
Targeted Delivery Strategies for Multiple Myeloma and Their Adverse Drug Reactions


Abstract
:
1. Introduction
2. Antibody–Drug Conjugates
2.1. Structure and Mechanism of ADCs
2.2. ADCs for MM Treatment
3. Peptide–Drug Conjugates
4. Aptamer–Drug Conjugates
4.1. Introduction to Aptamers and APDC Systems
4.2. Aptamer Delivery Systems in MM
5. Nanoparticle Drug Delivery Systems
5.1. Liposomes
5.2. Micelles
5.3. Polymeric Nanoparticles
5.4. Inorganic Nanoparticles
6. Prevention and Management of Adverse Events
6.1. Hematologic Toxicities
6.2. Ocular Disorders
6.3. Peripheral Neuropathy
6.4. Infusion-Related Reactions
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. N. Engl. J. Med. 2004, 351, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yu, Q.; Wei, G.; Wang, L.; Huang, Y.; Hu, K.; Hu, Y.; Huang, H. Measuring the global, regional, and national burden of multiple myeloma from 1990 to 2019. BMC Cancer 2021, 21, 606. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Green, D.J.; Kwok, M.; Lee, S.; Coffey, D.G.; Holmberg, L.A.; Tuazon, S.; Gopal, A.K.; Libby, E.N. Diagnosis and Management of Multiple Myeloma: A Review. JAMA 2022, 327, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.S.; Kaufman, J.L.; Dhodapkar, M.V.; Hofmeister, C.C.; Almaula, D.K.; Heffner, L.T.; Gupta, V.A.; Boise, L.H.; Lonial, S.; Nooka, A.K. Long-Term Follow-Up Results of Lenalidomide, Bortezomib, and Dexamethasone Induction Therapy and Risk-Adapted Maintenance Approach in Newly Diagnosed Multiple Myeloma. J. Clin. Oncol. 2020, 38, 1928–1937. [Google Scholar] [CrossRef]
- Orlowski, R.Z.; Moreau, P.; Niesvizky, R.; Ludwig, H.; Oriol, A.; Chng, W.J.; Goldschmidt, H.; Yang, Z.; Kimball, A.S.; Dimopoulos, M. Carfilzomib-Dexamethasone Versus Bortezomib-Dexamethasone in Relapsed or Refractory Multiple Myeloma: Updated Overall Survival, Safety, and Subgroups. Clin. Lymphoma Myeloma Leuk. 2019, 19, 522–530.e1. [Google Scholar] [CrossRef] [PubMed]
- Naymagon, L.; Abdul-Hay, M. Novel agents in the treatment of multiple myeloma: A review about the future. J. Hematol. Oncol. 2016, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Richardson, P.G.; Anderson, K.C. Promising therapies in multiple myeloma. Blood 2015, 126, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Y.; Gu, H.; Dong, M.; Cai, Z. Emerging agents and regimens for multiple myeloma. J. Hematol. Oncol. 2020, 13, 150. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Treatment of multiple myeloma. Nat. Rev. Clin. Oncol. 2011, 8, 479–491. [Google Scholar] [CrossRef]
- Kang, C. Teclistamab: First Approval. Drugs 2022, 82, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Meng, F.; Zhong, Z. Emerging targeted drug delivery strategies toward ovarian cancer. Adv. Drug Deliv. Rev. 2021, 178, 113969. [Google Scholar] [CrossRef]
- Jiang, Y.; Lin, W.; Zhu, L. Targeted Drug Delivery for the Treatment of Blood Cancers. Molecules 2022, 27, 1310. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Torchilin, V.P. Drug targeting. Eur. J. Pharm. Sci. 2000, 11 (Suppl. S2), S81–S91. [Google Scholar] [CrossRef]
- Goldberg, H.A.; Warner, K.J.; Li, M.C.; Hunter, G.K. Binding of bone sialoprotein, osteopontin and synthetic polypeptides to hydroxyapatite. Connect. Tissue Res. 2001, 42, 25–37. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- Yu, B.; Liu, D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J. Hematol. Oncol. 2019, 12, 94. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Saito, G.; Swanson, J.A.; Lee, K.-D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: Role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 2003, 55, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Bounds, D.; Paterson, J.; Herledan, G.; Sully, K.; Seestaller-Wehr, L.M.; Fieles, W.E.; Tunstead, J.; McCahon, L.; Germaschewski, F.M.; et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br. J. Haematol. 2016, 174, 911–922. [Google Scholar] [CrossRef]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.; Lam, H.P.J.; Kirkham, Z.; Popat, R. Antibody drug conjugates for the treatment of multiple myeloma. Am. J. Hematol. 2023, 98 (Suppl. S2), S22–S34. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Hungria, V.T.M.; Radinoff, A.; Delimpasi, S.; Mikala, G.; Masszi, T.; Li, J.; Capra, M.; Maiolino, A.; Pappa, V.; et al. Efficacy and safety of single-agent belantamab mafodotin versus pomalidomide plus low-dose dexamethasone in patients with relapsed or refractory multiple myeloma (DREAMM-3): A phase 3, open-label, randomised study. Lancet Haematol. 2023, 10, e801–e812. [Google Scholar] [CrossRef]
- Hungria, V.; Robak, P.; Hus, M.; Zherebtsova, V.; Ward, C.; Ho, P.J.; Ribas de Almeida, A.C.; Hajek, R.; Kim, K.; Grosicki, S.; et al. Belantamab Mafodotin, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2024. ahead of print. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Beksac, M.; Pour, L.; Delimpasi, S.; Vorobyev, V.; Quach, H.; Spicka, I.; Radocha, J.; Robak, P.; Kim, K.; et al. Belantamab Mafodotin, Pomalidomide, and Dexamethasone in Multiple Myeloma. N. Engl. J. Med. 2024. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Vazquez, V.; Ko, J.; Breunig, C.; Baumann, A.; Giesen, N.; Pálfi, A.; Müller, C.; Lutz, C.; Hechler, T.; Kulke, M.; et al. HDP-101, an Anti-BCMA Antibody-Drug Conjugate, Safely Delivers Amanitin to Induce Cell Death in Proliferating and Resting Multiple Myeloma Cells. Mol. Cancer Ther. 2021, 20, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, C.D.; Larsson, K.-M.; Kornberg, R.D. The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by alpha-amanitin. Mol. Cell 2008, 30, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Kaufman, J.L.; Richard, S.; Grosicki, S.; Takacs, I.; Strassz, A.; Pahl, A.M.; Michael, T.; Last, A.; Szaboki, H.; et al. Hdp-101, an Anti-BCMA Antibody-Drug Conjugate with a Novel Payload Amanitin in Patients with Relapsed Multiple Myeloma, Initial Findings of the First in Human Study. Blood 2023, 142, 3334. [Google Scholar] [CrossRef]
- Kumar, S.K.; Migkou, M.; Bhutani, M.; Spencer, A.; Ailawadhi, S.; Kalff, A.; Walcott, F.; Pore, N.; Gibson, D.; Wang, F. Phase 1, first-in-human study of MEDI2228, a BCMA-targeted ADC in patients with relapsed/refractory multiple myeloma. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Xing, L.; Lin, L.; Yu, T.; Li, Y.; Cho, S.-F.; Liu, J.; Wen, K.; Hsieh, P.A.; Kinneer, K.; Munshi, N.; et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia 2020, 34, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Wang, S.; Liu, J.; Yu, T.; Chen, H.; Wen, K.; Li, Y.; Lin, L.; Hsieh, P.A.; Cho, S.-F.; et al. BCMA-Specific ADC MEDI2228 and Daratumumab Induce Synergistic Myeloma Cytotoxicity via IFN-Driven Immune Responses and Enhanced CD38 Expression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Cornell, R.F.; Landgren, O.; Ailawadhi, S.; Higgins, J.P.; Willert, E.K.; Waltzman, R.; Lin, J.; Zhang, Y.; Lublinsky, A.R. A phase 1 first-in-human study of the anti-CD38 dimeric fusion protein TAK-169 for the treatment of patients (pts) with relapsed or refractory multiple myeloma (RRMM) who are proteasome inhibitor (PI)-and immunomodulatory drug (IMiD)-refractory, including Pts relapsed/refractory (R/R) or naïve to daratumumab (dara). Blood 2019, 134, 1867. [Google Scholar]
- Vogl, D.T.; Kaufman, J.L.; Holstein, S.A.; Atrash, S.; Nadeem, O.; Janakiram, M.; Suryanarayan, K.; Liu, Y.; Collins, S.; Parot, X.; et al. Modakafusp Alfa (TAK-573), an Immunocytokine, Shows Clinical Activity in Patients with Relapsed/Refractory Multiple Myeloma; Updated Results from a First-in-Human Phase 1 Study. Blood 2021, 138, 898. [Google Scholar] [CrossRef]
- Stewart, A.K.; Krishnan, A.Y.; Singhal, S.; Boccia, R.V.; Patel, M.R.; Niesvizky, R.; Chanan-Khan, A.A.; Ailawadhi, S.; Brumm, J.; Mundt, K.E.; et al. Phase I study of the anti-FcRH5 antibody-drug conjugate DFRF4539A in relapsed or refractory multiple myeloma. Blood Cancer J. 2019, 9, 17. [Google Scholar] [CrossRef]
- Kelly, K.R.; Ailawadhi, S.; Siegel, D.S.; Heffner, L.T.; Somlo, G.; Jagannath, S.; Zimmerman, T.M.; Munshi, N.C.; Madan, S.; Chanan-Khan, A.; et al. Indatuximab ravtansine plus dexamethasone with lenalidomide or pomalidomide in relapsed or refractory multiple myeloma: A multicentre, phase 1/2a study. Lancet Haematol. 2021, 8, e794–e807. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Niesvizky, R.; Stadtmauer, E.A.; Chanan-Khan, A.; Siegel, D.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 163, 478–486. [Google Scholar] [CrossRef]
- Skaletskaya, A.; Setiady, Y.Y.; Park, P.U.; Lutz, R.J. Lorvotuzumab mertansine (IMGN901) immune effector activity and its effect on human NK cells. Cancer Res. 2011, 71, 770. [Google Scholar] [CrossRef]
- Sherbenou, D.W.; Aftab, B.T.; Su, Y.; Behrens, C.R.; Wiita, A.; Logan, A.C.; Acosta-Alvear, D.; Hann, B.C.; Walter, P.; Shuman, M.A.; et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J. Clin. Investig. 2016, 126, 4640–4653. [Google Scholar] [CrossRef]
- Johnson, M.; El-Khoueiry, A.; Hafez, N.; Lakhani, N.; Mamdani, H.; Rodon, J.; Sanborn, R.E.; Garcia-Corbacho, J.; Boni, V.; Stroh, M.; et al. Phase I, First-in-Human Study of the Probody Therapeutic CX-2029 in Adults with Advanced Solid Tumor Malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 4521–4530. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; O’ Donovan, D.H.; Ölwegård Halvarsson, M.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide-drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Dhillon, S. Melphalan Flufenamide (Melflufen): First Approval. Drugs 2021, 81, 963–969. [Google Scholar] [CrossRef]
- Chauhan, D.; Ray, A.; Viktorsson, K.; Spira, J.; Paba-Prada, C.; Munshi, N.; Richardson, P.; Lewensohn, R.; Anderson, K.C. In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells. Clin. Cancer Res. 2013, 19, 3019–3031. [Google Scholar] [CrossRef]
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. Melflufen and Dexamethasone in Heavily Pretreated Relapsed and Refractory Multiple Myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef]
- Flanagan, K.; Kumari, R.; Miettinen, J.J.; Haney, S.L.; Varney, M.L.; Williams, J.T.; Majumder, M.M.; Suvela, M.; Slipicevic, A.; Lehmann, F.; et al. The Peptide-Drug Conjugate Melflufen Modulates the Unfolded Protein Response of Multiple Myeloma and Amyloidogenic Plasma Cells and Induces Cell Death. Hemasphere 2022, 6, e687. [Google Scholar] [CrossRef]
- Gebraad, A.; Ohlsbom, R.; Miettinen, J.J.; Emeh, P.; Pakarinen, T.-K.; Manninen, M.; Eskelinen, A.; Kuismanen, K.; Slipicevic, A.; Lehmann, F.; et al. Growth Response and Differentiation of Bone Marrow-Derived Mesenchymal Stem/Stromal Cells in the Presence of Novel Multiple Myeloma Drug Melflufen. Cells 2022, 11, 1574. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Bringhen, S.; Voorhees, P.; Plesner, T.; Mellqvist, U.-H.; Reeves, B.; Paba-Prada, C.; Zubair, H.; Byrne, C.; Chauhan, D.; et al. Melflufen plus dexamethasone in relapsed and refractory multiple myeloma (O-12-M1): A multicentre, international, open-label, phase 1-2 study. Lancet Haematol. 2020, 7, e395–e407. [Google Scholar] [CrossRef] [PubMed]
- Schjesvold, F.H.; Dimopoulos, M.-A.; Delimpasi, S.; Robak, P.; Coriu, D.; Legiec, W.; Pour, L.; Špička, I.; Masszi, T.; Doronin, V.; et al. Melflufen or pomalidomide plus dexamethasone for patients with multiple myeloma refractory to lenalidomide (OCEAN): A randomised, head-to-head, open-label, phase 3 study. Lancet Haematol. 2022, 9, e98–e110. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Efebera, Y.A.; Hájek, R.; Straub, J.; Maisnar, V.; Eveillard, J.-R.; Karlin, L.; Mateos, M.-V.; Oriol, A.; Ribrag, V.; et al. ANCHOR: Melflufen plus dexamethasone and daratumumab or bortezomib in relapsed/refractory multiple myeloma: Final results of a phase I/IIa study. Haematologica 2024, 109, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Pour, L.; Szarejko, M.; Bila, J.; Schjesvold, F.H.; Spicka, I.; Maisnar, V.; Jurczyszyn, A.; Grudeva-Popova, Z.; Hájek, R.; Usenko, G.; et al. Efficacy and safety of melflufen plus daratumumab and dexamethasone in relapsed/refractory multiple myeloma: Results from the randomized, open-label, phase III LIGHTHOUSE study. Haematologica 2024, 109, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, A.D.; Davies, D.R.; Janjic, N. Embracing proteins: Structural themes in aptamer-protein complexes. Curr. Opin. Struct. Biol. 2016, 36, 122–132. [Google Scholar] [CrossRef]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T.; Feinsod, M.; Guyer, D.R. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef]
- Catuogno, S.; Di Martino, M.T.; Nuzzo, S.; Esposito, C.L.; Tassone, P.; de Franciscis, V. An Anti-BCMA RNA Aptamer for miRNA Intracellular Delivery. Mol. Ther. Nucleic Acids 2019, 18, 981–990. [Google Scholar] [CrossRef]
- Wen, J.; Tao, W.; Hao, S.; Iyer, S.P.; Zu, Y. A unique aptamer-drug conjugate for targeted therapy of multiple myeloma. Leukemia 2016, 30, 987–991. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, H.; Zhou, W.; Sun, S.; Zeng, Y.; Zhang, H.; Liang, L.; Xiao, X.; Song, J.; Ye, M.; et al. Targeting c-met receptor tyrosine kinase by the DNA aptamer SL1 as a potential novel therapeutic option for myeloma. J. Cell. Mol. Med. 2018, 22, 5978–5990. [Google Scholar] [CrossRef]
- Seckinger, A.; Meissner, T.; Moreaux, J.; Depeweg, D.; Hillengass, J.; Hose, K.; Rème, T.; Rösen-Wolff, A.; Jauch, A.; Schnettler, R.; et al. Clinical and prognostic role of annexin A2 in multiple myeloma. Blood 2012, 120, 1087–1094. [Google Scholar] [CrossRef]
- D’Souza, S.; Kurihara, N.; Shiozawa, Y.; Joseph, J.; Taichman, R.; Galson, D.L.; Roodman, G.D. Annexin II interactions with the annexin II receptor enhance multiple myeloma cell adhesion and growth in the bone marrow microenvironment. Blood 2012, 119, 1888–1896. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, Y.; Zeng, Y.; Peng, M.; Li, H.; Sun, S.; Ma, B.; Wang, Y.; Ye, M.; Liu, J. Screening and characterization of an Annexin A2 binding aptamer that inhibits the proliferation of myeloma cells. Biochimie 2018, 151, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Paiva, B.; Shi, J.; Park, J.; Manier, S.; Takagi, S.; Massoud, M.; Perilla-Glen, A.; Aljawai, Y.; Huynh, D.; et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep. 2017, 19, 218–224. [Google Scholar] [CrossRef]
- de la Puente, P.; Azab, A.K. Nanoparticle delivery systems, general approaches, and their implementation in multiple myeloma. Eur. J. Haematol. 2017, 98, 529–541. [Google Scholar] [CrossRef]
- Guimarães, D.; Cavaco-Paulo, A.; Nogueira, E. Design of liposomes as drug delivery system for therapeutic applications. Int. J. Pharm. 2021, 601, 120571. [Google Scholar] [CrossRef] [PubMed]
- Cibeira, M.T.; Rozman, M.; Segarra, M.; Lozano, E.; Rosiñol, L.; Cid, M.C.; Filella, X.; Bladé, J. Bone marrow angiogenesis and angiogenic factors in multiple myeloma treated with novel agents. Cytokine 2008, 41, 244–253. [Google Scholar] [CrossRef]
- Fan, F.; Malvestiti, S.; Vallet, S.; Lind, J.; Garcia-Manteiga, J.M.; Morelli, E.; Jiang, Q.; Seckinger, A.; Hose, D.; Goldschmidt, H.; et al. JunB is a key regulator of multiple myeloma bone marrow angiogenesis. Leukemia 2021, 35, 3509–3525. [Google Scholar] [CrossRef]
- Lum, B.L.; Svec, J.M.; Torti, F.M. Doxorubicin: Alteration of dose scheduling as a means of reducing cardiotoxicity. Drug Intell. Clin. Pharm. 1985, 19, 259–264. [Google Scholar] [CrossRef]
- Kanwal, U.; Irfan Bukhari, N.; Ovais, M.; Abass, N.; Hussain, K.; Raza, A. Advances in nano-delivery systems for doxorubicin: An updated insight. J. Drug Target. 2018, 26, 296–310. [Google Scholar] [CrossRef]
- Li, M.; Shi, F.; Fei, X.; Wu, S.; Wu, D.; Pan, M.; Luo, S.; Gu, N.; Dou, J. PEGylated long-circulating liposomes deliver homoharringtonine to suppress multiple myeloma cancer stem cells. Exp. Biol. Med. 2017, 242, 996–1004. [Google Scholar] [CrossRef]
- Deshantri, A.K.; Fens, M.H.; Ruiter, R.W.J.; Metselaar, J.M.; Storm, G.; van Bloois, L.; Varela-Moreira, A.; Mandhane, S.N.; Mutis, T.; Martens, A.C.M.; et al. Liposomal dexamethasone inhibits tumor growth in an advanced human-mouse hybrid model of multiple myeloma. J. Control. Release 2019, 296, 232–240. [Google Scholar] [CrossRef]
- Metselaar, J.; Lammers, T.; Boquoi, A.; Fenk, R.; Testaquadra, F.; Schemionek, M.; Kiessling, F.; Isfort, S.; Wilop, S.; Crysandt, M. A phase I first-in-man study to investigate the pharmacokinetics and safety of liposomal dexamethasone in patients with progressive multiple myeloma. Drug Deliv. Transl. Res. 2023, 13, 915–923. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, T.; Gao, J. Apoptotic Vesicles of MSCs: The Natural Therapeutic Agents and Bio-Vehicles for Targeting Drug Delivery. Small 2023, 19, e2301671. [Google Scholar] [CrossRef]
- Cao, Z.; Li, P.; Li, Y.; Zhang, M.; Hao, M.; Li, W.; Mao, X.; Mo, L.; Yang, C.; Ding, X.; et al. Encapsulation of Nano-Bortezomib in Apoptotic Stem Cell-Derived Vesicles for the Treatment of Multiple Myeloma. Small 2023, 19, e2301748. [Google Scholar] [CrossRef]
- Omstead, D.T.; Mejia, F.; Sjoerdsma, J.; Kim, B.; Shin, J.; Khan, S.; Wu, J.; Kiziltepe, T.; Littlepage, L.E.; Bilgicer, B. In vivo evaluation of CD38 and CD138 as targets for nanoparticle-based drug delivery in multiple myeloma. J. Hematol. Oncol. 2020, 13, 145. [Google Scholar] [CrossRef]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Alhallak, K.; Sun, J.; Wasden, K.; Guenthner, N.; O’Neal, J.; Muz, B.; King, J.; Kohnen, D.; Vij, R.; Achilefu, S.; et al. Nanoparticle T-cell engagers as a modular platform for cancer immunotherapy. Leukemia 2021, 35, 2346–2357. [Google Scholar] [CrossRef]
- Bae, J.; Parayath, N.; Ma, W.; Amiji, M.; Munshi, N.; Anderson, K.C. BCMA peptide-engineered nanoparticles enhance induction and function of antigen-specific CD8(+) cytotoxic T lymphocytes against multiple myeloma: Clinical applications. Leukemia 2020, 34, 210–223. [Google Scholar] [CrossRef]
- Hari, S.K.; Gauba, A.; Shrivastava, N.; Tripathi, R.M.; Jain, S.K.; Pandey, A.K. Polymeric micelles and cancer therapy: An ingenious multimodal tumor-targeted drug delivery system. Drug Deliv. Transl. Res. 2023, 13, 135–163. [Google Scholar] [CrossRef]
- Chen, R.; Yang, J.; Mao, Y.; Zhao, X.; Cheng, R.; Deng, C.; Zhong, Z. Antibody-Mediated Nanodrug of Proteasome Inhibitor Carfilzomib Boosts the Treatment of Multiple Myeloma. Biomacromolecules 2023, 24, 5371–5380. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef]
- Smadja-Joffe, F.; Legras, S.; Girard, N.; Li, Y.; Delpech, B.; Bloget, F.; Morimoto, K.; Le Bousse-Kerdiles, C.; Clay, D.; Jasmin, C.; et al. CD44 and hyaluronan binding by human myeloid cells. Leuk. Lymphoma 1996, 21, 407–420, color plates following 528. [Google Scholar] [CrossRef]
- Carvalho, A.M.; Teixeira, R.; Novoa-Carballal, R.; Pires, R.A.; Reis, R.L.; Pashkuleva, I. Redox-Responsive Micellar Nanoparticles from Glycosaminoglycans for CD44 Targeted Drug Delivery. Biomacromolecules 2018, 19, 2991–2999. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.; Cheng, R.; Zhong, Z. A6 Peptide-Tagged Core-Disulfide-Cross-Linked Micelles for Targeted Delivery of Proteasome Inhibitor Carfilzomib to Multiple Myeloma In Vivo. Biomacromolecules 2020, 21, 2049–2059. [Google Scholar] [CrossRef]
- Gu, Z. Targeted Bortezomib Therapy of Multiple Myeloma by Hyaluronic Acid-Shelled and Core-Disulfide-Crosslinked Micelles. Master’s Thesis, Soochow University, Suzhou, China, 2018. [Google Scholar]
- Gu, Z.; Wang, X.; Cheng, R.; Cheng, L.; Zhong, Z. Hyaluronic acid shell and disulfide-crosslinked core micelles for in vivo targeted delivery of bortezomib for the treatment of multiple myeloma. Acta Biomater. 2018, 80, 288–295. [Google Scholar] [CrossRef]
- Piotrowicz, R.S.; Damaj, B.B.; Hachicha, M.; Incardona, F.; Howell, S.B.; Finlayson, M. A6 peptide activates CD44 adhesive activity, induces FAK and MEK phosphorylation, and inhibits the migration and metastasis of CD44-expressing cells. Mol. Cancer Ther. 2011, 10, 2072–2082. [Google Scholar] [CrossRef]
- Kortuem, K.M.; Stewart, A.K. Carfilzomib. Blood 2013, 121, 893–897. [Google Scholar] [CrossRef]
- Georgoulis, V.; Haidich, A.B.; Bougioukas, K.I.; Hatzimichael, E. Efficacy and safety of carfilzomib for the treatment of multiple myeloma: An overview of systematic reviews. Crit. Rev. Oncol. Hematol. 2022, 180, 103842. [Google Scholar] [CrossRef]
- Varela-Moreira, A.; van Straten, D.; van Leur, H.F.; Ruiter, R.W.J.; Deshantri, A.K.; Hennink, W.E.; Fens, M.; Groen, R.W.J.; Schiffelers, R.M. Polymeric micelles loaded with carfilzomib increase tolerability in a humanized bone marrow-like scaffold mouse model. Int. J. Pharm. X 2020, 2, 100049. [Google Scholar] [CrossRef]
- Wang, F.; Younis, M.; Luo, Y.; Zhang, L.; Yuan, L. Iguratimod-encapsulating PLGA-NPs induce human multiple myeloma cell death via reactive oxygen species and Caspase-dependent signalling. Int. Immunopharmacol. 2021, 95, 107532. [Google Scholar] [CrossRef]
- Yu, N.; Zhang, Y.; Li, J.; Gu, W.; Yue, S.; Li, B.; Meng, F.; Sun, H.; Haag, R.; Yuan, J.; et al. Daratumumab Immunopolymersome-Enabled Safe and CD38-Targeted Chemotherapy and Depletion of Multiple Myeloma. Adv. Mater. 2021, 33, e2007787. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Qu, Y.; Chu, B.; Wei, X.; Chen, Y.; Yang, Y.; Hu, D.; Huang, J.; Wang, F.; Chen, M.; Zheng, Y.; et al. Cancer-Cell-Biomimetic Nanoparticles for Targeted Therapy of Multiple Myeloma Based on Bone Marrow Homing. Adv. Mater. 2022, 34, e2107883. [Google Scholar] [CrossRef]
- Rajasagi, M.; Vitacolonna, M.; Benjak, B.; Marhaba, R.; Zöller, M. CD44 promotes progenitor homing into the thymus and T cell maturation. J. Leukoc. Biol. 2009, 85, 251–261. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, Z.; Zhao, J.J.; Calimeri, T.; Meng, J.; Hideshima, T.; Fulciniti, M.; Kang, Y.; Ficarro, S.B.; Tai, Y.T.; et al. The Cyclophilin A-CD147 complex promotes the proliferation and homing of multiple myeloma cells. Nat. Med. 2015, 21, 572–580. [Google Scholar] [CrossRef]
- Cui, H.Y.; Wang, S.J.; Miao, J.Y.; Fu, Z.G.; Feng, F.; Wu, J.; Yang, X.M.; Chen, Z.N.; Jiang, J.L. CD147 regulates cancer migration via direct interaction with Annexin A2 and DOCK3-β-catenin-WAVE2 signaling. Oncotarget 2016, 7, 5613–5629. [Google Scholar] [CrossRef]
- Guimarães, P.P.G.; Figueroa-Espada, C.G.; Riley, R.S.; Gong, N.; Xue, L.; Sewastianik, T.; Dennis, P.S.; Loebel, C.; Chung, A.; Shepherd, S.J.; et al. In vivo bone marrow microenvironment siRNA delivery using lipid-polymer nanoparticles for multiple myeloma therapy. Proc. Natl. Acad. Sci. USA 2023, 120, e2215711120. [Google Scholar] [CrossRef]
- Figueroa-Espada, C.G.; Guimarães, P.P.G.; Riley, R.S.; Xue, L.; Wang, K.; Mitchell, M.J. siRNA Lipid-Polymer Nanoparticles Targeting E-Selectin and Cyclophilin A in Bone Marrow for Combination Multiple Myeloma Therapy. Cell. Mol. Bioeng. 2023, 16, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Leone, E.; Amodio, N.; Foresta, U.; Lionetti, M.; Pitari, M.R.; Cantafio, M.E.; Gullà, A.; Conforti, F.; Morelli, E.; et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: In vitro and in vivo evidence. Clin. Cancer Res. 2012, 18, 6260–6270. [Google Scholar] [CrossRef] [PubMed]
- Zarone, M.R.; Misso, G.; Grimaldi, A.; Zappavigna, S.; Russo, M.; Amler, E.; Di Martino, M.T.; Amodio, N.; Tagliaferri, P.; Tassone, P.; et al. Evidence of novel miR-34a-based therapeutic approaches for multiple myeloma treatment. Sci. Rep. 2017, 7, 17949. [Google Scholar] [CrossRef]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutiérrez, N.C. Molecular Mechanisms of p53 Deregulation in Cancer: An Overview in Multiple Myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef]
- Cosco, D.; Cilurzo, F.; Maiuolo, J.; Federico, C.; Di Martino, M.T.; Cristiano, M.C.; Tassone, P.; Fresta, M.; Paolino, D. Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Sci. Rep. 2015, 5, 17579. [Google Scholar] [CrossRef]
- Dutta, D.; Liu, J.; Wen, K.; Kurata, K.; Fulciniti, M.; Gulla, A.; Hideshima, T.; Anderson, K.C. BCMA-targeted bortezomib nanotherapy improves therapeutic efficacy, overcomes resistance, and modulates the immune microenvironment in multiple myeloma. Blood Cancer J. 2023, 13, 184. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Liu, H.; Shen, H.; Meng, N.; Qi, X.; Ding, K.; Song, J.; Fu, R.; Ding, D.; et al. BSA-AIE Nanoparticles with Boosted ROS Generation for Immunogenic Cell Death Immunotherapy of Multiple Myeloma. Adv. Mater. 2023, 35, e2208692. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- El-Shershaby, H.M.; Farrag, N.S.; Ebeid, N.H.; Moustafa, K.A. Radiolabeling and cytotoxicity of monoclonal antibody Isatuximab functionalized silver nanoparticles on the growth of multiple myeloma. Int. J. Pharm. 2022, 624, 122019. [Google Scholar] [CrossRef] [PubMed]
- Reimer, P.; Weissleder, R.; Lee, A.S.; Wittenberg, J.; Brady, T.J. Receptor imaging: Application to MR imaging of liver cancer. Radiology 1990, 177, 729–734. [Google Scholar] [CrossRef]
- Smith, B.; Li, Y.; Fields, T.; Tucker, M.; Staskiewicz, A.; Wong, E.; Ma, H.; Mao, H.; Wang, X. Tumor integrin targeted theranostic iron oxide nanoparticles for delivery of caffeic acid phenethyl ester: Preparation, characterization, and anti-myeloma activities. Front. Pharmacol. 2024, 15, 1325196. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, J.; Chen, D.; Chen, J.; Xiong, F.; Zhang, H.; Zhang, Y.; Gu, N.; Dou, J. Paclitaxel-Fe3O4 nanoparticles inhibit growth of CD138− CD34− tumor stem-like cells in multiple myeloma-bearing mice. Int. J. Nanomed. 2013, 8, 1439–1449. [Google Scholar] [CrossRef]
- Zhang, W.; Qiao, L.; Wang, X.; Senthilkumar, R.; Wang, F.; Chen, B. Inducing cell cycle arrest and apoptosis by dimercaptosuccinic acid modified Fe3O4 magnetic nanoparticles combined with nontoxic concentration of bortezomib and gambogic acid in RPMI-8226 cells. Int. J. Nanomed. 2015, 10, 3275–3289. [Google Scholar] [CrossRef]
- Takamatsu, Y.; Sunami, K.; Hata, H.; Nagafuji, K.; Choi, I.; Higuchi, M.; Uozumi, K.; Masaki, Y.; Tamura, K. A phase I study of bortezomib in combination with doxorubicin and intermediate-dose dexamethasone (iPAD therapy) for relapsed or refractory multiple myeloma. Int. J. Hematol. 2010, 92, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, Y.; Sunami, K.; Muta, T.; Morimoto, H.; Miyamoto, T.; Higuchi, M.; Uozumi, K.; Hata, H.; Tamura, K. Bortezomib, doxorubicin and intermediate-dose dexamethasone (iPAD) therapy for relapsed or refractory multiple myeloma: A multicenter phase 2 study. Int. J. Hematol. 2013, 98, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Pfister, F.; Dörrie, J.; Schaft, N.; Buchele, V.; Unterweger, H.; Carnell, L.R.; Schreier, P.; Stein, R.; Kubánková, M.; Guck, J.; et al. Human T cells loaded with superparamagnetic iron oxide nanoparticles retain antigen-specific TCR functionality. Front. Immunol. 2023, 14, 1223695. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, D.; Yin, X.; Ding, S.; Shen, M.; Zhang, R.; Wang, Y.; Xu, R. Zinc oxide nanoparticles induce human multiple myeloma cell death via reactive oxygen species and Cyt-C/Apaf-1/Caspase-9/Caspase-3 signaling pathway in vitro. Biomed. Pharmacother. 2020, 122, 109712. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yin, X.; Lyu, C.; Wang, J.; Liu, K.; Cui, S.; Ding, S.; Wang, Y.; Wang, J.; Guo, D.; et al. Zinc Oxide Nanoparticles Trigger Autophagy in the Human Multiple Myeloma Cell Line RPMI8226: An In Vitro Study. Biol. Trace Elem. Res. 2024, 202, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Revia, R.A.; Stephen, Z.R.; Zhang, M. Theranostic Nanoparticles for RNA-Based Cancer Treatment. Acc. Chem. Res. 2019, 52, 1496–1506. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Patra, C.R.; Verma, R.; Kumar, S.; Greipp, P.R.; Mukherjee, P. Gold Nanoparticles Inhibit the Proliferation of Multiple Myeloma Cells. Adv. Mater. 2007, 19, 711–716. [Google Scholar] [CrossRef]
- Masters, J.C.; Nickens, D.J.; Xuan, D.; Shazer, R.L.; Amantea, M. Clinical toxicity of antibody drug conjugates: A meta-analysis of payloads. Investig. New Drugs 2018, 36, 121–135. [Google Scholar] [CrossRef]
- Terpos, E.; Kleber, M.; Engelhardt, M.; Zweegman, S.; Gay, F.; Kastritis, E.; van de Donk, N.W.C.J.; Bruno, B.; Sezer, O.; Broijl, A.; et al. European Myeloma Network guidelines for the management of multiple myeloma-related complications. Haematologica 2015, 100, 1254–1266. [Google Scholar] [CrossRef]
- Blayney, D.W.; Schwartzberg, L. Chemotherapy-induced neutropenia and emerging agents for prevention and treatment: A review. Cancer Treat. Rev. 2022, 109, 102427. [Google Scholar] [CrossRef]
- Segal, B.H.; Freifeld, A.G. Antibacterial prophylaxis in patients with neutropenia. J. Natl. Compr. Canc Netw. 2007, 5, 235–242. [Google Scholar] [CrossRef]
- Baaten, C.C.F.M.J.; Moenen, F.C.J.I.; Henskens, Y.M.C.; Swieringa, F.; Wetzels, R.J.H.; van Oerle, R.; Heijnen, H.F.G.; Ten Cate, H.; Holloway, G.P.; Beckers, E.A.M.; et al. Impaired mitochondrial activity explains platelet dysfunction in thrombocytopenic cancer patients undergoing chemotherapy. Haematologica 2018, 103, 1557–1567. [Google Scholar] [CrossRef]
- Vadhan-Raj, S. Management of chemotherapy-induced thrombocytopenia: Current status of thrombopoietic agents. Semin. Hematol. 2009, 46 (Suppl. S2), S26–S32. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Nooka, A.K.; Thulasi, P.; Badros, A.Z.; Jeng, B.H.; Callander, N.S.; Potter, H.A.; Sborov, D.; Zaugg, B.E.; Popat, R.; et al. Management of belantamab mafodotin-associated corneal events in patients with relapsed or refractory multiple myeloma (RRMM). Blood Cancer J. 2021, 11, 103. [Google Scholar] [CrossRef]
- Farooq, A.V.; Degli Esposti, S.; Popat, R.; Thulasi, P.; Lonial, S.; Nooka, A.K.; Jakubowiak, A.; Sborov, D.; Zaugg, B.E.; Badros, A.Z.; et al. Corneal Epithelial Findings in Patients with Multiple Myeloma Treated with Antibody-Drug Conjugate Belantamab Mafodotin in the Pivotal, Randomized, DREAMM-2 Study. Ophthalmol. Ther. 2020, 9, 889–911. [Google Scholar] [CrossRef]
- Munawar, U.; Theuersbacher, J.; Steinhardt, M.J.; Zhou, X.; Han, S.; Nerreter, S.; Vogt, C.; Kurian, S.; Keller, T.; Regensburger, A.-K.; et al. Soluble B-cell maturation antigen in lacrimal fluid as a potential biomarker and mediator of keratopathy in multiple myeloma. Haematologica 2024. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Ailawadhi, S.; Kelly, K.R.; Vescio, R.A.; Jagannath, S.; Wolf, J.; Gharibo, M.; Sher, T.; Bojanini, L.; Kirby, M.; Chanan-Khan, A. A Phase I Study to Assess the Safety and Pharmacokinetics of Single-agent Lorvotuzumab Mertansine (IMGN901) in Patients with Relapsed and/or Refractory CD-56-positive Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Best, R.L.; LaPointe, N.E.; Azarenko, O.; Miller, H.; Genualdi, C.; Chih, S.; Shen, B.-Q.; Jordan, M.A.; Wilson, L.; Feinstein, S.C.; et al. Microtubule and tubulin binding and regulation of microtubule dynamics by the antibody drug conjugate (ADC) payload, monomethyl auristatin E (MMAE): Mechanistic insights into MMAE ADC peripheral neuropathy. Toxicol. Appl. Pharmacol. 2021, 421, 115534. [Google Scholar] [CrossRef]
- Kågedal, M.; Samineni, D.; Gillespie, W.R.; Lu, D.; Fine, B.M.; Girish, S.; Li, C.; Jin, J.Y. Time-to-Event Modeling of Peripheral Neuropathy: Platform Analysis of Eight Valine-Citrulline-Monomethylauristatin E Antibody-Drug Conjugates. CPT Pharmacometrics Syst. Pharmacol. 2019, 8, 606–615. [Google Scholar] [CrossRef]
- Fu, Z.; Gao, C.; Wu, T.; Wang, L.; Li, S.; Zhang, Y.; Shi, C. Peripheral neuropathy associated with monomethyl auristatin E-based antibody-drug conjugates. iScience 2023, 26, 107778. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.R.; Aronson, J.K. Adverse drug reactions: Definitions, diagnosis, and management. Lancet 2000, 356, 1255–1259. [Google Scholar] [CrossRef]
- Roselló, S.; Blasco, I.; Fabregat, L.G.; Cervantes, A.; Jordan, K.; Committee, E.G. Management of infusion reactions to systemic anticancer therapy: ESMO Clinical Practice Guidelines. Ann. Oncol. 2017, 28, iv100–iv118. [Google Scholar] [CrossRef] [PubMed]
- Vultaggio, A.; Castells, M.C. Hypersensitivity reactions to biologic agents. Immunol. Allergy Clin. N. Am. 2014, 34, 615–632. [Google Scholar] [CrossRef]
- Barroso, A.; Estevinho, F.; Hespanhol, V.; Teixeira, E.; Ramalho-Carvalho, J.; Araújo, A. Management of infusion-related reactions in cancer therapy: Strategies and challenges. ESMO Open 2024, 9, 102922. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyak, A.; Shokeen, M.; Achilefu, S. Nanotherapeutics for multiple myeloma. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1526. [Google Scholar] [CrossRef]
- Iannazzo, D.; Ettari, R.; Giofrè, S.; Eid, A.H.; Bitto, A. Recent Advances in Nanotherapeutics for Multiple Myeloma. Cancers 2020, 12, 3144. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
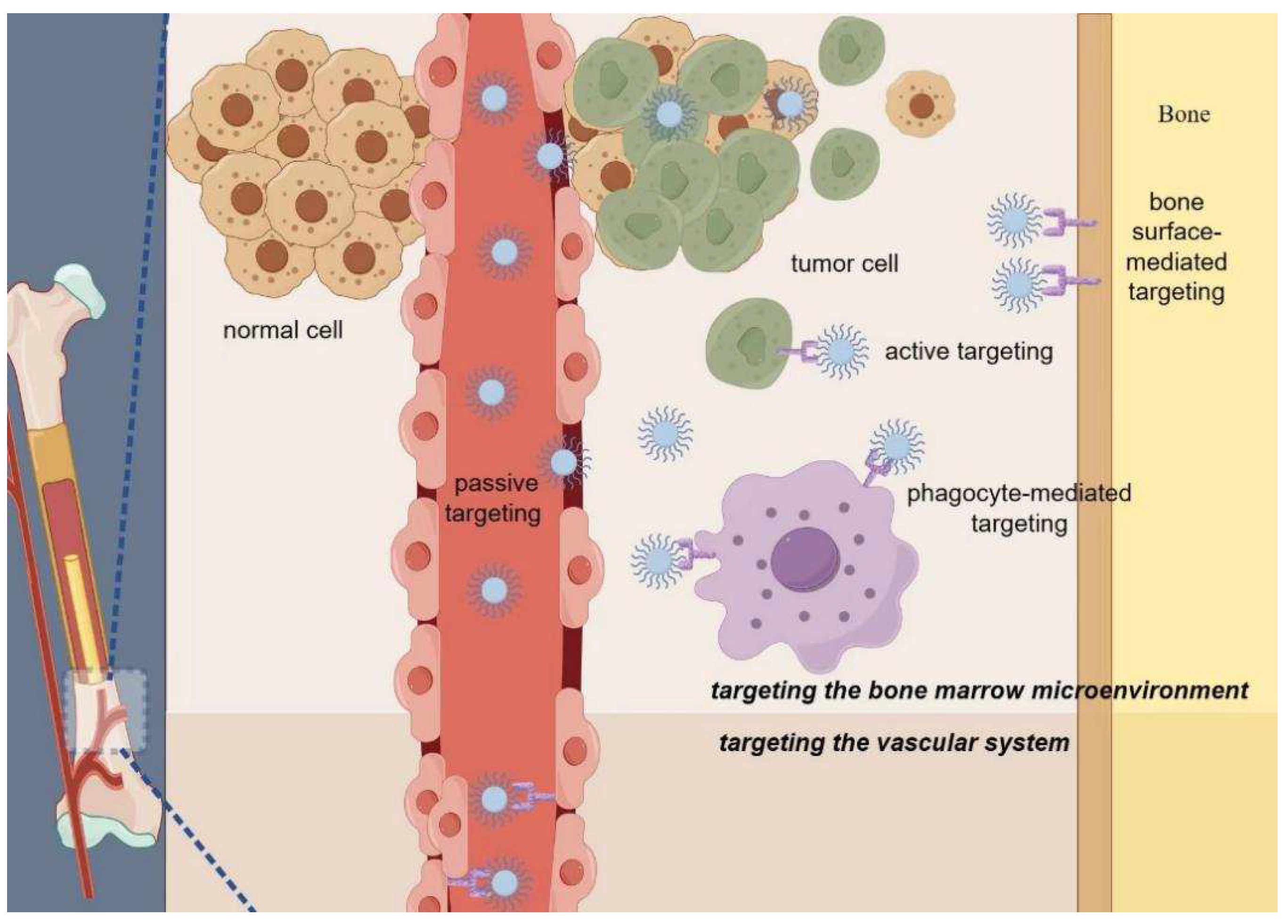
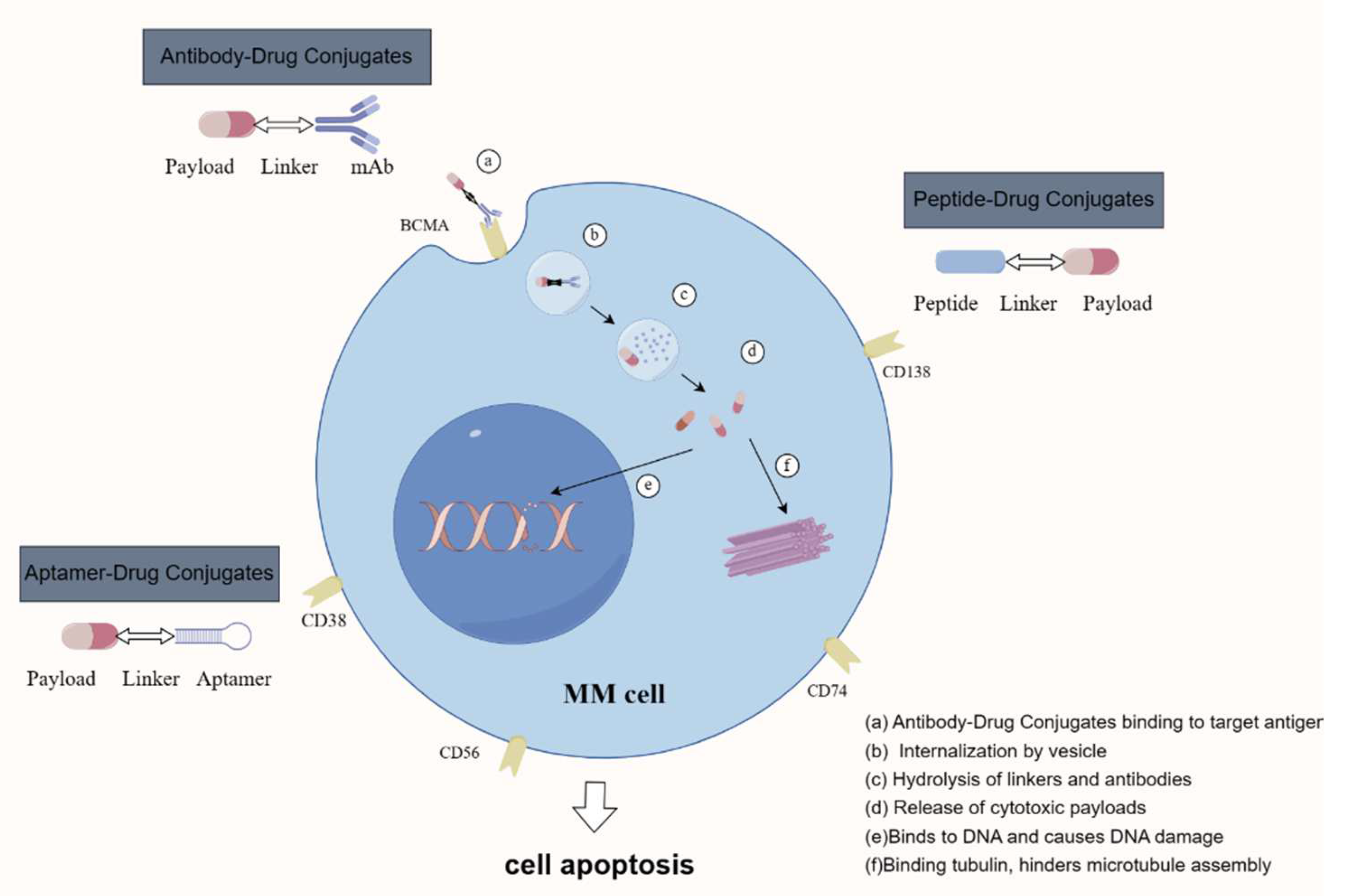
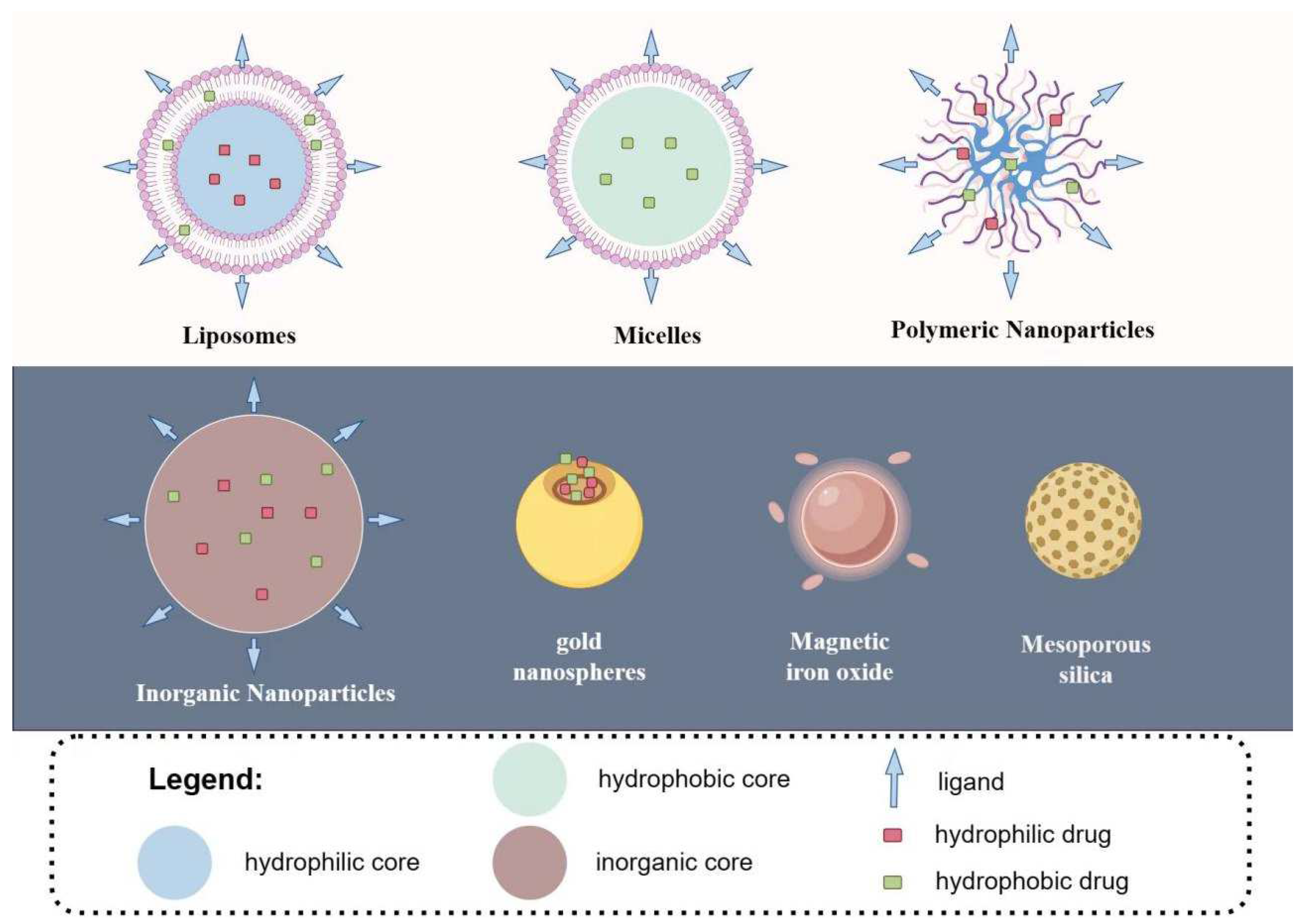

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC | Target | Payload | Linker | Mechanism of Action | Adverse Event | Clinical Status |
|---|---|---|---|---|---|---|
| TAK-169 | CD38 | Shiga-like toxin-A subunit (SLTA) | / | Enzymatic inactivation of cell ribosomes [38] | Myocarditis | Phase 1 (NCT04017130, terminated) |
| TAK-573 (Modakafusp Alfa) | CD38 | attenuated interferon alpha-2b (IFNα2b) | / | Direct anti-proliferative effects, and induce immune cell activation [39] | Infusion-related reactions (IRR), Hematologic toxicity | Phase 1/2 (NCT05590377, active, not recruiting) |
| DFRF4539A | FcRH5 | MMAE | protease-labile linker (MC-VC-PABC) | Disruption of microtubule networks [40] | Anemia, fatigue, peripheral neuropathy | Phase 1 (NCT01432353, completed) |
| Indatuximab ravtansine (BT062) | CD138 | DM4 | disulphide bonds (SPDB) | Disruption of microtubule networks [41] | Diarrhea and fatigue | Phase 1/2a (NCT01638936, completed) |
| Milatuzumab doxorubicin (hLL1-DOX) | CD74 | doxorubicin | acid-labile linker (hydrazone) | Preventing DNA replication and increasing double-strand breaks [42] | / | Phase 1/2 (NCT01101594, terminated) |
| Lorvotuzumab mertansine (IMGN901) | CD56 | DM1 | Disulphide bonds (SPP) | Disruption of microtubule networks [43] | peripheral neuropathy | Phase 1 (NCT00991562, completed) |
| FOR46 | CD46 | MMAE | protease-cleavable linker (mcvcpab) | Disruption of microtubule networks [44] | / | Phase 1 (NCT03650491, completed) |
| CX-2029 | CD71 | MMAE | protease-cleavable linker | Disruption of microtubule networks [45] | Hematologic toxicity, IRR | Preclinical |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Wang, H.; Xiong, S.; Liu, J.; Sun, S. Targeted Delivery Strategies for Multiple Myeloma and Their Adverse Drug Reactions. Pharmaceuticals 2024, 17, 832. https://doi.org/10.3390/ph17070832
Li S, Wang H, Xiong S, Liu J, Sun S. Targeted Delivery Strategies for Multiple Myeloma and Their Adverse Drug Reactions. Pharmaceuticals. 2024; 17(7):832. https://doi.org/10.3390/ph17070832
Chicago/Turabian StyleLi, Shuting, Hongjie Wang, Shijun Xiong, Jing Liu, and Shuming Sun. 2024. "Targeted Delivery Strategies for Multiple Myeloma and Their Adverse Drug Reactions" Pharmaceuticals 17, no. 7: 832. https://doi.org/10.3390/ph17070832
APA StyleLi, S., Wang, H., Xiong, S., Liu, J., & Sun, S. (2024). Targeted Delivery Strategies for Multiple Myeloma and Their Adverse Drug Reactions. Pharmaceuticals, 17(7), 832. https://doi.org/10.3390/ph17070832