
Approaches to Reducing Normal Tissue Radiation from Radiolabeled Antibodies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Dissociation of Radionuclides
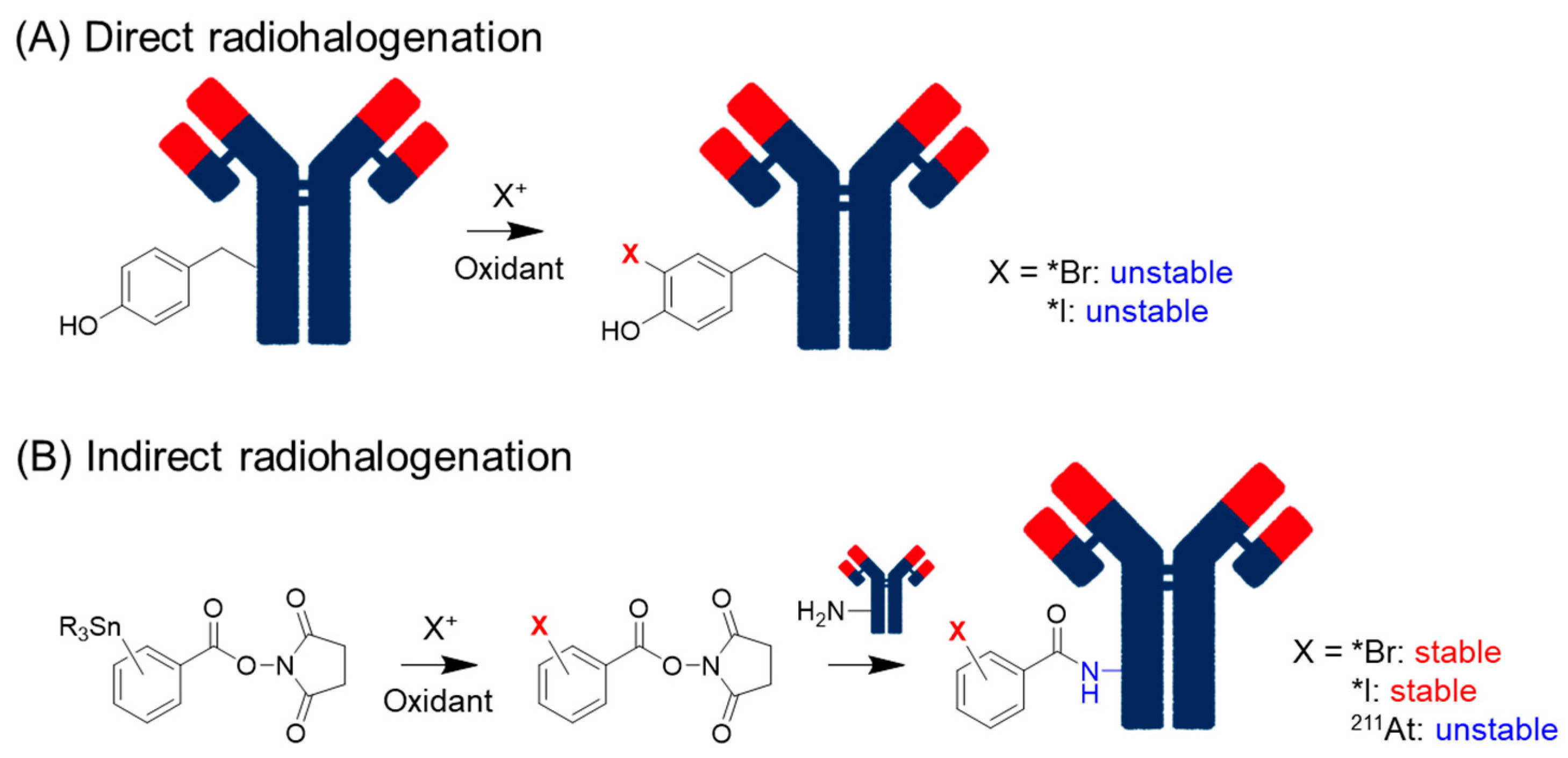
2.1. Dissociation of Radiohalogens
2.1.1. Radioiodine and Radiobromine
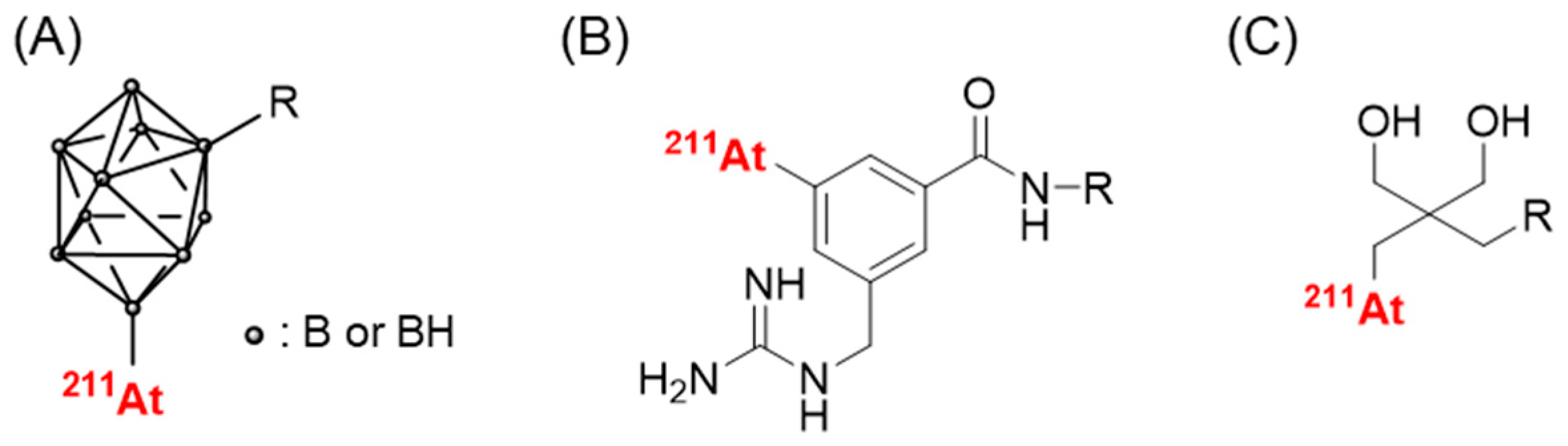
2.1.2. Astatine
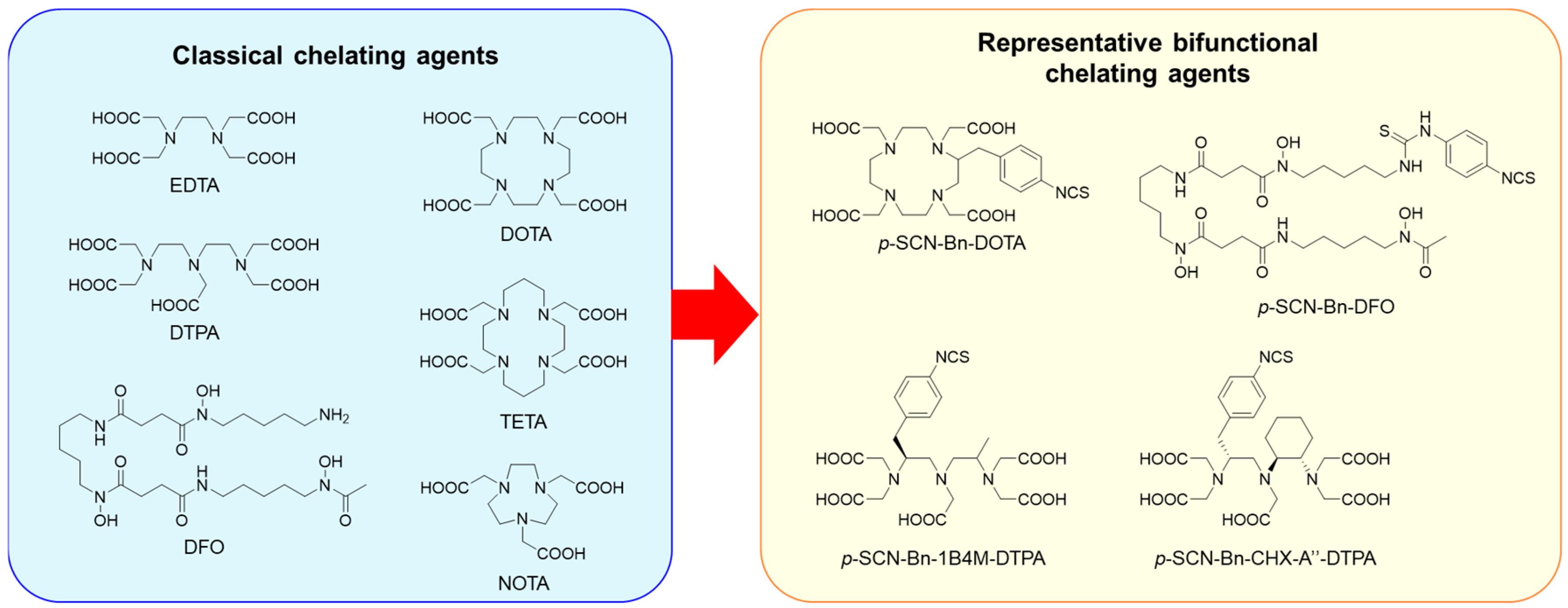
2.2. Dissociation of Radiometals
2.2.1. Representative Choices of the BFCs for Preparing Radiometal-Labeled Antibodies
2.2.2. Preparation of 89Zr-Labeled Antibodies
2.2.3. Preparation of 64Cu-Labeled Antibodies
2.2.4. Preparation of 225Ac-Labeled Antibodies
2.3. Dissociation of Daughter Radionuclides
2.3.1. Administration of Agents to Prevent Renal Toxicity
2.3.2. Strategies for Minimizing the Side Toxicity Caused by Daughter Radionuclides
3. Slow Blood Clearance
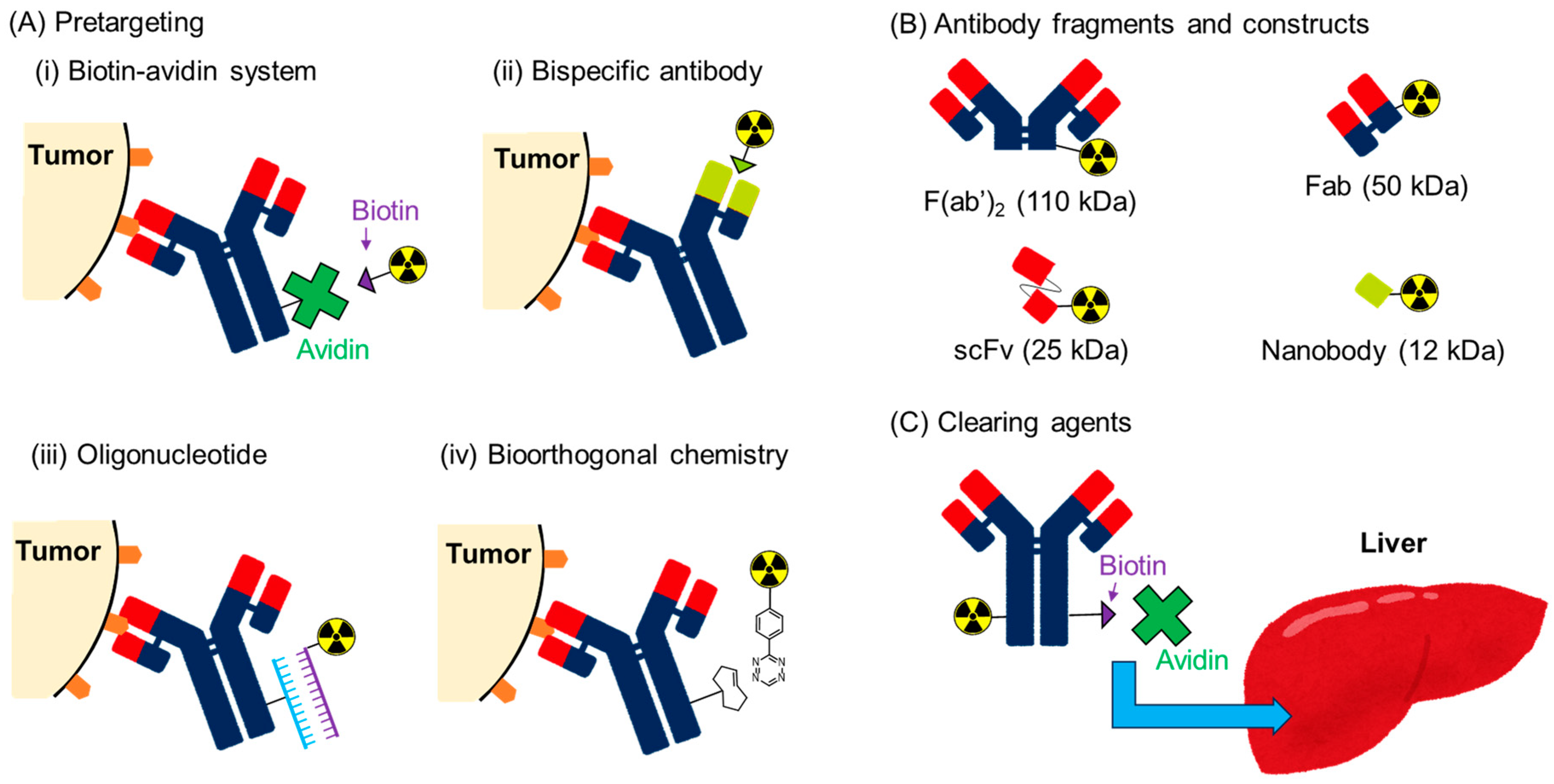
3.1. Pretargeting System
3.1.1. Biotin-Avidin System
3.1.2. Bispecific Antibody
3.1.3. Oligonucleotide
3.1.4. Bioorthogonal Chemistry
3.2. Antibody Fragments and Constructs
3.3. Clearing Agents
4. Accumulations in the Liver and Spleen
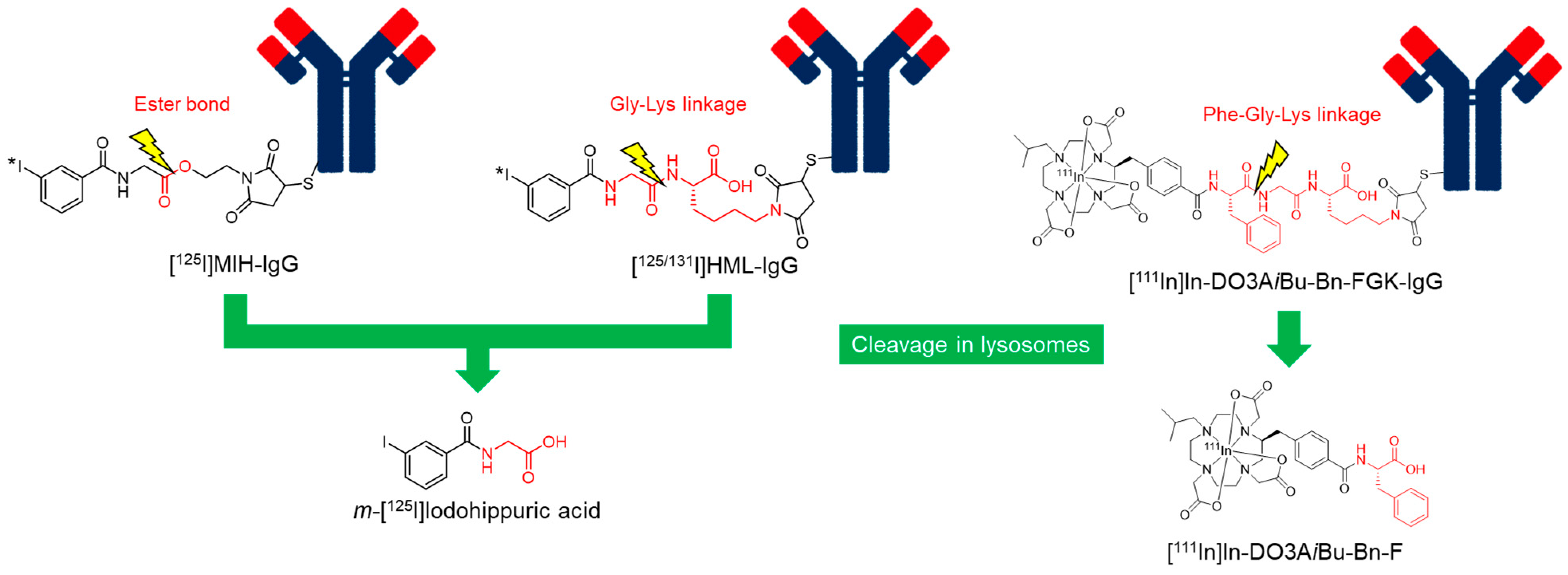
4.1. Use of an Ester Bond as the Cleavable Linkage
4.2. Use of a Peptide Linkage as the Cleavable Linkage
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Venktaraman, G.; Jain, M.; Senapati, S.; Garg, P.K.; Batra, S.K. Recent trends in antibody-based oncologic imaging. Cancer Lett. 2012, 315, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.-K.V.; Press, O.W. Radioimmunotherapy of human tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef]
- Moek, K.L.; Giesen, D.; Kok, I.C.; de Groot, D.J.A.; Jalving, M.; Fehrmann, R.S.; Lub-de Hooge, M.N.; Brouwers, A.H.; de Vries, E.G. Theranostics using antibodies and antibody-related therapeutics. J. Nucl. Med. 2017, 58, 83S–90S. [Google Scholar] [CrossRef] [PubMed]
- Kasbollah, A.; Eu, P.; Cowell, S.; Deb, P. Review on production of 89Zr in a medical cyclotron for PET radiopharmaceuticals. J. Nucl. Med. Technol. 2013, 41, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Teulon, I.; Lozza, C.; Pelegrin, A.; Vives, E.; Pouget, J.-P. General overview of radioimmunotherapy of solid tumors. Immunotherapy 2013, 5, 467–487. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M. Targeted therapy of cancer with radiolabeled antibodies. J. Nucl. Med. 2002, 43, 693–713. [Google Scholar] [PubMed]
- Wiseman, G.A.; White, C.A.; Witzig, T.E.; Gordon, L.I.; Emmanouilides, C.; Raubitschek, A.; Janakiraman, N.; Gutheil, J.; Schilder, R.J.; Spies, S. Radioimmunotherapy of relapsed non-Hodgkin’s lymphoma with zevalin, a 90Y-labeled anti-CD20 monoclonal antibody. Clin. Cancer Res. 1999, 5, 3281s–3286s. [Google Scholar]
- Wiseman, G.A.; White, C.A.; Stabin, M.; Dunn, W.L.; Erwin, W.; Dahlbom, M.; Raubitschek, A.; Karvelis, K.; Schultheiss, T.; Witzig, T.E. Phase I/II 90Y-Zevalin (yttrium-90 ibritumomab tiuxetan, IDEC-Y2B8) radioimmunotherapy dosimetry results in relapsed or refractory non-Hodgkin’s lymphoma. Eur. J. Nucl. Med. 2000, 27, 766–777. [Google Scholar] [CrossRef]
- Rutar, F.J.; Augustine, S.C.; Kaminski, M.S.; Wahl, R.L.; Siegel, J.A.; Colcher, D. Feasibility and safety of outpatient Bexxar® therapy (tositumomab and iodine I 131 tositumomab) for non-Hodgkin’s lymphoma based on radiation doses to family members. Clin. Lymphoma 2001, 2, 164–172. [Google Scholar] [CrossRef]
- Horning, S.J.; Younes, A.; Jain, V.; Kroll, S.; Lucas, J.; Podoloff, D.; Goris, M. Efficacy and safety of tositumomab and iodine-131 tositumomab (Bexxar) in B-cell lymphoma, progressive after rituximab. J. Clin. Oncol. 2005, 23, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’sullivan, J.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-targeted a-radiation therapy of metastatic castration-resistant prostate cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef] [PubMed]
- Makvandi, M.; Dupis, E.; Engle, J.W.; Nortier, F.M.; Fassbender, M.E.; Simon, S.; Birnbaum, E.R.; Atcher, R.W.; John, K.D.; Rixe, O. Alpha-emitters and targeted alpha therapy in oncology: From basic science to clinical investigations. Target. Oncol. 2018, 13, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Eychenne, R.; Chérel, M.; Haddad, F.; Guérard, F.; Gestin, J.-F. Overview of the most promising radionuclides for targeted alpha therapy: The “hopeful eight”. Pharmaceutics 2021, 13, 906. [Google Scholar] [CrossRef]
- Robertson, A.K.; Ramogida, C.F.; Schaffer, P.; Radchenko, V. Development of 225Ac radiopharmaceuticals: TRIUMF perspectives and experiences. Curr. Radiopharm. 2018, 11, 156–172. [Google Scholar] [CrossRef]
- De Kruijff, R.M.; Wolterbeek, H.T.; Denkova, A.G. A critical review of alpha radionuclide therapy—How to deal with recoiling daughters? Pharmaceuticals 2015, 8, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Singh Jaggi, J.; Kappel, B.J.; McDevitt, M.R.; Sgouros, G.; Flombaum, C.D.; Cabassa, C.; Scheinberg, D.A. Efforts to control the errant products of a targeted in vivo generator. Cancer Res. 2005, 65, 4888–4895. [Google Scholar] [CrossRef]
- Olafsen, T.; Wu, A.M. Antibody vectors for imaging. Semin. Nucl. Med. 2010, 40, 167–181. [Google Scholar] [CrossRef]
- Fu, R.; Carroll, L.; Yahioglu, G.; Aboagye, E.O.; Miller, P.W. Antibody fragment and affibody immunoPET imaging agents: Radiolabelling strategies and applications. ChemMedChem 2018, 13, 2466–2478. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Venkatraman, G.; Batra, S.K. Optimization of radioimmunotherapy of solid tumors: Biological impediments and their modulation. Clin. Cancer Res. 2007, 13, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Hosseinimehr, S.J.; Tolmachev, V.; Orlova, A. Liver uptake of radiolabeled targeting proteins and peptides: Considerations for targeting peptide conjugate design. Drug Discov. Today 2012, 17, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Vivier, D.; Sharma, S.K.; Zeglis, B.M. Understanding the in vivo fate of radioimmunoconjugates for nuclear imaging. J. Labelled Compd. Radiopharmaceut. 2018, 61, 672–692. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Pressman, D.; Korngold, L. The in vivo localization of anti-Wagner-osteogenic-sarcoma antibodies. Cancer 1953, 6, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, L.; Antoch, G.; Jentzen, W.; Pink, R.; Knust, J.; Görges, R.; Müller, S.; Bockisch, A.; Debatin, J.; Brandau, W. Value of 124I-PET/CT in staging of patients with differentiated thyroid cancer. Eur. Radiol. 2004, 14, 2092–2098. [Google Scholar] [CrossRef] [PubMed]
- Bzowski, P.; Borys, D.; Gorczewski, K.; Chmura, A.; Daszewska, K.; Gorczewska, I.; Kastelik-Hryniewiecka, A.; Szydło, M.; d’Amico, A.; Sokół, M. Efficiency of 124I radioisotope production from natural and enriched tellurium dioxide using 124Te(p,xn)124I reaction. EJNMMI Phys. 2022, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Seevers, R.H.; Counsell, R.E. Radioiodination techniques for small organic molecules. Chem. Rev. 1982, 82, 575–590. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Narula, A.S. A method for the radiohalogenation of proteins resulting in decreased thyroid uptake of radioiodine. Appl. Radiat. Isot. 1987, 38, 1051–1055. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Hadley, S.W.; Hylarides, M.D.; Abrams, P.G.; Beaumier, P.A.; Morgan, A.C.; Reno, J.M.; Fritzberg, A.R. Development of a stable radioiodinating reagent to label monoclonal antibodies for radiotherapy of cancer. J. Nucl. Med. 1989, 30, 216–226. [Google Scholar]
- Vaidyanathan, G.; Zalutsky, M.R. Preparation of N-succinimidyl 3-[*I] iodobenzoate: An agent for the indirect radioiodination of proteins. Nat. Protoc. 2006, 1, 707–713. [Google Scholar] [CrossRef]
- Lovqvist, A.; Sundin, A.; Ahlstrom, H.; Carlsson, J.; Lundqvist, H. Pharmacokinetics and experimental PET imaging of a bromine-76-labeled monoclonal anti-CEA antibody. J. Nucl. Med. 1997, 38, 395. [Google Scholar]
- Bruehlmeier, M.; Roelcke, U.; Bläuenstein, P.; Missimer, J.; Schubiger, P.A.; Locher, J.T.; Pellikka, R.; Ametamey, S.M. Measurement of the extracellular space in brain tumors using 76Br-bromide and PET. J. Nucl. Med. 2003, 44, 1210–1218. [Google Scholar] [PubMed]
- Lövqvist, A.; Sundin, A.; Ahlström, H.; Carlsson, J.; Lundqvist, H. 76Br-labeled monoclonal anti-CEA antibodies for radioimmuno positron emission tomography. Nucl. Med. Biol. 1995, 22, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Höglund, J.; Tolmachev, V.; Orlova, A.; Lundqvist, H.; Sundin, A. Optimized indirect 76Br-bromination of antibodies using N-succinimidyl para-[76Br] bromobenzoate for radioimmuno PET. Nucl. Med. Biol. 2000, 27, 837–843. [Google Scholar] [CrossRef]
- Aaij, C.; Tschroots, W.; Lindner, L.; Feltkamp, T. The preparation of astatine labelled proteins. Appl. Radiat. Isot. 1975, 26, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Wilbur, D.S. Enigmatic astatine. Nat. Chem. 2013, 5, 246. [Google Scholar] [CrossRef]
- Garg, P.K.; Harrison, C.L.; Zalutsky, M.R. Comparative tissue distribution in mice of the α-emitter 211At and 131I as labels of a monoclonal antibody and F(ab′)2 fragment. Cancer Res. 1990, 50, 3514–3520. [Google Scholar]
- Hadley, S.W.; Wilbur, D.S.; Gray, M.A.; Atcher, R.W. Astatine-211 labeling of an antimelanoma antibody and its Fab fragment using N-succinimidyl p-[211At] astatobenzoate: Comparisons in vivo with the p-[125I] iodobenzoyl conjugate. Bioconj. Chem. 1991, 2, 171–179. [Google Scholar] [CrossRef]
- Larsen, R.H.; Slade, S.; Zalutsky, M.R. Blocking [211At] astatide accumulation in normal tissues: Preliminary evaluation of seven potential compounds. Nucl. Med. Biol. 1998, 25, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Foulon, C.F.; Alston, K.L.; Zalutsky, M.R. Astatine-211-labeled biotin conjugates resistant to biotinidase for use in pretargeted radioimmunotherapy. Nucl. Med. Biol. 1998, 25, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Guérard, F.; Gestin, J.-F.; Brechbiel, M.W. Production of [211At]-astatinated radiopharmaceuticals and applications in targeted α-particle therapy. Cancer Biother. Radiopharm. 2013, 28, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Teze, D.; Sergentu, D.-C.; Kalichuk, V.; Barbet, J.; Deniaud, D.; Galland, N.; Maurice, R.; Montavon, G. Targeted radionuclide therapy with astatine-211: Oxidative dehalogenation of astatobenzoate conjugates. Sci. Rep. 2017, 7, 2579. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Chyan, M.-K.; Hamlin, D.K.; Vessella, R.L.; Wedge, T.J.; Hawthorne, M.F. Reagents for astatination of biomolecules. 2. Conjugation of anionic boron cage pendant groups to a protein provides a method for direct labeling that is stable to in vivo deastatination. Bioconj. Chem. 2007, 18, 1226–1240. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Chyan, M.-K.; Hamlin, D.K.; Nguyen, H.; Vessella, R.L. Reagents for Astatination of Biomolecules. 5. Evaluation of hydrazone linkers in 211At-and 125I-labeled closo-decaborate(2-) conjugates of Fab’ as a means of decreasing kidney retention. Bioconj. Chem. 2011, 22, 1089–1102. [Google Scholar] [CrossRef]
- Choi, J.; Vaidyanathan, G.; Koumarianou, E.; Kang, C.M.; Zalutsky, M.R. Astatine-211 labeled anti-HER2 5F7 single domain antibody fragment conjugates: Radiolabeling and preliminary evaluation. Nucl. Med. Biol. 2018, 56, 10–20. [Google Scholar] [CrossRef]
- Ogawa, K.; Takeda, T.; Mishiro, K.; Toyoshima, A.; Shiba, K.; Yoshimura, T.; Shinohara, A.; Kinuya, S.; Odani, A. Radiotheranostics Coupled between an At-211-Labeled RGD Peptide and the Corresponding Radioiodine-Labeled RGD Peptide. ACS Omega 2019, 4, 4584–4591. [Google Scholar] [CrossRef]
- Mease, R.C.; Kang, C.M.; Kumar, V.; Banerjee, S.R.; Minn, I.; Brummet, M.; Gabrielson, K.L.; Feng, Y.; Park, A.; Kiess, A.P. An improved 211At-labeled agent for PSMA-targeted α-therapy. J. Nucl. Med. 2022, 63, 259–267. [Google Scholar] [CrossRef]
- Ohshima, Y.; Suzuki, H.; Hanaoka, H.; Sasaki, I.; Watanabe, S.; Haba, H.; Arano, Y.; Tsushima, Y.; Ishioka, N.S. Preclinical evaluation of new α-radionuclide therapy targeting LAT1: 2-[211At]astato-α-methyl-L-phenylalanine in tumor-bearing model. Nucl. Med. Biol. 2020, 90–91, 15–22. [Google Scholar] [CrossRef]
- Suzuki, H.; Kaizuka, Y.; Tatsuta, M.; Tanaka, H.; Washiya, N.; Shirakami, Y.; Ooe, K.; Toyoshima, A.; Watabe, T.; Teramoto, T. Neopentyl glycol as a scaffold to provide radiohalogenated theranostic pairs of high in vivo stability. J. Med. Chem. 2021, 64, 15846–15857. [Google Scholar] [CrossRef] [PubMed]
- Berdal, M.; Gouard, S.; Eychenne, R.; Marionneau-Lambot, S.; Croyal, M.; Faivre-Chauvet, A.; Chérel, M.; Gaschet, J.; Gestin, J.-F.; Guérard, F. Investigation on the reactivity of nucleophilic radiohalogens with arylboronic acids in water: Access to an efficient single-step method for the radioiodination and astatination of antibodies. Chem. Sci. 2021, 12, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Meares, C.; Goodwin, D. Linking radiometals to proteins with bifunctional chelating agents. J. Protein Chem. 1984, 3, 215–228. [Google Scholar] [CrossRef]
- Brechbiel, M.W. Bifunctional chelates for metal nuclides. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 166. [Google Scholar]
- Zarschler, K.; Kubeil, M.; Stephan, H. Establishment of two complementary in vitro assays for radiocopper complexes achieving reliable and comparable evaluation of in vivo stability. RSC Adv. 2014, 4, 10157–10164. [Google Scholar] [CrossRef]
- Linder, M. Ceruloplasmin and other copper binding components of blood plasma and their functions: An update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, D.; Cornelissen, B. Emerging chelators for nuclear imaging. Curr. Opin. Chem. Biol. 2021, 63, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Wilson, J.J. Advancing chelation strategies for large metal ions for nuclear medicine applications. Acc. Chem. Res. 2022, 55, 904–915. [Google Scholar] [CrossRef]
- Deshpande, S.; Subramanian, R.; McCall, M.; DeNardo, S.J.; DeNardo, G.; Meares, C. Metabolism of indium chelates attached to monoclonal antibody: Minimal transchelation of indium from benzyl-EDTA chelate in vivo. J. Nucl. Med. 1990, 31, 218–224. [Google Scholar]
- Meares, C.; Moi, M.; Diril, H.; Kukis, D.; McCall, M.; Deshpande, S.; DeNardo, S.; Snook, D.; Epenetos, A. Macrocyclic chelates of radiometals for diagnosis and therapy. Br. J. Cancer Suppl. 1990, 10, 21. [Google Scholar]
- Cabbiness, D.K.; Margerum, D.W. Macrocyclic effect on the stability of copper (II) tetramine complexes. J. Am. Chem. Soc. 1969, 91, 6540–6541. [Google Scholar] [CrossRef]
- Cooper, M.S.; Sabbah, E.; Mather, S.J. Conjugation of chelating agents to proteins and radiolabeling with trivalent metallic isotopes. Nat. Protoc. 2006, 1, 314–317. [Google Scholar] [CrossRef]
- De León-Rodríguez, L.M.; Kovacs, Z. The synthesis and chelation chemistry of DOTA−peptide conjugates. Bioconj. Chem. 2008, 19, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Sosabowski, J.K.; Mather, S.J. Conjugation of DOTA-like chelating agents to peptides and radiolabeling with trivalent metallic isotopes. Nat. Protoc. 2006, 1, 972–976. [Google Scholar] [CrossRef]
- Brechbiel, M.W.; Gansow, O.A. Backbone-substituted DTPA ligands for yttrium-90 radioimmunotherapy. Bioconj. Chem. 1991, 2, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Kobayashi, H.; Sun, B.; Yoo, T.; Paik, C.; Gansow, O.; Carrasquillo, J.; Pastan, I.; Brechbiel, M. Stereochemical influence on the stability of radio-metal complexes in vivo. Synthesis and evaluation of the four stereoisomers of 2-(p-nitrobenzyl)-trans-CyDTPA. Biorg. Med. Chem. 1997, 5, 1925–1934. [Google Scholar] [CrossRef]
- Price, E.W.; Cawthray, J.F.; Bailey, G.A.; Ferreira, C.L.; Boros, E.; Adam, M.J.; Orvig, C. H4octapa: An acyclic chelator for 111In radiopharmaceuticals. J. Am. Chem. Soc. 2012, 134, 8670–8683. [Google Scholar] [CrossRef]
- Bailey, G.A.; Price, E.W.; Zeglis, B.M.; Ferreira, C.L.; Boros, E.; Lacasse, M.J.; Patrick, B.O.; Lewis, J.S.; Adam, M.J.; Orvig, C. H2azapa: A versatile acyclic multifunctional chelator for 67Ga, 64Cu, 111In, and 177Lu. Inorg. Chem. 2012, 51, 12575–12589. [Google Scholar] [CrossRef] [PubMed]
- Spreckelmeyer, S.; Ramogida, C.F.; Rousseau, J.; Arane, K.; Bratanovic, I.; Colpo, N.; Jermilova, U.; Dias, G.M.; Dude, I.; Jaraquemada-Pelaez, M.d.G. p-NO2–Bn–H4neunpa and H4neunpa–Trastuzumab: Bifunctional Chelator for Radiometalpharmaceuticals and 111In Immuno-Single Photon Emission Computed Tomography Imaging. Bioconj. Chem. 2017, 28, 2145–2159. [Google Scholar] [CrossRef]
- Deri, M.A.; Zeglis, B.M.; Francesconi, L.C.; Lewis, J.S. PET imaging with 89Zr: From radiochemistry to the clinic. Nucl. Med. Biol. 2013, 40, 3–14. [Google Scholar] [CrossRef]
- Guérard, F.; Lee, Y.-S.; Tripier, R.; Szajek, L.P.; Deschamps, J.R.; Brechbiel, M.W. Investigation of Zr (IV) and 89Zr (IV) complexation with hydroxamates: Progress towards designing a better chelator than desferrioxamine B for immuno-PET imaging. Chem. Commun. 2013, 49, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- Heskamp, S.; Raavé, R.; Boerman, O.; Rijpkema, M.; Goncalves, V.; Denat, F. 89Zr-immuno-positron emission tomography in oncology: State-of-the-art 89Zr radiochemistry. Bioconj. Chem. 2017, 28, 2211–2223. [Google Scholar] [CrossRef] [PubMed]
- Abou, D.S.; Ku, T.; Smith-Jones, P.M. In vivo biodistribution and accumulation of 89Zr in mice. Nucl. Med. Biol. 2011, 38, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.P.; Divilov, V.; Bander, N.H.; Smith-Jones, P.M.; Larson, S.M.; Lewis, J.S. 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J. Nucl. Med. 2010, 51, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, N.B.; Pandya, D.N.; Wadas, T.J. Recent advances in zirconium-89 chelator development. Molecules 2018, 23, 638. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Lu, X.; Janisse, J.; Muzik, O.; Shields, A.F. PET of human prostate cancer xenografts in mice with increased uptake of 64CuCl2. J. Nucl. Med. 2006, 47, 1649–1652. [Google Scholar] [PubMed]
- Mirick, G.R.; O’Donnell, R.T.; DeNardo, S.J.; Shen, S.; Meares, C.F.; DeNardo, G.L. Transfer of copper from a chelated 67Cu-antibody conjugate to ceruloplasmin in lymphoma patients. Nucl. Med. Biol. 1999, 26, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.V. Molecular imaging with copper-64. J. Inorg. Biochem. 2004, 98, 1874–1901. [Google Scholar] [CrossRef]
- Gutfilen, B.; Souza, S.A.; Valentini, G. Copper-64: A real theranostic agent. Drug Des. Devel. Ther. 2018, 12, 3235–3245. [Google Scholar] [CrossRef]
- Gaware, V. Ceruloplasmin its role and significance: A review. Int. J. Biomed. Res. 2010, 1, 153–162. [Google Scholar] [CrossRef]
- Bass, L.A.; Wang, M.; Welch, M.J.; Anderson, C.J. In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconj. Chem. 2000, 11, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Shokeen, M.; Anderson, C.J. Molecular imaging of cancer with copper-64 radiopharmaceuticals and positron emission tomography (PET). Acc. Chem. Res. 2009, 42, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Kurihara, H.; Yonemori, K.; Tsuda, H.; Suzuki, J.; Kono, Y.; Honda, N.; Kodaira, M.; Yamamoto, H.; Yunokawa, M. 64Cu-DOTA-trastuzumab PET imaging in patients with HER2-positive breast cancer. J. Nucl. Med. 2013, 54, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Ping Li, W.; Meyer, L.A.; Capretto, D.A.; Sherman, C.D.; Anderson, C.J. Receptor-binding, biodistribution, and metabolism studies of 64Cu-DOTA-cetuximab, a PET-imaging agent for epidermal growth-factor receptor-positive tumors. Cancer Biother. Radiopharm. 2008, 23, 158–171. [Google Scholar] [PubMed]
- Paudyal, B.; Paudyal, P.; Oriuchi, N.; Hanaoka, H.; Tominaga, H.; Endo, K. Positron emission tomography imaging and biodistribution of vascular endothelial growth factor with 64Cu-labeled bevacizumab in colorectal cancer xenografts. Cancer Sci. 2011, 102, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, V.; Dearling, J.L.; Treves, S.T.; Packard, A.B. Measurement of the rate of copper (II) exchange for 64Cu complexes of bifunctional chelators. Inorg. Chim. Acta 2012, 393, 318–323. [Google Scholar] [CrossRef]
- Zhang, Y.; Hong, H.; Engle, J.W.; Bean, J.; Yang, Y.; Leigh, B.R.; Barnhart, T.E.; Cai, W. Positron emission tomography imaging of CD105 expression with a 64Cu-labeled monoclonal antibody: NOTA is superior to DOTA. PLoS ONE 2011, 6, e28005. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Sun, X.; Niu, W.; Weisman, G.R.; Wong, E.H.; Rheingold, A.L.; Anderson, C.J. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J. Med. Chem. 2004, 47, 1465–1474. [Google Scholar] [CrossRef]
- Knighton, R.C.; Troadec, T.; Mazan, V.; Le Saëc, P.; Marionneau-Lambot, S.; Le Bihan, T.; Saffon-Merceron, N.; Le Bris, N.; Chérel, M.; Faivre-Chauvet, A. Cyclam-based chelators bearing phosphonated pyridine pendants for 64Cu-PET imaging: Synthesis, physicochemical studies, radiolabeling, and bioimaging. Inorg. Chem. 2021, 60, 2634–2648. [Google Scholar] [CrossRef]
- Pazderová, L.; David, T.; Hlinová, V.; Plutnar, J.; Kotek, J.; Lubal, P.; Kubíček, V.; Hermann, P. Cross-Bridged Cyclam with Phosphonate and Phosphinate Pendant Arms: Chelators for Copper Radioisotopes with Fast Complexation. Inorg. Chem. 2020, 59, 8432–8443. [Google Scholar] [CrossRef]
- Morgan, K.A.; Rudd, S.E.; Noor, A.; Donnelly, P.S. Theranostic Nuclear Medicine with Gallium-68, Lutetium-177, Copper-64/67, Actinium-225, and Lead-212/203 Radionuclides: Focus Review. Chem. Rev. 2023, 123, 12004–12035. [Google Scholar] [CrossRef] [PubMed]
- Comba, P.; Kubeil, M.; Pietzsch, J.; Rudolf, H.; Stephan, H.; Zarschler, K. Bispidine dioxotetraaza macrocycles: A new class of bispidines for 64Cu PET imaging. Inorg. Chem. 2014, 53, 6698–6707. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.R.; Ma, D.; Simon, J.; Frank, R.K.; Scheinberg, D.A. Design and synthesis of 225Ac radioimmunopharmaceuticals. Appl. Radiat. Isot. 2002, 57, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Maguire, W.F.; McDevitt, M.R.; Smith-Jones, P.M.; Scheinberg, D.A. Efficient 1-step radiolabeling of monoclonal antibodies to high specific activity with 225Ac for α-particle radioimmunotherapy of cancer. J. Nucl. Med. 2014, 55, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Thiele, N.A.; Brown, V.; Kelly, J.M.; Amor-Coarasa, A.; Jermilova, U.; MacMillan, S.N.; Nikolopoulou, A.; Ponnala, S.; Ramogida, C.F.; Robertson, A.K. An eighteen-membered macrocyclic ligand for actinium-225 targeted alpha therapy. Angew. Chem. Int. Ed. 2017, 56, 14712–14717. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.; Jaggi, J.; O’donoghue, J.; Ruan, S.; McDevitt, M.; Larson, S.; Scheinberg, D.; Humm, J. Renal uptake of bismuth-213 and its contribution to kidney radiation dose following administration of actinium-225-labeled antibody. Phys. Med. Biol. 2011, 56, 721. [Google Scholar] [CrossRef] [PubMed]
- Mirzadeh, S.; Kumar, K.; Gansow, O.A. The chemical fate of 212Bi-DOTA formed by β- decay of 212Pb (DOTA)2−. Radiochim. Acta 1993, 60, 1–10. [Google Scholar] [CrossRef]
- Jaggi, J.S.; Seshan, S.V.; McDevitt, M.R.; Sgouros, G.; Hyjek, E.; Scheinberg, D.A. Mitigation of radiation nephropathy after internal α-particle irradiation of kidneys. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 1503–1512. [Google Scholar] [CrossRef]
- Poty, S.; Francesconi, L.C.; McDevitt, M.R.; Morris, M.J.; Lewis, J.S. α-emitters for radiotherapy: From basic radiochemistry to clinical studies—Part 2. J. Nucl. Med. 2018, 59, 1020–1027. [Google Scholar] [CrossRef]
- Meredith, R.; Torgue, J.; Shen, S.; Fisher, D.R.; Banaga, E.; Bunch, P.; Morgan, D.; Fan, J.; Straughn, J.M. Dose escalation and dosimetry of first-in-human α radioimmunotherapy with 212Pb-TCMC-trastuzumab. J. Nucl. Med. 2014, 55, 1636–1642. [Google Scholar] [CrossRef]
- McDevitt, M.R.; Ma, D.; Lai, L.T.; Simon, J.; Borchardt, P.; Frank, R.K.; Wu, K.; Pellegrini, V.; Curcio, M.J.; Miederer, M. Tumor therapy with targeted atomic nanogenerators. Science 2001, 294, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Altai, M.; Membreno, R.; Cook, B.; Tolmachev, V.; Zeglis, B.M. Pretargeted imaging and therapy. J. Nucl. Med. 2017, 58, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, D.; Meares, C.; McCall, M.; McTigue, M.; Chaovapong, W.; Levy, R.; Starnes, C. Pre-targeted immunoscintigraphy with chimeric antibodies. J. Nucl. Med. 1987, 28, 561. [Google Scholar]
- Goodwin, D.; Meares, C.; McCall, M.; McTigue, M.; Chaovapong, W. An avidin-biotin chelate system for imaging tumor. J. Nucl. Med. 1987, 28, 129. [Google Scholar]
- Sakahara, H.; Saga, T. Avidin–biotin system for delivery of diagnostic agents. Adv. Drug Del. Rev. 1999, 37, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Livaniou, E.; Evangelatos, G.P.; Ithakissios, D.S. Radioiodinated biotin derivatives for in vitro radioassays. J. Nucl. Med. 1987, 28, 1430–1434. [Google Scholar] [PubMed]
- Hymes, J.; Wolf, B. Biotinidase and its roles in biotin metabolism. Clin. Chim. Acta 1996, 255, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wilbur, D.S.; Hamlin, D.K.; Pathare, P.M.; Weerawarna, S.A. Biotin reagents for antibody pretargeting. Synthesis, radioiodination, and in vitro evaluation of water soluble, biotinidase resistant biotin derivatives. Bioconj. Chem. 1997, 8, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Foulon, C.F.; Alston, K.L.; Zalutsky, M.R. Synthesis and preliminary biological evaluation of (3-iodobenzoyl) norbiotinamide and ((5-iodo-3-pyridinyl) carbonyl) norbiotinamide: Two radioiodinated biotin conjugates with improved stability. Bioconj. Chem. 1997, 8, 179–186. [Google Scholar] [CrossRef]
- Magnani, P.; Paganelli, G.; Modorati, G.; Zito, F.; Songini, C.; Sudati, F.; Koch, P.; Maecke, H.R.; Brancato, R.; Siccardi, A.G. Quantitative comparison of direct antibody labeling and tumor pretargeting in uveal melanoma. J. Nucl. Med. 1996, 37, 967–971. [Google Scholar]
- Verhoeven, M.; Seimbille, Y.; Dalm, S.U. Therapeutic applications of pretargeting. Pharmaceutics 2019, 11, 434. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, D.A.; Meares, C.F.; McCall, M.J.; McTigue, M.; Chaovapong, W. Pre-targeted immunoscintigraphy of murine tumors with indium-111-labeled bifunctional haptens. J. Nucl. Med. 1988, 29, 226–234. [Google Scholar] [PubMed]
- Goodwin, D.A.; Meares, C.F. Advances in pretargeting biotechnology. Biotechnol. Adv. 2001, 19, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Janevik-Ivanovska, E.; Gautherot, E.; Hillairet de Boisferon, M.; Cohen, M.; Milhaud, G.; Tartar, A.; Rostene, W.; Barbet, J.; Gruaz-Guyon, A. Bivalent hapten-bearing peptides designed for iodine-131 pretargeted radioimmunotherapy. Bioconj. Chem. 1997, 8, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Doussal, J.-M.L.; Gruaz-Guyon, A.; Martin, M.; Gautherot, E.; Delaage, M.; Barbet, J. Targeting of indium 111-labeled bivalent hapten to human melanoma mediated by bispecific monoclonal antibody conjugates: Imaging of tumors hosted in nude mice. Cancer Res. 1990, 50, 3445–3452. [Google Scholar] [PubMed]
- Gautherot, E.; Rouvier, E.; Daniel, L.; Loucif, E.; Bouhou, J.; Manetti, C.; Martin, M.; Le Doussal, J.-M.; Barbet, J. Pretargeted radioimmunotherapy of human colorectal xenografts with bispecific antibody and 131I-labeled bivalent hapten. J. Nucl. Med. 2000, 41, 480–487. [Google Scholar] [PubMed]
- Sharkey, R.M.; van Rij, C.M.; Karacay, H.; Rossi, E.A.; Frielink, C.; Regino, C.; Cardillo, T.M.; McBride, W.J.; Chang, C.-H.; Boerman, O.C. A new tri-Fab bispecific antibody for pretargeting Trop-2–expressing epithelial cancers. J. Nucl. Med. 2012, 53, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Schoffelen, R.; Boerman, O.C.; Goldenberg, D.M.; Sharkey, R.M.; van Herpen, C.M.; Franssen, G.M.; McBride, W.J.; Chang, C.-H.; Rossi, E.A.; van der Graaf, W.T. Development of an imaging-guided CEA-pretargeted radionuclide treatment of advanced colorectal cancer: First clinical results. Br. J. Cancer 2013, 109, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Bodet-Milin, C.; Faivre-Chauvet, A.; Carlier, T.; Rauscher, A.; Bourgeois, M.; Cerato, E.; Rohmer, V.; Couturier, O.; Drui, D.; Goldenberg, D.M. Immuno-PET using anticarcinoembryonic antigen bispecific antibody and 68Ga-labeled peptide in metastatic medullary thyroid carcinoma: Clinical optimization of the pretargeting parameters in a first-in-human trial. J. Nucl. Med. 2016, 57, 1505–1511. [Google Scholar] [CrossRef]
- Zhou, Y.; Penny, H.L.; Kroenke, M.A.; Bautista, B.; Hainline, K.; Chea, L.S.; Parnes, J.; Mytych, D.T. Immunogenicity assessment of bispecific antibody-based immunotherapy in oncology. J. Immunother. Cancer 2022, 10, e004225. [Google Scholar] [CrossRef]
- Rousseau, C.; Goldenberg, D.M.; Colombié, M.; Sébille, J.-C.; Meingan, P.; Ferrer, L.; Baumgartner, P.; Cerato, E.; Masson, D.; Campone, M. Initial clinical results of a novel immuno-PET theranostic probe in human epidermal growth factor receptor 2–negative breast cancer. J. Nucl. Med. 2020, 61, 1205–1211. [Google Scholar] [CrossRef]
- Touchefeu, Y.; Bailly, C.; Frampas, E.; Eugène, T.; Rousseau, C.; Bourgeois, M.; Bossard, C.; Faivre-Chauvet, A.; Rauscher, A.; Masson, D. Promising clinical performance of pretargeted immuno-PET with anti-CEA bispecific antibody and gallium-68-labelled IMP-288 peptide for imaging colorectal cancer metastases: A pilot study. Eur. J. Nucl. Med. Mol. Imag. 2021, 48, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Rusckowski, M.; Qu, T.; Chang, F.; Hnatowich, D.J. Pretargeting using peptide nucleic acid. Cancer 1997, 80, 2699–2705. [Google Scholar] [CrossRef]
- Liu, G.; Mang’era, K.; Liu, N.; Gupta, S.; Rusckowski, M.; Hnatowich, D.J. Tumor pretargeting in mice using 99mTc-labeled morpholino, a DNA analog. J. Nucl. Med. 2002, 43, 384–391. [Google Scholar] [PubMed]
- Schubert, M.; Bergmann, R.; Förster, C.; Sihver, W.; Vonhoff, S.; Klussmann, S.; Bethge, L.; Walther, M.; Schlesinger, J.; Pietzsch, J. Novel tumor pretargeting system based on complementary L-configured oligonucleotides. Bioconj. Chem. 2017, 28, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Zarschler, K.; Pietzsch, H.-J.; Stephan, H.; Gasser, G. New insights into the pretargeting approach to image and treat tumours. Chem. Soc. Rev. 2016, 45, 6415–6431. [Google Scholar] [CrossRef] [PubMed]
- Blackman, M.L.; Royzen, M.; Fox, J.M. Tetrazine ligation: Fast bioconjugation based on inverse-electron-demand Diels− Alder reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. [Google Scholar] [CrossRef] [PubMed]
- Rossin, R.; Verkerk, P.R.; van den Bosch, S.M.; Vulders, R.C.; Verel, I.; Lub, J.; Robillard, M.S. In vivo chemistry for pretargeted tumor imaging in live mice. Angew. Chem. Int. Ed. 2010, 49, 3375–3378. [Google Scholar] [CrossRef] [PubMed]
- Rossin, R.; Van Den Bosch, S.M.; Ten Hoeve, W.; Carvelli, M.; Versteegen, R.M.; Lub, J.; Robillard, M.S. Highly reactive trans-cyclooctene tags with improved stability for Diels–Alder chemistry in living systems. Bioconj. Chem. 2013, 24, 1210–1217. [Google Scholar] [CrossRef]
- Rondon, A.; Degoul, F. Antibody pretargeting based on bioorthogonal click chemistry for cancer imaging and targeted radionuclide therapy. Bioconj. Chem. 2019, 31, 159–173. [Google Scholar] [CrossRef]
- Rondon, A.; Rouanet, J.; Degoul, F. Radioimmunotherapy in oncology: Overview of the last decade clinical trials. Cancers 2021, 13, 5570. [Google Scholar] [CrossRef] [PubMed]
- Akizawa, H.; Uehara, T.; Arano, Y. Renal uptake and metabolism of radiopharmaceuticals derived from peptides and proteins. Adv. Drug Del. Rev. 2008, 60, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Yoo, T.M.; Kim, I.S.; Kim, M.-K.; Le, N.; Webber, K.O.; Pastan, I.; Paik, C.H.; Eckelman, W.C.; Carrasquillo, J.A. L-lysine effectively blocks renal uptake of 125I-or 99mTc-labeled anti-Tac disulfide-stabilized Fv fragment. Cancer Res. 1996, 56, 3788–3795. [Google Scholar]
- Kobayashi, H.; Le, N.; Kim, I.-s.; Kim, M.-K.; Pie, J.-E.; Drumm, D.; Paik, D.S.; Waldmann, T.A.; Paik, C.H.; Carrasquillo, J.A. The pharmacokinetic characteristics of glycolated humanized anti-Tac Fabs are determined by their isoelectric points. Cancer Res. 1999, 59, 422–430. [Google Scholar] [PubMed]
- Arano, Y. Renal brush border strategy: A developing procedure to reduce renal radioactivity levels of radiolabeled polypeptides. Nucl. Med. Biol. 2021, 92, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Arano, Y.; Fujioka, Y.; Akizawa, H.; Ono, M.; Uehara, T.; Wakisaka, K.; Nakayama, M.; Sakahara, H.; Konishi, J.; Saji, H. Chemical design of radiolabeled antibody fragments for low renal radioactivity levels. Cancer Res. 1999, 59, 128–134. [Google Scholar]
- Uehara, T.; Koike, M.; Nakata, H.; Hanaoka, H.; Iida, Y.; Hashimoto, K.; Akizawa, H.; Endo, K.; Arano, Y. Design, synthesis, and evaluation of [188Re]organorhenium-labeled antibody fragments with renal enzyme-cleavable linkage for low renal radioactivity levels. Bioconj. Chem. 2007, 18, 190–198. [Google Scholar] [CrossRef]
- Suzuki, C.; Uehara, T.; Kanazawa, N.; Wada, S.; Suzuki, H.; Arano, Y. Preferential cleavage of a tripeptide linkage by enzymes on renal brush border membrane to reduce renal radioactivity levels of radiolabeled antibody fragments. J. Med. Chem. 2018, 61, 5257–5268. [Google Scholar] [CrossRef]
- Uehara, T.; Kanazawa, N.; Suzuki, C.; Mizuno, Y.; Suzuki, H.; Hanaoka, H.; Arano, Y. Renal handling of 99mTc-labeled antibody fab fragments with a linkage cleavable by enzymes on brush border membrane. Bioconj. Chem. 2020, 31, 2618–2627. [Google Scholar] [CrossRef]
- Uehara, T.; Yokoyama, M.; Suzuki, H.; Hanaoka, H.; Arano, Y. Gallium-67/68-labeled antibody fragments for immuno-SPECT/PET show low renal radioactivity without loss of tumor uptake. Clin. Cancer Res. 2018, 24, 3309–3316. [Google Scholar] [CrossRef]
- Suzuki, H.; Kise, S.; Kaizuka, Y.; Watanabe, R.; Sugawa, T.; Furukawa, T.; Fujii, H.; Uehara, T. Copper-64-labeled antibody fragments for immuno-PET/radioimmunotherapy with low renal radioactivity levels and amplified tumor-kidney ratios. ACS Omega 2021, 6, 21556–21562. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Araki, M.; Tatsugi, K.; Ichinohe, K.; Uehara, T.; Arano, Y. Reduction of the Renal Radioactivity of 111In-DOTA-Labeled Antibody Fragments with a Linkage Cleaved by the Renal Brush Border Membrane Enzymes. J. Med. Chem. 2023, 66, 8600–8613. [Google Scholar] [CrossRef] [PubMed]
- Yudistiro, R.; Hanaoka, H.; Katsumata, N.; Yamaguchi, A.; Tsushima, Y. Bevacizumab radioimmunotherapy (RIT) with accelerated blood clearance using the avidin chase. Mol. Pharm. 2018, 15, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, T.L.; Brinkley, J.; Banks, S.; Vohra, N.; Englert, Z.P.; Zervos, E.E. The benefits of liver resection for non-colorectal, non-neuroendocrine liver metastases: A systematic review. Langenbecks Arch. Surg. 2014, 399, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Arano, Y.; Mukai, T.; Uezono, T.; Wakisaka, K.; Motonari, H.; Akizawa, H.; Taoka, Y.; Yokoyama, A. A biological method to evaluate bifunctional chelating agents to label antibodies with metallic radionuclides. J. Nucl. Med. 1994, 35, 890–898. [Google Scholar] [PubMed]
- Mukai, T.; Namba, S.; Arano, Y.; Ono, M.; Fujioka, Y.; Uehara, T.; Ogawa, K.; Konishi, J.; Saji, H. Synthesis and evaluation of a monoreactive DOTA derivative for indium-111-based residualizing label to estimate protein pharmacokinetics. J. Pharm. Pharmacol. 2002, 54, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Arano, Y.; Wakisaka, K.; Ohmono, Y.; Uezono, T.; Akizawa, H.; Nakayama, M.; Sakahara, H.; Tanaka, C.; Konishi, J.; Yokoyama, A. Assessment of radiochemical design of antibodies using an ester bond as the metabolizable linkage: Evaluation of maleimidoethyl 3-(tri-n-butylstannyl) hippurate as a radioiodination reagent of antibodies for diagnostic and therapeutic applications. Bioconj. Chem. 1996, 7, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Arano, Y.; Wakisaka, K.; Akizawa, H.; Ono, M.; Kawai, K.; Nakayama, M.; Sakahara, H.; Konishi, J.; Saji, H. Assessment of the radiochemical design of antibodies with a metabolizable linkage for target-selective radioactivity delivery. Bioconj. Chem. 1998, 9, 497–506. [Google Scholar] [CrossRef]
- Antczak, C.; Jaggi, J.S.; LeFave, C.V.; Curcio, M.J.; McDevitt, M.R.; Scheinberg, D.A. Influence of the linker on the biodistribution and catabolism of actinium-225 self-immolative tumor-targeted isotope generators. Bioconj. Chem. 2006, 17, 1551–1560. [Google Scholar] [CrossRef]
- Suzuki, H.; Matsukawa, M.; Madokoro, R.; Terasaka, Y.; Kannaka, K.; Uehara, T. Reduction of the hepatic radioactivity levels of [111In]In-DOTA–labeled antibodies via cleavage of a linkage metabolized in lysosomes. Nucl. Med. Biol. 2024; in press. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, H.; Kannaka, K.; Uehara, T. Approaches to Reducing Normal Tissue Radiation from Radiolabeled Antibodies. Pharmaceuticals 2024, 17, 508. https://doi.org/10.3390/ph17040508
Suzuki H, Kannaka K, Uehara T. Approaches to Reducing Normal Tissue Radiation from Radiolabeled Antibodies. Pharmaceuticals. 2024; 17(4):508. https://doi.org/10.3390/ph17040508
Chicago/Turabian StyleSuzuki, Hiroyuki, Kento Kannaka, and Tomoya Uehara. 2024. "Approaches to Reducing Normal Tissue Radiation from Radiolabeled Antibodies" Pharmaceuticals 17, no. 4: 508. https://doi.org/10.3390/ph17040508
APA StyleSuzuki, H., Kannaka, K., & Uehara, T. (2024). Approaches to Reducing Normal Tissue Radiation from Radiolabeled Antibodies. Pharmaceuticals, 17(4), 508. https://doi.org/10.3390/ph17040508