


Unveiling the Potential of Ent-Kaurane Diterpenoids: Multifaceted Natural Products for Drug Discovery

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
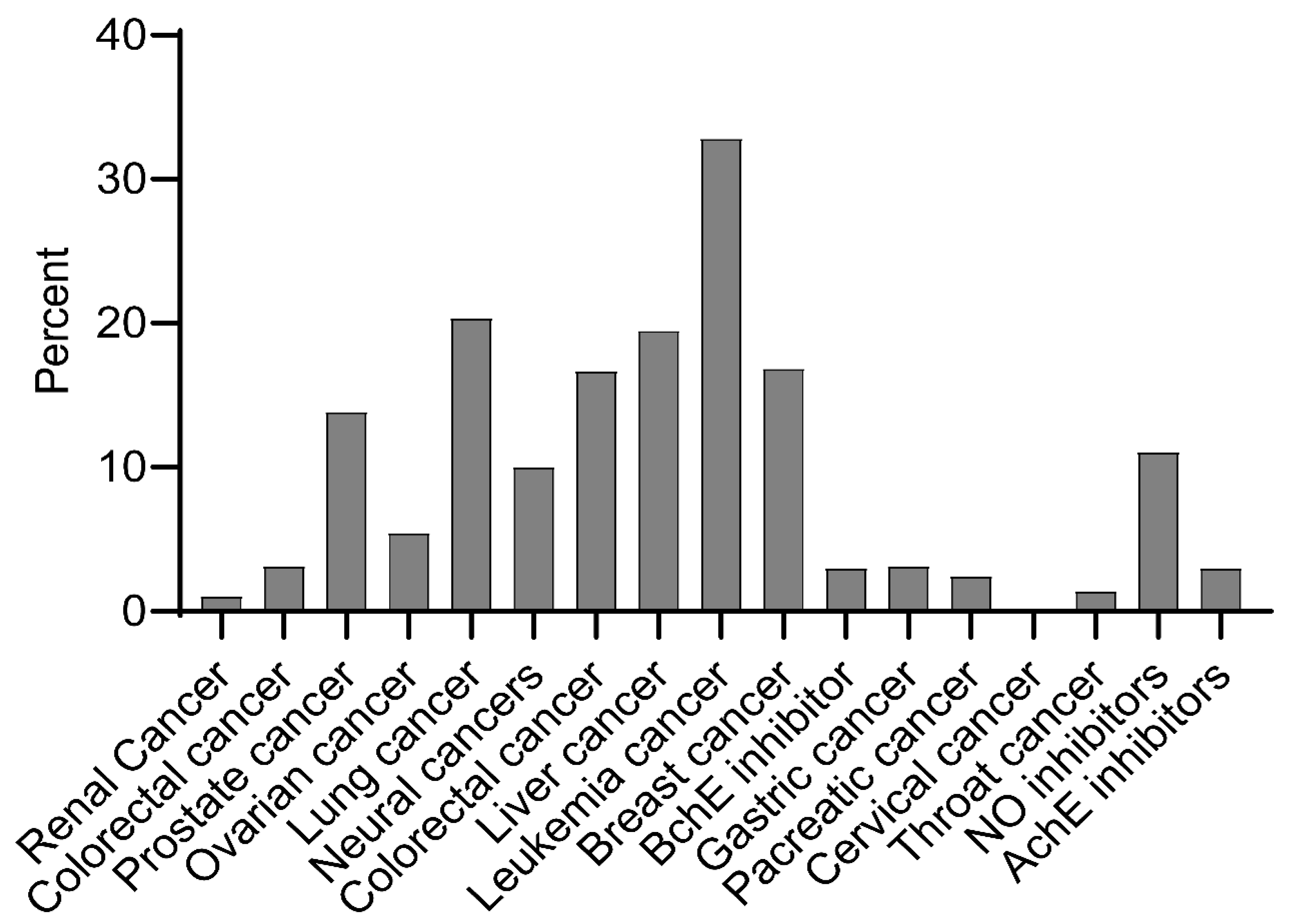
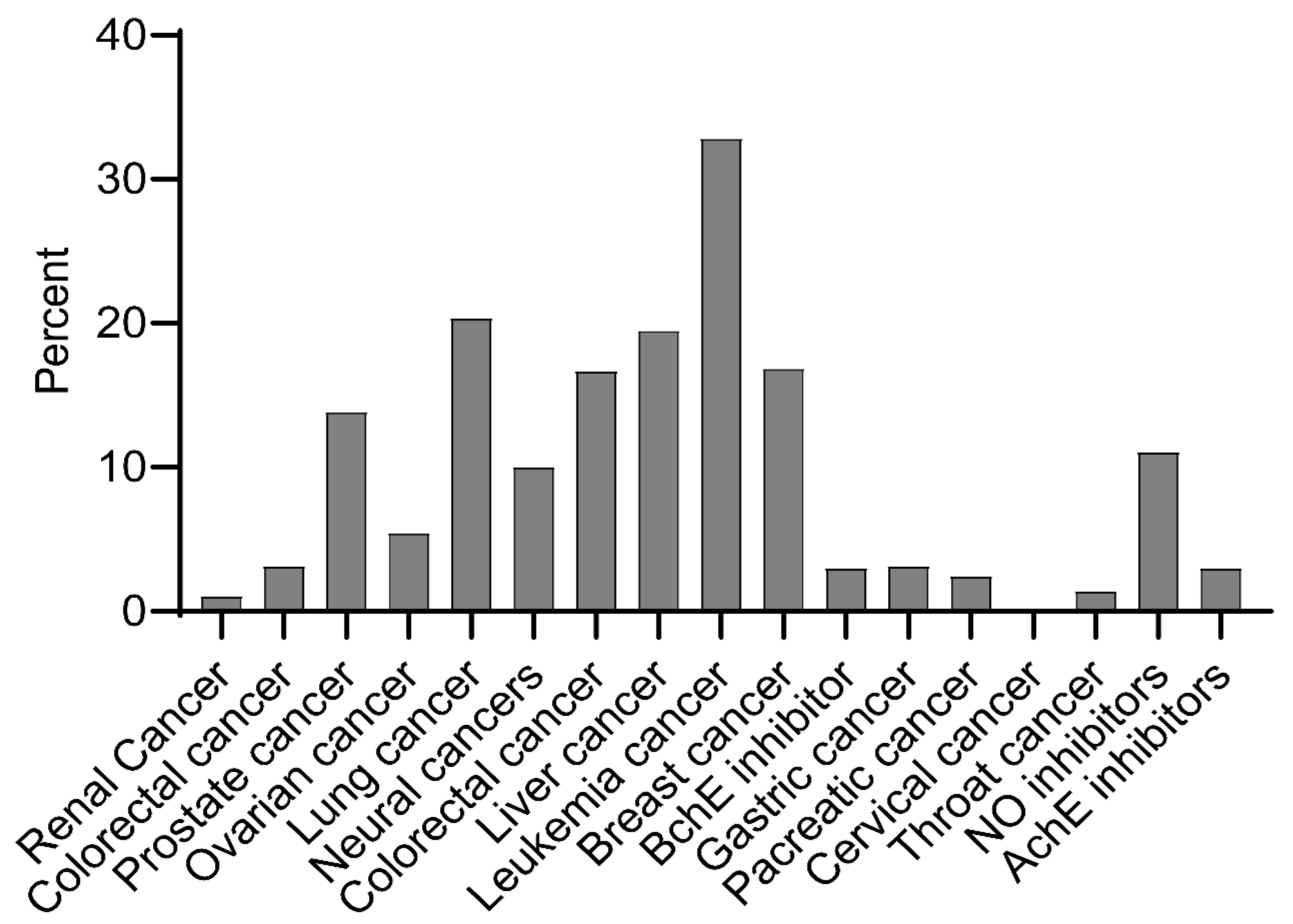
2.1. Bioactivity of Ent-Kaurane Diterpenoids
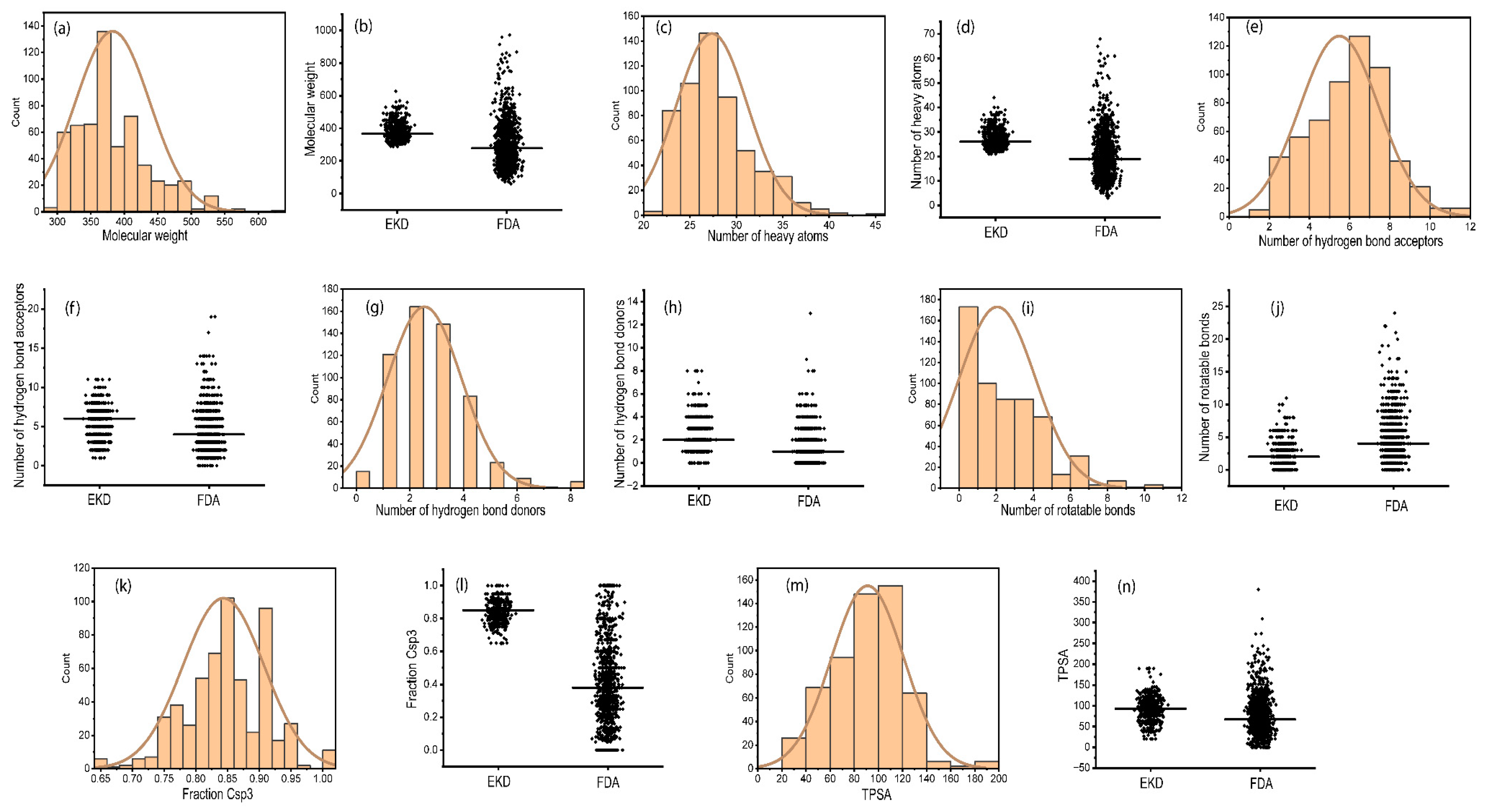
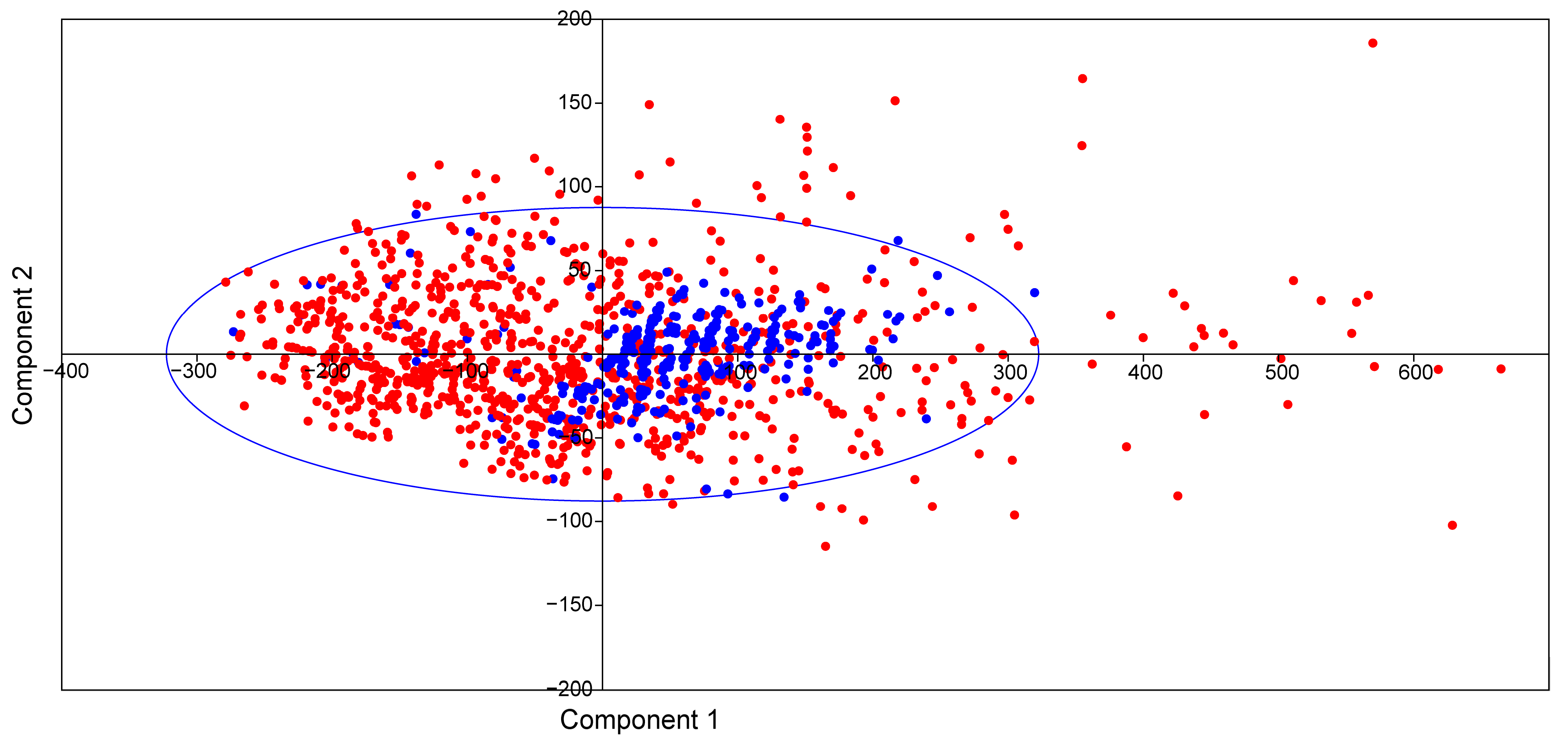
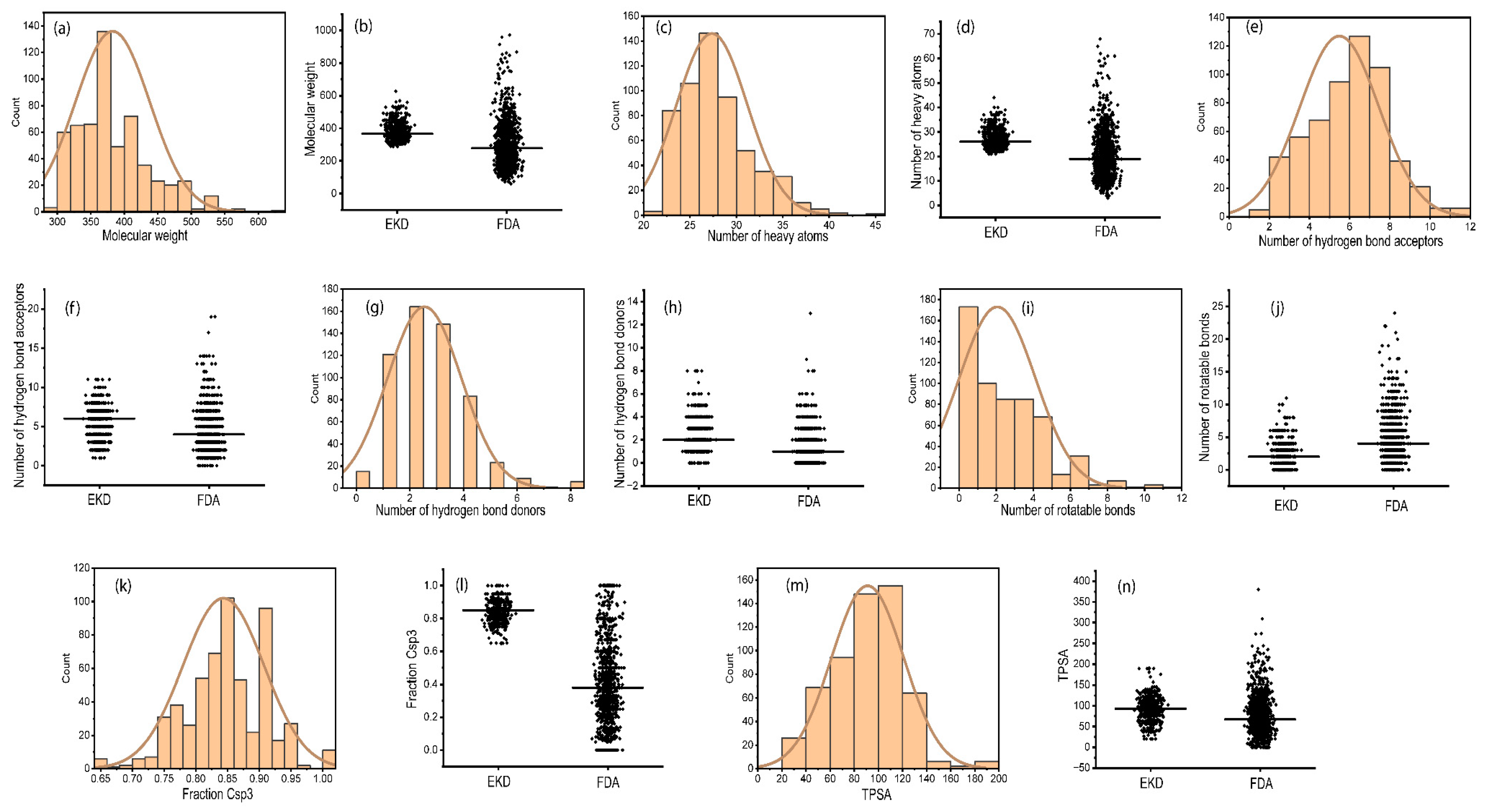
2.2. Physicochemical Properties of Ent-Kaurane Diterpenoids
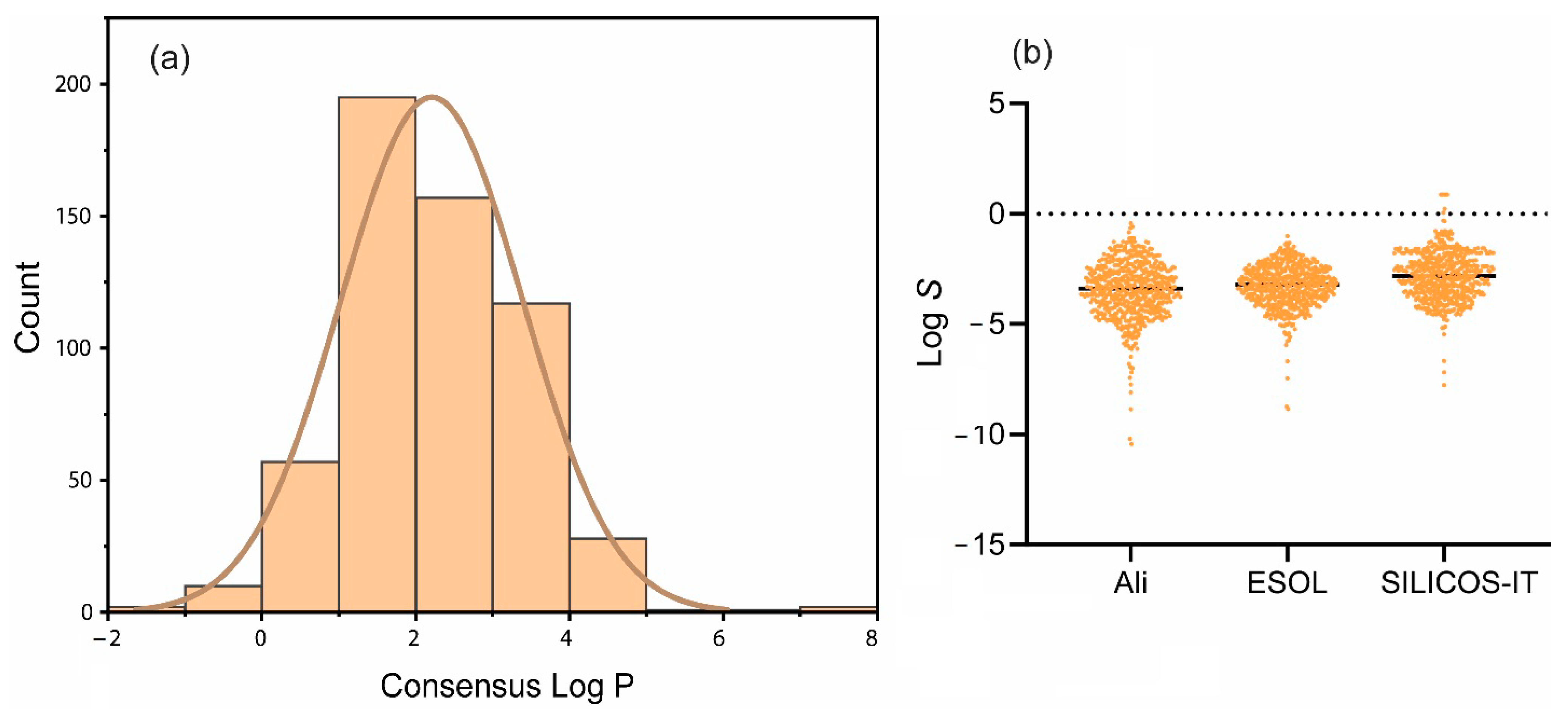
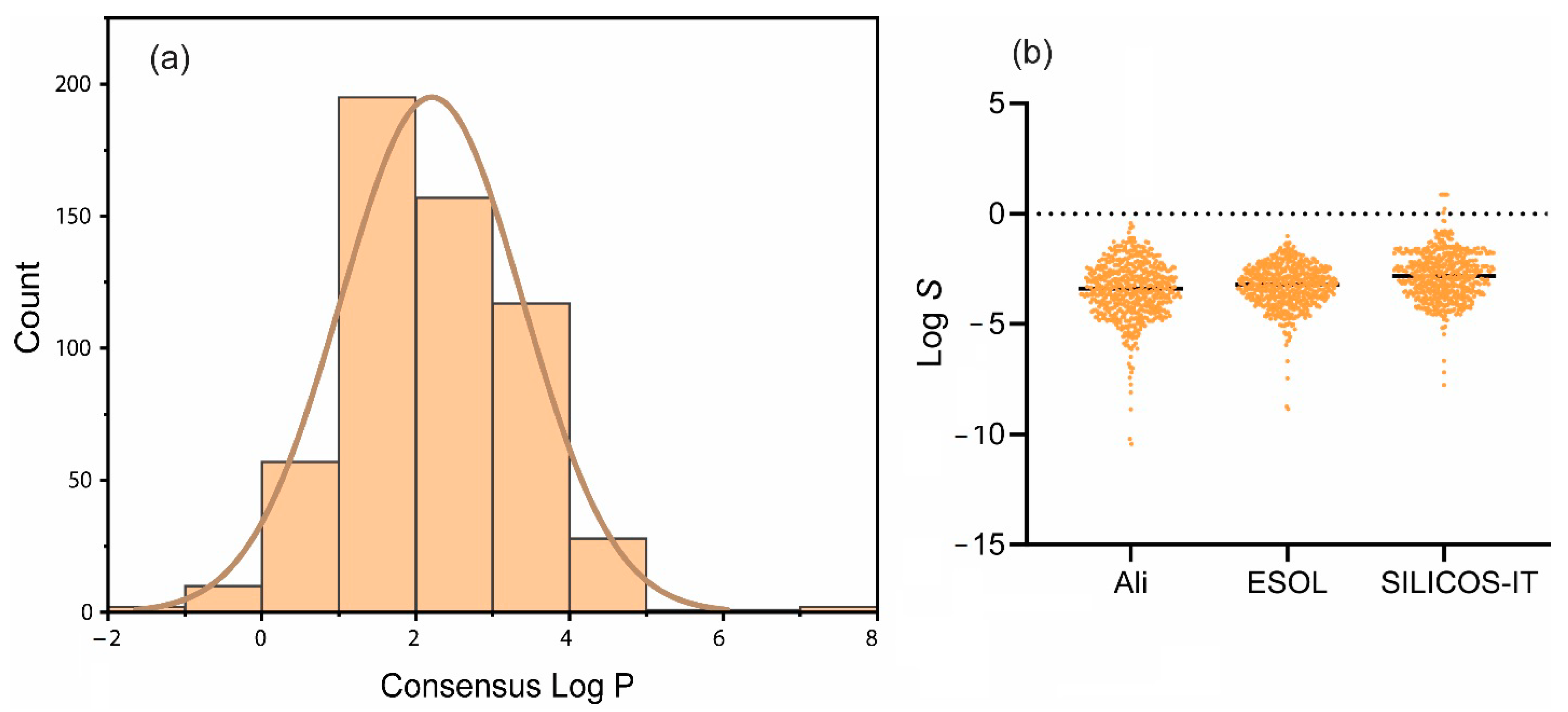
2.3. Lipophilicity and Water Solubility of Ent-Kaurane Diterpenoids
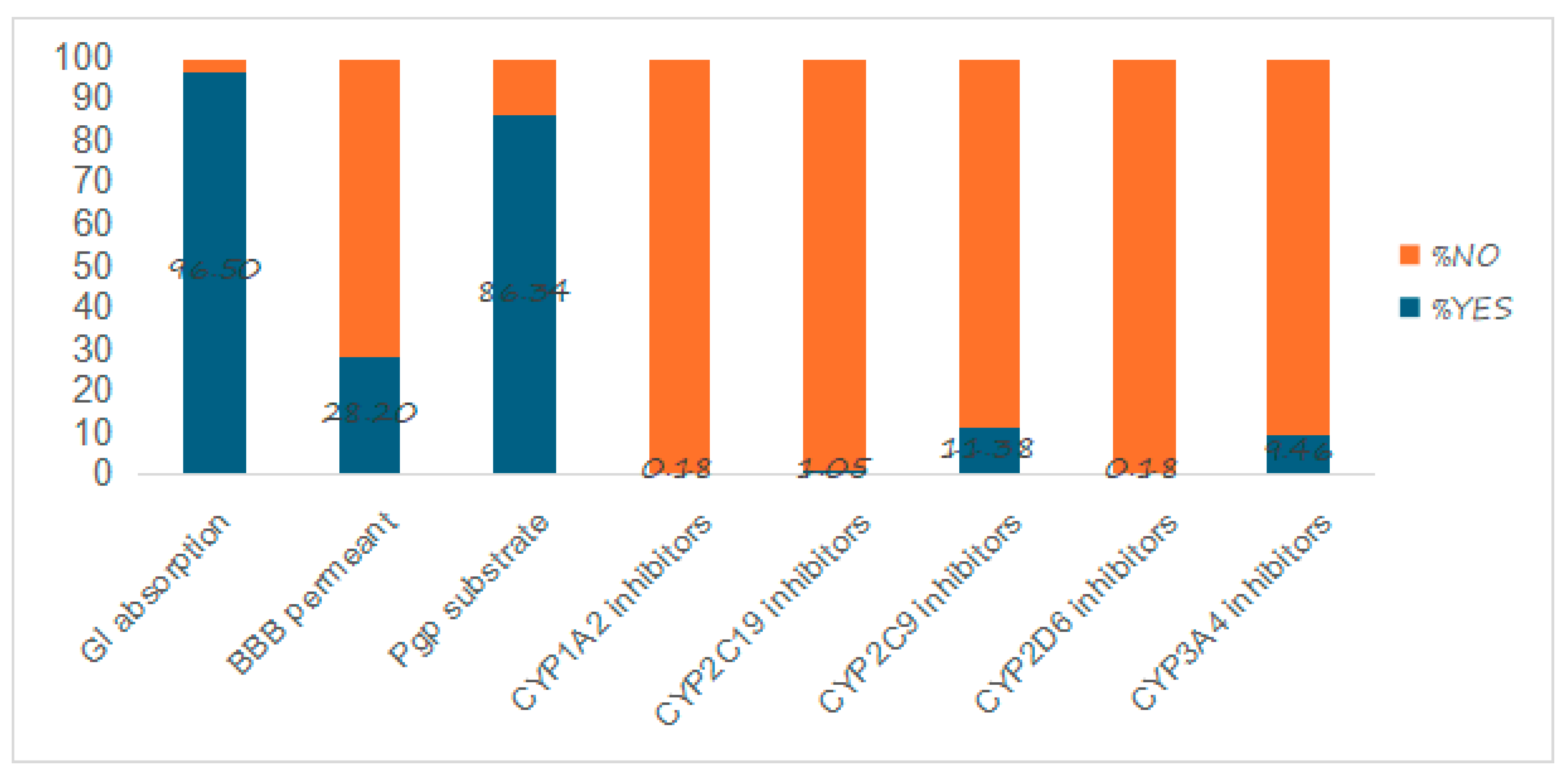
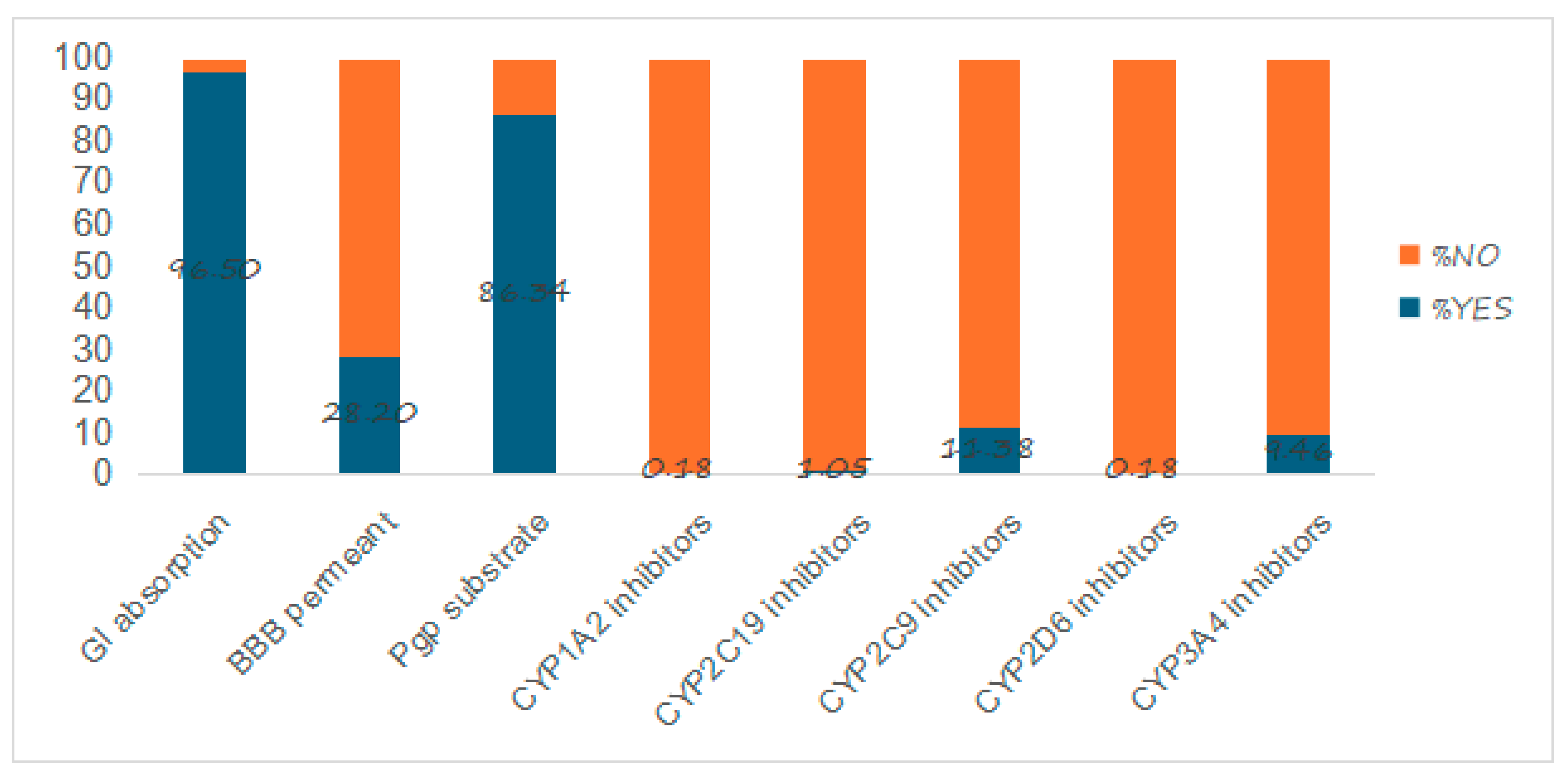
2.4. Pharmacokinetic Properties
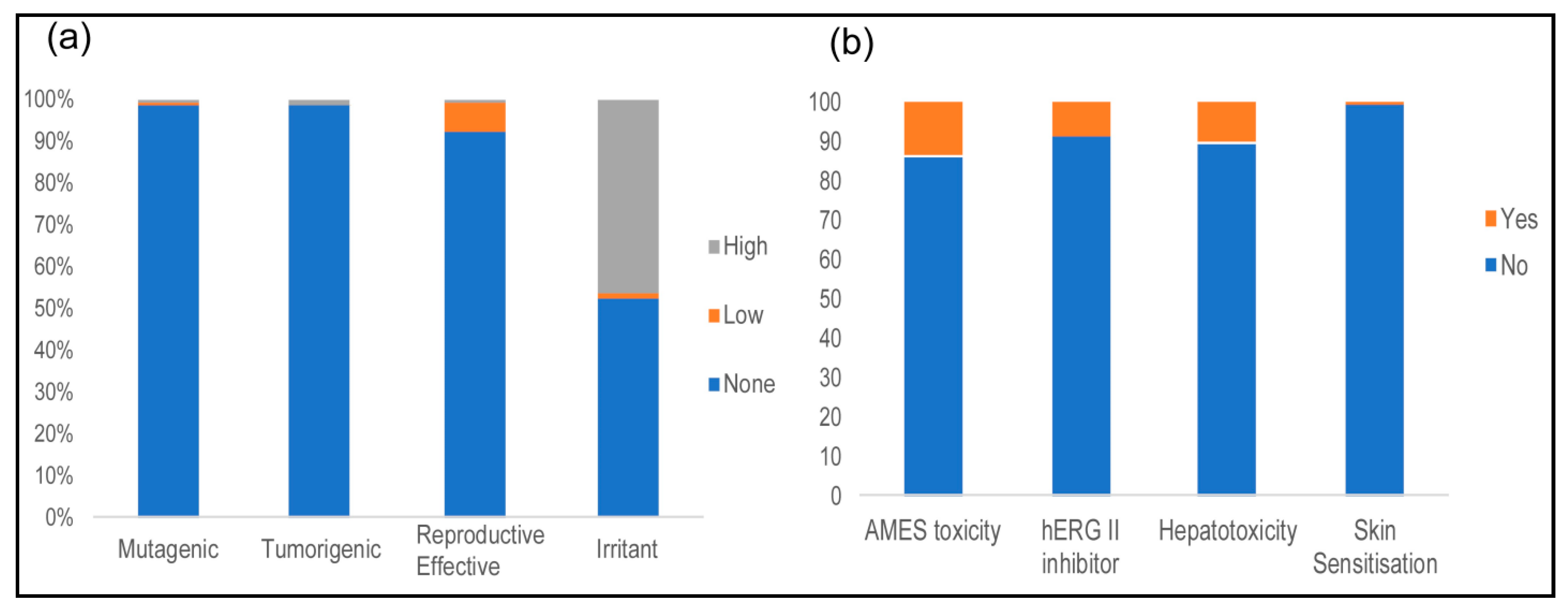
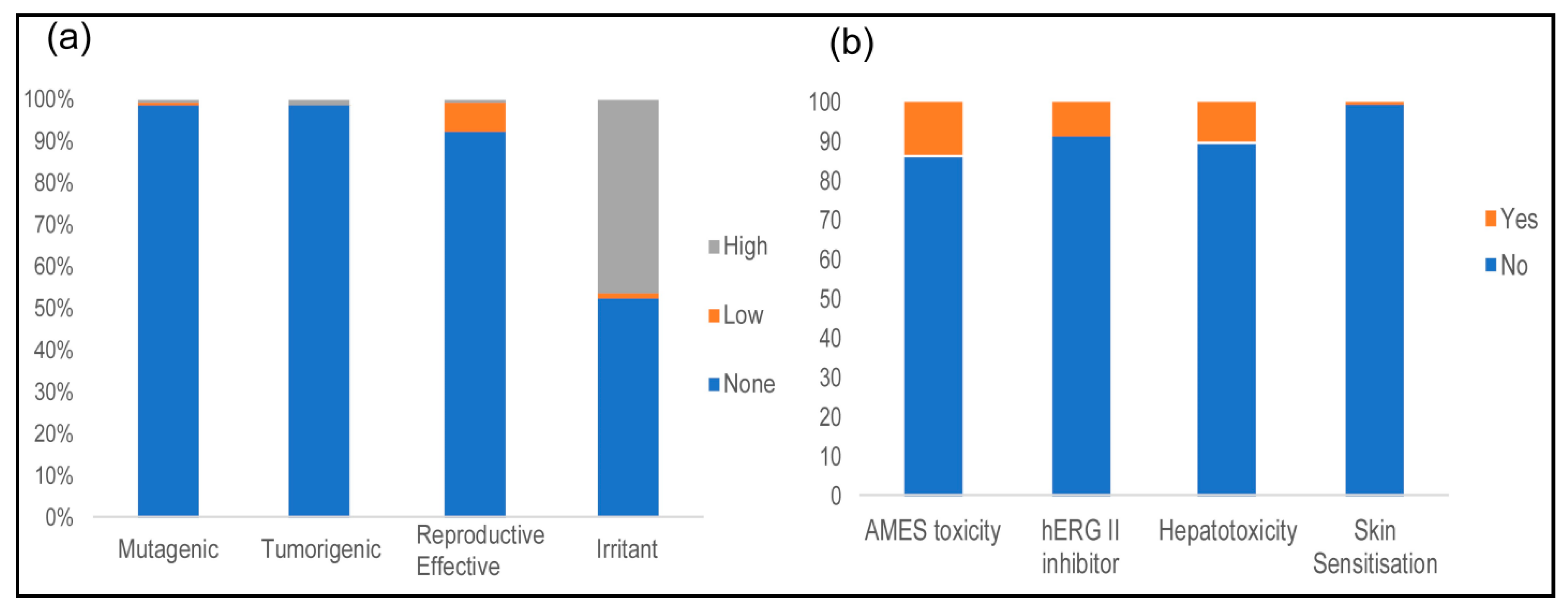
2.5. Toxicological Properties
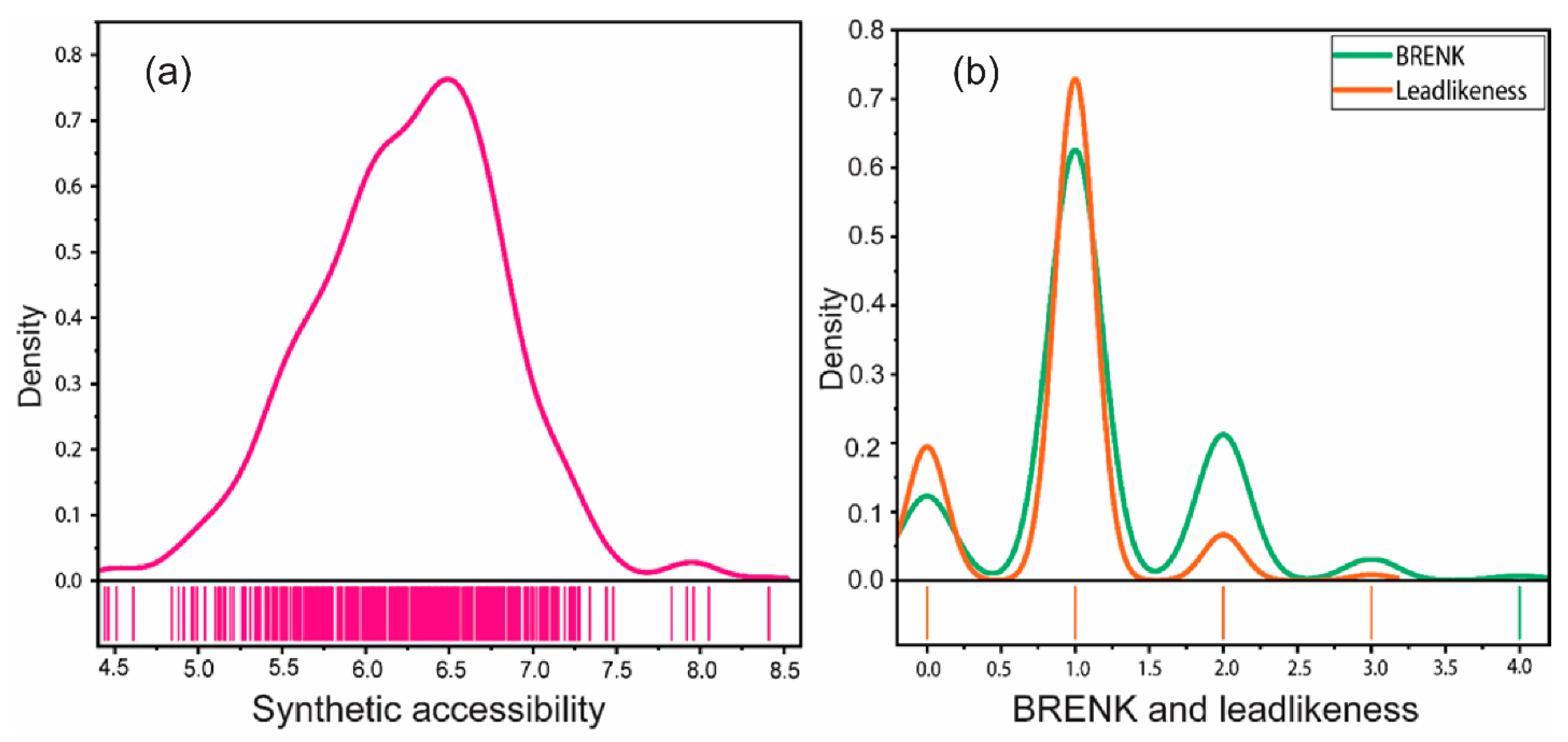
2.6. Synthetic Accessibility, PAINS, BRENK, and Lead-likeness
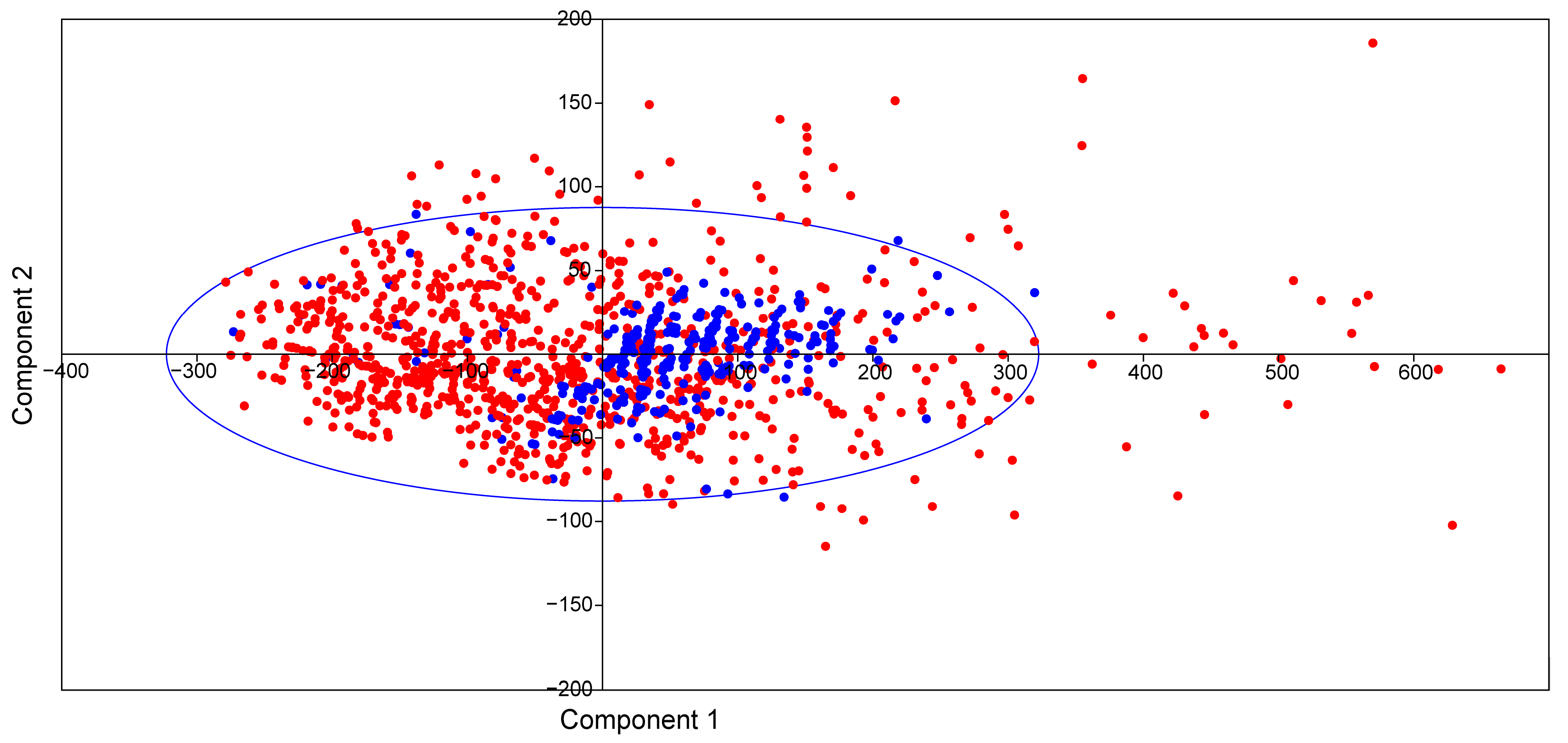
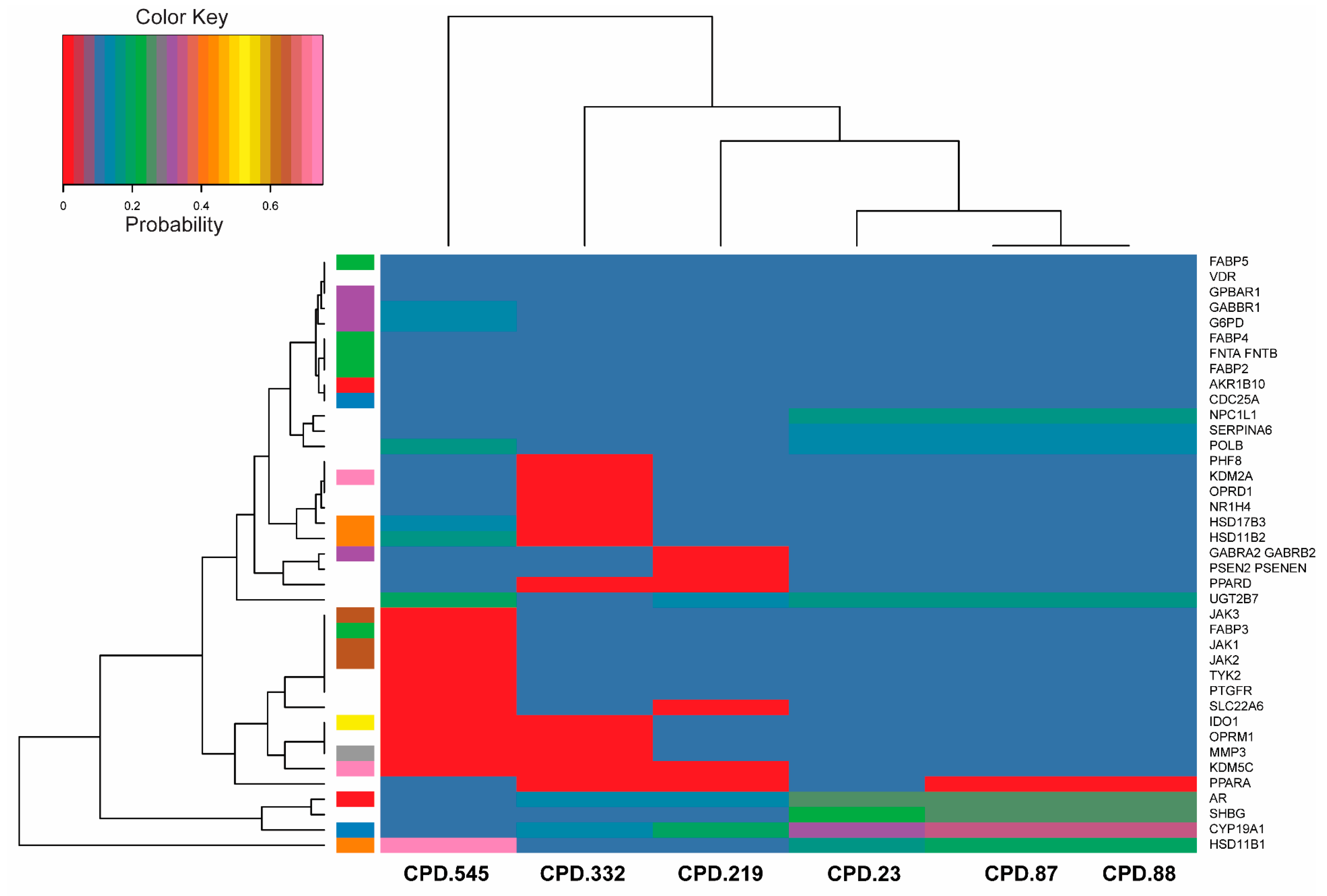
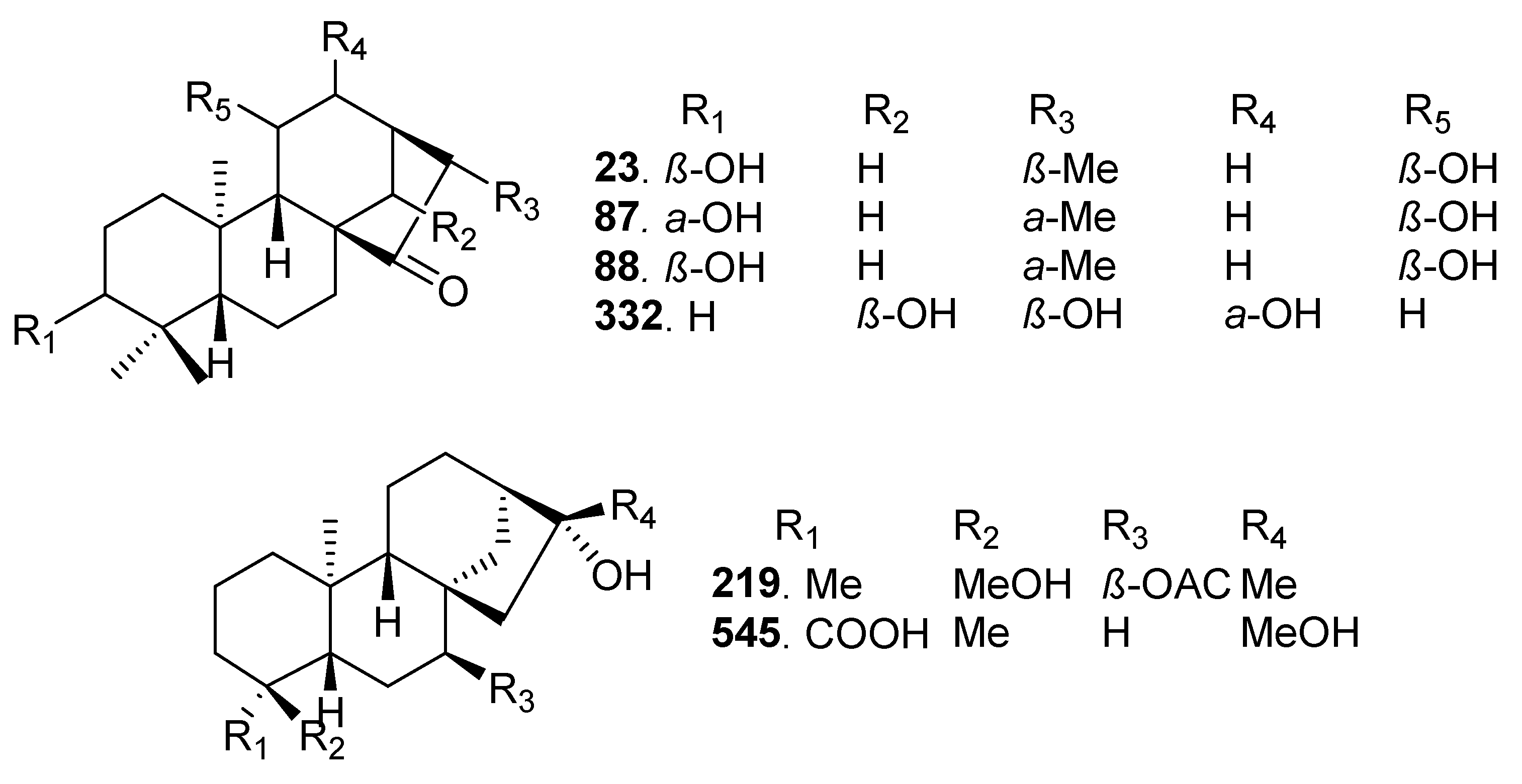
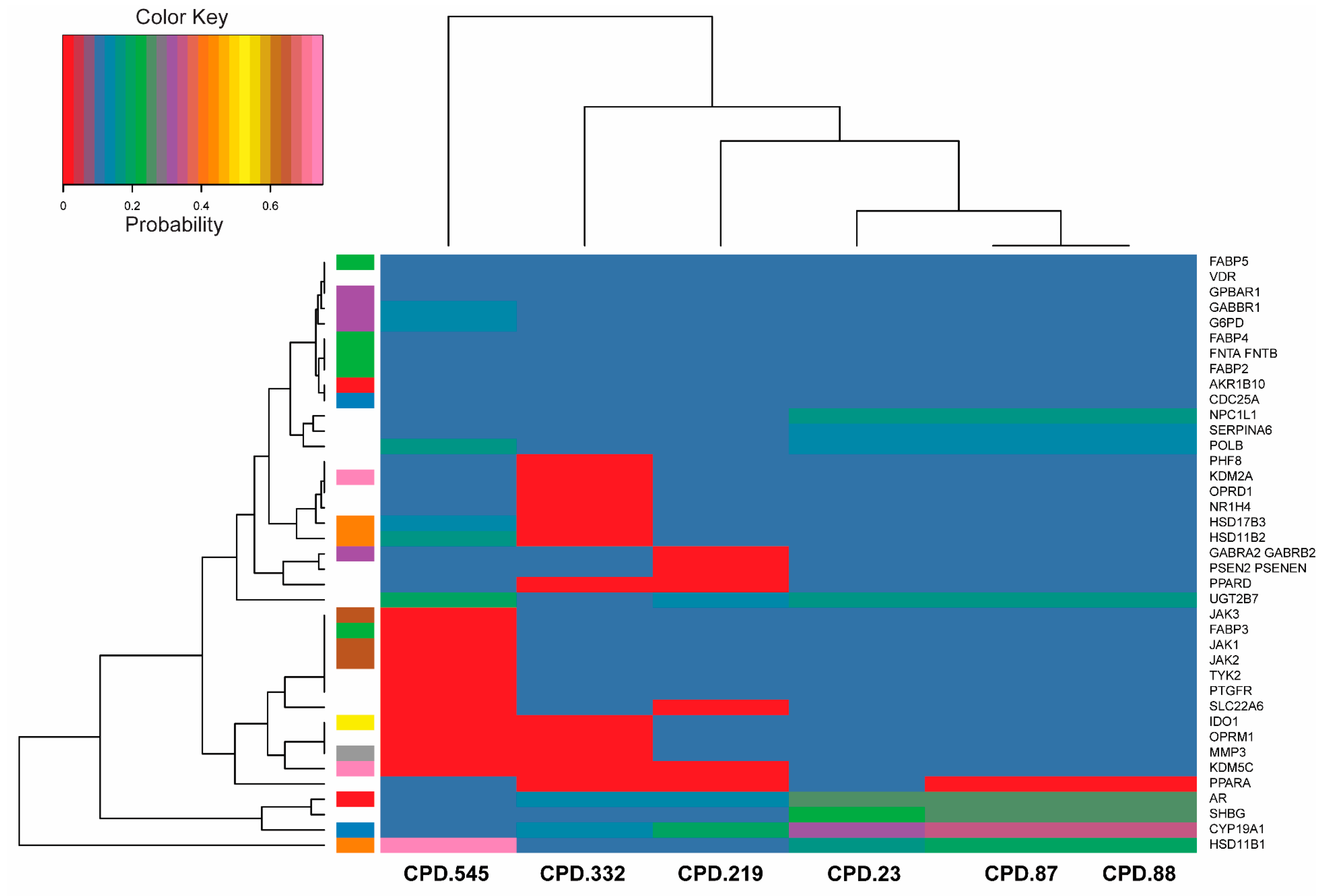
2.7. Bioactivity Scores and Macromolecular Targets of Selected Molecules
3. Discussion
3.1. Sources and Bioactivity of Ent-Kaurane Diterpenoids
3.2. Physicochemical Properties
3.3. Lipophilicity and Water Solubilities
3.4. Pharmacokinetic Properties
3.5. Toxicological Properties
3.6. Medicinal Chemistry: Synthetic Accessibility, PAINS, BRENK, and Lead-likeness
3.7. Macromolecular Targets of Selected Molecules
4. Materials and Methods
4.1. Accessing Ent-Kaurane Diterpenoids and FDA-Approved Drugs
4.2. Physicochemical and ADME Properties of Ent-Kaurane Diterpenoids
4.3. Toxicological Properties of Ent-Kaurane Diterpenoids
4.4. Selection of Molecules and Prediction of Their Macromolecular Targets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Malik, S.; Brudzyńska, P.; Khan, M.R.; Sytar, O.; Makhzoum, A.; Sionkowska, A. Natural Plant-Derived Compounds in Food and Cosmetics: A Paradigm of Shikonin and Its Derivatives. Materials 2023, 16, 4377. [Google Scholar] [CrossRef]
- Newman, D.J.; Phil, D. Natural products and drug discovery. Natl. Sci. Rev. 2022, 9, nwac206. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K. The role of natural products as sources of therapeutic agents for innovative drug discovery. Compr. Pharmacol. 2022, 408, 408–422. [Google Scholar] [CrossRef]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminformat. 2020, 12, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, W.; Wang, J.; Ma, D. A Convergent Route to ent-Kaurane Diterpenoids: Total Synthesis of Lungshengenin D and 1α,6α-Diacetoxy-ent-kaura-9(11),16-dien-12,15-dione. J. Am. Chem. Soc. 2017, 139, 2932–2935. [Google Scholar] [CrossRef]
- Yang, J.; Wang, W.-G.; Wu, H.-Y.; Du, X.; Li, X.-N.; Li, Y.; Pu, J.-X.; Sun, H.-D. Bioactive Enmein-Type ent-Kaurane Diterpenoids from Isodon phyllostachys. J. Nat. Prod. 2015, 79, 132–140. [Google Scholar] [CrossRef]
- Zhao, X.; Cacherat, B.; Hu, Q.; Ma, D. Natural Product Reports Recent advances in the synthesis of ent-kaurane diterpenoids. 2022, 39, 119–138. 39. [CrossRef]
- Lee, J.E.; Thuy, N.T.T.; Lee, Y.; Cho, N.; Yoo, H.M. An Antiproliferative ent-Kaurane Diterpene Isolated from the Roots of Mallotus japonicus Induced Apoptosis in Leukemic Cells. Nat. Prod. Commun. 2020, 15, 1934578X19897496. [Google Scholar] [CrossRef]
- Deng, Z.; Chen, J.; Geng, C. Bioorganic Chemistry α -glucosidase inhibitory activity. Bioorg. Chem. 2020, 95, 103571. [Google Scholar]
- Kimani, N.M.; Backhaus, S.; Matasyoh, J.C.; Kaiser, M.; Herrmann, F.C.; Schmidt, T.J.; Langer, K. Preparation of sesquiterpene lactone-loaded PLA nanoparticles and evaluation of their antitrypanosomal activity. Molecules 2019, 24, 2110. [Google Scholar] [CrossRef]
- Kimani, N.M.; Matasyoh, J.C.; Kaiser, M.; Brun, R.; Schmidt, T.J. Antiprotozoal sesquiterpene lactones and other constituents from Tarchonanthus camphoratus and Schkuhria pinnata. J. Nat. Prod. 2018, 81, 124–130. [Google Scholar] [CrossRef]
- Kimani, N.M.; Ochieng, C.O.; Ogutu, M.D.; Yamo, K.O.; Onyango, J.O.; Santos, C.B.R. Inhibition Kinetics and Theoretical Studies on Zanthoxylum chalybeum Engl. Dual Inhibitors of α-Glucosidase and α-Amylase. J. Xenobiotics 2023, 13, 102–120. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Parveen, A.; Parveen, B.; Parveen, R.; Ahmad, S. Challenges and guidelines for clinical trial of herbal drugs. J. Pharm. Bioallied Sci. 2015, 7, 329. [Google Scholar]
- Cui, W.; Aouidate, A.; Wang, S.; Yu, Q.; Li, Y.; Yuan, S. Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 2020, 11, 542271. [Google Scholar] [CrossRef]
- Hingorani, A.D.; Kuan, V.; Finan, C.; Kruger, F.A.; Gaulton, A.; Chopade, S.; Sofat, R.; MacAllister, R.J.; Overington, J.P.; Hemingway, H.; et al. Improving the odds of drug development success through human genomics: Modelling study. Sci. Rep. 2019, 9, 18911. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.; Schnider, P.; Mattei, P.; Kansy, M. Pharmacological promiscuity: Dependence on compound properties and target specificity in a set of recent Roche compounds. ChemMedChem Chem. Enabling Drug Discov. 2009, 4, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Hersey, A.; Montanari, D.; Overington, J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discov. 2011, 10, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Luker, T.; Alcaraz, L.; Chohan, K.K.; Blomberg, N.; Brown, D.S.; Butlin, R.J.; Elebring, T.; Griffin, A.M.; Guile, S.; St-Gallay, S.; et al. Strategies to improve in vivo toxicology outcomes for basic candidate drug molecules. Bioorg. Med. Chem. Lett. 2011, 21, 5673–5679. [Google Scholar] [CrossRef] [PubMed]
- Bucar, F.; Wube, A.; Schmid, M. Natural product isolation–how to get from biological material to pure compounds. Nat. Prod. Rep. 2013, 30, 525–545. [Google Scholar] [CrossRef]
- Pham, J.V.; Yilma, M.A.; Feliz, A.; Majid, M.T.; Maffetone, N.; Walker, J.R.; Kim, E.; Cho, H.J.; Reynolds, J.M.; Song, M.C.; et al. A review of the microbial production of bioactive natural products and biologics. Front. Microbiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, S.; Spohn, M.S.; Schäberle, T.F. Bioactive natural products from Bacteroidetes. Nat. Prod. Rep. 2022, 39, 1045–1065. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.-T. Medicinal Chemistry of Bioactive Natural Products; Wiley Online Library: Hoboken, NJ, USA, 2006. [Google Scholar]
- Ojuka, P.; Kimani, N.M.; Apollo, S.; Nyariki, J.; Ramos, R.S.; Santos, C.B.R. Phytochemistry of plants in the genus Vepris: A review and in silico analysis of their ADMET properties. S. Afr. J. Bot. 2023, 157, 106–114. [Google Scholar] [CrossRef]
- Wekesa, E.N.; Kimani, N.M.; Kituyi, S.N.; Omosa, L.K.; Santos, C.B.R. Therapeutic potential of the genus Zanthoxylum phytochemicals: A theoretical ADME/Tox analysis. S. Afr. J. Bot. 2023, 162, 129–141. [Google Scholar] [CrossRef]
- Simoben, C.V.; Qaseem, A.; Moumbock, A.F.A.; Telukunta, K.K.; Günther, S.; Sippl, W.; Ntie-Kang, F. Pharmacoinformatic Investigation of Medicinal Plants from East Africa. Mol. Inform. 2020, 39, 2000163. [Google Scholar] [CrossRef]
- Sun, H.D.; Xu, Y.L.; Jiang, B. Diterpenoids from Isodon species. Nat. Prod. Rep. 2001, 23, 673–698. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; An, L.; Song, Z.; Li, S.; Wang, H.; Wang, C.; Zhang, J.; Tuerhong, M.; Abudukeremu, M.; Li, D.; et al. Anti-Inflammatory ent -Kaurane Diterpenoids from Isodon serra. no. Table 2; American Chemical Society and American Society of Pharmacognosy: Washington, DC, USA, 2019; pp. 2–11. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, Y.; Zhao, P.; Zhou, L.; Wang, X.; Huang, X. ent -Kaurane Diterpenoids with Neuroprotective Properties from Corn Silk (Zea mays). J. Nat. Prod. 2017, 7, 1225–1234. [Google Scholar] [CrossRef]
- García, P.A.; De Oliveira, A.B.; Batista, R. Occurrence, Biological Activities and Synthesis of Kaurane Diterpenes and their Glycosides. Molecules 2007, 12, 455–483. [Google Scholar] [CrossRef]
- Sun, Y.; Qiao, Y.; Liu, Y.; Zhou, J.; Wang, X.; Zheng, H.; Xu, Z.; Zhang, J.; Zhou, Y.; Qian, L.; et al. Redox Biology ent -Kaurane diterpenoids induce apoptosis and ferroptosis through targeting redox resetting to overcome cisplatin resistance. Redox Biol. 2021, 43, 101977. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.H.; Meng, L.L.; Liu, X.T.; Shu, P.F.; Yuan, C.; An, X.T.; Jia, T.X.; Yang, Q.Q.; Zhen, X.; Fan, C.A. Asymmetric Divergent Synthesis of ent-Kaurane-, ent-Atisane-, ent-Beyerane-, ent-Trachylobane-, and ent-Gibberellane-type Diterpenoids. J. Am. Chem. Soc. 2023, 145, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hong, B.; Hu, D.; Kadonaga, Y.; Tang, R.; Lei, X. Protecting-Group-Free Syntheses of ent-Kaurane Diterpenoids: [3+2+1] Cycloaddition/Cycloalkenylation Approach. J. Am. Chem. Soc. 2020, 142, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Cherney, E.C.; Green, J.C.; Baran, P.S. Synthesis of ent-Kaurane and Beyerane Diterpenoids via Controlled Fragmentations of Overbred Intermediates. Angew. Chem. Int. Ed. Engl. 2013, 52, 9019–9022. [Google Scholar] [CrossRef]
- Hu, Y.; Li, X.N.; Ma, Z.J.; Puno, P.T.; Zhao, Y.; Zhao, Y.; Xiao, Y.Z.; Zhang, W.; Liu, J.P. Synthesis of Novel ent-Kaurane-Type Diterpenoid Derivatives Effective for Highly Aggressive Tumor Cells. Molecules 2018, 23, 3216. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Guo, Y.; Gao, Y.; Wang, S.; Wang, X.; Xie, Z.; Niu, H.; Chang, W.; Liu, L.; Yuan, H.; et al. ent-kaurane diterpenoids from Chinese liverworts and their antitumor activities through Michael addition as detected in situ by a fluorescence probe. J. Med. Chem. 2015, 58, 3944–3956. [Google Scholar] [CrossRef]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: Review of clinical drug–drug interaction studies. Clin. Pharmacokinet. 2020, 59, 699–714. [Google Scholar] [CrossRef]
- Murray, C.W.; Erlanson, D.A.; Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H.; Richmond, N.J. Validity of Ligand Efficiency Metrics. ACS Med. Chem. Lett. 2014, 5, 616–618. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, J.T.S.; Feghali, R.; Ribeiro, R.P.; Ramos, M.J.; Fernandes, P.A. The importance of intramolecular hydrogen bonds on the translocation of the small drug piracetam through a lipid bilayer. RSC Adv. 2020, 11, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.; Millan, D.S.; Perez, M.; Wakenhut, F.; Whitlock, G.A. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. Medchemcomm 2011, 2, 669–674. [Google Scholar] [CrossRef]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.Z.; Xu, X. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.W. Hydrogen-Bond Donors in Drug Design. J. Med. Chem. 2022, 65, 14261–14275. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Bojarska, J.; Remko, M.; Breza, M.; Madura, I.D.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. A Supramolecular Approach to Structure-Based Design with A Focus on Synthons Hierarchy in Ornithine-Derived Ligands: Review, Synthesis, Experimental and in Silico Studies. Molecules 2020, 25, 1135. [Google Scholar] [CrossRef] [PubMed]
- Bryant, M.J.; Black, S.N.; Blade, H.; Docherty, R.; Maloney, A.G.P.; Taylor, S.C. The CSD Drug Subset: The Changing Chemistry and Crystallography of Small Molecule Pharmaceuticals. J. Pharm. Sci. 2019, 108, 1655–1662. [Google Scholar] [CrossRef]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef]
- Yan, A.; Gasteiger, J. Prediction of aqueous solubility of organic compounds based on a 3D structure representation. J. Chem. Inf. Comput. Sci. 2003, 43, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.; Doerksen, R.J. Topological Polar Surface Area: A Useful Descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, O.; Bernkop-Schnürch, A. Lipophilic peptide character–What oral barriers fear the most. J. Control. Release 2017, 255, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Flitsch, S.L.; Grigalunas, M.; Leeson, P.D.; Quinn, R.J.; Turner, N.J.; Waldmann, H. The time and place for nature in drug discovery. JACS Au 2022, 2, 2400–2416. [Google Scholar] [CrossRef] [PubMed]
- Wardecki, D.; Dołowy, M.; Bober-Majnusz, K. Assessment of Lipophilicity Parameters of Antimicrobial and Immunosuppressive Compounds. Molecules 2023, 28, 2820. [Google Scholar] [CrossRef] [PubMed]
- Tsaioun, K.; Blaauboer, B.J.; Hartung, T. Evidence-based absorption, distribution, metabolism, excretion (ADME) and its interplay with alternative toxicity methods. ALTEX-Alternatives Anim. Exp. 2016, 33, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, H.; Handral, H.K.; Cha, S.; Nguyen, D.T.P.; Cai, J.; Cao, T.; Wu, C.; Kang, L. Enhancement of skin delivery of drugs using proposome depends on drug lipophilicity. Pharmaceutics 2021, 13, 1457. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, R.E.; Atatreh, N.; Ghattas, M.A. Using filters in virtual screening: A comprehensive guide to minimize errors and maximize efficiency. Annu. Rep. Med. Chem. 2022, 59, 99–136. [Google Scholar] [CrossRef]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, E.; Thorn, H.; Tannergren, C. In silico modeling of gastrointestinal drug absorption: Predictive performance of three physiologically based absorption models. Mol. Pharm. 2016, 13, 1763–1778. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, A.; Alexiou, A.; Bilgrami, A.L.; Kamal, M.A.; Ashraf, G.M. DeePred-BBB: A blood brain barrier permeability prediction model with improved accuracy. Front. Neurosci. 2022, 16, 858126. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.M.; Zhao, Z.; Zhu, D. Blood-Brain Barrier (BBB) permeability and transport measurement in vitro and in vivo. Permeability Barrier Methods Protoc. 2021, 2367, 105–122. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Mohammad, A.S.; Adkins, C.E.; Lockman, P.R. Molecular determinants of blood–brain barrier permeation. Ther. Deliv. 2015, 6, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Bors, L.A.; Erdő, F. Overcoming the blood–brain barrier. challenges and tricks for CNS drug delivery. Sci. Pharm. 2019, 87, 6. [Google Scholar] [CrossRef]
- Hunter, J.; Hirst, B.H. Intestinal secretion of drugs. The role of P-glycoprotein and related drug efflux systems in limiting oral drug absorption. Adv. Drug Deliv. Rev. 1997, 25, 129–157. [Google Scholar] [CrossRef]
- Mai, Y.; Dou, L.; Yao, Z.; Madla, C.M.; Gavins, F.K.H.; Taherali, F.; Yin, H.; Orlu, M.; Murdan, S.; Basit, A.W. Quantification of P-Glycoprotein in the gastrointestinal tract of humans and rodents: Methodology, gut region, sex, and species matter. Mol. Pharm. 2021, 18, 1895–1904. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef]
- Veith, H.; Southall, N.; Huang, R.; James, T.; Fayne, D.; Artemenko, N.; Shen, M.; Inglese, J.; Austin, C.P.; Lloyd, D.G. Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 2009, 27, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Ononamadu, C.; Ibrahim, A. Molecular docking and prediction of ADME/drug-likeness properties of potentially active antidiabetic compounds isolated from aqueous-methanol extracts of Gymnema sylvestre and Combretum micranthum. BioTechnologia 2021, 102, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Polinsky, A. Lead-likeness and drug-likeness. In The Practice of Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2008; pp. 244–254. [Google Scholar]
- Hann, M.M.; Oprea, T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Unnisa, A.; Abouzied, A.S.; Baratam, A.; Lakshmi, K.N.V.C.; Hussain, T.; Kunduru, R.D.; Banu, H.; Fatima, S.B.; Hussian, A.; Selvarajan, K.K. Design, synthesis, characterization, computational study and in-vitro antioxidant and anti-inflammatory activities of few novel 6-aryl substituted pyrimidine azo dyes. Arab. J. Chem. 2020, 13, 8638–8649. [Google Scholar] [CrossRef]
- Emon, N.U.; Alam, S.; Rudra, S.; Riya, S.R.; Paul, A.; Hossen, S.M.M.; Kulsum, U.; Ganguly, A. Antidepressant, anxiolytic, antipyretic, and thrombolytic profiling of methanol extract of the aerial part of Piper nigrum: In vivo, in vitro, and in silico approaches. Food Sci. Nutr. 2021, 9, 833–846. [Google Scholar] [CrossRef]
- Czajka-Oraniec, I.; Simpson, E.R. Aromatase research and its clinical significance. Endokrynol. Pol. 2010, 61, 126–134. [Google Scholar] [PubMed]
- Kupczyk, D.; Bilski, R.; Kozakiewicz, M.; Studzińska, R.; Kędziora-Kornatowska, K.; Kosmalski, T.; Pedrycz-Wieczorska, A.; Głowacka, M. 11β-HSD As a new target in pharmacotherapy of metabolic diseases. Int. J. Mol. Sci. 2022, 23, 8984. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | Compound | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
|---|---|---|---|---|---|---|---|
| 1 | 23 | 0.43 | 0.36 | −0.42 | 0.72 | 0.28 | 0.69 |
| 2 | 87 | 0.43 | 0.36 | −0.42 | 0.72 | 0.28 | 0.69 |
| 3 | 88 | 0.43 | 0.36 | −0.42 | 0.72 | 0.28 | 0.69 |
| 4 | 219 | 0.28 | 0.25 | −0.23 | 0.79 | 0.31 | 0.53 |
| 5 | 332 | 0.19 | 0.19 | −0.18 | 0.64 | 0.21 | 0.57 |
| 6 | 545 | 0.37 | 0.20 | −0.19 | 0.62 | 0.20 | 0.53 |
| Activity | Probability of Pharmacological Activity (Pa) | |||
|---|---|---|---|---|
| Compounds 23, 87, and 88 | Compound 219 | Compound 332 | Compound 545 | |
| Antineoplastic | 0.946 | 0.957 | 0.969 | 0.712 |
| Acylcarnitine hydrolase inhibitor | 0.888 | 0.845 | 0.863 | NA |
| Testosterone 17beta-dehydrogenase (NADP+) inhibitor | 0.874 | 0.827 | 0.846 | 0.780 |
| Apoptosis agonist | 0.836 | 0.729 | 0.735 | NA |
| Alkylacetylglycerophosphatase inhibitor | 0.824 | 0.757 | 0.785 | NA |
| Alkenylglycerophosphocholine hydrolase inhibitor | 0.824 | 0.746 | 0.782 | 0.780 |
| CYP2J substrate | 0.815 | 0.832 | 0.849 | 0.702 |
| Phosphatase inhibitor | 0.780 | NA | NA | NA |
| Antineoplastic (lung cancer) | 0.777 | 0.858 | 0.899 | NA |
| Caspase 8 stimulant | 0.754 | 0.701 | 0.723 | 0.761 |
| Oxidoreductase inhibitor | 0.754 | NA | NA | NA |
| Antinociceptive | 0.742 | NA | NA | NA |
| Caspase 3 stimulant | 0.746 | NA | NA | NA |
| Transcription factor NF kappa B stimulant | 0.738 | 0.713 | 0.725 | 0.746 |
| Transcription factor stimulant | 0.738 | 0.713 | 0.725 | 0.746 |
| Polarization stimulant | 0.732 | 0.748 | NA | NA |
| Antileukemic | 0.731 | NA | 0.759 | NA |
| CYP3A4 inducer | 0.733 | NA | NA | NA |
| Gluconate 2-dehydrogenase (acceptor) inhibitor | 0.750 | 0.763 | NA | NA |
| CYP2J2 substrate | 0.740 | NA | NA | NA |
| CYP3A inducer | 0.720 | NA | NA | NA |
| Myc inhibitor | 0.711 | NA | NA | NA |
| Glyceryl-ether monooxygenase inhibitor | 0.711 | NA | NA | NA |
| Immunosuppressant | 0.701 | NA | NA | NA |
| Antineoplastic (breast cancer) | NA | NA | 0.740 | NA |
| DNA polymerase I inhibitor | NA | NA | NA | 0.716 |
| Chemoprotective | NA | NA | NA | 0.879 |
| Hepatoprotectant | NA | NA | NA | 0.879 |
| Hepatic disorder treatment | NA | NA | NA | 0.942 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kibet, S.; Kimani, N.M.; Mwanza, S.S.; Mudalungu, C.M.; Santos, C.B.R.; Tanga, C.M. Unveiling the Potential of Ent-Kaurane Diterpenoids: Multifaceted Natural Products for Drug Discovery. Pharmaceuticals 2024, 17, 510. https://doi.org/10.3390/ph17040510
Kibet S, Kimani NM, Mwanza SS, Mudalungu CM, Santos CBR, Tanga CM. Unveiling the Potential of Ent-Kaurane Diterpenoids: Multifaceted Natural Products for Drug Discovery. Pharmaceuticals. 2024; 17(4):510. https://doi.org/10.3390/ph17040510
Chicago/Turabian StyleKibet, Shadrack, Njogu M. Kimani, Syombua S. Mwanza, Cynthia M. Mudalungu, Cleydson B. R. Santos, and Chrysantus M. Tanga. 2024. "Unveiling the Potential of Ent-Kaurane Diterpenoids: Multifaceted Natural Products for Drug Discovery" Pharmaceuticals 17, no. 4: 510. https://doi.org/10.3390/ph17040510