

Study of the Lipophilicity and ADMET Parameters of New Anticancer Diquinothiazines with Pharmacophore Substituents



Abstract
:
1. Introduction
2. Results
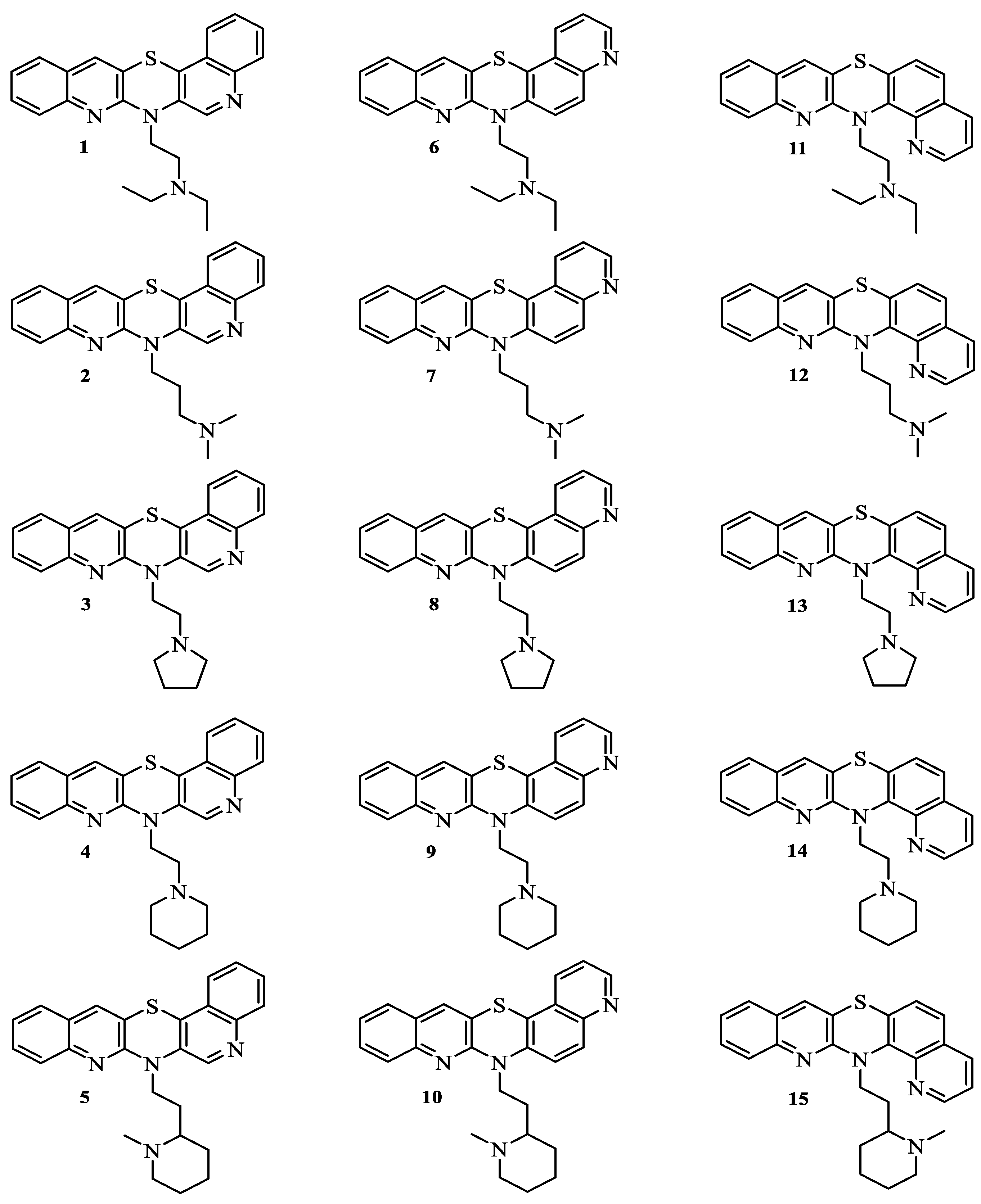
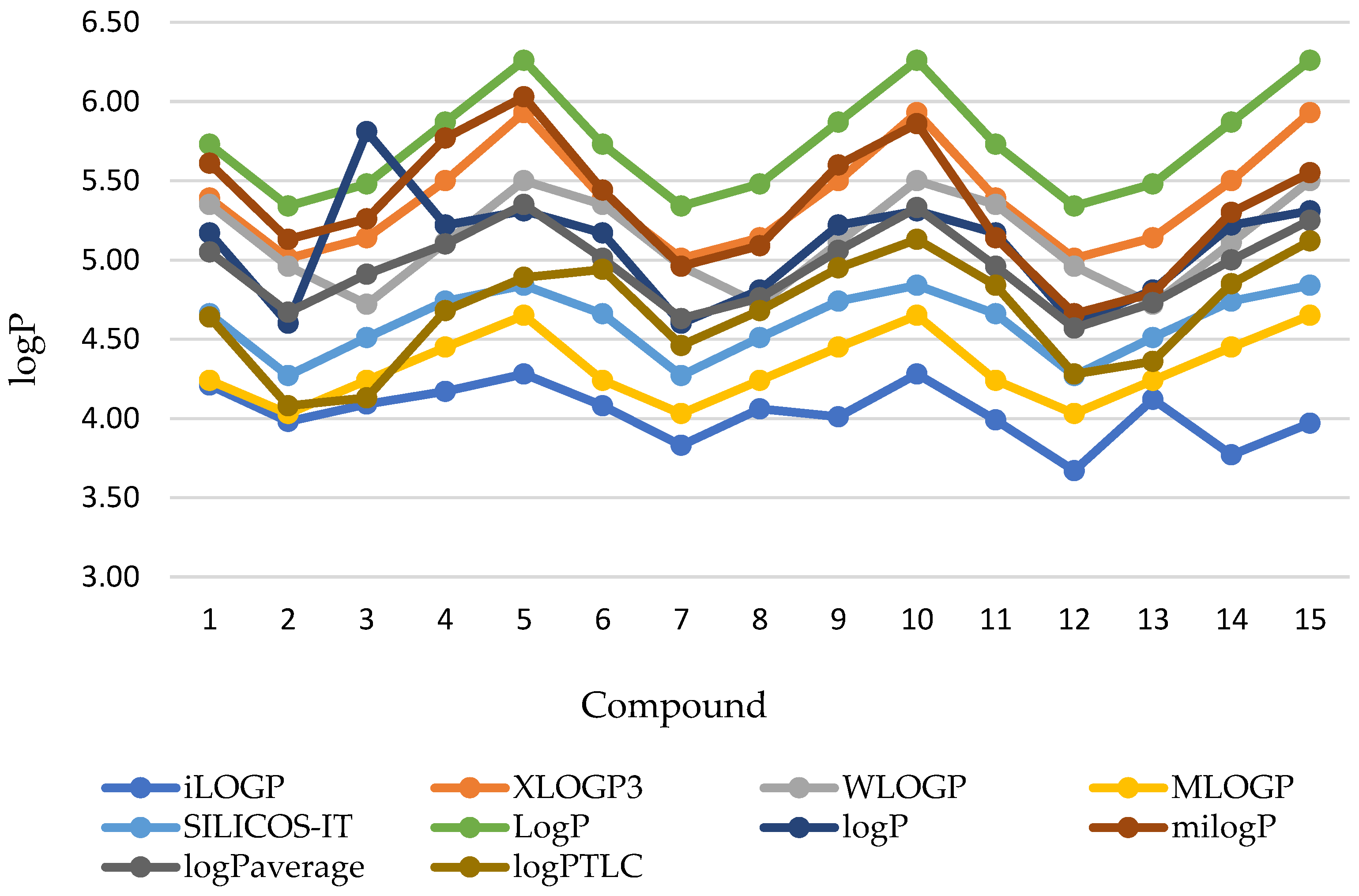
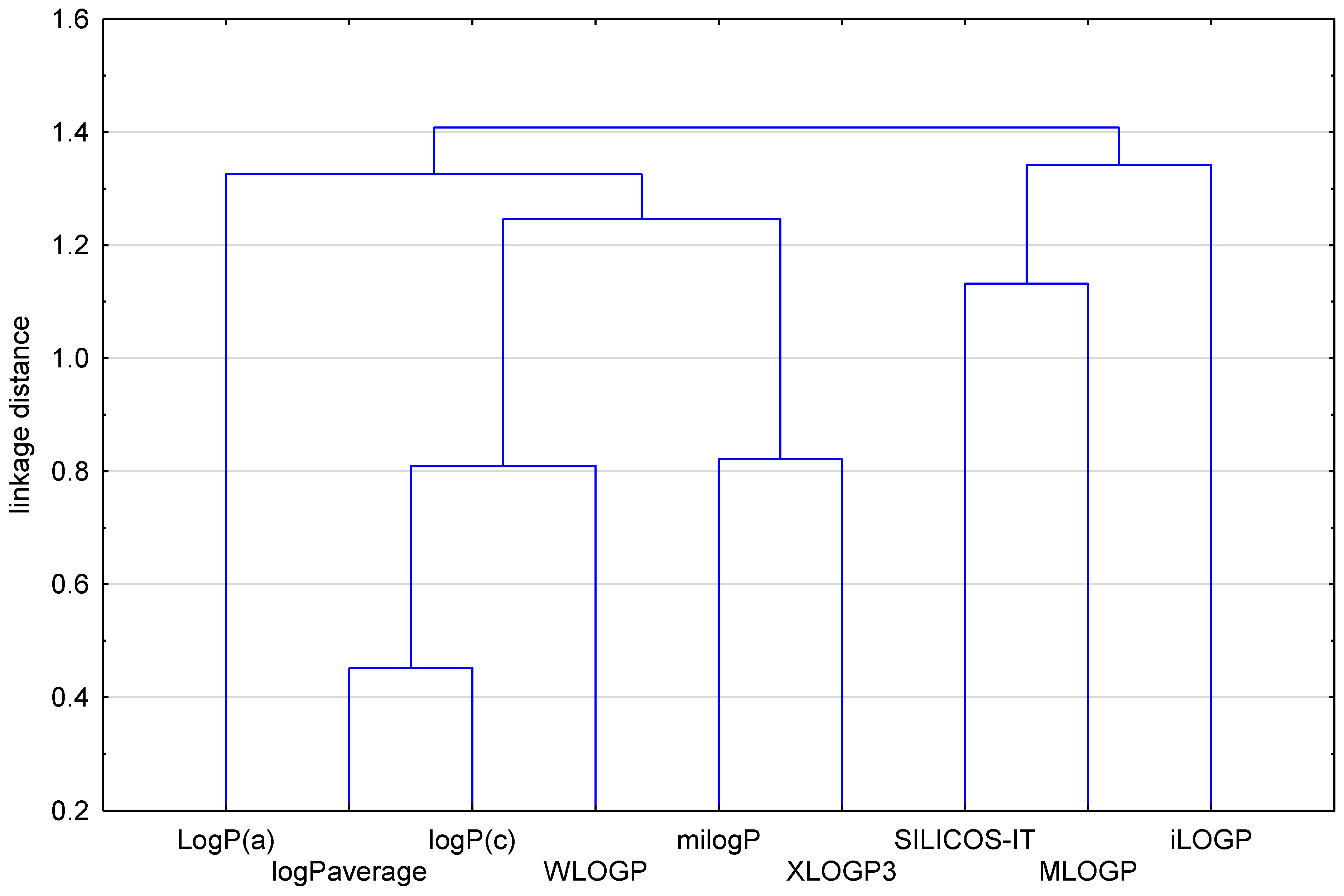
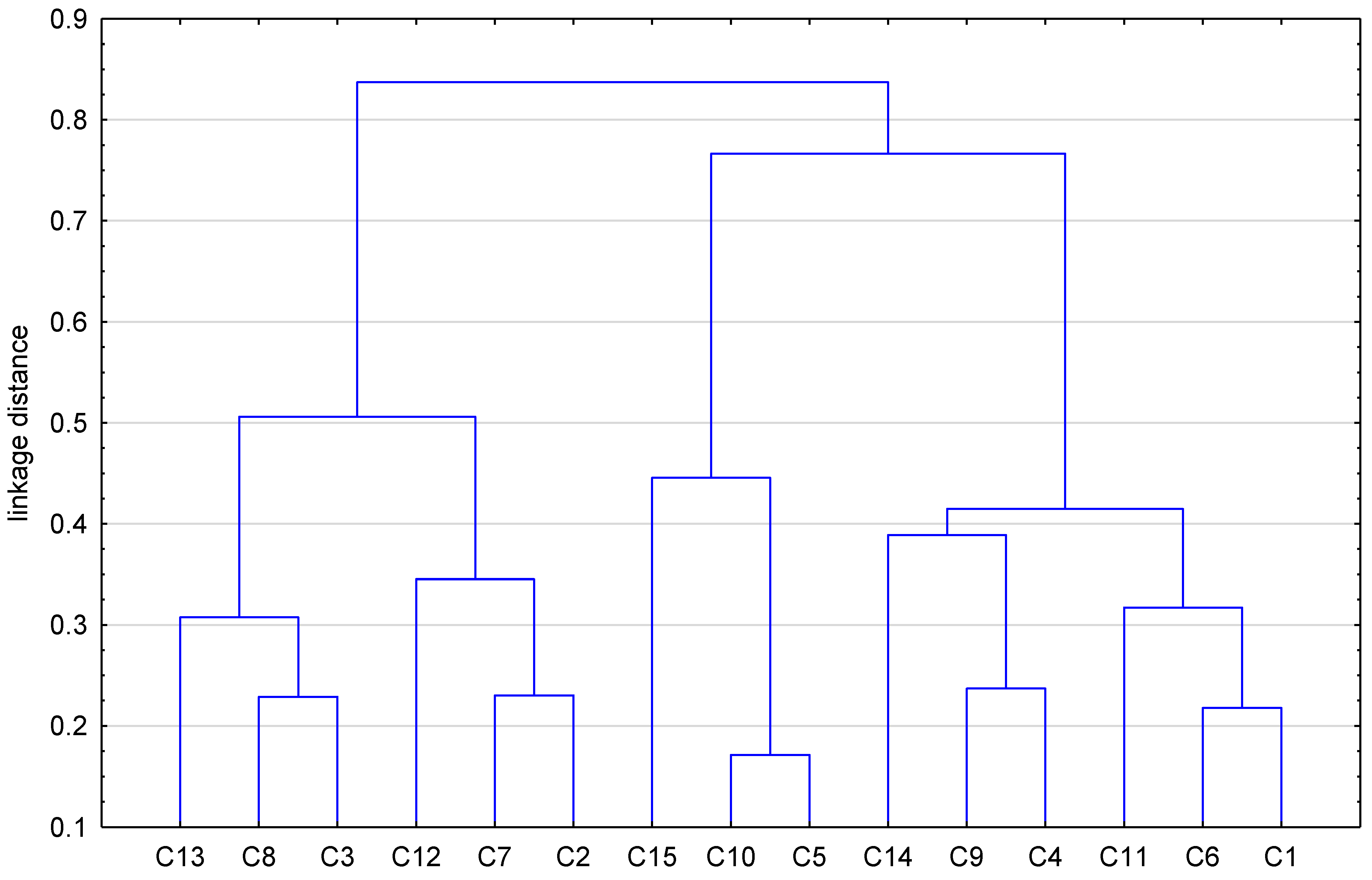
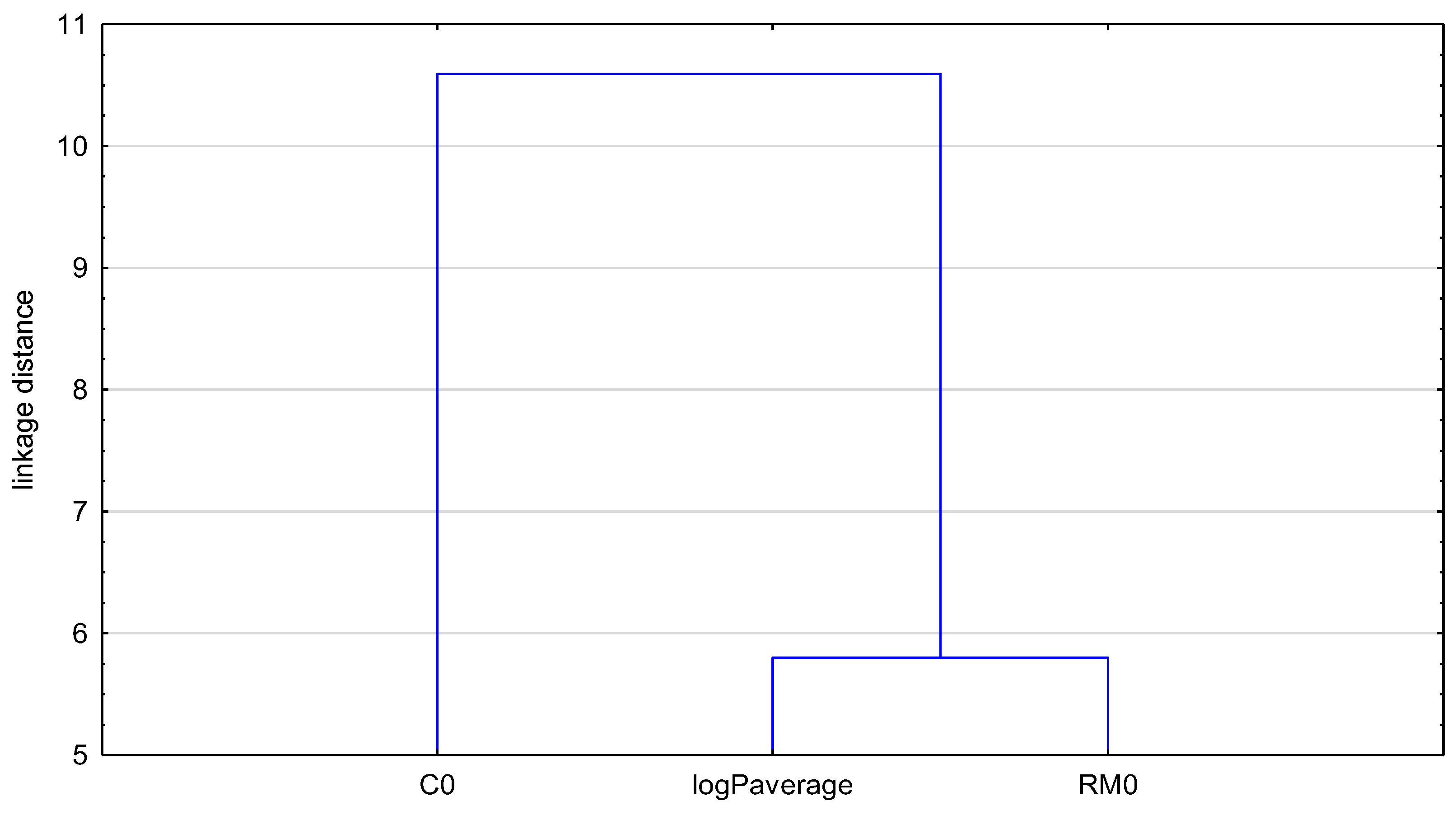
2.1. Lipophilicity Studies
2.2. Molecular Descriptors
2.3. In Silico ADME Prediction
2.3.1. Absorption
2.3.2. Distribution
2.3.3. Excretion and Toxicity
3. Discussion
In Silico ADME Prediction
4. Materials and Methods
4.1. Solvents and Reference Standards
4.2. RP-TLC Analysis
4.3. In Silico Calculated Descriptors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Ohlow, M.J.; Moosmann, B. Foundation Review: Phenothiazine: The seven lives of pharmacology’s first lead structure. Drug Discov. Today 2011, 16, 119–131. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Armistead, G.; Rosa, C.A.; Anderson, A.; Patil, R.; Cornett, E.M.; Murnane, K.S.; Kaye, A.M.; Kaye, A.D. Phenothiazines and their evolving roles in clinical practice: A narrative review. Health Psychol. Res. 2022, 10, 38930. [Google Scholar] [CrossRef]
- Posso, M.C.; Domingues, F.C.; Ferreira, S.; Silvestre, S. Development of phenothiazine hybrids with potential medicinal interest: A Review. Molecules 2022, 27, 276. [Google Scholar] [CrossRef]
- Pluta, K.; Morak-Młodawska, B.; Jeleń, M. Recent progress in biological activities of synthesized phenothiazines. Eur. J. Med. Chem. 2011, 46, 3179–3189. [Google Scholar] [CrossRef]
- Spengler, G.; Csonka, Á.; Molnár, J.; Amaral, L. The anticancer activity of the old neuroleptic phenothiazine-type drug thioridazine. Anticancer Res. 2016, 36, 5701–5706. [Google Scholar] [CrossRef]
- Zubas, A.; Ghinet, A.; Farce, A.; Dubois, J.; Bîcu, E. Phenothiazineand carbazole-cyanochalcones as dual inhibitors of tubulin polymerization and human farnesyltransferase. Pharmaceuticals 2023, 16, 888. [Google Scholar] [CrossRef]
- Mosnaim, A.D.; Ranade, V.V.; Wolf, M.E.; Puente, J.; Valenzuela, A.M. Phenothiazine molecule provides the basic chemical structure for various classes of pharmacotherapeutic agents. Am. J. Ther. 2006, 13, 261–273. [Google Scholar] [CrossRef]
- Chuang, S.T.; Papp, H.; Kuczmog, A.; Eells, R.; Condor Capcha, J.M.; Shehadeh, L.A.; Jakab, F.; Buchwald, P. Methylene blue is a nonspecific protein–protein interaction inhibitor with potential for repurposing as an antiviral for COVID-19. Pharmaceuticals 2022, 15, 621. [Google Scholar] [CrossRef]
- Boyd-Kimball, D.; Gonczy, K.; Lewis, B.; Mason, T.; Siliko, N.; Wolfe, J. Classics in chemical neuroscience: Chlorpromazine. ACS Chem. Neurosci. 2019, 10, 79–88. [Google Scholar] [CrossRef]
- Ginex, T.; Vazquez, J.; Gilbert, E.; Herrero, E.; Luque, F.J. Lipophilicity in drug design: An overview of lipophilicity descriptors in 3D-QSAR studies. Future Med. Chem. 2019, 11, 1177–1193. [Google Scholar] [CrossRef]
- Johnson, T.W.; Dress, K.R.; Edwards, M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef]
- Johnson, T.W.; Gallego, R.A.; Edwards, M.P. Lipophilic efficiency as an important metric in drug design. J. Med. Chem. 2018, 61, 6401–6420. [Google Scholar] [CrossRef]
- Ditzinger, F.; Price, D.J.; Ilie, A.R.; Köhl, N.J.; Jankovic, S.; Tsakiridou, G.; Aleandri, S.; Kalantzi, L.; Holm, R.; Nair, A.; et al. Lipophilicity and hydrophobicity considerations in bioenabling oral formulations approaches—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 464–482. [Google Scholar] [CrossRef]
- Leeson, P.D.; Davis, A.M. Time-related differences in the physical property profiles of oral drugs. J. Med. Chem. 2004, 47, 6338–6348. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Lipidic prodrug approach for improved oral drug delivery and therapy. Med. Res. Rev. 2019, 39, 579–607. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Profiling drug-like properties in discovery research. Curr. Opin. Chem. Biol. 2003, 7, 402–408. [Google Scholar] [CrossRef]
- Dulsat, J.; López-Nieto, B.; Estrada-Tejedor, R.; Borrell, J.I. Evaluation of free online ADMET tools for academic or small biotech environments. Molecules 2023, 28, 776. [Google Scholar] [CrossRef]
- Rutkowska, E.; Pająk, K.; Jóźwiak, K. Lipophilicity—Methods of determination and its role in medicinal chemistry. Acta Pol. Pharm. Drug Res. 2013, 70, 3–18. [Google Scholar]
- OECD. Test No. 117: Partition Coefficient (n-Octanol/Water), HPLC Method; OECD Publishing: Paris, France, 2022. [Google Scholar]
- Roman, I.P.; Mastromichali, A.; Tyrovola, K.; Canals, A.; Psillakis, E. Rapid determination of octanol-water partition coefficient using vortex-assisted liquid-liquid microextraction. J. Chromatogr. A 2014, 1330, 1–5. [Google Scholar] [CrossRef]
- Eadsforth, C.V.; Moser, P. Assessment of reverse-phase chromatographic methods for determining partition coefficients. Chemosphere 1983, 12, 1459–1475. [Google Scholar] [CrossRef]
- Soares, J.X.; Santos, Á.; Fernandes, C.; Pinto, M.M.M. Liquid chromatography on the different methods for the determination of lipophilicity: An essential analytical tool in medicinal chemistry. Chemosensors 2022, 10, 340. [Google Scholar] [CrossRef]
- Jeleń, M.; Pluta, K.; Latocha, M.; Morak-Młodawska, B.; Suwińska, K.; Kuśmierz, D. Evaluation of angularly condensed diquinothiazines as potential anticancer agents. Bioorg. Chem. 2019, 87, 810–820. [Google Scholar] [CrossRef]
- Skonieczna, M.; Kasprzycka, A.; Jeleń, M.; Morak-Młodawska, B. Tri- and pentacyclic azaphenothiazine as pro-apoptotic agents in lung carcinoma with a protective potential to healthy cell lines. Molecules 2022, 27, 5255. [Google Scholar] [CrossRef]
- Available online: http://swissadme.ch (accessed on 10 January 2024).
- Available online: https://biosig.unimelb.edu.au/pkcsm/prediction (accessed on 5 January 2024).
- Available online: https://www.molinspiration.com/services/logp.html (accessed on 5 January 2024).
- ChemDraw: ChemDraw Ultra, Version 22.2.0; CambridgeSoft, PerkinElmer Informatics: Austin, TX, USA, 2022.
- Bodor, N.; Gabanyi, Z.; Wong, C.K. A new method for the estimation of partition coefficient. J. Am. Chem. Soc. 1989, 111, 3783–3786. [Google Scholar] [CrossRef]
- Mannhold, R.; Cruciani, G.; Dross, K.; Rekker, R. Multivariate analysis of experimental and computational descriptors of molecular lipophilicity. J. Comput. Mol. Des. 1998, 12, 573–581. [Google Scholar] [CrossRef]
- Brooke, D.; Dobbs, J.; Williams, N. Octanol:water partition coefficients (P): Measurement, estimation, and interpretation, particularly for chemicals with P > 105. Ecotoxicol. Environ. Saf. 1986, 11, 251–260. [Google Scholar] [CrossRef]
- Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal absorption study: Challenges and absorption enhancement strategies in improving oral drug delivery. Pharmaceuticals 2022, 15, 975. [Google Scholar] [CrossRef]
- Tsakovska, I.; Pajeva, I.; Al Sharif, M.; Alov, P.; Fioravanzo, E.; Kovarich, S.; Worth, A.P.; Richarz, A.N.; Yang, C.; Mostrag-Szlichtyng, A.; et al. Quantitative structure-skin permeability relationships. Toxicology 2017, 387, 27–42. [Google Scholar] [CrossRef]
- Gombar, V.K.; Hall, S.D. Quantitative structure−activity relationship models of clinical pharmacokinetics: Clearance and volume of distribution. J. Chem. Inf. Model. 2013, 53, 948–957. [Google Scholar] [CrossRef]
- Poulin, P.; Theil, F.P. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J. Pharm. Sci. 2002, 91, 129–156. [Google Scholar] [CrossRef]
- Mulpuru, V.; Mishra, N. In silico prediction of fraction unbound in human plasma from chemical fingerprint using automated machine learning. ACS Omega 2021, 10, 6791–6797. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Horde, G.W.; Gupta, V. Drug Clearance. 20 June 2023. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar] [PubMed]
- Das, A.; Kumar, S.; Persoons, L.; Daelemans, D.; Schols, D.; Alici, H.; Tahtaci, H.; Karki, S.S. Synthesis, in silico ADME, molecular docking and in vitro cytotoxicity evaluation of stilbene linked 1,2,3-triazoles. Heliyon 2021, 7, e05893. [Google Scholar] [CrossRef]
- Goetz, G.H.; Shalaeva, M. Leveraging chromatography based physicochemical properties for efficient drug design. ADMET DMPK 2018, 6, 85. [Google Scholar] [CrossRef]
- Kaliszan, R. QSRR: Quantitative Structure-(Chromatographic) Retention Relationships. Chem. Rev. 2007, 107, 3212–3246. [Google Scholar] [CrossRef]
- Kempińska, D.; Chmiel, T.; Kot-Wasik, A.; Mróz, A.; Mazerska, Z.; Namie’snik, J. State of the art and prospects of methods for determination of lipophilicity of chemical compounds. Trends Anal. Chem. 2019, 113, 54–73. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Drug-Like Properties; Academic Press: New York, NY, USA, 2008. [Google Scholar]
- Petrauskas, A.A.; Kolovanov, E.A. ACD/Log P method description. Perspect. Drug Discov. Des. 2000, 19, 99–116. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lobo, S. Is there enough focus on lipophilicity in drug discovery? Expert Opin. Drug Discov. 2020, 15, 261–263. [Google Scholar] [CrossRef]
- Pantaleao, S.Q.; Fernandes, P.O.; Goncalves, J.E.; Maltarollo, V.G.; Honorio, K.M. Recent advances in the prediction of pharmacokinetics properties in drug design studies: A review. Chem. Med. Chem. 2022, 17, e202100542. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem. Med. Chem. 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Šegan, S.; Penjišević, J.; Šukalović, V.; Andrić, D.; Milojković-Opsenica, D.; Kostić-Rajačić, S. Investigation of lipophilicity and pharmacokinetic properties of 2-(methoxy)phenylpiperazine dopamine D2 ligands. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1124, 146–153. [Google Scholar] [CrossRef]
- Moriguchi, I.; Shuichi, H.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Moriguchi, I.; Shuichi, H.; Nakagome, I.; Hirano, H. Comparison of reliability of log P values for drugs calculated by several methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef]
- Stanisz, A. Przystępny Kurs Statystyki z Zastosowaniem STATISTICA PL na Przykładach Medycyny; Analizy Wielowymiarowe; Stat Soft Polska: Kraków, Poland, 2007; Volume 3. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. of Compound | iLOGP | XLOGP3 | WLOGP | MLOGP | SILICOS-IT | LogP a | milogP b | logP c | logPaverage |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4.21 | 5.39 | 5.35 | 4.24 | 4.66 | 5.73 | 5.61 | 5.17 | 5.05 (±0.60) |
| 2 | 3.98 | 5.01 | 4.96 | 4.03 | 4.27 | 5.34 | 5.13 | 4.60 | 4.67 (±0.52) |
| 3 | 4.09 | 5.14 | 4.72 | 4.24 | 4.51 | 5.48 | 5.26 | 4.81 | 4.91 (±0.61) |
| 4 | 4.17 | 5.50 | 5.11 | 4.45 | 4.74 | 5.87 | 5.77 | 5.22 | 5.10 (±0.61) |
| 5 | 4.28 | 5.93 | 5.50 | 4.65 | 4.84 | 6.26 | 6.03 | 5.31 | 5.35 (±0.71) |
| 6 | 4.08 | 5.39 | 5.35 | 4.24 | 4.66 | 5.73 | 5.44 | 5.17 | 5.01 (±0.61) |
| 7 | 3.83 | 5.01 | 4.96 | 4.03 | 4.27 | 5.34 | 4.96 | 4.60 | 4.63 (±0.53) |
| 8 | 4.06 | 5.14 | 4.72 | 4.24 | 4.51 | 5.48 | 5.09 | 4.81 | 4.76 (±0.48) |
| 9 | 4.01 | 5.50 | 5.11 | 4.45 | 4.74 | 5.87 | 5.60 | 5.22 | 5.06 (±0.63) |
| 10 | 4.28 | 5.93 | 5.50 | 4.65 | 4.84 | 6.26 | 5.86 | 5.31 | 5.33 (±0.69) |
| 11 | 3.99 | 5.39 | 5.35 | 4.24 | 4.66 | 5.73 | 5.14 | 5.17 | 4.96 (±0.60) |
| 12 | 3.67 | 5.01 | 4.96 | 4.03 | 4.27 | 5.34 | 4.66 | 4.60 | 4.57 (±0.55) |
| 13 | 4.12 | 5.14 | 4.72 | 4.24 | 4.51 | 5.48 | 4.79 | 4.81 | 4.73 (±0.45) |
| 14 | 3.77 | 5.50 | 5.11 | 4.45 | 4.74 | 5.87 | 5.30 | 5.22 | 5.00 (±0.66) |
| 15 | 3.97 | 5.93 | 5.50 | 4.65 | 4.84 | 6.26 | 5.55 | 5.31 | 5.25 (±0.74) |
| No. of Compound | RM0 | b | r | C0 |
|---|---|---|---|---|
| 1 | 3.45 | −4.77 | 0.9932 | 0.7233 |
| 2 | 3.01 | −4.09 | 0.9961 | 0.7359 |
| 3 | 3.05 | −4.23 | 0.9916 | 0.7210 |
| 4 | 3.48 | −4.89 | 0.9953 | 0.7117 |
| 5 | 3.64 | −5.14 | 0.9980 | 0.7082 |
| 6 | 3.68 | −4.85 | 0.9951 | 0.7588 |
| 7 | 3.31 | −4.34 | 0.9938 | 0.7627 |
| 8 | 3.48 | −4.60 | 0.9941 | 0.7565 |
| 9 | 3.69 | −4.88 | 0.9962 | 0.7562 |
| 10 | 3.83 | −5.04 | 0.9962 | 0.7599 |
| 11 | 3.60 | −4.75 | 0.9927 | 0.7579 |
| 12 | 3.17 | −4.25 | 0.9935 | 0.7458 |
| 13 | 3.23 | −4.21 | 0.9907 | 0.7672 |
| 14 | 3.61 | −4.69 | 0.9952 | 0.7697 |
| 15 | 3.82 | −5.01 | 0.9942 | 0.7625 |
| Lipophilicity Parameters | Standards | ||||
|---|---|---|---|---|---|
| I | II | III | IV | V | |
| logPlit | 1.21 [30] | 1.87 [31] | 3.18 [31] | 4.45 [31] | 6.38 [32] |
| RM0 | 0.78 | 1.16 | 2.51 | 3.33 | 4.69 |
| -b | 0.0162 | 0.0247 | 0.0328 | 0.0412 | 0.0564 |
| r | 0.9923 | 0.9937 | 0.9971 | 0.9982 | 0.9977 |
| logPTLC | 1.21 | 1.70 | 3.43 | 4.49 | 6.24 |
| No. of Compound | logPTLC | No. of Compound | logPTLC | No. of Compound | logPTLC |
|---|---|---|---|---|---|
| 1 | 4.64 | 6 | 4.94 | 11 | 4.84 |
| 2 | 4.08 | 7 | 4.46 | 12 | 4.28 |
| 3 | 4.13 | 8 | 4.68 | 13 | 4.36 |
| 4 | 4.68 | 9 | 4.95 | 14 | 4.85 |
| 5 | 4.89 | 10 | 5.13 | 15 | 5.12 |
| No. of Compound | Molar Mass (M) [g/mol] | Molar Volume (VM) [cm3] | Molar Refractivity (RefM) [cm3/mol] | Surface Area [Å] |
|---|---|---|---|---|
| 1 | 400.55 | 322.1 | 121.077 | 175.12 |
| 2 | 386.52 | 305.6 | 116.446 | 168.75 |
| 3 | 398.54 | 304.6 | 119.121 | 174.11 |
| 4 | 412.56 | 322.4 | 123.722 | 180.48 |
| 5 | 426.59 | 342.3 | 128.258 | 186.84 |
| 6 | 400.55 | 322.1 | 121.077 | 175.12 |
| 7 | 386.52 | 305.6 | 116.446 | 168.75 |
| 8 | 398.54 | 304.6 | 119.121 | 174.11 |
| 9 | 412.56 | 322.4 | 123.722 | 180.48 |
| 10 | 426.59 | 342.3 | 128.258 | 186.84 |
| 11 | 400.55 | 322.1 | 121.077 | 175.12 |
| 12 | 386.52 | 305.6 | 116.446 | 168.75 |
| 13 | 398.54 | 304.6 | 119.121 | 174.11 |
| 14 | 412.56 | 322.4 | 123.722 | 180.48 |
| 15 | 426.59 | 342.3 | 128.258 | 186.84 |
| Predicted Parameter | Compound No. | ||||
|---|---|---|---|---|---|
| 1, 6, 11 | 2, 7, 12 | 3, 8, 13 | 4, 9, 14 | 5, 10, 15 | |
| Physicochemical Properties | |||||
| Num. heavy atoms | 29 | 28 | 29 | 30 | 31 |
| Num. arom. heavy atoms | 20 | 20 | 20 | 20 | 20 |
| Hydrogen bond acceptors | 3 | 3 | 3 | 3 | 3 |
| Hydrogen bond donors | 0 | 0 | 0 | 0 | 0 |
| Number of rotatable bonds | 5 | 4 | 3 | 3 | 3 |
| Topological polar surface area [Å2] | 57.56 | 57.56 | 57.56 | 57.56 | 57.56 |
| Drug-Likeness Prediction | |||||
| Rule of Lipinski | + | + | + | + | + |
| Rule of Ghose | + | + | + | −(MR>130) | −(MR>130) |
| Rule of Veber | + | + | + | + | + |
| Rule of Egan | + | + | + | + | + |
| Rule of Muegge | −(XLOGP3>5) | −(XLOGP3>5) | −(XLOGP3>5) | −(XLOGP3>5) | −(XLOGP3>5) |
| Bioavailability | |||||
| Bioactivity score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| No. of Compound | Water Solubility [log mol/L] | Caco-2 Permeability [log Papp in 10−6 cm/s] | Intestinal Absorption [% Absorbed] | Skin Permeability [log Kp] |
|---|---|---|---|---|
| 1 | −5.871 | 1.003 | 92.241 | −2.697 |
| 2 | −5.783 | 1.028 | 92.931 | −2.701 |
| 3 | −4.551 | 1.145 | 92.725 | −2.733 |
| 4 | −4.660 | 1.143 | 92.337 | −2.733 |
| 5 | −5.820 | 1.021 | 92.309 | −2.711 |
| 6 | −5.793 | 1.032 | 95.220 | −2.694 |
| 7 | −5.737 | 1.057 | 96.401 | −2.700 |
| 8 | −4.517 | 1.226 | 94.349 | −2.723 |
| 9 | −4.652 | 1.224 | 93.960 | −2.723 |
| 10 | −5.829 | 1.049 | 95.779 | −2.712 |
| 11 | −5.373 | 1.043 | 95.329 | −2.691 |
| 12 | −5.263 | 1.068 | 96.510 | −2.697 |
| 13 | −3.965 | 1.211 | 93.469 | −2.744 |
| 14 | −4.061 | 1.209 | 93.080 | −2.743 |
| 15 | −5.329 | 1.060 | 95.888 | −2.709 |
| No. of Compound | VDss [log L/kg] | Unbound Fraction [Fu] | BBB Permeability [log BB] | CNS Permeability [log PS] |
|---|---|---|---|---|
| 1 | 0.986 | 0.259 | 0.478 | −1.464 |
| 2 | 0.864 | 0.253 | 0.483 | −1.402 |
| 3 | 0.868 | 0.193 | 0.424 | −1.394 |
| 4 | 0.923 | 0.189 | 0.437 | −1.381 |
| 5 | 1.041 | 0.255 | 0.535 | −1.422 |
| 6 | 1.178 | 0.268 | 0.537 | −1.493 |
| 7 | 1.062 | 0.262 | 0.484 | −1.478 |
| 8 | 1.269 | 0.206 | 0.559 | −1.318 |
| 9 | 1.328 | 0.200 | 0.572 | −1.304 |
| 10 | 1.244 | 0.257 | 0.536 | −1.498 |
| 11 | 1.332 | 0.267 | 0.387 | −1.475 |
| 12 | 1.200 | 0.261 | 0.357 | −1.483 |
| 13 | 1.170 | 0.200 | 0.425 | −1.370 |
| 14 | 1.222 | 0.196 | 0.438 | −1.356 |
| 15 | 1.368 | 0.262 | 0.410 | −1.503 |
| No. of Compound | Total Clearance [log ml/min/kg] | Max. Tolerated Dose [log mg/kg/day] | Oral Rat Acute Toxicity [mol/kg] | Oral Rat Chronic Toxicity [log mg/kg bw/day] | Tetrahymena pyriformis Toxicity [log μg/L] | Minnow Toxicity [log mM] |
|---|---|---|---|---|---|---|
| 1 | 0.672 | 0.672 | 2.277 | 0.449 | 0.299 | 1.983 |
| 2 | 0.526 | 0.647 | 2.272 | 0.490 | 0.300 | 1.944 |
| 3 | 0.824 | 0.537 | 2.734 | 0.827 | 0.291 | −0.279 |
| 4 | 0.779 | 0.550 | 2.756 | 0.848 | 0.291 | −0.396 |
| 5 | 0.599 | 0.568 | 2.363 | 0.542 | 0.291 | 1.736 |
| 6 | 0.714 | 0.293 | 2.361 | 0.773 | 0.327 | 0.423 |
| 7 | 0.584 | 0.258 | 2.312 | 0.807 | 0.331 | 0.597 |
| 8 | 0.832 | 0.211 | 3.157 | 1.074 | 0.300 | −0.872 |
| 9 | 0.787 | 0.228 | 3.184 | 1.096 | 0.299 | −0.989 |
| 10 | 0.657 | 0.188 | 2.413 | 0.606 | 0.304 | 0.389 |
| 11 | 0.707 | 0.594 | 2.513 | 0.590 | 0.299 | 0.542 |
| 12 | 0.562 | 0.557 | 2.485 | 0.610 | 0.300 | 0.460 |
| 13 | 0.826 | 0.543 | 2.949 | 1.020 | 0.292 | −0.848 |
| 14 | 0.781 | 0.557 | 2.972 | 1.041 | 0.291 | −0.965 |
| 15 | 0.634 | 0.483 | 2.588 | 0.663 | 0.291 | 0.252 |
| Compounds | Lipophilicity Parameter | Equation | r |
|---|---|---|---|
| 1–5 | iLOGP | iLOGP = 0.3877 RM0 + 2.8566 | 0.9403 |
| 1–15 | XLOGP3 | XLOGP3 = 1.0578 RM0 + 1.7233 | 0.8426 |
| 1–5 | XLOGP3 = 1.1972RM0 + 1.4122 | 0.9384 | |
| 6–10 | XLOGP3 = 1.6538 RM0 − 0.5565 | 0.9427 | |
| 11–15 | XLOGP3 = 1.2383 RM0 + 1.0773 | 0.9577 | |
| 1–15 | WLOGP | WLOGP = 0.8350 RM0 + 2.2304 | 0.7683 |
| 1–5 | WLOGP = 0.9794 RM0 + 1.8705 | 0.8868 | |
| 11–15 | WLOGP = 0.9985 RM0 + 1.6473 | 0.8919 | |
| 1–15 | MLOGP | MLOGP = 0.6500 RM0 + 2.0664 | 0.7840 |
| 6–10 | MLOGP = 1.0545 RM0 + 0.5279 | 0.9101 | |
| 11–15 | MLOGP = 0.7511 RM0 + 1.7035 | 0.8795 | |
| 1–15 | SILCOS-IT | SILCOS-IT = 0.6630 RM0 + 2.3034 | 0.8486 |
| 1–5 | SILCOS-IT = 0.7442 RM0 + 2.1287 | 0.9374 | |
| 6–10 | SILCOS-IT = 1.0799 RM0 + 0.7184 | 0.9892 | |
| 11–15 | SILCOS-IT = 0.7588 RM0 + 1.9588 | 0.9432 | |
| 1–15 | LogP | LogP = 1.0697 RM0 + 2.0243 | 0.8484 |
| 1–5 | LogP = 1.2120 RM0 + 1.7050 | 0.9460 | |
| 6–10 | LogP = 1.6739 RM0 − 0.2866 | 0.9500 | |
| 11–15 | LogP = 1.2499 RM0 + 1.3788 | 0.9624 | |
| 6–10 | logP = 1.4693 RM0 − 0.2644 | 0.9877 | |
| 11–15 | logP = 1.0652 RM0 + 1.3086 | 0.9714 | |
| 1–15 | milogP | milogP = 0.9755 RM0 + 1.9610 | 0.6499 |
| 1–5 | milogP = 1.2903 RM0 + 1.2684 | 0.9813 | |
| 6–10 | milogP = 1.7585 RM0 − 0.9371 | 0.9725 | |
| 11–15 | milogP = 1.3037 RM0 + 0.5431 | 0.9867 | |
| 1–15 | logPaverage | logPaverage = 0.7369 RM0 + 2.4017 | 0.7854 |
| 1–5 | logPaverage = 0.8307 RM0 + 2.2530 | 0.9292 | |
| 6–10 | logPaverage = 1.3129 RM0 + 0.2343 | 0.9791 | |
| 11–15 | logPaverage = 0.9300 RM0 + 1.6601 | 0.9819 |
| Compounds | Molecular Descriptor | Equation | r |
|---|---|---|---|
| 1–15 | Molar mass | M = 42.779 RM0 + 256.508 | 0.8000 |
| 1–5 | M = 47.859 RM0 + 245.773 | 0.8807 | |
| 6–10 | M = 68.869 RM0 + 157.161 | 0.9216 | |
| 11–15 | M = 49.570 RM0 + 232.152 | 0.8999 | |
| 1–15 | Molar volume | VM = 45.782 RM0 + 160.711 | 0.8444 |
| 1–5 | VM = 52.381 RM0 + 145.180 | 0.9518 | |
| 6–10 | VM = 69.514 RM0 + 69.288 | 0.9182 | |
| 11–15 | VM = 53.970 RM0 + 131.262 | 0.9674 | |
| 1–15 | Molar refractivity | RefM = 13.202 RM0 + 75.913 | 0.8311 |
| 1–5 | RefM = 14.871 RM0 + 72.263 | 0.9213 | |
| 6–10 | RefM = 20.942 RM0 + 46.375 | 0.9434 | |
| 11–15 | RefM = 15.363 RM0 + 68.170 | 0.9389 | |
| 1–15 | Surface area | Surface area = 19.960 RM0 + 109.882 | 0.8017 |
| 1–5 | Surface area = 21.667 RM0 + 105.000 | 0.8831 | |
| 6–10 | Surface area = 31.142 RM0 + 65.010 | 0.9229 | |
| 11–15 | Surface area = 22.437 RM0 + 98.843 | 0.9021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimoszek, D.; Jeleń, M.; Dołowy, M.; Morak-Młodawska, B. Study of the Lipophilicity and ADMET Parameters of New Anticancer Diquinothiazines with Pharmacophore Substituents. Pharmaceuticals 2024, 17, 725. https://doi.org/10.3390/ph17060725
Klimoszek D, Jeleń M, Dołowy M, Morak-Młodawska B. Study of the Lipophilicity and ADMET Parameters of New Anticancer Diquinothiazines with Pharmacophore Substituents. Pharmaceuticals. 2024; 17(6):725. https://doi.org/10.3390/ph17060725
Chicago/Turabian StyleKlimoszek, Daria, Małgorzata Jeleń, Małgorzata Dołowy, and Beata Morak-Młodawska. 2024. "Study of the Lipophilicity and ADMET Parameters of New Anticancer Diquinothiazines with Pharmacophore Substituents" Pharmaceuticals 17, no. 6: 725. https://doi.org/10.3390/ph17060725