

Unveiling the Multifaceted Capabilities of Endophytic Aspergillus flavus Isolated from Annona squamosa Fruit Peels against Staphylococcus Isolates and HCoV 229E—In Vitro and In Silico Investigations
 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Isolation and Identification of Endophytic Fungi
2.2. Metabolic Profiling of the Ethyl Acetate Extract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound | Chemical Class | Molecular Formula | [M-H]− | Abundance | M. Weight | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Heptelidic acid | Sesquiterpene | C15H20O5 | 279 | 26.8% | 280 | [38,39] |
| 2 | Ferulic acid | Phenolic | C10H10O4 | 193 | 25.3% | 194 | [40,41] |
| 3 | Oleic acid | Fatty acid | C18H34O2 | 281 | 23.2% | 282 | [42] |
| 4 | Paxilline | Diterpene indole polycyclic alkaloid | C27H33NO4 | 432 | 8.3% | 435 | [43,44] |
| 5 | Indole | Alkaloid | C8H7N | 116 | 7.4% | 117 | [45] |
| 6 | Orientin | Flavonoid | C21H20O11 | 446 | 6.4% | 447 | [46] |
| 7 | Kojic acid | Pyrone | C6H6O4 | 141 | 2% | 142 | [47,48] |
2.3. Antimicrobial Potential
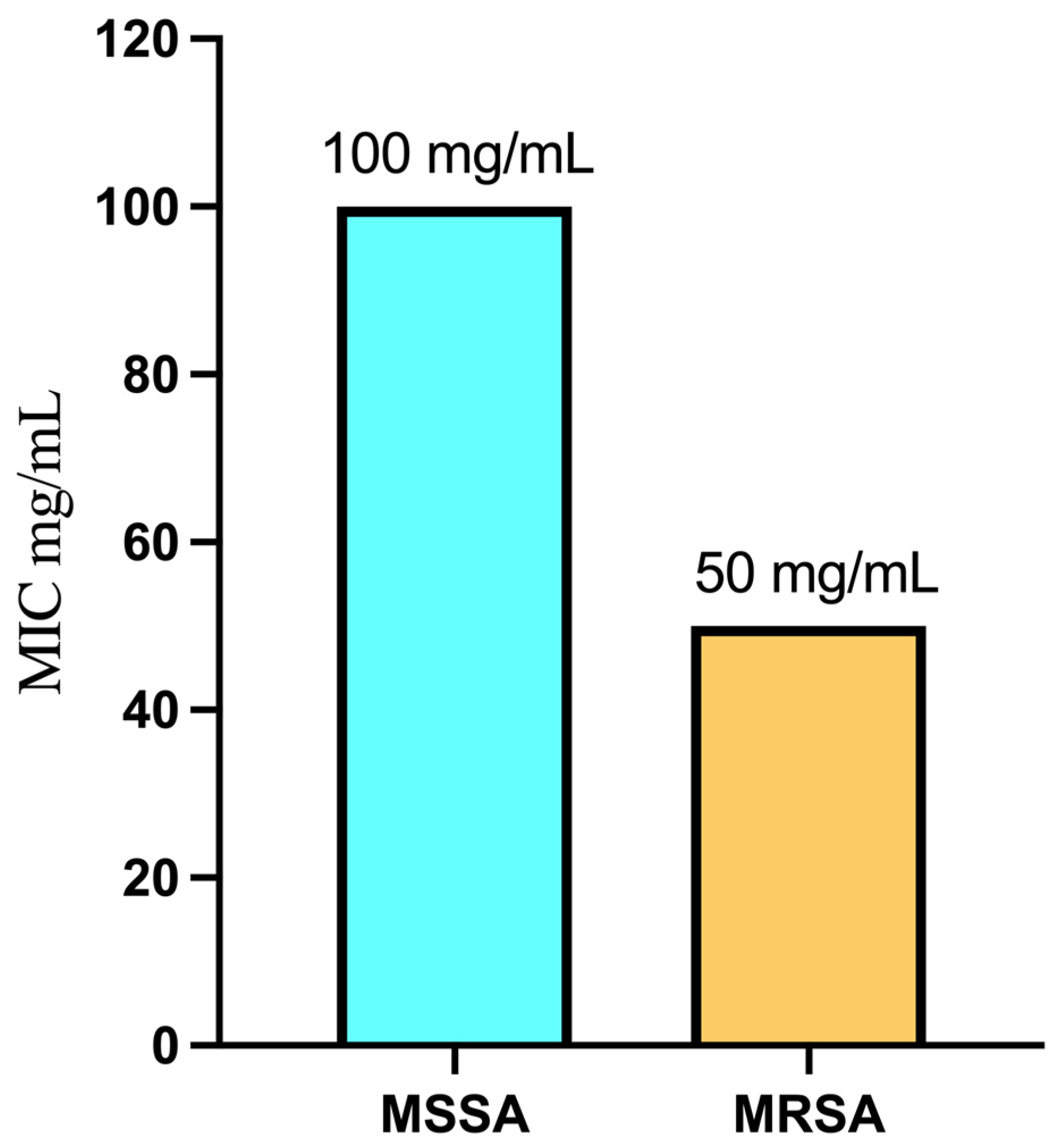
2.4. Minimum Inhibitory Concentration
2.5. Antibiofilm Activity/Anti-Adhesion
2.5.1. Prevention of Cell Attachment
2.5.2. Evaluating Biofilm Mass Destruction
2.6. Antiviral Activity of Crude Extract
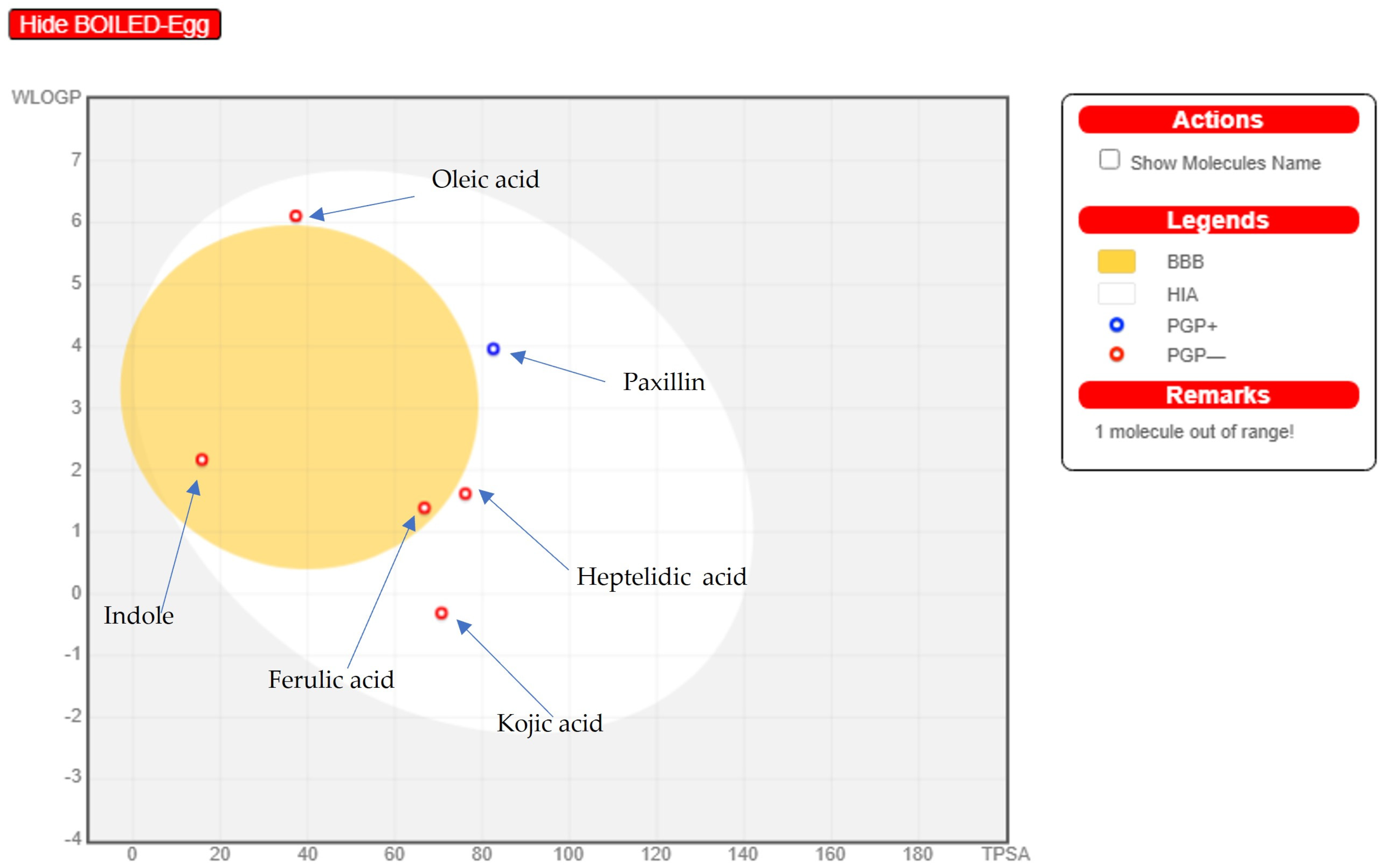
2.7. Online Software Swiss ADME Prediction (Boiled Egg Method and Lipinski’s Rule of Five)
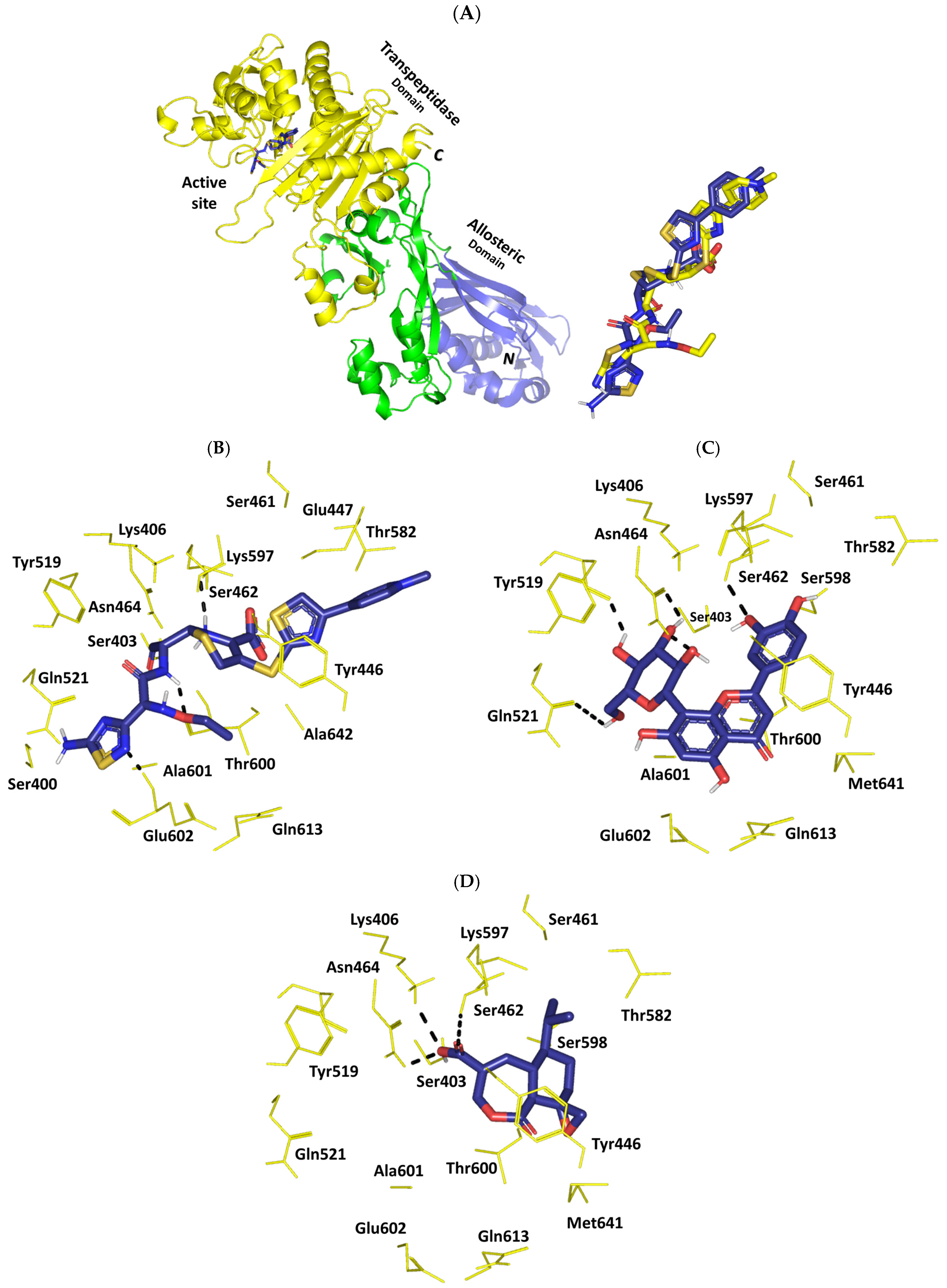
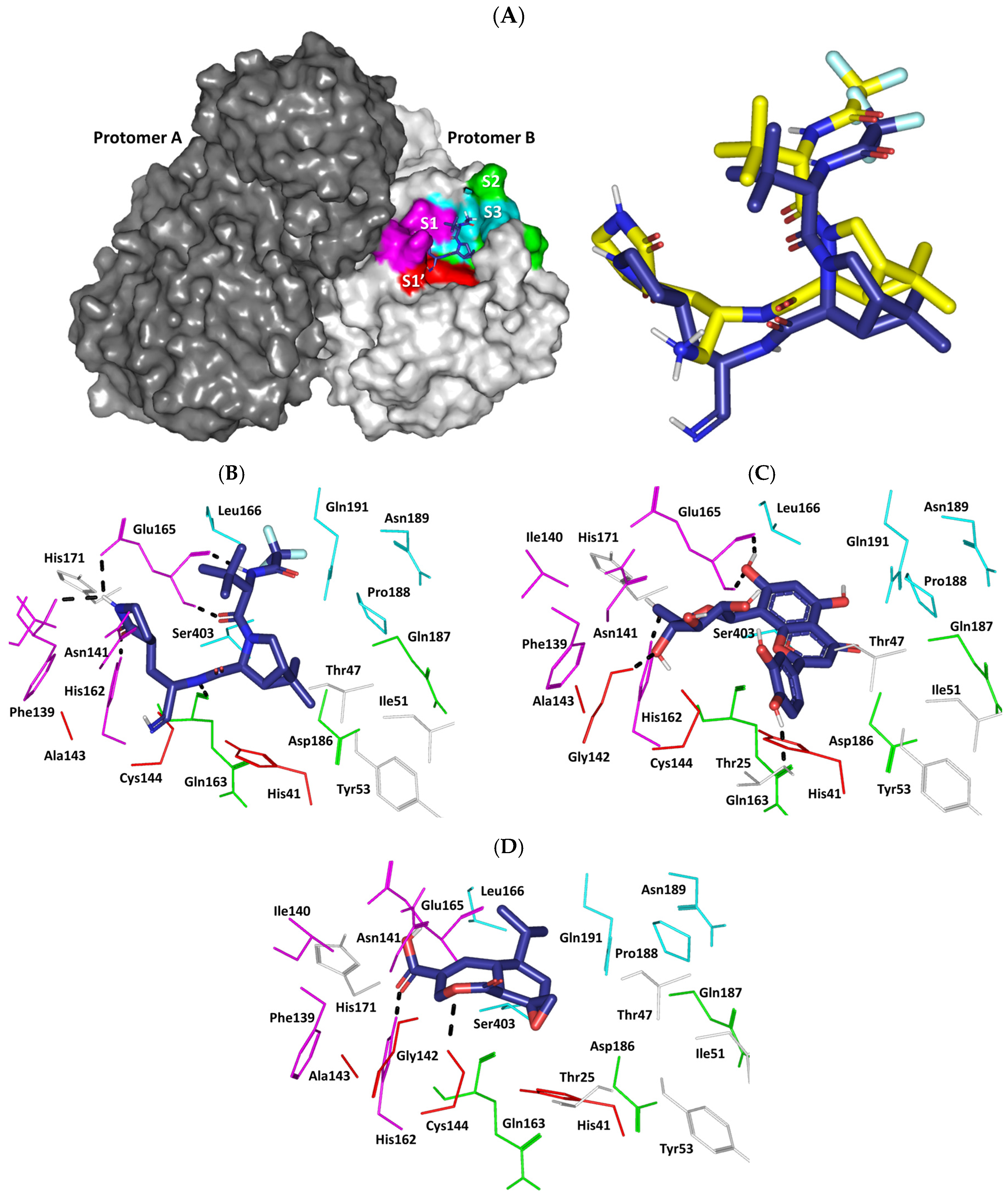
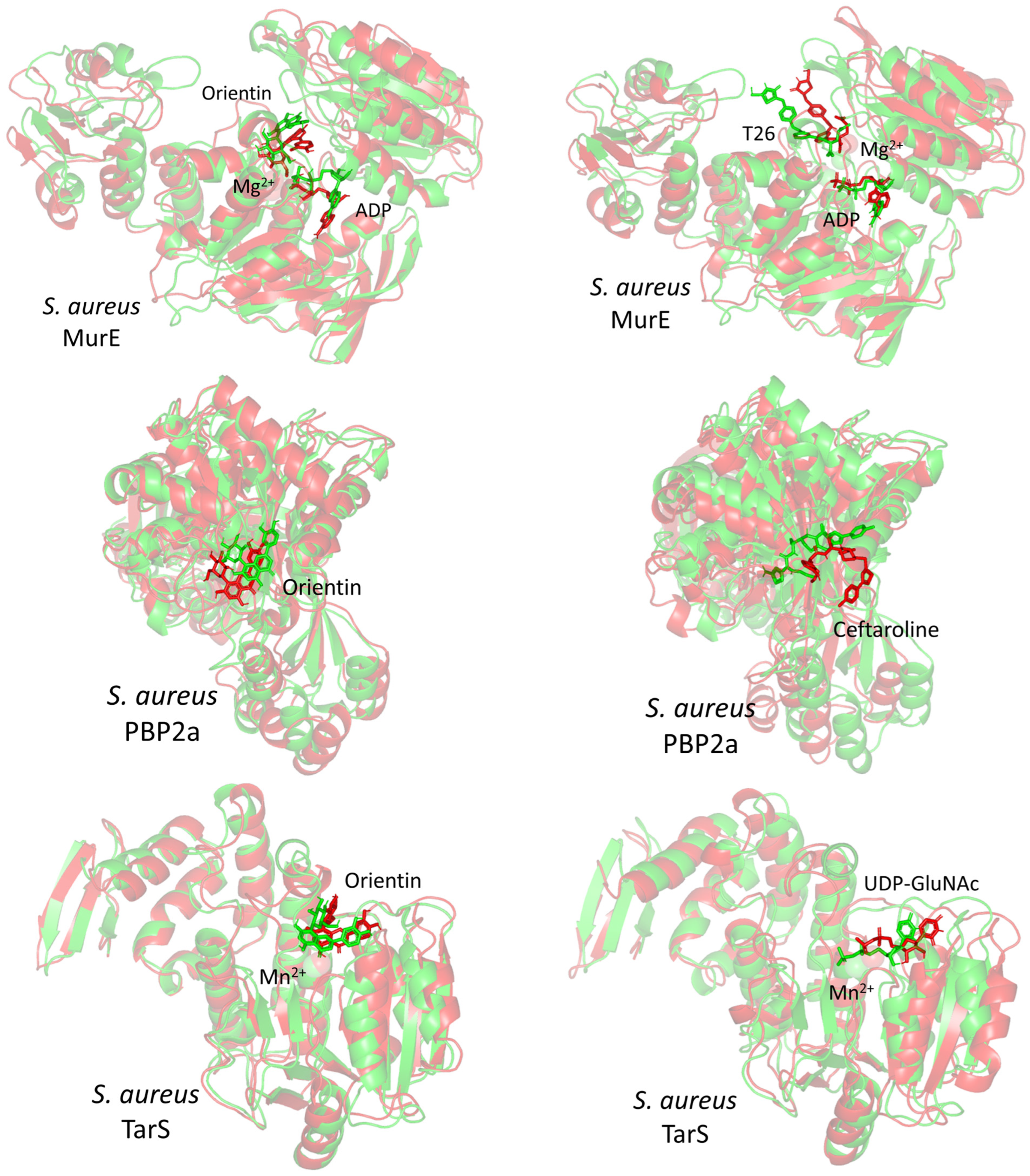
2.8. In Silico Investigation: Molecular Docking Simulation
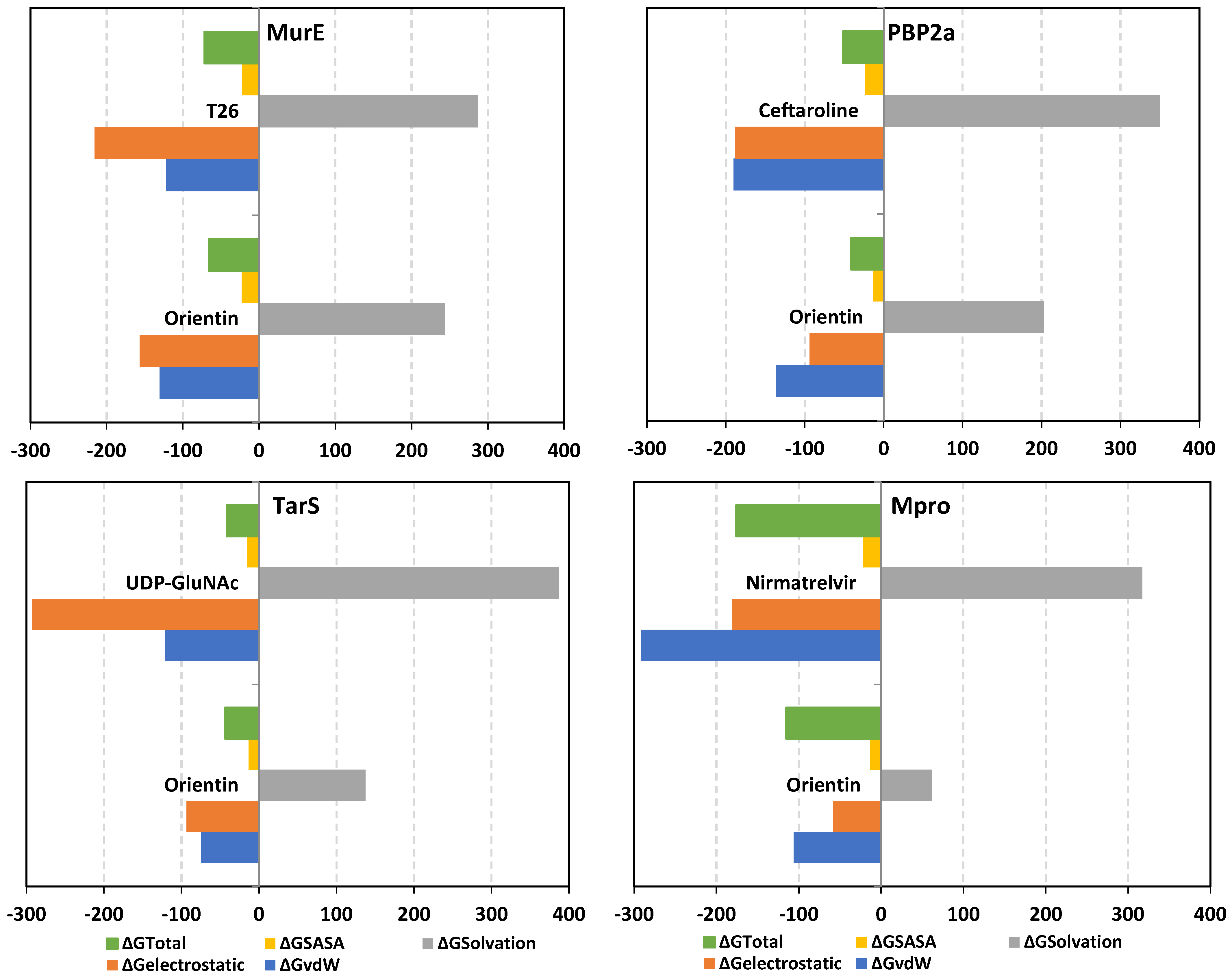
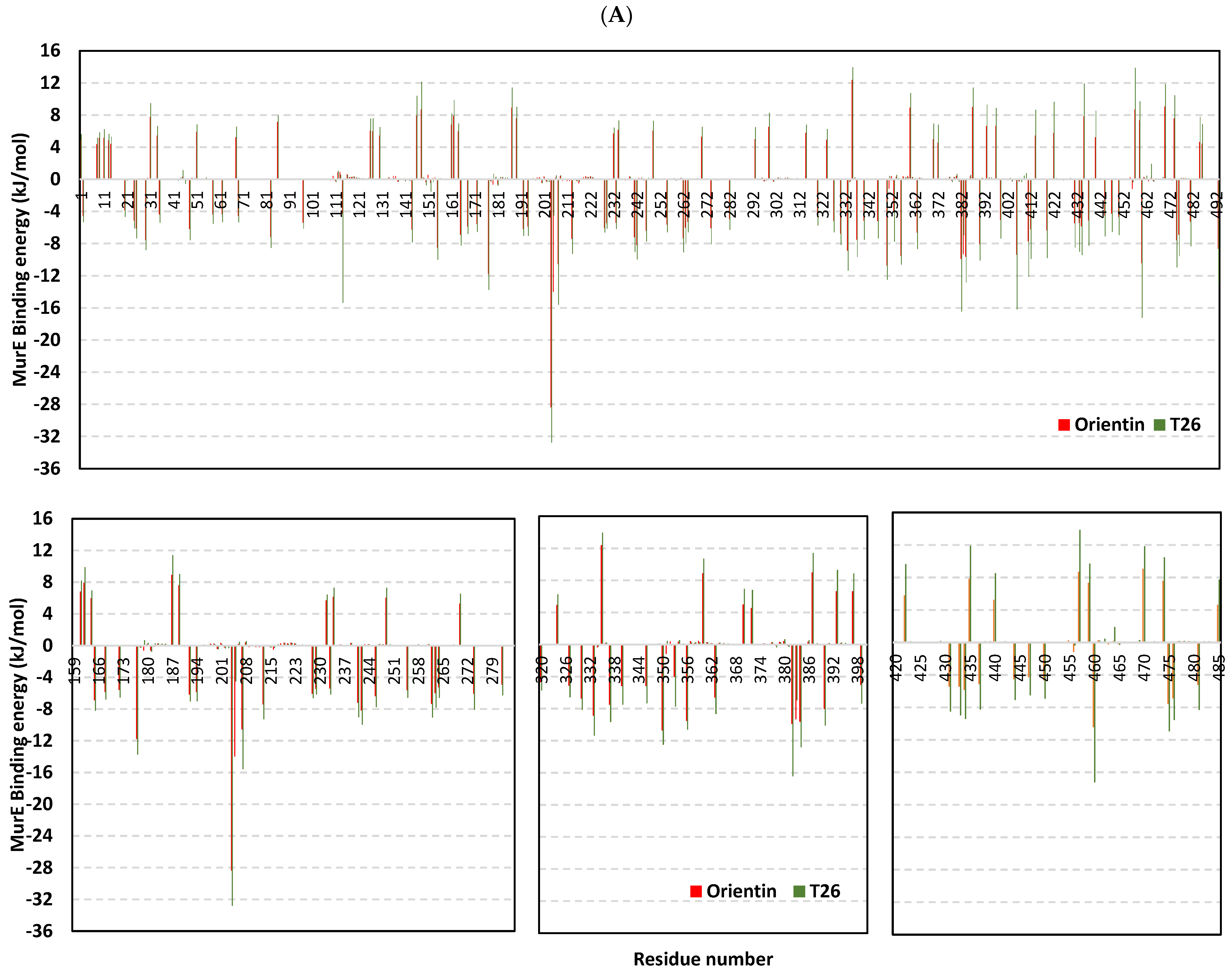
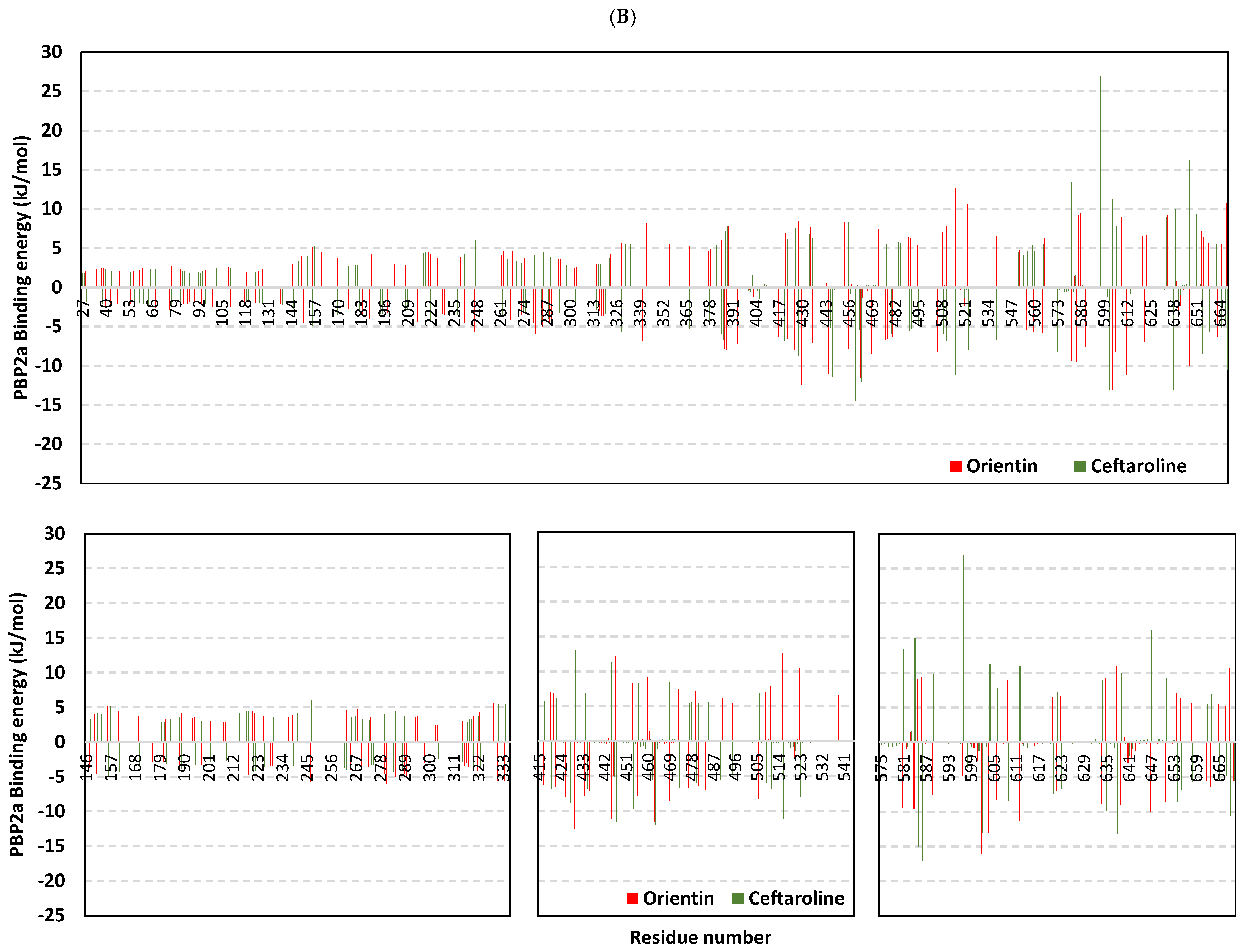
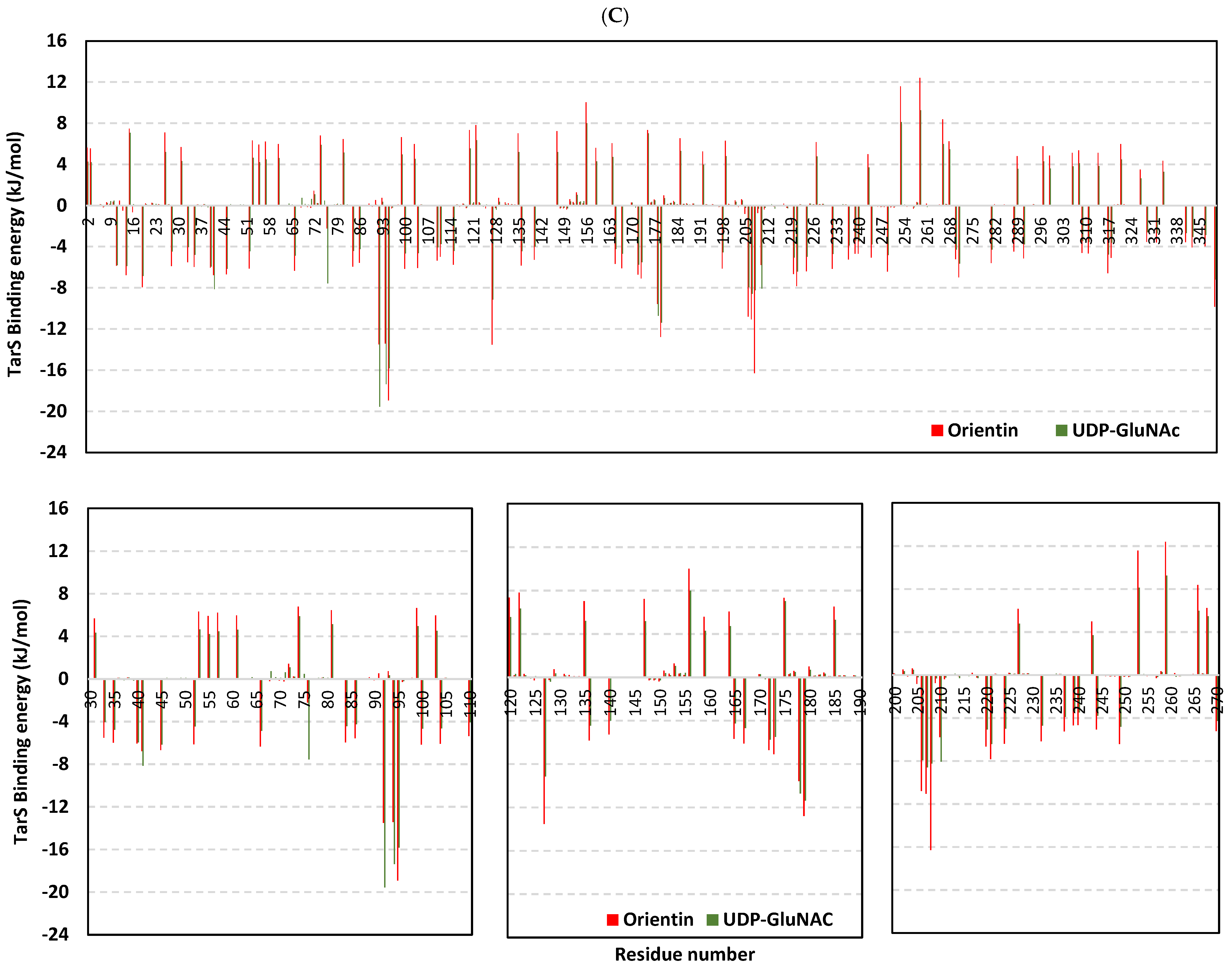
2.9. Molecular Dynamics Studies
3. Materials and Methods
3.1. Plant Material
3.2. Isolation of the Endophytic Fungi
3.3. Morphology of Fungi and Microscopic Analysis
3.4. Identification of Fungi Using a Molecular Approach
3.5. Fermentation in Solid-State Media and Extraction of the Fungi Metabolites
3.6. Preparation of Ethyl Acetate Fungal Extract
3.7. Liquid Chromatography-Mass Spectrometry Analysis (LC/MS)
3.8. Antimicrobial Screening
3.9. Determination of Minimum Inhibitory Concentrations
3.10. Antibiofilm Screening
3.10.1. Inhibition of Biofilm Formation–Prevention of Initial Bacterial Cell Attachment
3.10.2. Inhibition of Development of Pre-formed Biofilms–Assessment of Destruction of Biofilm Mass
3.10.3. The Crystal Violet Staining Assay
3.11. Antiviral Activity
3.12. Research on ADME (Absorption, Distribution, Metabolism, and Excretion) and Pharmacokinetics
3.13. In Silico Studies (Molecular Docking-Coupled Dynamics Simulations)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fathallah, N.; Raafat, M.M.; Issa, M.Y.; Abdel-Aziz, M.M.; Bishr, M.; Abdelkawy, M.A.; Salama, O. Bio-guided fractionation of prenylated benzaldehyde derivatives as potent antimicrobial and antibiofilm from Ammi majus L. fruits-associated Aspergillus amstelodami. Molecules 2019, 24, 4118. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.; El-Shokry, M.; Ismail, G.; Hafez, S.; El-Kholy, A.; Attia, E.; Talaat, M. Methicillin-resistant Staphylococcus aureus recovered from healthcare-and community-associated infections in Egypt. Int. J. Bacteriol. 2016, 2016, 5751785. [Google Scholar] [CrossRef]
- Kandeel, A.; Fahim, M.; Deghedy, O.; Roshdy, W.H.; Khalifa, M.K.; El Shesheny, R.; Kandeil, A.; Wagdy, S.; Naguib, A.; Afifi, S. Multicenter study to describe viral etiologies, clinical profiles, and outcomes of hospitalized children with severe acute respiratory infections, Egypt 2022. Sci. Rep. 2023, 13, 21860. [Google Scholar] [CrossRef] [PubMed]
- Hasöksüz, M.; Kilic, S.; Saraç, F. Coronaviruses and SARS-CoV-2. Turk. J. Med. Sci. 2020, 50, 549–556. [Google Scholar] [CrossRef]
- Santhoshkumar, R.; Kumar, N.S. Phytochemical analysis and antimicrobial activities of Annona squamosa (L) leaf extracts. J. Pharmacogn. Phytochem. 2016, 5, 128–131. [Google Scholar]
- Elkady, W.M.; Raafat, M.M.; Abdel-Aziz, M.M.; Al-Huqail, A.A.; Ashour, M.L.; Fathallah, N. Endophytic Fungus from Opuntia ficus-indica: A Source of Potential Bioactive Antimicrobial Compounds against Multidrug-Resistant Bacteria. Plants 2022, 11, 1070. [Google Scholar] [CrossRef]
- Singab, A.N.B.; Elkhawas, Y.A.; Al-Sayed, E.; Elissawy, A.M.; Fawzy, I.M.; Mostafa, N.M. Antimicrobial activities of metabolites isolated from endophytic Aspergillus flavus of Sarcophyton ehrenbergi supported by in-silico study and NMR spectroscopy. Fungal Biol. Biotechnol. 2023, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Khattak, S.U.; Lutfullah, G.; Iqbal, Z.; Ahmad, J.; Rehman, I.U.; Shi, Y.; Ikram, S. Aspergillus flavus originated pure compound as a potential antibacterial. BMC Microbiol. 2021, 21, 322. [Google Scholar] [CrossRef]
- Nair, D.N.; Padmavathy, S. Impact of endophytic microorganisms on plants, environment and humans. Sci. World J. 2014, 2014, 250693. [Google Scholar] [CrossRef]
- Rodriguez, R.; White, J., Jr.; Arnold, A.; Redman, A.R.A. Fungal endophytes: Diversity and functional roles. New Phytol. 2009, 182, 314–330. [Google Scholar] [CrossRef]
- Chang, Y.; Xia, X.; Sui, L.; Kang, Q.; Lu, Y.; Li, L.; Liu, W.; Li, Q.; Zhang, Z. Endophytic colonization of entomopathogenic fungi increases plant disease resistance by changing the endophytic bacterial community. J. Basic Microbiol. 2021, 61, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Mengistu, A.A. Endophytes: Colonization, behaviour, and their role in defense mechanism. Int. J. Microbiol. 2020, 2020, 6927219. [Google Scholar] [CrossRef]
- Busby, P.E.; Ridout, M.; Newcombe, G. Fungal endophytes: Modifiers of plant disease. Plant Mol. Biol. 2016, 90, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.A.; El-Gengaihi, S.; Enein, A.; Hassan, E.M.; Ahmed, O.; Asker, M. Chemical constituents and antimicrobial activity of different Annona species cultivated in Egypt. J. Chem. Pharm. Res 2016, 8, 261–271. [Google Scholar]
- Nguyen, T.T.; Tran, P.N.T.; Phan, H.T. Evaluation of anti-inflammatory effect of fruit peel extracts of Annona squamosa L. on mouse models of rheumatoid arthritis. J. Microbiol. Biotechnol. Food Sci. 2021, 11, e2075. [Google Scholar]
- Vikas, B.; Anil, S.; Remani, P. Cytotoxicity profiling of Annona squamosa in cancer cell lines. Asian Pac. J. Cancer Prev. APJCP 2019, 20, 2831. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Arif, M.; Rahman, M.A.; Mujahid, M. Hepatoprotective and antioxidant activities of Annona squamosa seed extract against alcohol-induced liver injury in Sprague Dawley rats. Drug Chem. Toxicol. 2020, 43, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Tomar, R.S.; Sisodia, S.S. Antidiabetic activity of Annona squamosa L. in experimental induced diabetic rats. Int. J. Pharm. Biol. Arch. 2012, 3, 1492–1495. [Google Scholar]
- Intaranongpai, J.; Chavasiri, W.; Gritsanapan, W. Anti-head lice effect of Annona squamosa seeds. S. Asian J. Trop. Med. Public Health 2006, 37, 532. [Google Scholar]
- Pandey, N.; Barve, D. Phytochemical and pharmacological review on Annona squamosa Linn. Int. J. Res. Pharm. Biomed. Sci. 2011, 2, 1404–1412. [Google Scholar]
- Chowdhury, S.S.; Tareq, A.M.; Tareq, S.M.; Farhad, S.; Sayeed, M.A. Screening of antidiabetic and antioxidant potential along with phytochemicals of Annona genus: A review. Future J. Pharm. Sci. 2021, 7, 144. [Google Scholar] [CrossRef]
- Zaghlol, A.A.; Kandil, Z.A.; Yousif, M.F.; EL-Dine, R.S.; Elkady, W.M. Unveiling the anti-cancer potential of Euphorbia greenwayi: Cytotoxicity, cell migration, and identification of its chemical constituents. Future J. Pharm. Sci. 2024, 10, 24. [Google Scholar] [CrossRef]
- El-Chaghaby, G.A.; Ahmad, A.F.; Ramis, E.S. Evaluation of the antioxidant and antibacterial properties of various solvents extracts of Annona squamosa L. leaves. Arab. J. Chem. 2014, 7, 227–233. [Google Scholar] [CrossRef]
- Yunita, M.N.; Raharjo, A.P.; Wibawati, P.A.; Agustono, B. Antiviral activity of ethanolic extract of srikaya seeds (Annona squamosa L.) against avian influenza virus. Indian Vet. J. 2019, 96, 26–29. [Google Scholar]
- Ola, A.R.; Soa, C.A.; Sugi, Y.; Cunha, T.; Belli, H.; Lalel, H. Antimicrobial metabolite from the endophytic fungi Aspergillus flavus isolated from Sonneratia alba, a mangrove plant of Timor-Indonesia. Rasayan J. Chem. 2020, 13, 377–381. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, J.-Y.; Sun, S.-F.; Li, Y.; Qu, J.; Liu, H.-T.; Liu, Y.-b. Sesquiterpenes from an endophytic Aspergillus flavus. J. Nat. Prod. 2019, 82, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Patil, R.; Maheshwari, V. Biological activities and identification of bioactive metabolite from endophytic Aspergillus flavus L7 isolated from Aegle marmelos. Curr. Microbiol. 2015, 71, 39–48. [Google Scholar] [CrossRef]
- Lin, M.; Dianese, J. A coconut-agar medium for rapid detection of aflatoxin production by Aspergillus spp. Phytopathology 1976, 66, 1466–1469. [Google Scholar] [CrossRef]
- Rao, K.R.; Vipin, A.; Venkateswaran, G. Molecular profile of non-aflatoxigenic phenotype in native strains of Aspergillus flavus. Arch. Microbiol. 2020, 202, 1143–1155. [Google Scholar] [CrossRef]
- Mowaka, S.; Ayoub, B.M. Comparative study between UHPLC-UV and UPLC-MS/MS methods for determination of alogliptin and metformin in their pharmaceutical combination. Die Pharm.-Int. J. Pharm. Sci. 2017, 72, 67–72. [Google Scholar]
- Tanaka, Y.; Shiomi, K.; Kamei, K.; Sugoh-Hagino, M.; Enomoto, Y.; Fang, F.; Yamaguchi, Y.; Masuma, R.; Zhang, C.G.; Zhang, X.W. Antimalarial activity of radicicol, heptelidic acid and other fungal metabolites. J. Antibiot. 1998, 51, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Kodama, K.; Furuya, K.; Takahashi, S.; Haneishi, T.; Takiguchi, Y.; Arai, M. A new sesquiterpene antibiotic, heptelidic acid producing organisms, fermentation, isolation and characterization. J. Antibiot. 1980, 33, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Lee, C.-H. Heptelidic acid, a sesquiterpene lactone, inhibits etoposide-induced apoptosis in human leukemia U937 cells. J. Microbiol. Biotechnol. 2009, 19, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, P.G.; Santiago, G.M.P.; da Silva, F.E.F.; de Araújo, A.C.J.; de Oliveira, C.R.T.; Freitas, P.R.; Rocha, J.E.; de Araújo Neto, J.B.; da Silva, M.M.C.; Tintino, S.R. Ferulic acid derivatives inhibiting Staphylococcus aureus tetK and MsrA efflux pumps. Biotechnol. Rep. 2022, 34, e00717. [Google Scholar] [CrossRef] [PubMed]
- Ibitoye, O.; Ajiboye, T. Ferulic acid potentiates the antibacterial activity of quinolone-based antibiotics against Acinetobacter baumannii. Microb. Pathog. 2019, 126, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Batista, R. Uses and potential applications of ferulic acid. In Ferulic Acid: Antioxidant Properties, Uses and Potential Health Benefits; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2014; pp. 39–70. [Google Scholar]
- Deakin, N.O.; Turner, C.E. Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol. Biol. Cell 2011, 22, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Cho, J.-Y.; Lee, Y.G.; Lee, H.J.; Lim, S.-I.; Lee, S.-Y.; Nam, Y.-D.; Moon, J.-H. Furan, phenolic, and heptelidic acid derivatives produced by Aspergillus oryzae. Food Sci. Biotechnol. 2016, 25, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Kovač, T.; Borišev, I.; Kovač, M.; Lončarić, A.; Čačić Kenjerić, F.; Djordjevic, A.; Strelec, I.; Ezekiel, C.N.; Sulyok, M.; Krska, R. Impact of fullerol C60(OH)24 nanoparticles on the production of emerging toxins by Aspergillus flavus. Sci. Rep. 2020, 10, 725. [Google Scholar] [CrossRef]
- Todokoro, T.; Negoro, H.; Kotaka, A.; Hata, Y.; Ishida, H. Aspergillus oryzae FaeA is responsible for the release of ferulic acid, a precursor of off-odor 4-vinylguaiacol in sake brewing. J. Biosci. Bioeng. 2022, 133, 140–145. [Google Scholar] [CrossRef]
- Faulds, C.; Williamson, G. Release of ferulic acid from wheat bran by a ferulic acid esterase (FAE-III) from Aspergillus niger. Appl. Microbiol. Biotechnol. 1995, 43, 1082–1087. [Google Scholar] [CrossRef]
- Amaike, S.; Keller, N.P. Aspergillus flavus. Annu. Rev. Phytopathol. 2011, 49, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Monahan, B.J.; Tkacz, J.S.; Scott, B. Indole-diterpene gene cluster from Aspergillus flavus. Appl. Environ. Microbiol. 2004, 70, 6875–6883. [Google Scholar] [CrossRef]
- Nicholson, M.J.; Koulman, A.; Monahan, B.J.; Pritchard, B.L.; Payne, G.A.; Scott, B. Identification of two aflatrem biosynthesis gene loci in Aspergillus flavus and metabolic engineering of Penicillium paxilli to elucidate their function. Appl. Environ. Microbiol. 2009, 75, 7469–7481. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.J.; Dorner, J.W.; Springer, J.P.; Cox, R.H. Indole metabolites from a strain of Aspergillus flavus. J. Agric. Food Chem. 1981, 29, 293–295. [Google Scholar] [CrossRef]
- Adione, N.M.; Onyeka, I.P.; Abba, C.C.; Okoye, N.N.; Okolo, C.C.; Eze, P.M.; Umeokoli, B.O.; Anyanwu, O.O.; Okoye, F.B.C. Detection, isolation and identification of more bioactive compounds from Fusarium equiseti, an endophytic fungus isolated from Ocimum gratissimum. GSC Biolog. Pharm. Sci. 2022, 20, 130–140. [Google Scholar] [CrossRef]
- Ola, A.R.; Metboki, G.; Lay, C.S.; Sugi, Y.; Rozari, P.D.; Darmakusuma, D.; Hakim, E.H. Single production of kojic acid by Aspergillus flavus and the revision of flufuran. Molecules 2019, 24, 4200. [Google Scholar] [CrossRef]
- de Caldas Felipe, M.T.; do Nascimento Barbosa, R.; Bezerra, J.D.P.; de Souza-Motta, C.M. Production of kojic acid by Aspergillus species: Trends and applications. Fungal Biol. Rev. 2023, 45, 100313. [Google Scholar]
- Bhattacharya, A.; Chakraverty, R. The pharmacological properties of Annona squamosa Linn: A Review. Int. J. Pharm. Eng. 2016, 4, 692–699. [Google Scholar]
- Clegg, J.; Soldaini, E.; McLoughlin, R.M.; Rittenhouse, S.; Bagnoli, F.; Phogat, S. Staphylococcus aureus vaccine research and development: The past, present and future, including novel therapeutic strategies. Front. Immunol. 2021, 12, 705360. [Google Scholar] [CrossRef]
- Howden, B.P.; Giulieri, S.G.; Wong Fok Lung, T.; Baines, S.L.; Sharkey, L.K.; Lee, J.Y.; Hachani, A.; Monk, I.R.; Stinear, T.P. Staphylococcus aureus host interactions and adaptation. Nat. Rev. Microbiol. 2023, 21, 380–395. [Google Scholar] [CrossRef]
- Famuyide, I.M.; Aro, A.O.; Fasina, F.O.; Eloff, J.N.; McGaw, L.J. Antibacterial and antibiofilm activity of acetone leaf extracts of nine under-investigated south African Eugenia and Syzygium (Myrtaceae) species and their selectivity indices. BMC Complement. Altern. Med. 2019, 19, 141. [Google Scholar] [CrossRef] [PubMed]
- Sandasi, M.; Leonard, C.; Viljoen, A. The effect of five common essential oil components on Listeria monocytogenes biofilms. Food Control 2008, 19, 1070–1075. [Google Scholar] [CrossRef]
- Pasquereau, S.; Nehme, Z.; Haidar Ahmad, S.; Daouad, F.; Van Assche, J.; Wallet, C.; Schwartz, C.; Rohr, O.; Morot-Bizot, S.; Herbein, G. Resveratrol inhibits HCoV-229E and SARS-CoV-2 coronavirus replication in vitro. Viruses 2021, 13, 354. [Google Scholar] [CrossRef] [PubMed]
- Selick, H.E.; Beresford, A.P.; Tarbit, M.H. The emerging importance of predictive ADME simulation in drug discovery. Drug Discov. Today 2002, 7, 109–116. [Google Scholar] [CrossRef]
- Attique, S.A.; Hassan, M.; Usman, M.; Atif, R.M.; Mahboob, S.; Al-Ghanim, K.A.; Bilal, M.; Nawaz, M.Z. A molecular docking approach to evaluate the pharmacological properties of natural and synthetic treatment candidates for use against hypertension. Int. J. Environ. Res. Public Health 2019, 16, 923. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Maddiboyina, B.; Roy, H.; Ramaiah, M.; Sarvesh, C.N.; Kosuru, S.H.; Nakkala, R.K.; Nayak, B.S. Methicillin-resistant Staphylococcus aureus: Novel treatment approach breakthroughs. Bull. Natl. Res. Cent. 2023, 47, 95. [Google Scholar] [CrossRef]
- Monteiro, J.M.; Covas, G.; Rausch, D.; Filipe, S.R.; Schneider, T.; Sahl, H.G.; Pinho, M.G. The pentaglycine bridges of Staphylococcus aureus peptidoglycan are essential for cell integrity. Sci. Rep. 2019, 9, 5010. [Google Scholar] [CrossRef]
- Ruane, K.M.; Lloyd, A.J.; Fülöp, V.; Dowson, C.G.; Barreteau, H.; Boniface, A.; Dementin, S.; Blanot, D.; Mengin-Lecreulx, D.; Gobec, S.; et al. Specificity determinants for lysine incorporation in Staphylococcus aureus peptidoglycan as revealed by the structure of a MurE enzyme ternary complex. J. Biol. Chem. 2013, 288, 33439–33448. [Google Scholar] [CrossRef]
- Bouhss, A.; Trunkfield, A.E.; Bugg, T.D.; Mengin-Lecreulx, D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Rev. 2008, 32, 208–233. [Google Scholar] [CrossRef]
- Nikolaidis, I.; Favini-Stabile, S.; Dessen, A. Resistance to antibiotics targeted to the bacterial cell wall. Protein Sci. 2014, 23, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Stelitano, G.; Sammartino, J.C.; Chiarelli, L.R. Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis. Molecules 2020, 25, 1239. [Google Scholar] [CrossRef] [PubMed]
- Igler, C.; Rolff, J.; Regoes, R. Multi-step vs. single-step resistance evolution under different drugs, pharmacokinetics, and treatment regimens. eLife 2021, 10, e64116. [Google Scholar] [CrossRef] [PubMed]
- Sobhanifar, S.; Worrall, L.J.; King, D.T.; Wasney, G.A.; Baumann, L.; Gale, R.T.; Nosella, M.; Brown, E.D.; Withers, S.G.; Strynadka, N.C. Structure and Mechanism of Staphylococcus aureus TarS, the Wall Teichoic Acid β-glycosyltransferase Involved in Methicillin Resistance. PLoS Pathog. 2016, 12, e1006067. [Google Scholar] [CrossRef]
- Oku, Y.; Kurokawa, K.; Matsuo, M.; Yamada, S.; Lee, B.L.; Sekimizu, K. Pleiotropic roles of polyglycerolphosphate synthase of lipoteichoic acid in growth of Staphylococcus aureus cells. J. Bacteriol. 2009, 191, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Draing, C.; Sigel, S.; Deininger, S.; Traub, S.; Munke, R.; Mayer, C.; Hareng, L.; Hartung, T.; von Aulock, S.; Hermann, C. Cytokine induction by Gram-positive bacteria. Immunobiology 2008, 213, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Morath, S.; Geyer, A.; Hartung, T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J. Exp. Med. 2001, 193, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Kim, T.; Lester, E.; Deep, D.; Spiegel, D.A. Wall teichoic acids prevent antibody binding to epitopes within the cell wall of Staphylococcus aureus. ACS Chem. Biol. 2016, 11, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Cramton, S.E.; Götz, F.; Peschel, A. Key role of teichoic acid net charge in Staphylococcus aureus colonization of artificial surfaces. Infect. Immun. 2001, 69, 3423–3426. [Google Scholar] [CrossRef]
- Bera, A.; Biswas, R.; Herbert, S.; Kulauzovic, E.; Weidenmaier, C.; Peschel, A.; Götz, F. Influence of wall teichoic acid on lysozyme resistance in Staphylococcus aureus. J. Bacteriol. 2007, 189, 280–283. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, N.H.; Winstel, V.; Kurokawa, K.; Larsen, J.; An, J.H.; Khan, A.; Seong, M.Y.; Lee, M.J.; Andersen, P.S.; et al. Surface Glycopolymers Are Crucial for In Vitro Anti-Wall Teichoic Acid IgG-Mediated Complement Activation and Opsonophagocytosis of Staphylococcus aureus. Infect. Immun. 2015, 83, 4247–4255. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Xia, G.; Luhachack, L.G.; Campbell, J.; Meredith, T.C.; Chen, C.; Winstel, V.; Gekeler, C.; Irazoqui, J.E.; Peschel, A.; et al. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 18909–18914. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, W.; Zeng, P.; Feng, J.; Li, D.; Jing, Y.; Zhang, J.; Yin, X.; Li, J.; Ye, H.; et al. Structural basis of main proteases of HCoV-229E bound to inhibitor PF-07304814 and PF-07321332. Biochem. Biophys. Res. Commun. 2023, 657, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, X.; Zhang, Y.; Zhong, F.; Lin, C.; McCormick, P.J.; Jiang, F.; Luo, J.; Zhou, H.; Wang, Q.; et al. Crystal structure of SARS-CoV-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci. Bull. 2021, 66, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Pasquereau, S.; Galais, M.; Bellefroid, M.; Pachón Angona, I.; Morot-Bizot, S.; Ismaili, L.; Van Lint, C.; Herbein, G. Ferulic acid derivatives block coronaviruses HCoV-229E and SARS-CoV-2 replication in vitro. Sci. Rep. 2022, 12, 20309. [Google Scholar] [CrossRef] [PubMed]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- van der Hoek, L. Human coronaviruses: What do they cause? Antivir. Ther. 2007, 12 Pt B, 651–658. [Google Scholar] [CrossRef]
- Ren, Z.; Yan, L.; Zhang, N.; Guo, Y.; Yang, C.; Lou, Z.; Rao, Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles’ heel”: Current effective inhibitor targeting a 3C-like protease. Protein Cell 2013, 4, 248–250. [Google Scholar] [CrossRef]
- Gupta, S.; Lynn, A.M.; Gupta, V. Standardization of virtual-screening and post-processing protocols relevant to in-silico drug discovery. 3 Biotech 2018, 8, 504. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef] [PubMed]
- Tomašić, T.; Šink, R.; Zidar, N.; Fic, A.; Contreras-Martel, C.; Dessen, A.; Patin, D.; Blanot, D.; Müller-Premru, M.; Gobec, S.; et al. Dual Inhibitor of MurD and MurE Ligases from Escherichia coli and Staphylococcus aureus. ACS Med. Chem. Lett. 2012, 3, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.; Flouret, B.; Chantalat, L.; van Heijenoort, J.; Mengin-Lecreulx, D.; Dideberg, O. Crystal structure of UDP-N-acetylmuramoyl-L-alanyl-D-glutamate: Meso-diaminopimelate ligase from Escherichia coli. J. Biol. Chem. 2001, 276, 10999–11006. [Google Scholar] [CrossRef] [PubMed]
- Otero, L.H.; Rojas-Altuve, A.; Llarrull, L.I.; Carrasco-López, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. USA 2013, 110, 16808–16813. [Google Scholar] [CrossRef] [PubMed]
- Duplessis, C.; Crum-Cianflone, N.F. Ceftaroline: A New Cephalosporin with Activity against Methicillin-Resistant Staphylococcus aureus (MRSA). Clin. Med. Rev. Ther. 2011, 3, a2466. [Google Scholar] [PubMed]
- NCBI. PubChem Compound Summary for CID 155903259, Nirmatrelvir. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nirmatrelvir (accessed on 9 February 2024).
- Billones, J.; Bangalan, M. Structure-Based Discovery of Inhibitors Against MurE in Methicillin-Resistant Staphylococcus aureus. Orient. J. Chem. 2019, 35, 618–625. [Google Scholar] [CrossRef]
- Zouhir, A.; Jemli, S.; Omrani, R.; Kthiri, A.; Jridi, T.; Sebei, K. In Silico Molecular Analysis and Docking of Potent Antimicrobial Peptides against MurE Enzyme of Methicillin Resistant Staphylococcus aureus. Int. J. Pept. Res. Ther. 2021, 27, 1253–1263. [Google Scholar] [CrossRef]
- Hervin, V.; Roy, V.; Agrofoglio, L.A. Antibiotics and Antibiotic Resistance—Mur Ligases as an Antibacterial Target. Molecules 2023, 28, 8076. [Google Scholar] [CrossRef]
- Oselusi, S.O.; Fadaka, A.O.; Wyckoff, G.J.; Egieyeh, S.A. Computational Target-Based Screening of Anti-MRSA Natural Products Reveals Potential Multitarget Mechanisms of Action through Peptidoglycan Synthesis Proteins. ACS Omega 2022, 7, 37896–37906. [Google Scholar] [CrossRef]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of docking performance: Comparative data on docking algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef]
- Ghuysen, J.M. Serine beta-lactamases and penicillin-binding proteins. Annu. Rev. Microbiol. 1991, 45, 37–67. [Google Scholar] [CrossRef]
- Frère, J.M.; Page, M.G. Penicillin-binding proteins: Evergreen drug targets. Curr. Opin. Pharmacol. 2014, 18, 112–119. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [PubMed]
- Sobhanifar, S.; Worrall, L.J.; Gruninger, R.J.; Wasney, G.A.; Blaukopf, M.; Baumann, L.; Lameignere, E.; Solomonson, M.; Brown, E.D.; Withers, S.G.; et al. Structure and mechanism of Staphylococcus aureus TarM, the wall teichoic acid α-glycosyltransferase. Proc. Natl. Acad. Sci. USA 2015, 112, E576–E585. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, J.G.; Meredith, T.C.; Campbell, J.; Brown, S.; Suzuki, T.; Bollenbach, T.; Malhowski, A.J.; Kishony, R.; Gilmore, M.S.; Walker, S. Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus. ACS Chem. Biol. 2009, 4, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gill, C.J.; Lee, S.H.; Mann, P.; Zuck, P.; Meredith, T.C.; Murgolo, N.; She, X.; Kales, S.; Liang, L.; et al. Discovery of wall teichoic acid inhibitors as potential anti-MRSA β-lactam combination agents. Chem. Biol. 2013, 20, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Ragab, S.S.; Sweed, A.M.K.; Elrashedy, A.A.; Allayeh, A.K. Design, Synthesis, Antiviral Evaluation, and Molecular Dynamics Simulation Studies of New Spirocyclic Thiopyrimidinones as Anti HCoV-229E. Chem. Biodivers. 2022, 19, e202200632. [Google Scholar] [CrossRef]
- Li, J.; Lin, C.; Zhou, X.; Zhong, F.; Zeng, P.; McCormick, P.J.; Jiang, H.; Zhang, J. Structural Basis of Main Proteases of Coronavirus Bound to Drug Candidate PF-07304814. J. Mol. Biol. 2022, 434, 167706. [Google Scholar] [CrossRef]
- Li, J.; Lin, C.; Zhou, X.; Zhong, F.; Zeng, P.; Yang, Y.; Zhang, Y.; Yu, B.; Fan, X.; McCormick, P.J.; et al. Structural Basis of the Main Proteases of Coronavirus Bound to Drug Candidate PF-07321332. J. Virol. 2022, 96, e0201321. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Zaki, A.A.; Ashour, A.; Elhady, S.S.; Darwish, K.M.; Al-Karmalawy, A.A. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies. J. Tradit. Complement. Med. 2022, 12, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Elhady, S.S.; Abdelhameed, R.F.A.; Malatani, R.T.; Alahdal, A.M.; Bogari, H.A.; Almalki, A.J.; Mohammad, K.A.; Ahmed, S.A.; Khedr, A.I.M.; Darwish, K.M. Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors. Biology 2021, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Soltan, M.A.; Eldeen, M.A.; Elbassiouny, N.; Kamel, H.L.; Abdelraheem, K.M.; El-Gayyed, H.A.; Gouda, A.M.; Sheha, M.F.; Fayad, E.; Ali, O.A.A.; et al. In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi. Cells 2021, 10, 3014. [Google Scholar] [CrossRef] [PubMed]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure Of Biomolecules through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput.-Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Manandhar, A.; Blass, B.E.; Colussi, D.J.; Almi, I.; Abou-Gharbia, M.; Klein, M.L.; Elokely, K.M. Targeting SARS-CoV-2 M3CLpro by HCV NS3/4a Inhibitors: In Silico Modeling and In Vitro Screening. J. Chem. Inf. Model. 2021, 61, 1020–1032. [Google Scholar] [CrossRef]
- Almalki, A.J.; Ibrahim, T.S.; Elhady, S.S.; Hegazy, W.A.H.; Darwish, K.M. Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents. Pharmaceuticals 2022, 15, 110. [Google Scholar] [CrossRef]
- Benson, N.C.; Daggett, V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef]
- Singh, W.; Karabencheva-Christova, T.G.; Black, G.W.; Ainsley, J.; Dover, L.; Christov, C.Z. Conformational Dynamics, Ligand Binding and Effects of Mutations in NirE an S-Adenosyl-L-Methionine Dependent Methyltransferase. Sci. Rep. 2016, 6, 20107. [Google Scholar] [CrossRef]
- Fatriansyah, J.F.; Rizqillah, R.K.; Yandi, M.Y.; Fadilah; Sahlan, M. Molecular docking and dynamics studies on propolis sulabiroin-A as a potential inhibitor of SARS-CoV-2. J. King Saud Univ. Sci. 2022, 34, 101707. [Google Scholar] [CrossRef]
- de Souza, A.S.; Pacheco, B.D.C.; Pinheiro, S.; Muri, E.M.F.; Dias, L.R.S.; Lima, C.H.S.; Garrett, R.; de Moraes, M.B.M.; de Souza, B.E.G.; Puzer, L. 3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7. Bioorg. Med. Chem. Lett. 2019, 29, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N. Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization. Methods Mol. Biol. 2020, 2114, 257–268. [Google Scholar] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Serohijos, A.W.R.; Murphy, S.; Lucarelli, D.; Lofranco, L.L.; Feldman, A.; Shakhnovich, E.I. Minimalistic predictor of protein binding energy: Contribution of solvation factor to protein binding. Biophys. J. 2015, 108, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The normal-mode entropy in the MM/GBSA method: Effect of system truncation, buffer region, and dielectric constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.M.; Donaldson, S.H.; Song, J.; Cheng, C.-Y.; Lee, D.W.; Han, S.; Israelachvili, J.N. Correlating steric hydration forces with water dynamics through surface force and diffusion NMR measurements in a lipid–DMSO–H2O system. Proc. Natl. Acad. Sci. USA 2015, 112, 10708–10713. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Leach, A.R.; Kuntz, I.D. Ligand solvation in molecular docking. Proteins 1999, 34, 4–16. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, L.; Liu, J.; Liu, G. Bioisosteres in drug discovery: Focus on tetrazole. Future Med. Chem. 2020, 12, 91–93. [Google Scholar] [CrossRef]
- Behairy, M.Y.; Soltan, M.A.; Adam, M.S.; Refaat, A.M.; Ezz, E.M.; Albogami, S.; Fayad, E.; Althobaiti, F.; Gouda, A.M.; Sileem, A.E.; et al. Computational Analysis of Deleterious SNPs in NRAS to Assess Their Potential Correlation With Carcinogenesis. Front. Genet. 2022, 13, 872845. [Google Scholar] [CrossRef]
- Hazalin, N.A.; Ramasamy, K.; Lim, S.S.M.; Wahab, I.A.; Cole, A.L.; Abdul Majeed, A.B. Cytotoxic and antibacterial activities of endophytic fungi isolated from plants at the National Park, Pahang, Malaysia. BMC Complement. Altern. Med. 2009, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Hubka, V.; Kolařík, M.; Kubátová, A.; Peterson, S.W. Taxonomic revision of Eurotium and transfer of species to Aspergillus. Mycologia 2013, 105, 912–937. [Google Scholar] [CrossRef]
- Turan, C.; Nanni, I.M.; Brunelli, A.; Collina, M. New rapid DNA extraction method with Chelex from Venturia inaequalis spores. J. Microbiol. Methods 2015, 115, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.C.; Boyette, M.; Goforth, C.; Sperry, K.V.; Greene, S.R. Comparison of the Biolog OmniLog Identification System and 16S ribosomal RNA gene sequencing for accuracy in identification of atypical bacteria of clinical origin. J. Microbiol. Methods 2009, 79, 336–343. [Google Scholar] [CrossRef]
- Elissawy, A.M.; Ebada, S.S.; Ashour, M.L.; El-Neketi, M.; Ebrahim, W.; Singab, A.B. New secondary metabolites from the mangrove-derived fungus Aspergillus sp. AV-2. Phytochem. Lett. 2019, 29, 1–5. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; Raja, H.A.; El-Elimat, T.; Oberlies, N.H. Evaluation of culture media for the production of secondary metabolites in a natural products screening program. AMB Express 2013, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing: 20th Informational Supplement; CLSI Doc. M100-S20; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2010. [Google Scholar]
- Karamolah, K.S.; Mousavi, F.; Mahmoudi, H. Antimicrobial inhibitory activity of aqueous, hydroalcoholic and alcoholic extracts of leaves and stem of Daphne mucronata on growth of oral bacteria. GMS Hyg. Infect. Control 2017, 12, 301. [Google Scholar]
- Gonelimali, F.D.; Lin, J.; Miao, W.; Xuan, J.; Charles, F.; Chen, M.; Hatab, S.R. Antimicrobial properties and mechanism of action of some plant extracts against food pathogens and spoilage microorganisms. Front. Microbiol. 2018, 9, 1639. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.; Morales, C.R.; Castillo, S.; Leos-Rivas, C.; García-Becerra, L.; Martínez, D.M.O. Antibacterial and antibiofilm activity of methanolic plant extracts against nosocomial microorganisms. Evid.-Based Complement. Altern. Med. Ecam 2016, 2016, 1572697. [Google Scholar] [CrossRef]
- Djordjevic, D.; Wiedmann, M.; McLandsborough, L. Microtiter plate assay for assessment of Listeria monocytogenes biofilm formation. Appl. Environ. Microbiol. 2002, 68, 2950–2958. [Google Scholar] [CrossRef]
- Choi, H.J.; Song, J.H.; Park, K.S.; Kwon, D.H. Inhibitory effects of quercetin 3-rhamnoside on influenza A virus replication. Eur. J. Pharm. Sci. 2009, 37, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Donalisio, M.; Nana, H.M.; Ngono Ngane, R.A.; Gatsing, D.; Tiabou Tchinda, A.; Rovito, R.; Cagno, V.; Cagliero, C.; Boyom, F.F.; Rubiolo, P. In vitro anti-Herpes simplex virus activity of crude extract of the roots of Nauclea latifolia Smith (Rubiaceae). BMC Complement. Altern. Med. 2013, 13, 266. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R.; Balzarini, J.; Baba, M.; Snoeck, R.; Schols, D.; Herdewijn, P.; Desmyter, J.; De Clercq, E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 1988, 20, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Solving Software Challenges for Exascale. In Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS; Markidis, S., Laure, E., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
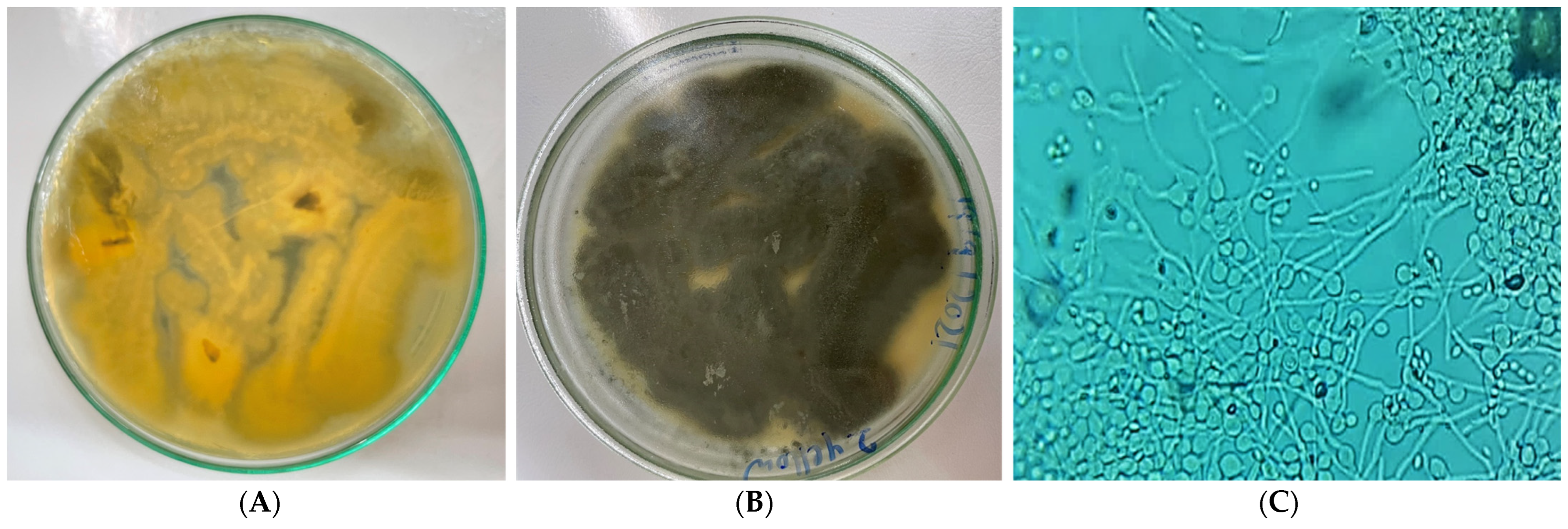









| Morphological Characters | Microscopic Characters | ||
|---|---|---|---|
| Surface | Yellowish-black | Hyphae | Thread-like septate branched |
| Margins | Entire | Conidia | Olive green (4 to 7 μm), roughened |
| Reverse side | Greenish-yellow | Phialides | uniseriate and biseriate phialides |
| Growth | Moderate | ||
| Elevations | Umbonate | ||
| Inhibition Zone Diameter (mm) | |||||
|---|---|---|---|---|---|
| Bacterial Strains | Negative Control | Positive Control | |||
| FEA | DMSO | Vancomycin | Gentamicin | Nystatin | |
| S. aureus ATCC 25923 (MSSA) | 15 ± 0.4 | 0 | 18 ± 0.2 | – | – |
| MRSA ATCC-700788 | 11 ± 0.7 | 0 | 13 ± 0.3 | – | – |
| E. coli ATCC 25922 | 0 | 0 | – | 19 ± 0.7 | – |
| P. aeruginosa ATCC9027 | 0 | 0 | – | 25 ± 1.1 | – |
| C. albicans ATCC 10231 | 0 | 0 | – | – | 15 ± 0.5 |
| No. | Compound | M. wt. | Lipophilicity Log Po/w(MLOGP) | Hydrogen Bond Donors | Hydrogen Bond Acceptors | No. of Rule Violations | Drug Likeness |
|---|---|---|---|---|---|---|---|
| Less than 500 g/mol | Less than 5 | Less than 5 | Less than 10 | Less than 2 Violations | Lipinski’s Rule Follows Rule | ||
| 1 | Heptelidic acid | 280 | 1.60 | 1 | 5 | 0 | Yes |
| 2 | Ferulic acid | 194 | 1.00 | 2 | 4 | 0 | Yes |
| 3 | Oleic acid | 282 | 4.57 | 1 | 2 | 0 | Yes |
| 4 | Paxilline | 435 | 2.58 | 3 | 4 | 0 | Yes |
| 5 | Indole | 117 | 1.57 | 1 | 0 | 0 | Yes |
| 6 | Orientin | 448 | −2.51 | 8 | 11 | 2 | No |
| 7 | Kojic acid | 142 | −1.69 | 2 | 4 | 0 | Yes |
| Compounds | Designated Targets | |||
|---|---|---|---|---|
| S. aureus MurE (PDB; 4c12) | S. aureus PBP2a (PDB; 3zg0) | S. aureus TarS (PDB; 5tzj) | HCoV-229E Mpro (PDB; 7yrz) | |
| Orientin | −51.75 | −49.69 | −37.48 | −50.15 |
| Heptelidic acid | −49.58 | −42.93 | −34.54 | −39.71 |
| Paxilline | −43.45 | −40.51 | −31.21 | −26.44 |
| Ferulic acid | −39.22 | −38.56 | −20.76 | −25.21 |
| Kojic acid | −22.18 | −14.11 | −13.23 | −19.49 |
| Oleic acid | −21.24 | −36.54 | −20.15 | −21.23 |
| Reference | −54.87 | −51.58 | −33.67 | −69.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fathallah, N.; Elkady, W.M.; Zahran, S.A.; Darwish, K.M.; Elhady, S.S.; Elkhawas, Y.A. Unveiling the Multifaceted Capabilities of Endophytic Aspergillus flavus Isolated from Annona squamosa Fruit Peels against Staphylococcus Isolates and HCoV 229E—In Vitro and In Silico Investigations. Pharmaceuticals 2024, 17, 656. https://doi.org/10.3390/ph17050656
Fathallah N, Elkady WM, Zahran SA, Darwish KM, Elhady SS, Elkhawas YA. Unveiling the Multifaceted Capabilities of Endophytic Aspergillus flavus Isolated from Annona squamosa Fruit Peels against Staphylococcus Isolates and HCoV 229E—In Vitro and In Silico Investigations. Pharmaceuticals. 2024; 17(5):656. https://doi.org/10.3390/ph17050656
Chicago/Turabian StyleFathallah, Noha, Wafaa M. Elkady, Sara A. Zahran, Khaled M. Darwish, Sameh S. Elhady, and Yasmin A. Elkhawas. 2024. "Unveiling the Multifaceted Capabilities of Endophytic Aspergillus flavus Isolated from Annona squamosa Fruit Peels against Staphylococcus Isolates and HCoV 229E—In Vitro and In Silico Investigations" Pharmaceuticals 17, no. 5: 656. https://doi.org/10.3390/ph17050656
APA StyleFathallah, N., Elkady, W. M., Zahran, S. A., Darwish, K. M., Elhady, S. S., & Elkhawas, Y. A. (2024). Unveiling the Multifaceted Capabilities of Endophytic Aspergillus flavus Isolated from Annona squamosa Fruit Peels against Staphylococcus Isolates and HCoV 229E—In Vitro and In Silico Investigations. Pharmaceuticals, 17(5), 656. https://doi.org/10.3390/ph17050656