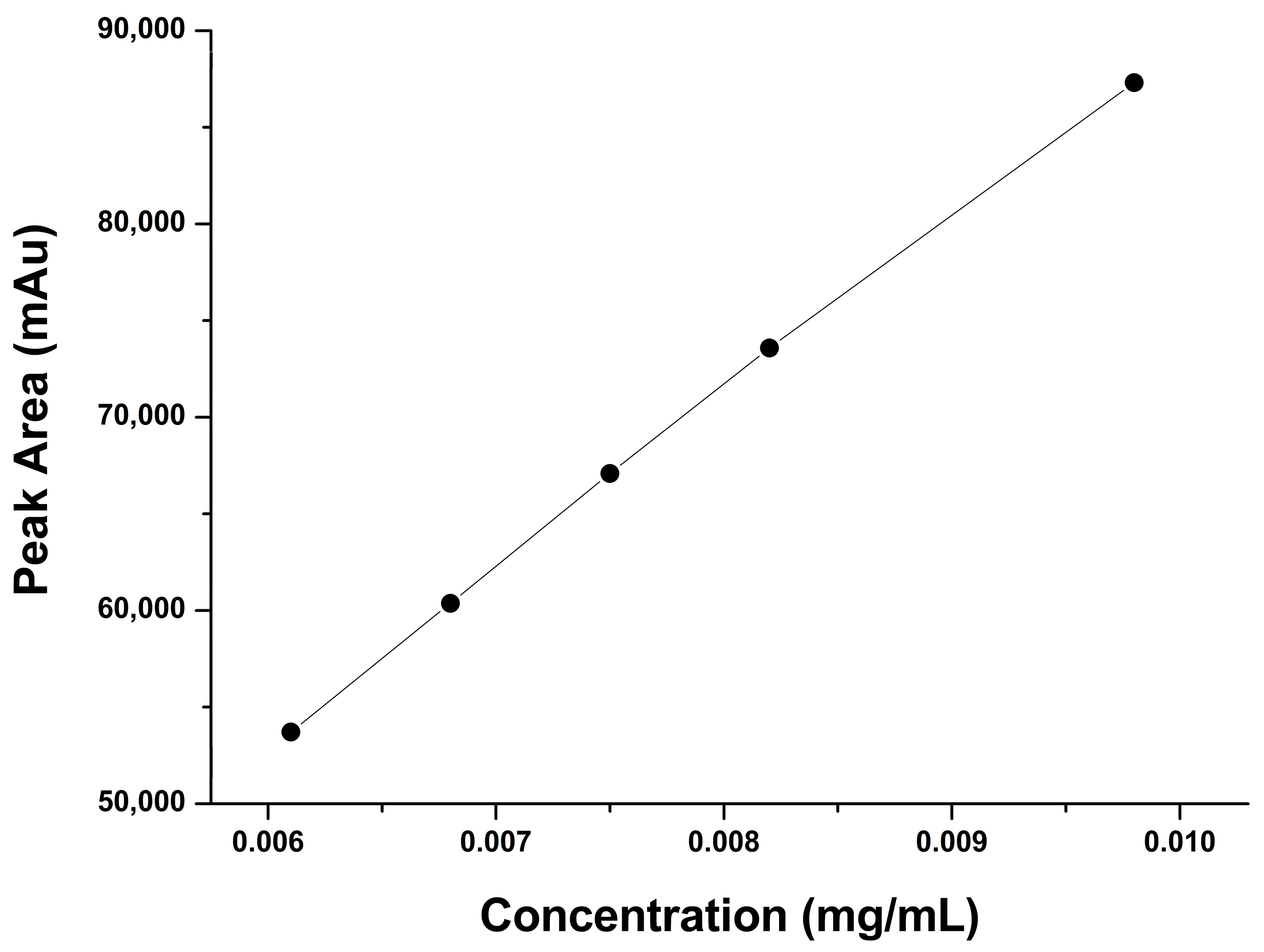
2.2.2. Linearity and Range
2.2.3. Accuracy and Recovery
2.3. Precision
2.3.1. Repeatability (System Precision)
2.3.2. Intermediate Precision (Ruggedness)
Within Day’s Variation
By Different Analyst
2.3.3. Robustness
2.4. Limit of Detection (LoD)
2.5. Limit of Quantification (LoQ)
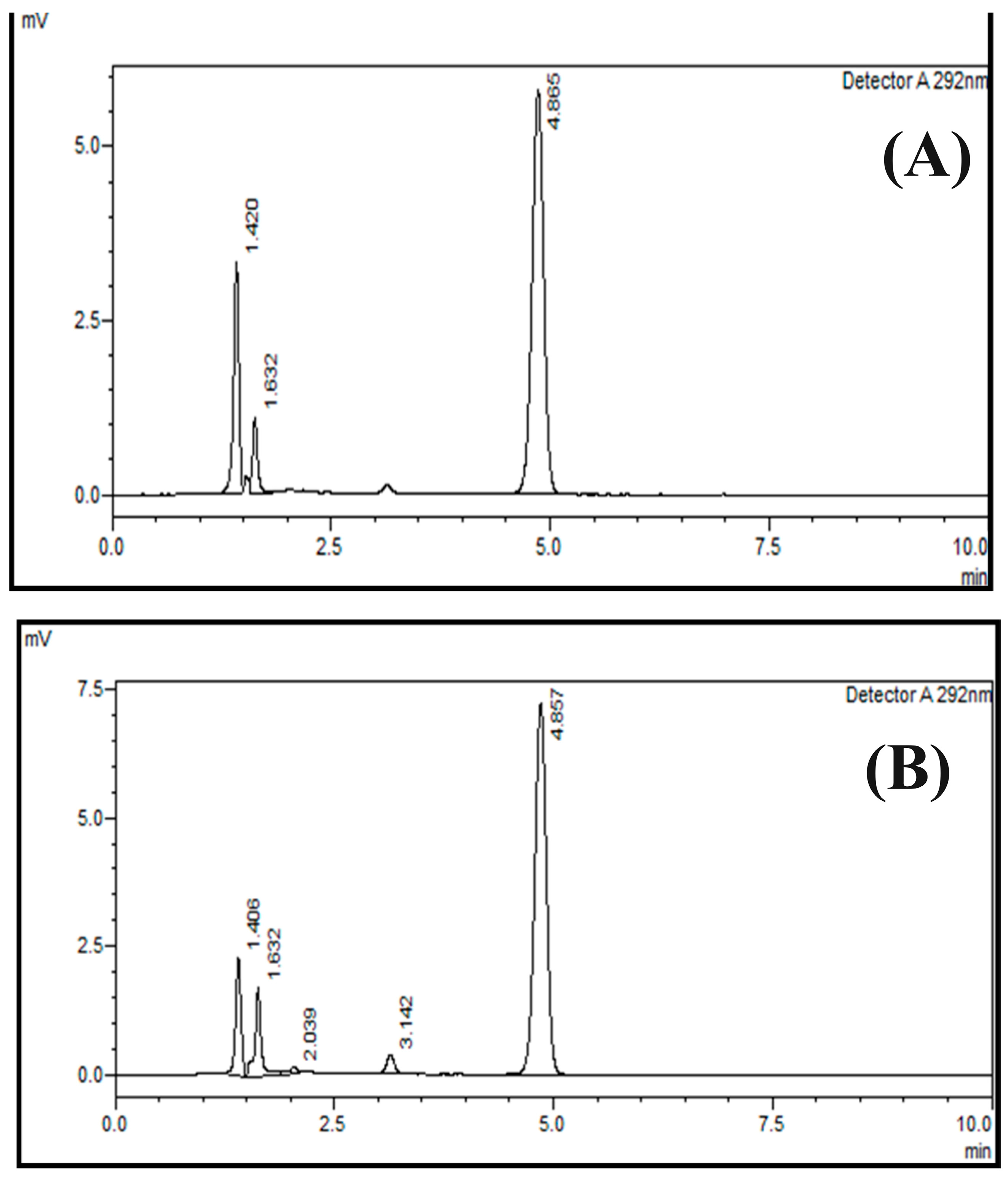
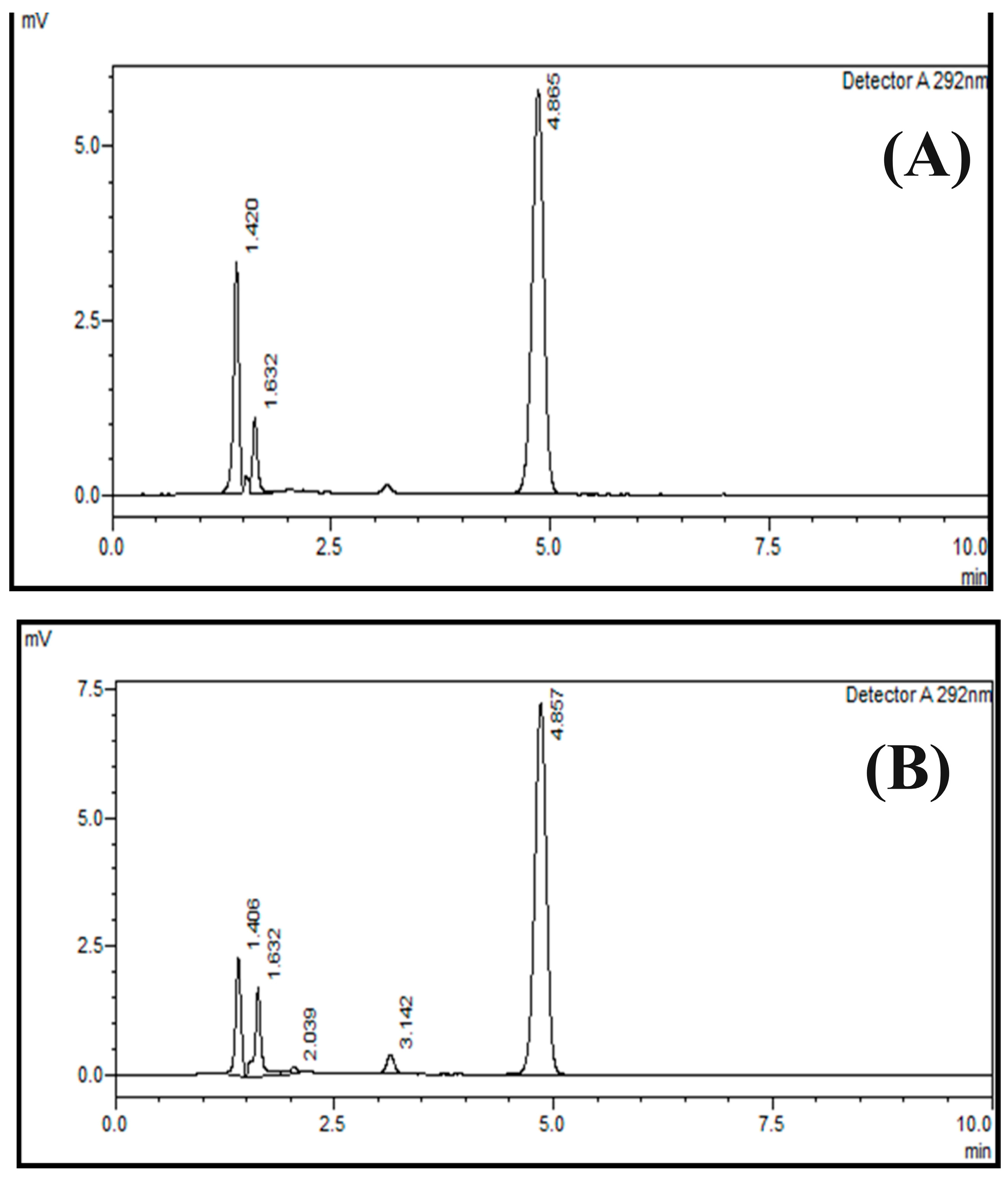
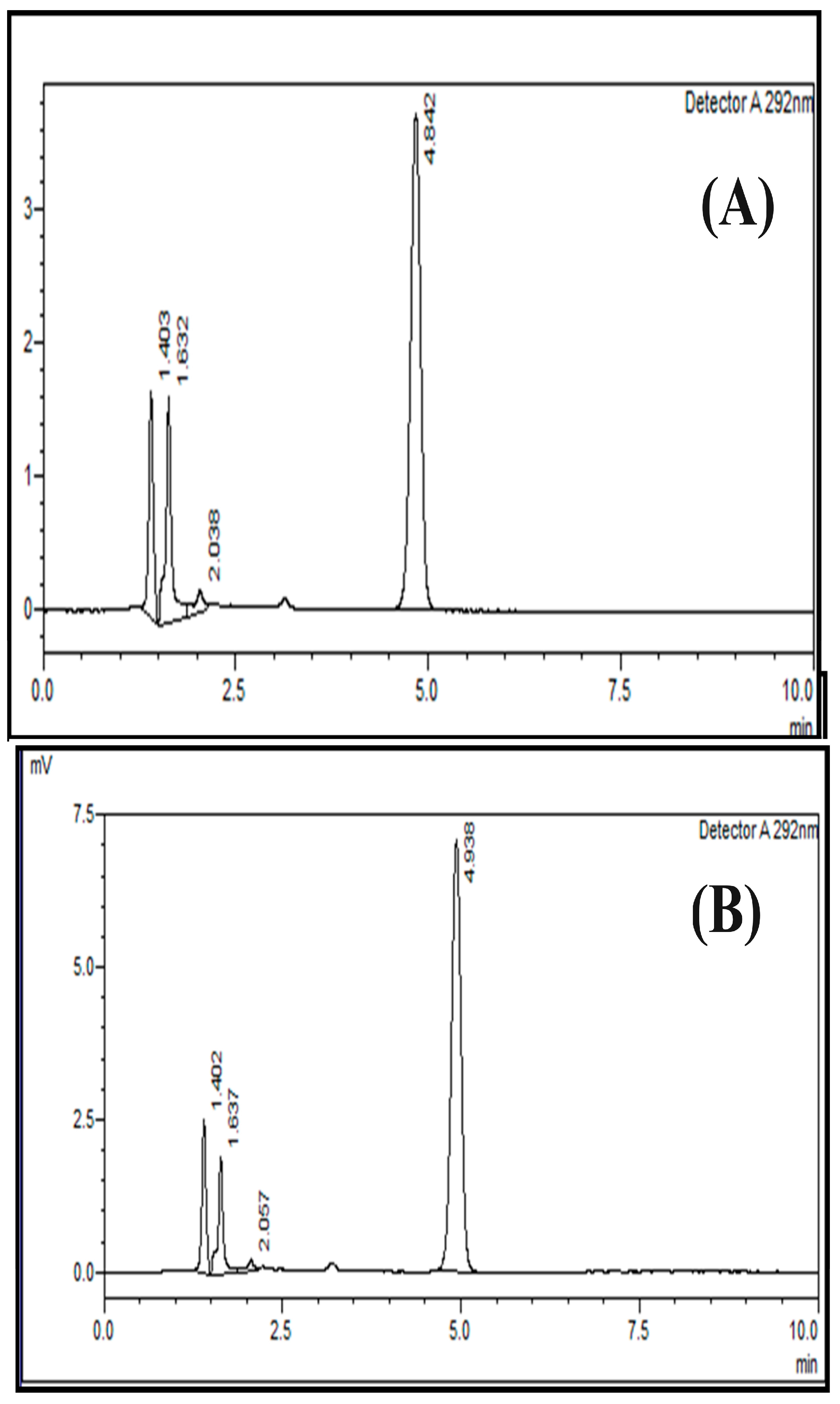
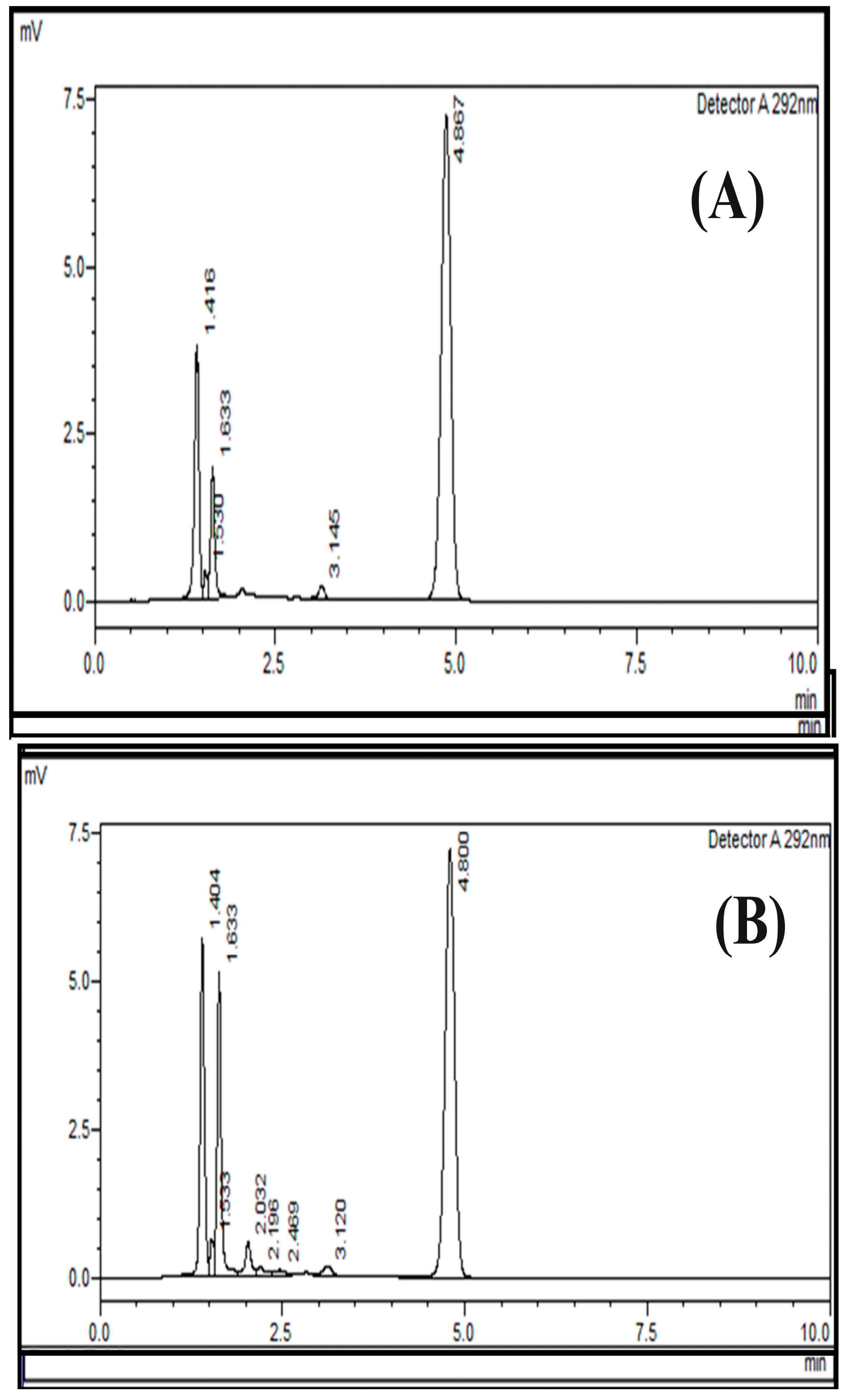
2.6. Specificity
2.7. Acceptance Criteria
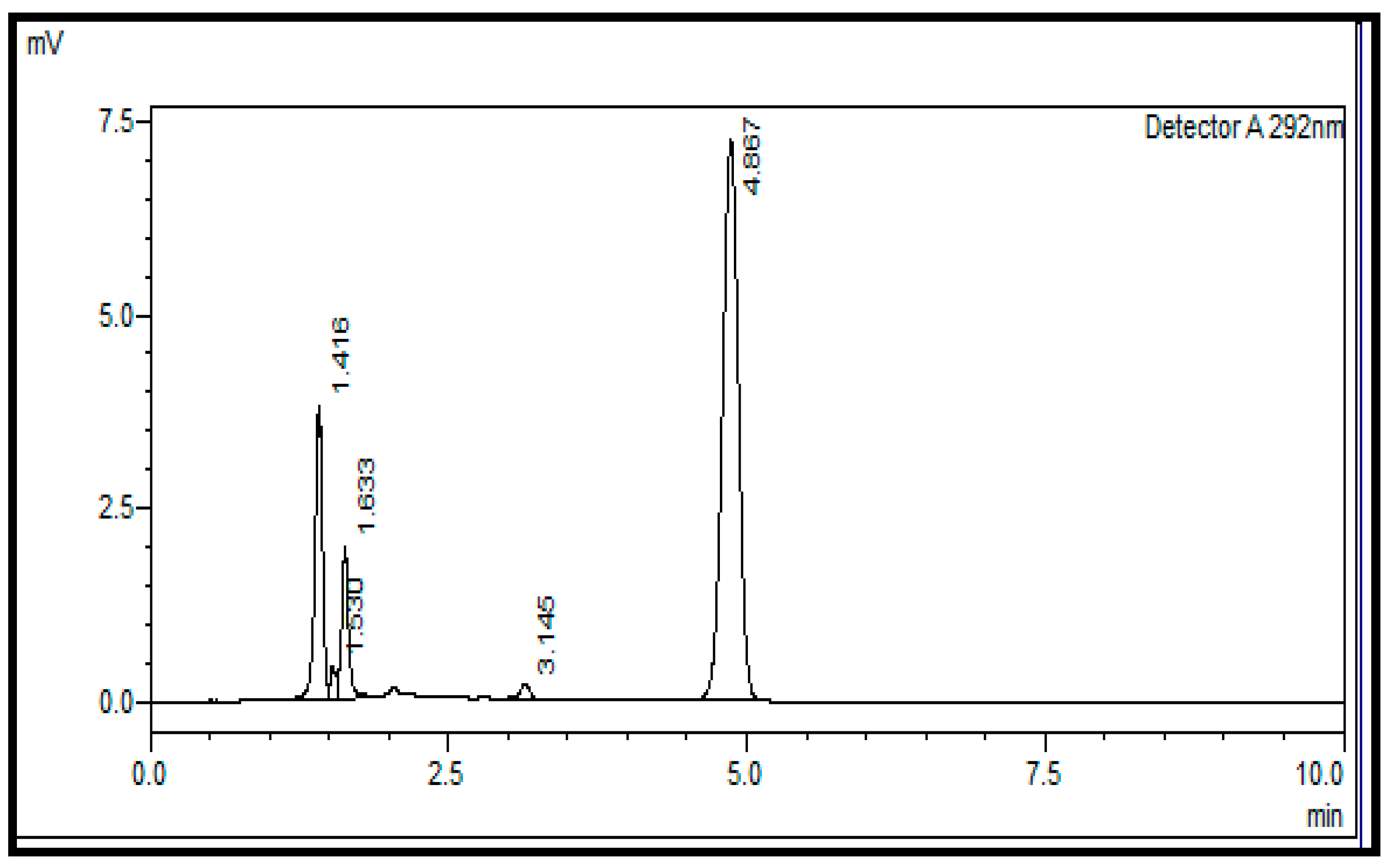
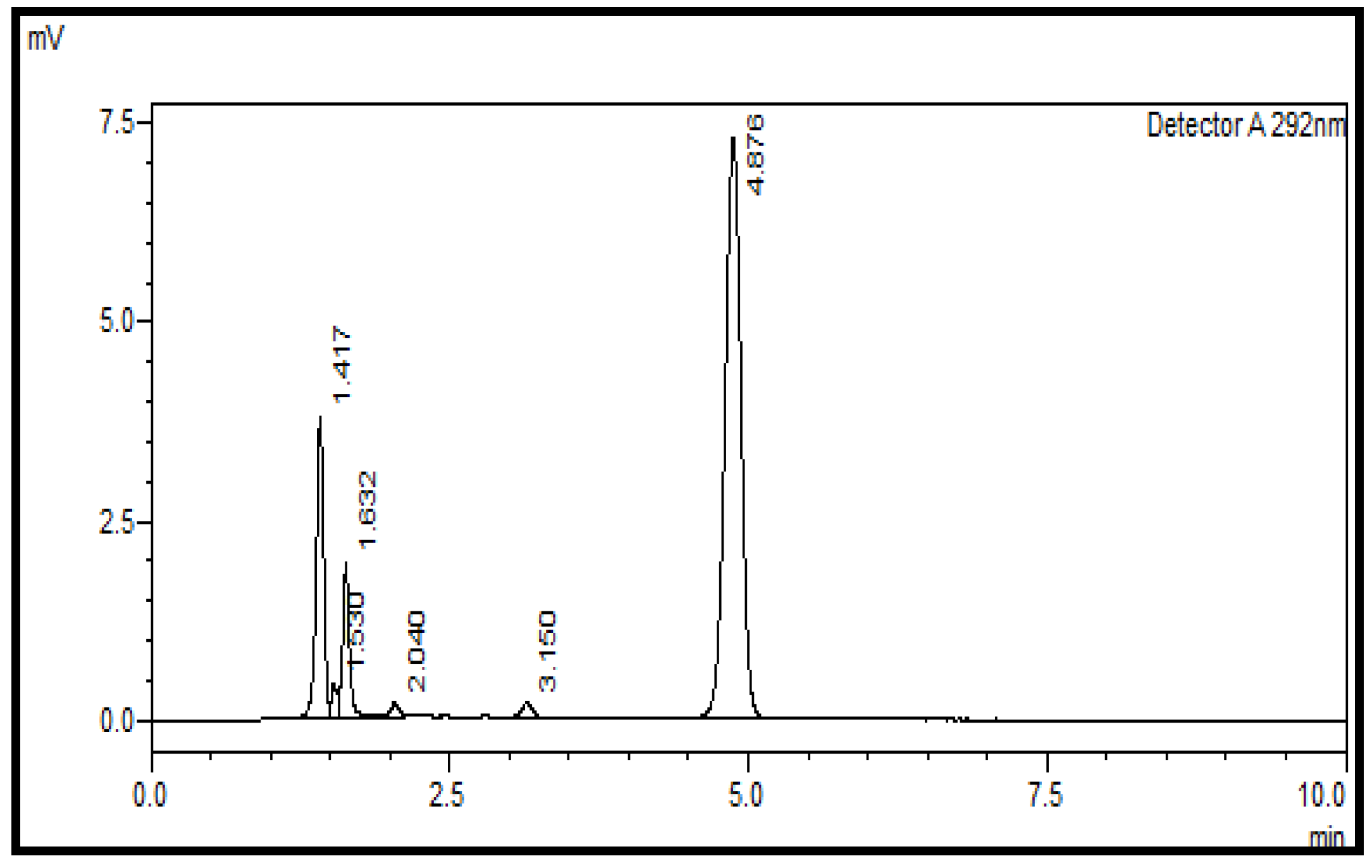
2.8. Application on Commercial Batches
2.9. Comparison of the Developed Method with Other Similar Methods
3. Materials and Methods
3.1. Selection of Dosage Form
3.2. Standard and Sample Preparation
3.3. HPLC Analysis
3.4. System Suitability Test
3.5. Method Validation
3.5.1. System Suitability
3.5.2. Linearity
3.5.3. Accuracy and Recovery
3.5.4. Precision
3.5.5. Intermediate Precision or Ruggedness
3.5.6. Robustness
3.6. Limit of Detection (LoD)
3.7. Limit of Quantification (LoQ)
3.8. Specificity
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siddiqui, M.R.; AlOthman, Z.A.; Rahman, N. Analytical techniques in pharmaceutical analysis: A review. Arab. J. Chem. 2017, 10, S1409–S1421. [Google Scholar] [CrossRef]
- Taraji, M.; Haddad, P.R.; Amos, R.I.J.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A. Rapid Method Development in Hydrophilic Interaction Liquid Chromatography for Pharmaceutical Analysis Using a Combination of Quantitative Structure–Retention Relationships and Design of Experiments. Anal. Chem. 2017, 89, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- D’atri, V.; Fekete, S.; Clarke, A.; Veuthey, J.-L.; Guillarme, D. Recent Advances in Chromatography for Pharmaceutical Analysis. Anal. Chem. 2019, 91, 210–239. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.A.H.; Kiffer, A.; Barrientos, R.C.; Losacco, G.L.; Singh, A.; Shchurik, V.; Wang, H.; Mangion, I.; Regalado, E.L. In Silico Method Development of Achiral and Chiral Tandem Column Reversed-phase Liquid Chromatography for Multicomponent Pharmaceutical Mixtures. Anal. Chem. 2022, 94, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Bonfilio, R.; De Araujo, M.B.; Salgado, H.R.N. Recent applications of analytical techniques for quantitative pharmaceutical analysis: A review. WSEAS Trans. Biol. Biomed. 2010, 7, 316–338. [Google Scholar]
- Makey, D.M.; Shchurik, V.; Wang, H.; Lhotka, H.R.; Stoll, D.R.; Vazhentsev, A.; Mangion, I.; Regalado, E.L.; Ahmad, I.A.H. Mapping the Separation Landscape in Two-Dimensional Liquid Chromatography: Blueprints for Efficient Analysis and Purification of Pharmaceuticals Enabled by Computer-Assisted Modeling. Anal. Chem. 2021, 93, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Suntornsuk, L. Recent advances of capillary electrophoresis in pharmaceutical analysis. Anal. Bioanal. Chem. 2010, 398, 29. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.; Ahmad, I.A.H.; DaSilva, J.; Figus, M.; Hullen, K.; Tsay, F.-R.; Makarov, A.A.; Mann, B.F.; Regalado, E.L. Mapping the Separation Landscape of Pharmaceuticals: Rapid and Efficient Scale-Up of Preparative Purifications Enabled by Computer-Assisted Chromatographic Method Development. Org. Process. Res. Dev. 2019, 23, 2678–2684. [Google Scholar] [CrossRef]
- Ali, A.; Alharthi, S.; Shad, S.; Al-Shaalan, N.H.; Iqbal, M. Preparation of polar embedded C18 stationary phase for efficient separation of peptides and proteins in high performance liquid chromatography. J. Chromatogr. A 2022, 1684, 463534. [Google Scholar] [CrossRef]
- Ali, A.; Alharthi, S.; Al-Shaalan, N.H.; Santali, E.Y. Development of Narrow-Bore C18 Column for Fast Separation of Peptides and Proteins in High-Performance Liquid Chromatography. Polymers 2022, 14, 2576. [Google Scholar] [CrossRef]
- Alharthi, S.; Ali, A.; Iqbal, M.; Ibrar, A.; Ahmad, B.; Nisa, S.; Mabood, F. Preparation of mixed-mode stationary phase for separation of peptides and proteins in high performance liquid chromatography. Sci. Rep. 2022, 12, 4061. [Google Scholar] [CrossRef] [PubMed]
- Iguiniz, M.; Heinisch, S. Two-dimensional liquid chromatography in pharmaceutical analysis. Instrumental aspects, trends and applications. J. Pharm. Biomed. Anal. 2017, 145, 482–503. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wang, Q.; Zhou, Y.; Shackman, J.G. Trapping mode two-dimensional liquid chromatography for quantitative low-level impurity enrichment in pharmaceutical development. J. Chromatogr. A 2023, 1700, 464043. [Google Scholar] [CrossRef] [PubMed]
- Jaag, S.; Shirokikh, M.; Lämmerhofer, M. Charge variant analysis of protein-based biopharmaceuticals using two-dimensional liquid chromatography hyphenated to mass spectrometry. J. Chromatogr. A 2021, 1636, 461786. [Google Scholar] [CrossRef]
- Alvarenga, L.; Ferreira, D.; Altekruse, D.; Menezes, J.; Lochmann, D. Tablet identification using near-infrared spectroscopy (NIRS) for pharmaceutical quality control. J. Pharm. Biomed. Anal. 2008, 48, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Anjum, I.; Jaffery, S.S.; Fayyaz, M.; Samoo, Z.; Anjum, S. The Role of Vitamin D in Brain Health: A Mini Literature Review. Cureus 2018, 10, e2960. [Google Scholar] [CrossRef] [PubMed]
- Maddock, J.; Zhou, A.; Cavadino, A.; Kuźma, E.; Bao, Y.; Smart, M.C.; Saum, K.-U.; Schöttker, B.; Engmann, J.; Kjærgaard, M.; et al. Vitamin D and cognitive function: A Mendelian randomisation study. Sci. Rep. 2017, 7, 13230. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.F.; Majeed, S.; Aslam, S.; Irfan, J.A. Vitamins: Key role players in boosting up immune response—A mini review. Vitam. Miner. 2017, 6, 2376. [Google Scholar] [CrossRef]
- Boucher, B.J. The problems of vitamin d insufficiency in older people. Aging Dis. 2012, 3, 313–329. [Google Scholar]
- Cui, X.; Gooch, H.; Groves, N.J.; Sah, P.; Burne, T.H.; Eyles, D.W.; McGrath, J.J. Vitamin D and the brain: Key questions for future research. J. Steroid Biochem. Mol. Biol. 2015, 148, 305–309. [Google Scholar] [CrossRef]
- Höck, A.D. Review: Vitamin D3 deficiency results in dysfunctions of immunity with severe fatigue and depression in a variety of diseases. In Vivo 2014, 28, 133–145. [Google Scholar] [PubMed]
- Di Molfetta, I.V.; Bordoni, L.; Gabbianelli, R.; Sagratini, G.; Alessandroni, L. Vitamin D and Its Role on the Fatigue Mitigation: A Narrative Review. Nutrients 2024, 16, 221. [Google Scholar] [CrossRef] [PubMed]
- Fabri, M.; Stenger, S.; Shin, D.M.; Yuk, J.M.; Liu, P.T.; Realegeno, S.; Lee, H.M.; Krutzik, S.R.; Schenk, M.; Sieling, P.A.; et al. Vitamin D is required for IFN-γ–mediated antimicrobial activity of human macrophages. Sci. Transl. Med. 2011, 3, 104ra102. [Google Scholar] [CrossRef] [PubMed]
- Bernicke, B.; Engelbogen, N.; Klein, K.; Franzenburg, J.; Borzikowsky, C.; Peters, C.; Janssen, O.; Junker, R.; Serrano, R.; Kabelitz, D. Analysis of the Seasonal Fluctuation of γδ T Cells and Its Potential Relation with Vitamin D(3). Cells 2022, 11, 1460. [Google Scholar] [CrossRef] [PubMed]
- Meloni, A.; Pistoia, L.; Vassalle, C.; Spasiano, A.; Fotzi, I.; Bagnato, S.; Putti, M.C.; Cossu, A.; Massei, F.; Giovangrossi, P.; et al. Low Vitamin D Levels Are Associated with Increased Cardiac Iron Uptake in Beta-Thalassemia Major. Diagnostics 2023, 13, 3656. [Google Scholar] [CrossRef] [PubMed]
- Mena-Bravo, A.; Calderón-Santiago, M.; de Castro, L.; Priego-Capote, F. Evaluation of short-term storage prior to analysis of vitamin D3 and metabolites in human serum by liquid chromatography coupled to tandem mass spectrometry. Talanta 2019, 198, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Ouweland, J.M.v.D. Analysis of vitamin D metabolites by liquid chromatography-tandem mass spectrometry. TrAC Trends Anal. Chem. 2016, 84, 117–130. [Google Scholar] [CrossRef]
- Schadt, H.S.; Gössl, R.; Seibel, N.; Aebischer, C.-P. Quantification of vitamin D3 in feed, food, and pharmaceuticals using high-performance liquid chromatography/tandem mass spectrometry. J. AOAC Int. 2012, 95, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Winters, D. Application of Ultra-performance liquid chromatography/tandem mass spectrometry for the measurement of vitamin D in foods and nutritional supplements. J. AOAC Int. 2011, 94, 211–223. [Google Scholar] [CrossRef]
- Huang, M.; LaLuzerne, P.; Winters, D.; Sullivan, D. Measurement of vitamin D in foods and nutritional supplements by liquid chromatography/tandem mass spectrometry. J. AOAC Int. 2009, 92, 1327–1335. [Google Scholar] [CrossRef]
- Bartolucci, G.; Giocaliere, E.; Boscaro, F.; Vannacci, A.; Gallo, E.; Pieraccini, G.; Moneti, G. Vitamin D3 quantification in a cod liver oil-based supplement. J. Pharm. Biomed. Anal. 2011, 55, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Al-Qadi, E.; Battah, A.; Hadidi, K. Development of high-performance liquid chromatographic method for vitamin D3 analysis in pharmaceutical preparation. Jordan J. Pharm. Sci. 2010, 3, 78–86. [Google Scholar]
- Gámiz-Gracia, L.; Jiménez-Carmona, M.M.; de Castro, M.D.L. Determination of vitamins D2 and D3 in pharmaceuticals by supercritical-fluid extraction and HPLC separation with UV detection. Chromatographia 2000, 51, 428–432. [Google Scholar] [CrossRef]
- Priya, S. Development and validation of RP-HPLC method for the estimation of vitamin D3 in liquid formulations. Res. Gate Pharm. Sci. 2012, 1, 5–7. [Google Scholar]
- Sarioglu, K.; Celebi, S.; Mutlu, M. A rapid method for determination of vitamins D2 and D3 in pharmaceutical preparations by HPLC. J. Liq. Chromatogr. Relat. Technol. 2001, 24, 973–982. [Google Scholar] [CrossRef]
- Jehangir, M.; Ahmed, M.; Shafiq, M.I.; Samad, A.; Haq, I.U. UHPLC-PDA Assay for Simultaneous Determination of Vitamin D3 and Menaquinone-7 in Pharmaceutical Solid Dosage Formulation. J. Anal. Methods Chem. 2017, 2017, 1208753. [Google Scholar] [CrossRef] [PubMed]
- Temova, Ž.; Roškar, R. Stability-Indicating HPLC–UV Method for Vitamin D3 Determination in Solutions, Nutritional Supplements and Pharmaceuticals. J. Chromatogr. Sci. 2016, 54, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Sohail, A.; Al Menhali, A.; Hisaindee, S.; Shah, I. An LC-MS/MS Method for Analysis of Vitamin D Metabolites and C3 Epimers in Mice Serum: Oral Supplementation Compared to UV Irradiation. Molecules 2021, 26, 5182. [Google Scholar] [CrossRef] [PubMed]
- Ertugrul, S.; Yucel, C.; Sertoglu, E.; Ozkan, Y.; Ozgurtas, T. Development and optimization of simultaneous determination of fat soluble vitamins by liquid chromatography tandem mass spectrometry. Chem. Phys. Lip. 2020, 230, 104932–104941. [Google Scholar] [CrossRef]
- Becze, A.; Fuss, V.L.B.; Scurtu, D.A.; Tomoaia-Cotisel, M.; Mocanu, A.; Cadar, O. Simultaneous Determination of Vitamins D3 (Calcitriol, Cholecalciferol) and K2 (Menaquinone-4 and Menaquinone-7) in Dietary Supplements by UHPLC. Molecules 2021, 26, 6982. [Google Scholar] [CrossRef]
- Peck, R.W. Precision Dosing: An Industry Perspective. Clin. Pharmacol. Ther. 2021, 109, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Guideline, I.H.T. Validation of Analytical Procedures: Text and Methodology Q2 (R1). In Proceedings of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 2005. [Google Scholar]
- Higashi, T.; Shimada, K.; Toyo’oka, T. Advances in determination of vitamin D related compounds in biological samples using liquid chromatography–mass spectrometry: A review. J. Chromatogr. B 2010, 878, 1654–1661. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration % | Conc. mg/mL | Mean Peak Area |
|---|---|---|
| 80 | 0.0061 | 53,707 |
| 90 | 0.0068 | 60,367 |
| 100 | 0.0075 | 67,078 |
| 110 | 0.0082 | 73,582 |
| 120 | 0.0098 | 80,315 |
| Conc. mg/mL | Peak Area | %RSD | Recovery (%) | Average Recovery (%) | |
|---|---|---|---|---|---|
| 50% | 0.00375 | 33,869 | 0.929 | 50.33 | 49.92% |
| 33,257 | 49.42 | ||||
| 33,669 | 50.03 | ||||
| 100% | 0.0075 | 66,838 | 0.1446 | 99.31 | 99.42% |
| 66,875 | 99.37 | ||||
| 67,021 | 99.58 |
| Days | Sample | Assay (%) | Average Assay (%) | STDV | % RSD |
|---|---|---|---|---|---|
| 1 | 1 | 99.26 | 99.33 | 0.059 | 0.059 |
| 2 | 99.37 | ||||
| 3 | 99.35 | ||||
| 2 | 1 | 98.68 | 98.92 | 0.221 | 0.223 |
| 2 | 99.11 | ||||
| 3 | 98.97 | ||||
| 3 | 1 | 98.91 | 98.54 | 0.390 | 0.396 |
| 2 | 98.13 | ||||
| 3 | 98.57 |
| B# | Sample | Peak Area of Sample | Av. Abs. of Sample | Peak Area of Standard | Av. Peak Area of Standard | Assay % | Average Assay % | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | ||||||
| 097 | 1 | 66,710 | 66,582 | 66,476 | 66,589 | 66,205 | 65,661 | 66,353 | 66,073 | 100.78 | 100.78% |
| 098 | 1 | 67,543 | 67,852 | 67,509 | 67,635 | 65,465 | 65,691 | 66,205 | 65,787 | 102.81 | 102.81% |
| 099 | 1 | 67,544 | 67,887 | 67,358 | 67,635 | 66,874 | 66,587 | 66,048 | 65,787 | 100.81 | 100.81% |
| Method Developed | Sample/Matrix | Coefficient of Determination (R2) | Recovery (%) | Limit of Detection | Limit of Quantification | Ref. |
|---|---|---|---|---|---|---|
| RP-UHPLC-PDA | Pharmaceutical | 0.998 | 98.97 | 0.24 μg mL−1 | 0.72 μg mL−1 | [36] |
| RP-HPLC–UV | Nutritional supplements | 0.999 | 98.3 | 0.019 mg/L | 0.057 mg/L | [37] |
| RP-UHPLC-UV | Dietary supplements | 0.999 | 82 | 0.04 µg/mL | 0.05 µg/mL | [40] |
| NP-HPLC-UV | Pharmaceutical | 0.999 | 99.42 | 0.0539 µg/mL | 0.1633 µg/mL | Current study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gohar, M.S.; Rahman, T.U.; Bahadur, A.; Ali, A.; Alharthi, S.; Al-Shaalan, N.H. Development and Validation of Novel HPLC Methods for Quantitative Determination of Vitamin D3 in Tablet Dosage Form. Pharmaceuticals 2024, 17, 505. https://doi.org/10.3390/ph17040505
Gohar MS, Rahman TU, Bahadur A, Ali A, Alharthi S, Al-Shaalan NH. Development and Validation of Novel HPLC Methods for Quantitative Determination of Vitamin D3 in Tablet Dosage Form. Pharmaceuticals. 2024; 17(4):505. https://doi.org/10.3390/ph17040505
Chicago/Turabian StyleGohar, Muhammad Saqib, Taj Ur Rahman, Ali Bahadur, Ashraf Ali, Sarah Alharthi, and Nora Hamad Al-Shaalan. 2024. "Development and Validation of Novel HPLC Methods for Quantitative Determination of Vitamin D3 in Tablet Dosage Form" Pharmaceuticals 17, no. 4: 505. https://doi.org/10.3390/ph17040505
APA StyleGohar, M. S., Rahman, T. U., Bahadur, A., Ali, A., Alharthi, S., & Al-Shaalan, N. H. (2024). Development and Validation of Novel HPLC Methods for Quantitative Determination of Vitamin D3 in Tablet Dosage Form. Pharmaceuticals, 17(4), 505. https://doi.org/10.3390/ph17040505