
Use of Extracellular Monomeric Ubiquitin as a Therapeutic Option for Major Depressive Disorder

 , ,
, ,  , , ,
, , ,  , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Neuroimmunoendocrine and Inflammation Alterations in MDD
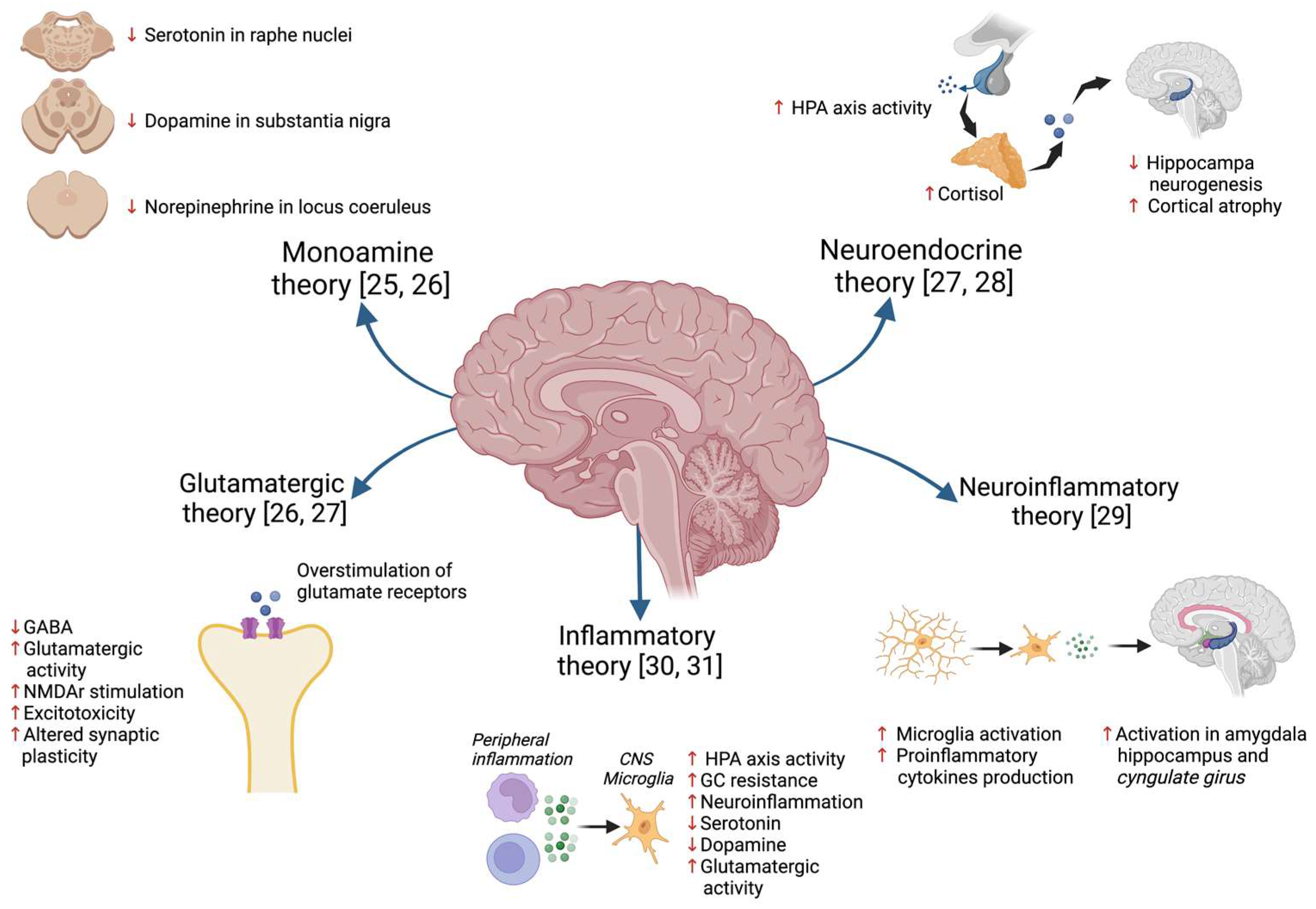
2.1. Hypothesis of MDD Pathophysiology
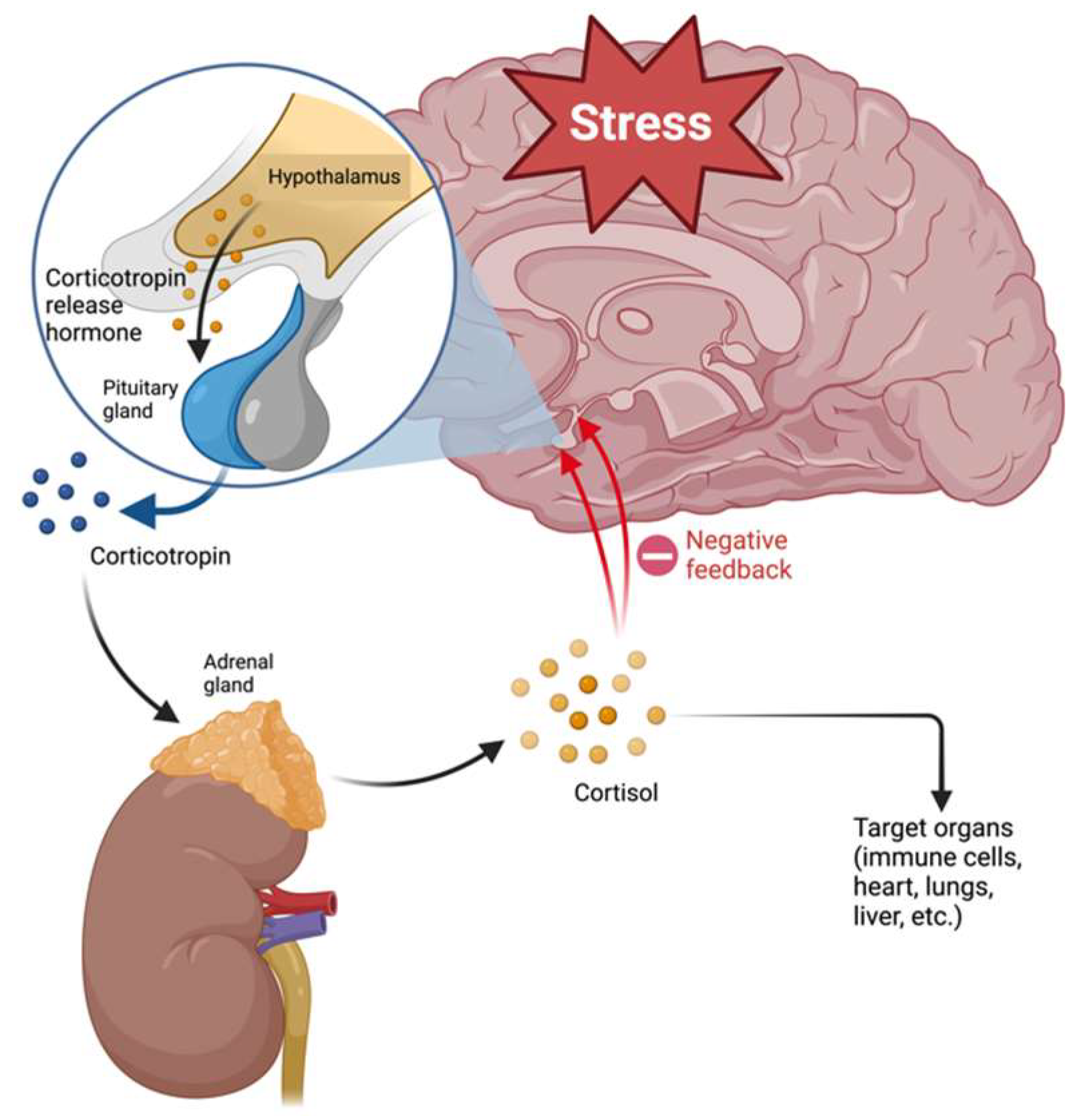
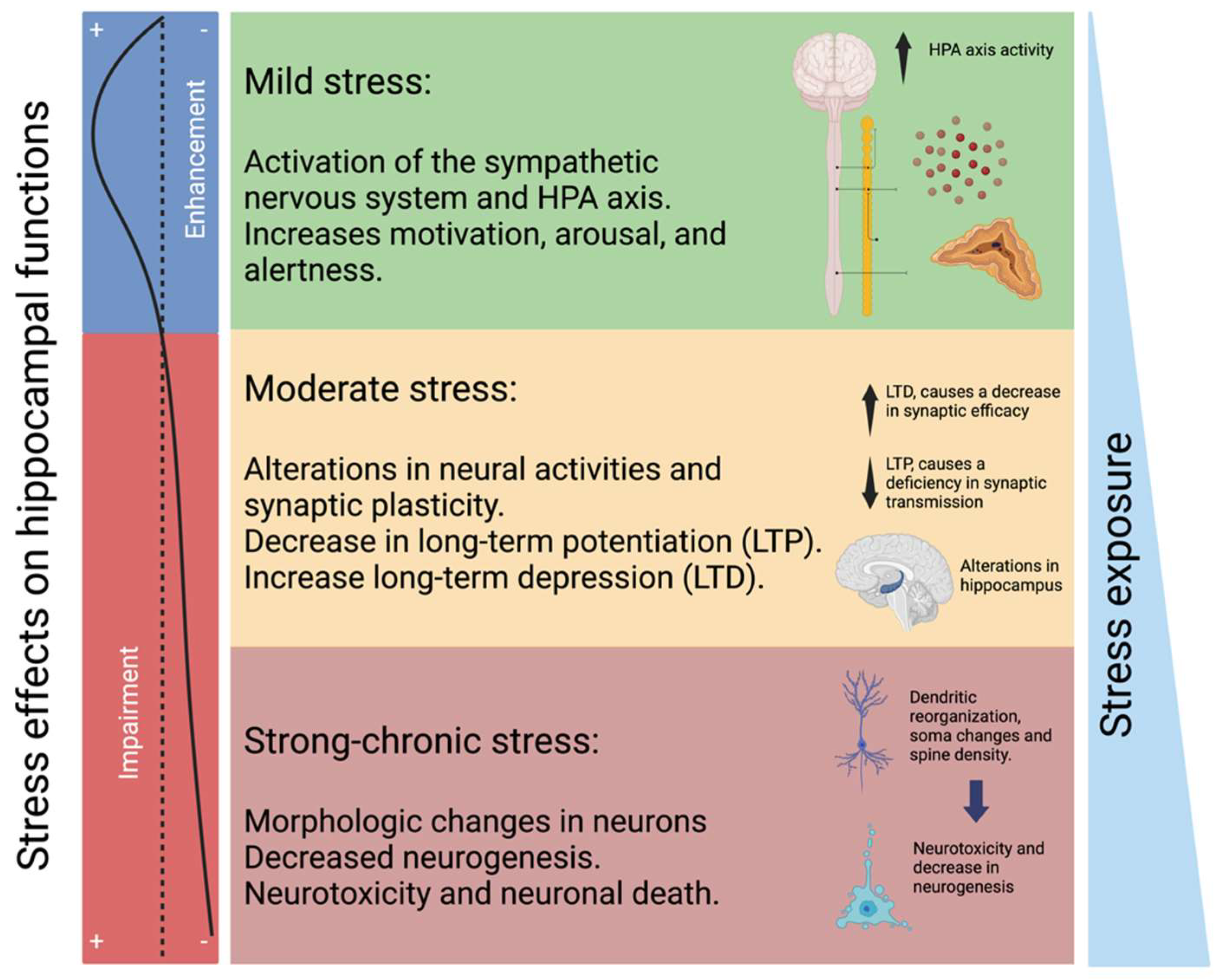
2.2. HPA Axis and Glucocorticoid Resistance
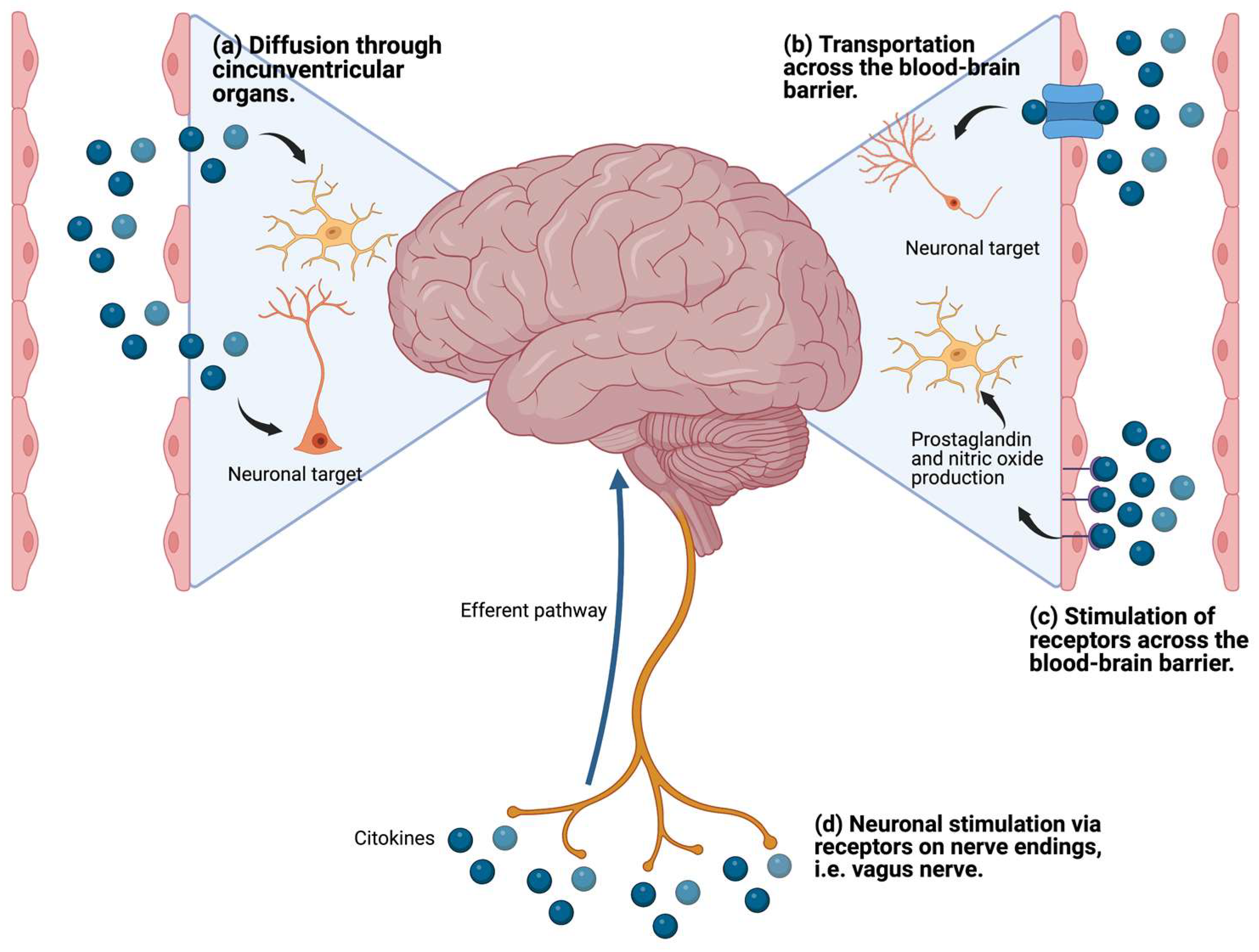
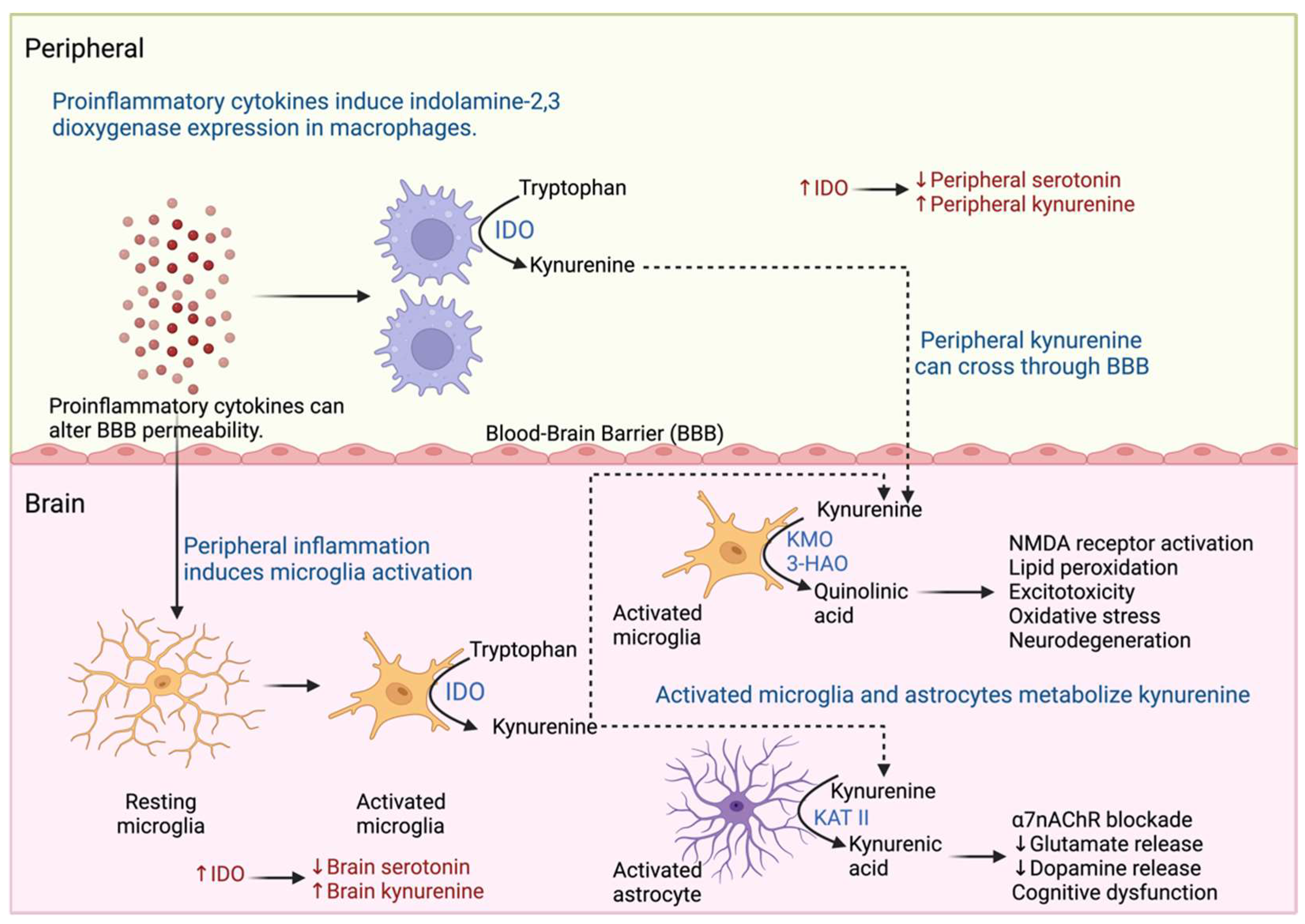
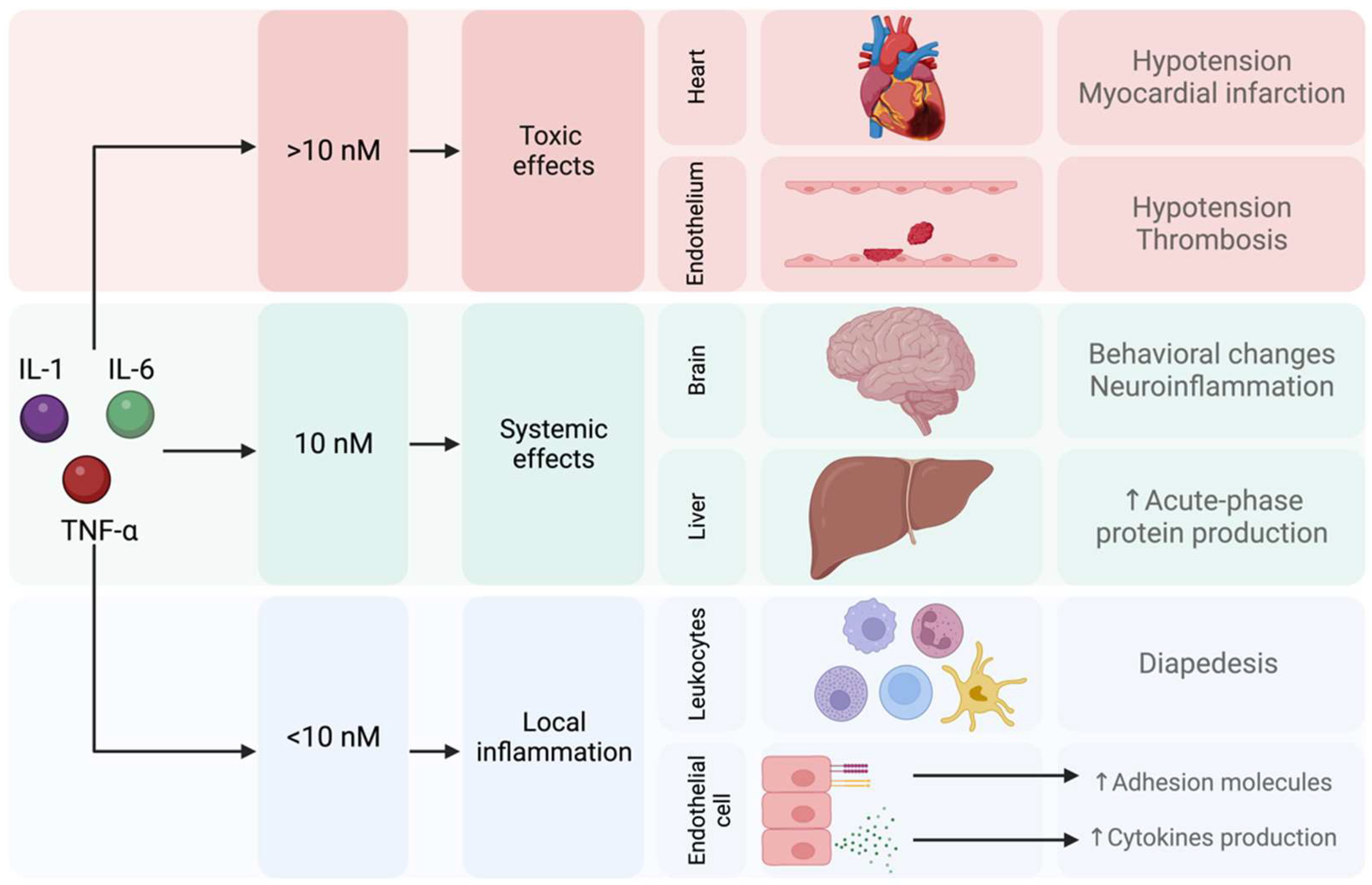
2.3. Proinflammatory Cytokines and Neurotransmitter Metabolism
3. Inflammation as a Therapeutic Target for MDD
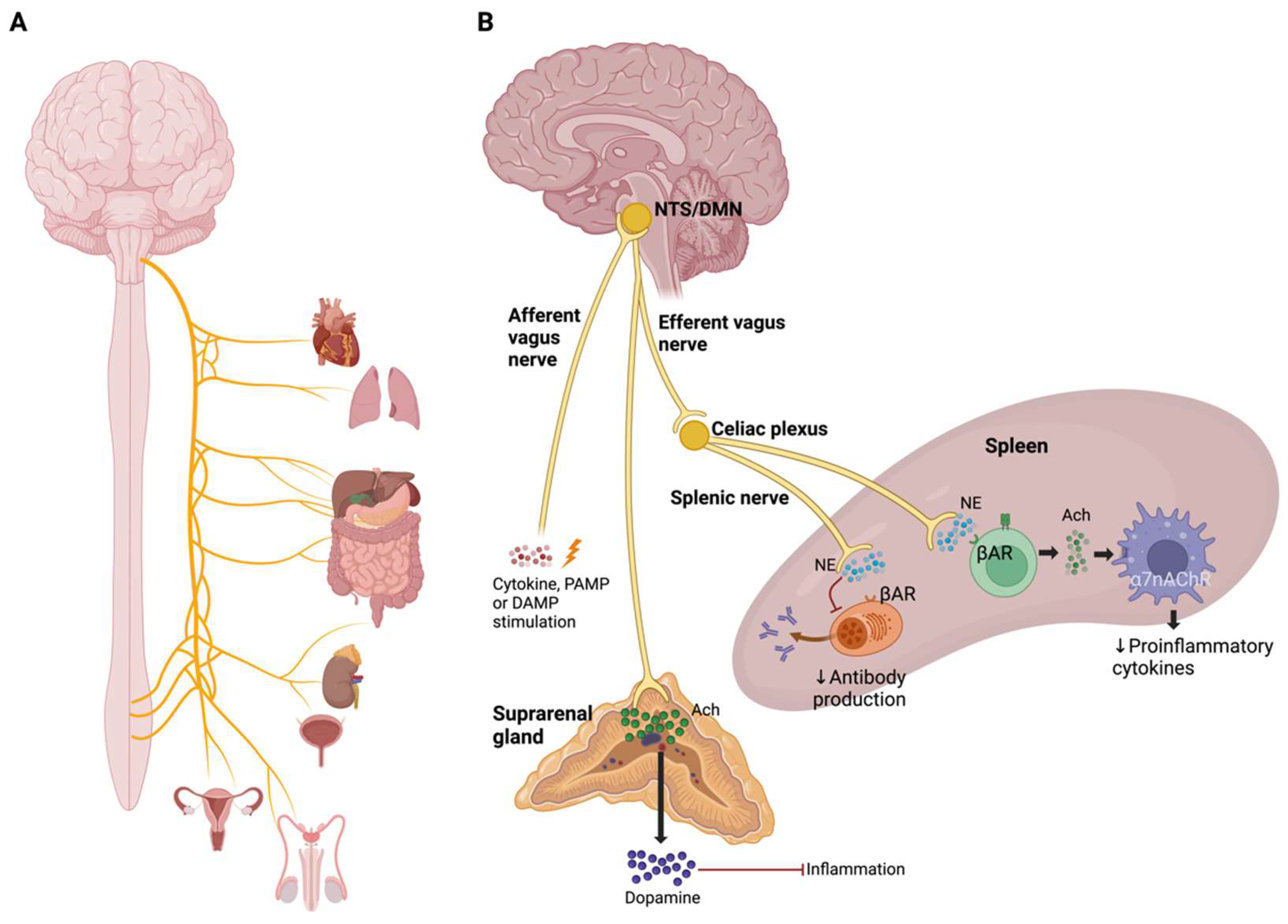
3.1. Cholinergic Anti-Inflammatory Pathway and Inflammatory Reflex
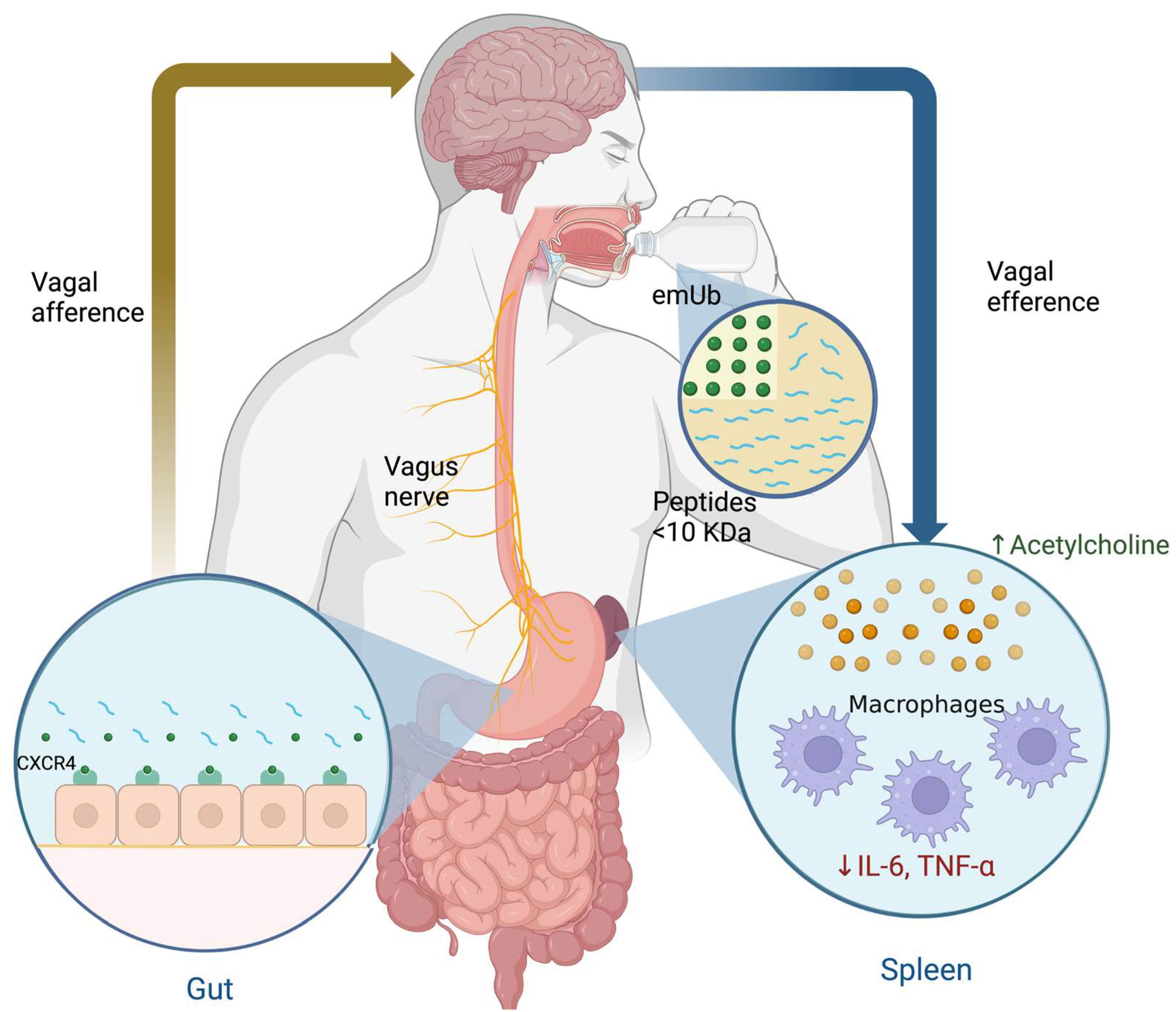
3.2. Extracellular Monomeric Ubiquitin (emUb)
4. Use of emUb as a Part of the Peptide Composition of the hDLE in MDD Treatment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marx, W.; Penninx, B.W.J.H.; Solmi, M.; Furukawa, T.A.; Firth, J.; Carvalho, A.F.; Berk, M. Major Depressive Disorder. Nat. Rev. Dis. Prim. 2023, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major Depressive Disorder. Nat. Rev. Dis. Prim. 2016, 2, 16065. [Google Scholar] [CrossRef] [PubMed]
- Lopizzo, N.; Chiavetto, L.B.; Cattane, N.; Plazzotta, G.; Tarazi, F.I.; Pariante, C.M.; Riva, M.A.; Cattaneo, A. Gene–Environment Interaction in Major Depression: Focus on Experience-Dependent Biological Systems. Front. Psychiatry 2015, 6, 68. [Google Scholar] [CrossRef]
- Moussavi, S.; Chatterji, S.; Verdes, E.; Tandon, A.; Patel, V.; Ustun, B. Depression, Chronic Diseases, and Decrements in Health: Results from the World Health Surveys. Lancet 2007, 370, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.K.; Barton, D.A. Depression and the Link with Cardiovascular Disease. Front. Psychiatry 2016, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Al-Khatib, Y.; Akhtar, M.A.; Kanawati, M.A.; Mucheke, R.; Mahfouz, M.; Al-Nufoury, M. Depression and Metabolic Syndrome: A Narrative Review. Cureus 2022, 14, e22153. [Google Scholar] [CrossRef] [PubMed]
- Sanches, A.; Costa, R.; Marcondes, F.K.; Cunha, T.S. Relationship among Stress, Depression, Cardiovascular and Metabolic Changes and Physical Exercise. Fisioter. Mov. 2016, 29, 23–36. [Google Scholar] [CrossRef]
- Ferrari, A. Global, Regional, and National Burden of 12 Mental Disorders in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Psychiatry 2022, 9, 137–150. [Google Scholar] [CrossRef]
- James, J.E. Mental Health. In The Health of Populations; Elsevier: Amsterdam, The Netherlands, 2016; pp. 429–464. ISBN 978-0-12-802812-4. [Google Scholar]
- Greenberg, P.E.; Fournier, A.A.; Sisitsky, T.; Simes, M.; Berman, R.; Koenigsberg, S.H.; Kessler, R.C. The Economic Burden of Adults with Major Depressive Disorder in the United States (2010 and 2018). Pharmacoeconomics 2021, 39, 653–665. [Google Scholar] [CrossRef]
- Bermudes, R.A. Rapid Response Therapies for Major Depression Could Dramatically Reduce Societal Cost. Psychiatr. News 2023, 58. [Google Scholar] [CrossRef]
- Karrouri, R.; Hammani, Z.; Otheman, Y.; Benjelloun, R. Major Depressive Disorder: Validated Treatments and Future Challenges. World J. Clin. Cases 2021, 9, 9350. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.S. Major Depressive Disorder; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780323581318. [Google Scholar]
- Sheffler, Z.M.; Patel, P.; Abdijadid, S. Antidepressants; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Sinyor, M.; Schaffer, A.; Levitt, A. The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Trial: A Review. Can. J. Psychiatry 2010, 55, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Gaynes, B.N.; Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Spencer, D.; Fava, M. The STAR*D Study: Treating Depression in the Real World. Cleve. Clin. J. Med. 2008, 75, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.H. A Review of Antidepressant Therapy in Primary Care: Current Practices and Future Directions. Prim. Care Companion J. Clin. Psychiatry 2013, 15, 23071. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.S.; Zizzi, F.B.; Cattaneo, A.; Comandini, A.; Di Dato, G.; Lubrano, E.; Pellicano, C.; Spallone, V.; Tongiani, S.; Torta, R. Management and Treatment of Patients With Major Depressive Disorder and Chronic Diseases: A Multidisciplinary Approach. Front. Psychol. 2020, 11, 542444. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Khullar, S.; Singh, M.; Kaur, G.; Mastana, S. Diabetes to Cardiovascular Disease: Is Depression the Potential Missing Link? Med. Hypotheses 2015, 84, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, N. Depression and Diabetes. Dialogues Clin. Neurosci. 2018, 20, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Tafet, G.E.; Nemeroff, C.B. The Links Between Stress and Depression: Psychoneuroendocrinological, Genetic, and Environmental Interactions. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 77–88. [Google Scholar] [CrossRef]
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural Basis of Major Depressive Disorder: Beyond Monoamine Hypothesis. Psychiatry Clin. Neurosci. 2018, 72, 3–12. [Google Scholar] [CrossRef]
- Seki, K.; Yoshida, S.; Jaiswal, M. Molecular Mechanism of Noradrenaline during the Stress-Induced Major Depressive Disorder. Neural Regen. Res. 2018, 13, 1159–1169. [Google Scholar] [CrossRef]
- Bruno, A.; Dolcetti, E.; Rizzo, F.R.; Fresegna, D.; Musella, A.; Gentile, A.; De Vito, F.; Caioli, S.; Guadalupi, L.; Bullitta, S.; et al. Inflammation-Associated Synaptic Alterations as Shared Threads in Depression and Multiple Sclerosis. Front. Cell. Neurosci. 2020, 14, 169. [Google Scholar] [CrossRef] [PubMed]
- Cosci, F.; Chouinard, G. The Monoamine Hypothesis of Depression Revisited: Could It Mechanistically Novel Antidepressant Strategies? In Neurobiology of Depression; Elsevier: Amsterdam, The Netherlands, 2019; pp. 63–73. ISBN 9780128133330. [Google Scholar]
- Musazzi, L.; Treccani, G.; Popoli, M. Glutamate Hypothesis of Depression and Its Consequences for Antidepressant Treatments. Expert Rev. Neurother. 2012, 12, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Onaolapo, A.Y.; Onaolapo, O.J. Glutamate and Depression: Reflecting a Deepening Knowledge of the Gut and Brain Effects of a Ubiquitous Molecule. World J. Psychiatry 2021, 11, 297. [Google Scholar] [CrossRef] [PubMed]
- Holsboer, F. Stress, Hypercortisolism and Corticosteroid Receptors in Depression: Implicatons for Therapy. J. Affect. Disord. 2001, 62, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Afridi, R.; Suk, K. Neuroinflammatory Basis of Depression: Learning From Experimental Models. Front. Cell. Neurosci. 2021, 15, 691067. [Google Scholar] [CrossRef] [PubMed]
- Alesci, S.; Martinez, P.E.; Kelkar, S.; Ilias, I.; Ronsaville, D.S.; Listwak, S.J.; Ayala, A.R.; Licinio, J.; Gold, H.K.; Kling, M.A.; et al. Major Depression Is Associated with Significant Diurnal Elevations in Plasma Interleukin-6 Levels, a Shift of Its Circadian Rhythm, and Loss of Physiological Complexity in Its Secretion: Clinical Implications. J. Clin. Endocrinol. Metab. 2005, 90, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Gałecki, P.; Talarowska, M. Inflammatory Theory of Depression. Psychiatr. Pol. 2018, 52, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.C.; Su, Y.P.; Su, K.P.; Chen, P.C. Recurrence of Depressive Disorders after Interferon-Induced Depression. Transl. Psychiatry 2017, 7, e1026. [Google Scholar] [CrossRef]
- Amodio, P.; De Toni, E.N.; Cavalletto, L.; Mapelli, D.; Bernardinello, E.; Del Piccolo, F.; Bergamelli, C.; Costanzo, R.; Bergamaschi, F.; Poma, S.Z.; et al. Mood, Cognition and EEG Changes during Interferon Alpha (Alpha-IFN) Treatment for Chronic Hepatitis C. J. Affect. Disord. 2005, 84, 93–98. [Google Scholar] [CrossRef]
- Salvador, A.F.; de Lima, K.A.; Kipnis, J. Neuromodulation by the Immune System: A Focus on Cytokines. Nat. Rev. Immunol. 2021, 21, 526–541. [Google Scholar] [CrossRef]
- Brebner, K.; Hayley, S.; Zacharko, R.; Merali, Z.; Anisman, H. Synergistic Effects of Interleukin-1beta, Interleukin-6, and Tumor Necrosis Factor-Alpha: Central Monoamine, Corticosterone, and Behavioral Variations. Neuropsychopharmacology 2000, 22, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Yabut, J.M.; Crane, J.D.; Green, A.E.; Keating, D.J.; Khan, W.I.; Steinberg, G.R. Emerging Roles for Serotonin in Regulating Metabolism: New Implications for an Ancient Molecule. Endocr. Rev. 2019, 40, 1092–1107. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From Inflammation to Sickness and Depression: When the Immune System Subjugates the Brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R. Cytokine, Sickness Behavior, and Depression. Immunol. Allergy Clin. N. Am. 2009, 29, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Capuron, L.; Miller, A.H. Cytokines Sing the Blues: Inflammation and the Pathogenesis of Depression. Trends Immunol. 2006, 27, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Cardani, A.N.; Yang, C.H.; Quinnies, K.M.; Crihfield, A.; Lynch, K.R.; Kipnis, J. Regulation of Learning and Memory by Meningeal Immunity: A Key Role for IL-4. J. Exp. Med. 2010, 207, 1067. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Brigas, H.C.; Temido-Ferreira, M.; Pousinha, P.A.; Regen, T.; Santa, C.; Coelho, J.E.; Marques-Morgado, I.; Valente, C.A.; Omenetti, S.; et al. Meningeal Γδ T Cell-Derived IL-17 Controls Synaptic Plasticity and Short-Term Memory. Sci. Immunol. 2019, 4, eaay5199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Rong, P.; Zhang, L.; He, H.; Zhou, T.; Fan, Y.; Mo, L.; Zhao, Q.; Han, Y.; Li, S.; et al. IL4-Driven Microglia Modulate Stress Resilience through BDNF-Dependent Neurogenesis. Sci. Adv. 2021, 7, 9888–9905. [Google Scholar] [CrossRef]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The Maternal Interleukin-17a Pathway in Mice Promotes Autism-like Phenotypes in Offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef]
- Filiano, A.J.; Xu, Y.; Tustison, N.J.; Marsh, R.L.; Baker, W.; Smirnov, I.; Overall, C.C.; Gadani, S.P.; Turner, S.D.; Weng, Z.; et al. Unexpected Role of Interferon-γ in Regulating Neuronal Connectivity and Social Behaviour. Nature 2016, 535, 425–429. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Hawksworth, O.A.; Woodruff, T.M. Complement: The Emerging Architect of the Developing Brain. Trends Neurosci. 2018, 41, 373–384. [Google Scholar] [CrossRef]
- Westacott, L.J.; Humby, T.; Haan, N.; Brain, S.A.; Bush, E.L.; Toneva, M.; Baloc, A.I.; Moon, A.L.; Reddaway, J.; Owen, M.J.; et al. Complement C3 and C3aR Mediate Different Aspects of Emotional Behaviours; Relevance to Risk for Psychiatric Disorder. Brain Behav. Immun. 2022, 99, 70–82. [Google Scholar] [CrossRef]
- Allswede, D.M.; Zheutlin, A.B.; Chung, Y.; Anderson, K.; Hultman, C.M.; Ingvar, M.; Cannon, T.D. Complement Gene Expression Correlates with Superior Frontal Cortical Thickness in Humans. Neuropsychopharmacology 2018, 43, 525–533. [Google Scholar] [CrossRef]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia Risk from Complex Variation of Complement Component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef]
- Mikulska, J.; Juszczyk, G.; Gawrońska-Grzywacz, M.; Herbet, M. Hpa Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation. Brain Sci. 2021, 11, 1298. [Google Scholar] [CrossRef]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603. [Google Scholar] [CrossRef]
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M.; Schatzberg, A.F. HPA Axis in Major Depression: Cortisol, Clinical Symptomatology, and Genetic Variation Predict Cognition. Mol. Psychiatry 2017, 22, 527. [Google Scholar] [CrossRef]
- Gomez, R.G.; Fleming, S.H.; Keller, J.; Flores, B.; Kenna, H.; DeBattista, C.; Solvason, B.; Schatzberg, A.F. The Neuropsychological Profile of Psychotic Major Depression and Its Relation to Cortisol. Biol. Psychiatry 2006, 60, 472–478. [Google Scholar] [CrossRef]
- Lupien, S.J.; Gillin, C.J.; Hauger, R.L. Working Memory Is More Sensitive than Declarative Memory to the Acute Effects of Corticosteroids: A Dose-Response Study in Humans. Behav. Neurosci. 1999, 113, 420–430. [Google Scholar] [CrossRef]
- Rock, P.L.; Roiser, J.P.; Riedel, W.J.; Blackwell, A.D. Cognitive Impairment in Depression: A Systematic Review and Meta-Analysis. Psychol. Med. 2014, 44, 2029–2040. [Google Scholar] [CrossRef]
- Kim, E.J.; Kim, J.J. Neurocognitive Effects of Stress: A Metaparadigm Perspective. Mol. Psychiatry 2023, 28, 2750–2763. [Google Scholar] [CrossRef]
- Kim, E.J.; Pellman, B.; Kim, J.J. Stress Effects on the Hippocampus: A Critical Review. Learn. Mem. 2015, 22, 411. [Google Scholar] [CrossRef]
- Pariante, C.M. Why Are Depressed Patients Inflamed? A Reflection on 20 Years of Research on Depression, Glucocorticoid Resistance and Inflammation. Eur. Neuropsychopharmacol. 2017, 27, 554–559. [Google Scholar] [CrossRef]
- Silverman, M.N.; Sternberg, E.M. Glucocorticoid Regulation of Inflammation and Its Functional Correlates: From HPA Axis to Glucocorticoid Receptor Dysfunction. Ann. N. Y. Acad. Sci. 2012, 1261, 55–63. [Google Scholar] [CrossRef]
- Cohen, S.; Janicki-Deverts, D.; Doyle, W.J.; Miller, G.E.; Frank, E.; Rabin, B.S.; Turner, R.B. Chronic Stress, Glucocorticoid Receptor Resistance, Inflammation, and Disease Risk. Proc. Natl. Acad. Sci. USA 2012, 109, 5995–5999. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Carvalho, L.A.; Pariante, C.M. Glucocorticoids, Cytokines and Brain Abnormalities in Depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 722–729. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The Anti-Inflammatory and Immunosuppressive Effects of Glucocorticoids, Recent Developments and Mechanistic Insights. Mol. Cell. Endocrinol. 2011, 335, 2. [Google Scholar] [CrossRef]
- Perrin, A.J.; Horowitz, M.A.; Roelofs, J.; Zunszain, P.A.; Pariante, C.M. Glucocorticoid Resistance: Is It a Requisite for Increased Cytokine Production in Depression? A Systematic Review and Meta-Analysis. Front. Psychiatry 2019, 10, 423. [Google Scholar] [CrossRef]
- Weber, M.D.; Godbout, J.P.; Sheridan, J.F. Repeated Social Defeat, Neuroinflammation, and Behavior: Monocytes Carry the Signal. Neuropsychopharmacology 2017, 42, 46–61. [Google Scholar] [CrossRef]
- Irwin, M.R.; Miller, A.H. Depressive Disorders and Immunity: 20 Years of Progress and Discovery. Brain Behav. Immun. 2007, 21, 374–383. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Monsalves-Alvarez, M.; Henriquez, S.; Llanos, M.N.; Troncoso, R. Glucocorticoid Resistance in Chronic Diseases. Steroids 2016, 115, 182–192. [Google Scholar] [CrossRef]
- Hernández, M.E.; Mendieta, D.; Martínez-Fong, D.; Loría, F.; Moreno, J.; Estrada, I.; Bojalil, R.; Pavón, L. Variations in Circulating Cytokine Levels during 52 Week Course of Treatment with SSRI for Major Depressive Disorder. Eur. Neuropsychopharmacol. 2008, 18, 917–924. [Google Scholar] [CrossRef]
- Himmerich, H.; Patsalos, O.; Lichtblau, N.; Ibrahim, M.A.A.; Dalton, B. Cytokine Research in Depression: Principles, Challenges, and Open Questions. Front. Psychiatry 2019, 10, 30. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine Networks in Neuroinflammation. Nat. Rev. Immunol. 2016, 17, 49–59. [Google Scholar] [CrossRef]
- Bauer, M.E. Accelerated Immunosenescence in Rheumatoid Arthritis: Impact on Clinical Progression. Immun. Ageing 2020, 17, 6. [Google Scholar] [CrossRef]
- Roohi, E.; Jaafari, N.; Hashemian, F. On Inflammatory Hypothesis of Depression: What Is the Role of IL-6 in the Middle of the Chaos? J. Neuroinflamm. 2021, 18, 45. [Google Scholar] [CrossRef]
- Corrigan, M.; O’Rourke, A.M.; Moran, B.; Fletcher, J.M.; Harkin, A. Inflammation in the Pathogenesis of Depression: A Disorder of Neuroimmune Origin. Neuronal Signal. 2023, 7, 20220054. [Google Scholar] [CrossRef]
- Herselman, M.F.; Bailey, S.; Bobrovskaya, L. The Effects of Stress and Diet on the “Brain–Gut” and “Gut–Brain” Pathways in Animal Models of Stress and Depression. Int. J. Mol. Sci. 2022, 23, 2013. [Google Scholar] [CrossRef]
- Miller, A.H. Mechanisms of Cytokine-Induced Behavioral Changes: Psychoneuroimmunology at the Translational Interface. Brain Behav. Immun. 2009, 23, 149–158. [Google Scholar] [CrossRef]
- Huang, Y.S.; Ogbechi, J.; Clanchy, F.I.; Williams, R.O.; Stone, T.W. IDO and Kynurenine Metabolites in Peripheral and CNS Disorders. Front. Immunol. 2020, 11, 493984. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology Meets Neuropsychopharmacology: Translational Implications of the Impact of Inflammation on Behavior. Neuropsychopharmacology 2012, 37, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Mithaiwala, M.N.; Santana-Coelho, D.; Porter, G.A.; O’connor, J.C. Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells 2021, 10, 1548. [Google Scholar] [CrossRef] [PubMed]
- Becerril-Villanueva, E.; Olvera-Alvarez, M.I.; Alvarez-Herrera, S.; Maldonado-García, J.L.; López-Torres, A.; Ramírez-Marroquín, O.A.; González-Ruiz, O.; Nogueira-Fernández, J.M.; Mendoza-Contreras, J.M.; Sánchez-García, H.O.; et al. Screening of SERT and P11 MRNA Levels in Airline Pilots: A Translational Approach. Front. Psychiatry 2022, 13, 859768. [Google Scholar] [CrossRef] [PubMed]
- Vancassel, S.; Capuron, L.; Castanon, N. Brain Kynurenine and BH4 Pathways: Relevance to the Pathophysiology and Treatment of Inflammation-Driven Depressive Symptoms. Front. Neurosci. 2018, 12, 499. [Google Scholar] [CrossRef] [PubMed]
- Hassamal, S. Chronic Stress, Neuroinflammation, and Depression: An Overview of Pathophysiological Mechanisms and Emerging Anti-Inflammatories. Front. Psychiatry 2023, 14, 1130989. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Toups, M.; Nemeroff, C.B. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron 2020, 107, 234–256. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of Depression with C-Reactive Protein, IL-1, and IL-6: A Meta-Analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef]
- Liu, Y.; Ho, R.C.M.; Mak, A. Interleukin (IL)-6, Tumour Necrosis Factor Alpha (TNF-α) and Soluble Interleukin-2 Receptors (SIL-2R) Are Elevated in Patients with Major Depressive Disorder: A Meta-Analysis and Meta-Regression. J. Affect. Disord. 2012, 139, 230–239. [Google Scholar] [CrossRef]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; de Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral Cytokine and Chemokine Alterations in Depression: A Meta-Analysis of 82 Studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Osimo, E.F.; Pillinger, T.; Rodriguez, I.M.; Khandaker, G.M.; Pariante, C.M.; Howes, O.D. Inflammatory Markers in Depression: A Meta-Analysis of Mean Differences and Variability in 5166 Patients and 5083 Controls. Brain Behav. Immun. 2020, 87, 901. [Google Scholar] [CrossRef]
- Min, X.; Wang, G.; Cui, Y.; Meng, P.; Hu, X.; Liu, S.; Wang, Y. Association between Inflammatory Cytokines and Symptoms of Major Depressive Disorder in Adults. Front. Immunol. 2023, 14, 1110775. [Google Scholar] [CrossRef] [PubMed]
- Harsanyi, S.; Kupcova, I.; Danisovic, L.; Klein, M. Selected Biomarkers of Depression: What Are the Effects of Cytokines and Inflammation? Int. J. Mol. Sci. 2023, 24, 578. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Halbreich, U.; Han, C.; Leonard, B.E.; Luo, H. Imbalance between Pro- and Anti-Inflammatory Cytokines, and between Th1 and Th2 Cytokines in Depressed Patients: The Effect of Electroacupuncture or Fluoxetine Treatment. Pharmacopsychiatry 2009, 42, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Leonard, B.E.; Steinbusch, H.W.M.; Kim, Y.K. Th1, Th2, and Th3 Cytokine Alterations in Major Depression. J. Affect. Disord. 2005, 88, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yu, K. Th1, Th2, and Th17 Cells and Their Corresponding Cytokines Are Associated with Anxiety, Depression, and Cognitive Impairment in Elderly Gastric Cancer Patients. Front. Surg. 2022, 9, 996680. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Fahimi, A. Immune and Neuroprotective Effects of Physical Activity on the Brain in Depression. Front. Neurosci. 2018, 12, 498. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Tu, H.; Chen, T. The Microbiota–Gut–Brain Axis in Depression: The Potential Pathophysiological Mechanisms and Microbiota Combined Antidepression Effect. Nutrients 2022, 14, 2081. [Google Scholar] [CrossRef]
- Chang, L.; Wei, Y.; Hashimoto, K. Brain–Gut–Microbiota Axis in Depression: A Historical Overview and Future Directions. Brain Res. Bull. 2022, 182, 44–56. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13, 2099. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rodriguez, E.M.; Watson, J.; Reyes, J.; Trivedi, M.; Beurel, E. Th17 Cells Sense Microbiome to Promote Depressive-like Behaviors. Microbiome 2023, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rodriguez, E.M.; Madorma, D.; O’Connor, G.; Mason, B.L.; Han, D.; Deo, S.K.; Oppenheimer, M.; Nemeroff, C.B.; Trivedi, M.H.; Daunert, S.; et al. Identification of a Signalling Mechanism by Which the Microbiome Regulates Th17 Cell-Mediated Depressive-like Behaviors in Mice. Am. J. Psychiatry 2020, 177, 974. [Google Scholar] [CrossRef] [PubMed]
- Rusch, J.A.; Layden, B.T.; Dugas, L.R. Signalling Cognition: The Gut Microbiota and Hypothalamic-Pituitary-Adrenal Axis. Front. Endocrinol. 2023, 14, 1130689. [Google Scholar] [CrossRef] [PubMed]
- Farzi, A.; Fröhlich, E.E.; Holzer, P. Gut Microbiota and the Neuroendocrine System. Neurotherapeutics 2018, 15, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, H.; Chen, X.; Zhang, Y.; Zhang, H.; Xie, P. Gut Microbiota and Its Metabolites in Depression: From Pathogenesis to Treatment. eBioMedicine 2023, 90, 104527. [Google Scholar] [CrossRef] [PubMed]
- Gujral, S.; Aizenstein, H.; Reynolds, C.F.; Butters, M.A.; Erickson, K.I. Exercise Effects on Depression: Possible Neural Mechanisms. Gen. Hosp. Psychiatry 2017, 49, 2. [Google Scholar] [CrossRef]
- Zhao, J.L.; Jiang, W.T.; Wang, X.; Cai, Z.D.; Liu, Z.H.; Liu, G.R. Exercise, Brain Plasticity, and Depression. CNS Neurosci. Ther. 2020, 26, 885. [Google Scholar] [CrossRef] [PubMed]
- Kohler, O.; Krogh, J.; Mors, O.; Eriksen Benros, M. Inflammation in Depression and the Potential for Anti-Inflammatory Treatment. Curr. Neuropharmacol. 2016, 14, 732–742. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The Role of Inflammation in Depression: From Evolutionary Imperative to Modern Treatment Target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Alvarez-Herrera, S.; Pavon, L. Interacciones Neuroendocrinoinmunológicas. In Inmunología Molecular, Celular y Traslacional; Pavón, L., Ed.; Wolters Kluwer: Mexico City, Mexico, 2021; pp. 227–242. ISBN 9788417949181. [Google Scholar]
- Fioranelli, M.; Roccia, M.G.; Flavin, D.; Cota, L. Regulation of Inflammatory Reaction in Health and Disease. Int. J. Mol. Sci. 2021, 22, 5277. [Google Scholar] [CrossRef]
- Balkan, B.; Pogun, S. Nicotinic Cholinergic System in the Hypothalamus Modulates the Activity of the Hypothalamic Neuropeptides during the Stress Resp. Curr. Neuropharmacol. 2017, 15, 371–387. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. Neural Regulation of Immunity: Molecular Mechanisms and Clinical Translation. Nat. Neurosci. 2017, 20, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, P.S.; Rosas-Ballina, M.; Levine, Y.A.; Tracey, K.J. Rethinking Inflammation: Neural Circuits in the Regulation of Immunity. Immunol. Rev. 2012, 248, 188–204. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The Vagus Nerve and the Inflammatory Reflex-Linking Immunity and Metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The Cholinergic Anti-Inflammatory Pathway: A Missing Link in Neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Vida, G.; Peña, G.; Deitch, E.A.; Ulloa, L. A7-Cholinergic Receptor Mediates Vagal Induction of Splenic Norepinephrine. J. Immunol. 2011, 186, 4340–4346. [Google Scholar] [CrossRef]
- Zhou, J.; Yan, J.; Liang, H.; Jiang, J. Epinephrine Enhances the Response of Macrophages under LPS Stimulation. Biomed. Res. Int. 2014, 2014, 254686. [Google Scholar] [CrossRef]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of a Self-Amplifying Catecholamine Loop Reduces Cytokine Release Syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef]
- Macias, A.E.; Guaní-Guerra, E. Transfer Factor: Myths and Facts. Arch. Med. Res. 2020, 51, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Islas-Weinstein, L.; Maldonado-García, J.L. Immunomodulatory Supplements; Elsevier Inc.: Amsterdam, The Netherlands, 2021; ISBN 9780128187319. [Google Scholar]
- Vallejo-Castillo, L.; Favari, L.; Vázquez-Leyva, S.; Mellado-Sánchez, G.; Macías-Palacios, Z.; López-Juárez, L.E.; Valencia-Flores, L.; Medina-Rivero, E.; Chacón-Salinas, R.; Pavón, L.; et al. Sequencing Analysis and Identification of the Primary Peptide Component of the Dialyzable Leukocyte Extract “Transferon Oral”: The Starting Point to Understand Its Mechanism of Action. Front. Pharmacol. 2020, 11, 569039. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Stolk, P.; De Bruin, M.L.; Leufkens, H.G.M.; Crommelin, D.J.A.; De Vlieger, J.S.B. The EU Regulatory Landscape of Non-Biological Complex Drugs (NBCDs) Follow-on Products: Observations and Recommendations. Eur. J. Pharm. Sci. 2019, 133, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rivero, E.; Merchand-Reyes, G.; Pavón, L.; Vázquez-Leyva, S.; Pérez-Sánchez, G.; Salinas-Jazmín, N.; Estrada-Parra, S.; Velasco-Velázquez, M.; Pérez-Tapia, S.M. Batch-to-Batch Reproducibility of TransferonTM. J. Pharm. Biomed. Anal. 2014, 88, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rivero, E.; Vallejo-Castillo, L.; Vázquez-Leyva, S.; Pérez-Sánchez, G.; Favari, L.; Velasco-Velázquez, M.; Estrada-Parra, S.; Pavón, L.; Pérez-Tapia, S.M. Physicochemical Characteristics of TransferonTM Batches. Biomed. Res. Int. 2016, 2016, 7935181. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Leyva, S.; Vallejo-Castillo, L.; López-Morales, C.A.; Herbert-Pucheta, J.E.; Zepeda-Vallejo, L.G.; Velasco-Velázquez, M.; Pavón, L.; Pérez-Tapia, S.M.; Medina-Rivero, E. Identity Profiling of Complex Mixtures of Peptide Products by Structural and Mass Mobility Orthogonal Analysis. Anal. Chem. 2019, 91, 14392–14400. [Google Scholar] [CrossRef] [PubMed]
- Herbert-Pucheta, J.E.; López-Morales, C.A.; Medina-Rivero, E.; Estrada-Parra, S.; Pérez-Tapia, S.M.; Zepeda-Vallejo, L.G. Consistency of a Dialyzable Leucocyte Extract Manufactured at GMP Facilities by Nuclear Magnetic Resonance Spectroscopy. J. Pharm. Biomed. Anal. 2021, 196, 113940. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, Functions, Mechanisms. Biochim. Biophys. Acta-Mol. Cell Res. 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, Z.J. The Novel Functions of Ubiquitination in Signaling. Curr. Opin. Cell Biol. 2004, 16, 119–126. [Google Scholar] [CrossRef]
- Mendoza-Salazar, I.; Fragozo, A.; González-Martínez, A.P.; Trejo-Martínez, I.; Arreola, R.; Pavón, L.; Almagro, J.C.; Vallejo-Castillo, L.; Aguilar-Alonso, F.A.; Pérez-Tapia, S.M. Almost 50 Years of Monomeric Extracellular Ubiquitin (EUb). Pharmaceuticals 2024, 17, 185. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin Signalling in Neurodegeneration: Mechanisms and Therapeutic Opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin Modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.M.; Laporte, H.M.; Albee, L.J.; Baker, T.A.; Bach, H.H.; Vana, P.G.; Evans, A.E.; Gamelli, R.L.; Majetschak, M. Ubiquitin Urine Levels in Burn Patients. J. Burn Care Res. 2017, 38, e133–e143. [Google Scholar] [CrossRef] [PubMed]
- Majetschak, M.; Krehmeier, U.; Bardenheuer, M.; Denz, C.; Quintel, M.; Voggenreiter, G.; Obertacke, U. Extracellular Ubiquitin Inhibits the TNF-Alpha Response to Endotoxin in Peripheral Blood Mononuclear Cells and Regulates Endotoxin Hyporesponsiveness in Critical Illness. Blood 2003, 101, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Majetschak, M. Extracellular Ubiquitin: Immune Modulator and Endogenous Opponent of Damage-Associated Molecular Pattern Molecules. J. Leukoc. Biol. 2011, 89, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Majetschak, M.; Cohn, S.M.; Nelson, J.A.; Burton, E.H.; Obertacke, U.; Proctor, K.G. Effects of Exogenous Ubiquitin in Lethal Endotoxemia. Surgery 2004, 135, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Romero, J.; Bach, H.H.; Strom, J.A.; Gamelli, R.L.; Majetschak, M. Effects of Exogenous Ubiquitin in a Polytrauma Model with Blunt Chest Trauma. Crit. Care Med. 2012, 40, 2376–2384. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Parra, S.; Nagaya, A.; Serrano, E.; Rodriguez, O.; Santamaria, V.; Ondarza, R.; Chavez, R.; Correa, B.; Monges, A.; Cabezas, R.; et al. Comparative Study of Transfer Factor and Acyclovir in the Treatment of Herpes Zoster. Int. J. Immunopharmacol. 1998, 20, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Jazmin, N.; Estrada-Parra, S.; Becerril-Garcia, M.A.; Limon-Flores, A.Y.; Vazquez-Leyva, S.; Medina-Rivero, E.; Pavon, L.; Velasco-Velazquez, M.A.; Perez-Tapia, S.M. Herpes Murine Model as a Biological Assay to Test Dialyzable Leukocyte Extracts Activity. J. Immunol. Res. 2015, 2015, 146305. [Google Scholar] [CrossRef]
- Muñoz, A.I.; Vallejo-Castillo, L.; Fragozo, A.; Vázquez-Leyva, S.; Pavón, L.; Pérez-Sánchez, G.; Soria-Castro, R.; Mellado-Sánchez, G.; Cobos-Marin, L.; Pérez-Tapia, S.M. Increased Survival in Puppies Affected by Canine Parvovirus Type II Using an Immunomodulator as a Therapeutic Aid. Sci. Rep. 2021, 11, 19864. [Google Scholar] [CrossRef]
- Muñoz, A.I.; Maldonado-García, J.L.; Fragozo, A.; Vallejo-Castillo, L.; Lucas-Gonzalez, A.; Trejo-Martínez, I.; Pavón, L.; Pérez-Sánchez, G.; Cobos-Marin, L.; Pérez-Tapia, S.M. Altered Neutrophil-to-Lymphocyte Ratio in Sepsis Secondary to Canine Parvoviral Enteritis Treated with and without an Immunomodulator in Puppies. Front. Vet. Sci. 2022, 9, 995443. [Google Scholar] [CrossRef] [PubMed]
- Saini, V.; Marchese, A.; Majetschak, M. CXC Chemokine Receptor 4 Is a Cell Surface Receptor for Extracellular Ubiquitin. J. Biol. Chem. 2010, 285, 15566–15576. [Google Scholar] [CrossRef]
- Saini, V.; Staren, D.M.; Ziarek, J.J.; Nashaat, Z.N.; Campbell, E.M.; Volkman, B.F.; Marchese, A.; Majetschak, M. The CXC Chemokine Receptor 4 Ligands Ubiquitin and Stromal Cell-Derived Factor-1α Function through Distinct Receptor Interactions. J. Biol. Chem. 2011, 286, 33466–33477. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Davis, J.D.; Staren, D.M.; Volkman, B.F.; Majetschak, M. CXC Chemokine Receptor 4 Signaling upon Co-Activation with Stromal Cell-Derived Factor-1α and Ubiquitin. Cytokine 2014, 65, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Xie, G.; Xiao, H.; Ding, F.; Bao, W.; Zhang, M. CXCR4 Knockdown Prevents Inflammatory Cytokine Expression in Macrophages by Suppressing Activation of MAPK and NF-ΚB Signaling Pathways. Cell Biosci. 2019, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Guyon, A. CXCL12 Chemokine and Its Receptors as Major Players in the Interactions between Immune and Nervous Systems. Front. Cell. Neurosci. 2014, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martínez, I.E.; Rodríguez, M.C.; Cerbón, M.; Ramos-Martínez, J.C.; Ramos-Martínez, E.G. Role of the Cholinergic Anti-Inflammatory Reflex in Central Nervous System Diseases. Int. J. Mol. Sci. 2021, 22, 13427. [Google Scholar] [CrossRef] [PubMed]
- Abarca-Castro, E.A.; Talavera-Peña, A.K.; Reyes-Lagos, J.J.; Becerril-Villanueva, E.; Pérez-Sanchez, G.; de la Peña, F.R.; Maldonado-García, J.L.; Pavón, L. Modulation of Vagal Activity May Help Reduce Neurodevelopmental Damage in the Offspring of Mothers with Pre-Eclampsia. Front. Immunol. 2023, 14, 1280334. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Sun, S.C. Ubiquitin Signaling in Immune Responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef]
- Liu, H.; Wilson, K.R.; Schriek, P.; Macri, C.; Blum, A.B.; Francis, L.; Heinlein, M.; Nataraja, C.; Harris, J.; Jones, S.A.; et al. Ubiquitination of MHC Class II Is Required for Development of Regulatory but Not Conventional CD4+ T Cells. J. Immunol. 2020, 205, 1207–1216. [Google Scholar] [CrossRef]
- Schriek, P.; Liu, H.; Ching, A.C.; Huang, P.; Gupta, N.; Wilson, K.R.; Tsai, M.H.; Yan, Y.; Macri, C.F.; Dagley, L.F.; et al. Physiological Substrates and Ontogeny-Specific Expression of the Ubiquitin Ligases MARCH1 and MARCH8. Curr. Res. Immunol. 2021, 2, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Uribe, A.P.; Valencia-Martínez, H.; Carballo-Uicab, G.; Vallejo-Castillo, L.; Medina-Rivero, E.; Chacón-Salinas, R.; Pavón, L.; Velasco-Velázquez, M.A.; Mellado-Sánchez, G.; Estrada-Parra, S.; et al. CD80 Expression Correlates with IL-6 Production in THP-1-like Macrophages Costimulated with LPS and Dialyzable Leukocyte Extract (Transferon®). J. Immunol. Res. 2019, 2019, 2198508. [Google Scholar] [CrossRef] [PubMed]
- Pavón, L.; Sandoval-López, G.; Eugenia Hernández, M.; Loría, F.; Estrada, I.; Pérez, M.; Moreno, J.; Ávila, U.; Leff, P.; Antón, B.; et al. Th2 Cytokine Response in Major Depressive Disorder Patients before Treatment. J. Neuroimmunol. 2006, 172, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.E.; Mendieta, D.; Pérez-Tapia, M.; Bojalil, R.; Estrada-Garcia, I.; Estrada-Parra, S.; Pavón, L. Effect of Selective Serotonin Reuptake Inhibitors and Immunomodulator on Cytokines Levels: An Alternative Therapy for Patients with Major Depressive Disorder. Clin. Dev. Immunol. 2013, 2013, 267871. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-García, J.L.; Pérez-Sánchez, G.; Becerril Villanueva, E.; Alvarez-Herrera, S.; Pavón, L.; Gutiérrez-Ospina, G.; López-Santiago, R.; Maldonado-Tapia, J.O.; Pérez-Tapia, S.M.; Moreno-Lafont, M.C. Behavioral and Neurochemical Shifts at the Hippocampus and Frontal Cortex Are Associated to Peripheral Inflammation in Balb/c Mice Infected with Brucella Abortus 2308. Microorganisms 2021, 9, 1937. [Google Scholar] [CrossRef]
- Maldonado-García, J.L.; Pérez-Sánchez, G.; Becerril-Villanueva, E.; Alvarez-Herrera, S.; Pavón, L.; Sánchez-Torres, L.; Gutiérrez-Ospina, G.; Girón-Pérez, M.I.; Damian-Morales, G.; Maldonado-Tapia, J.O.; et al. Imipramine Administration in Brucella Abortus 2308-Infected Mice Restores Hippocampal Serotonin Levels, Muscle Strength, and Mood, and Decreases Spleen CFU Count. Pharmaceuticals 2023, 16, 1525. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Mechanism of Action | Examples |
|---|---|---|
| Selective serotonin reuptake inhibitors (SSRIs) | Inhibit serotonin reuptake, thus increasing serotonin activity. | Citalopram Escitalopram Paroxetine Sertraline Fluoxetine Fluvoxamine |
| Serotonin–norepinephrine reuptake inhibitors (SNRIs) | Block serotonin and norepinephrine reuptake in the synaptic button, increasing postsynaptic receptors’ stimulation | Venlafaxine Desvenlafaxine Duloxetine Milnacipran Levomilnacipran |
| Atypical antidepressants | This group is characterized by different mechanisms of action, with the following examples: Bupropion inhibits dopamine and norepinephrine reuptake. Mirtazapine blocks α-2 adrenergic receptors on the cell bodies and nerve terminals and increases the release of norepinephrine into the synapse. Mirtazapine works by blocking alpha-2 adrenergic receptors on the cell bodies and nerve terminals, promoting the release of norepinephrine into the synapse, and in addition antagonizes 5-HT receptors | Bupropion Mirtazapine Agomelatine |
| Serotonin modulators | This group has different mechanisms of action on the serotonergic system. Trazodone acts upon postsynaptic serotonin 5-HT2A and 5-HT2C receptors and weakly inhibits presynaptic serotonin reuptake. In addition, it has additional postsynaptic alpha-adrenergic receptors and histamine receptors blocking activity. In addition, it blocks α-adrenergic receptors and histamine receptors in the postsynaptic button. Nefazodone antagonizes postsynaptic serotonin 5-HT2A receptors and inhibits presynaptic serotonin and norepinephrine reuptake. Vortioxetine acts as a 5-HT1A receptor agonist and a 5-HT3 and 5-HT7 receptor antagonist. | Nefazodone Trazodone Vilazodone Vortioxetine |
| Tricyclic antidepressants (TCAs) | Inhibit the reuptake of norepinephrine and serotonin at the presynaptic neuronal membrane. | Amitriptyline Clomipramine Doxepin Imipramine Trimipramine Desipramine Nortriptyline Protriptyline Maprotiline Amoxapine |
| Monoamine oxidase inhibitors (MAOIs) | Inhibit the monoamine oxidase enzyme responsible for catabolizing serotonin, norepinephrine, and dopamine. | Selegiline Moclobemide Tranylcypromine Isocarboxazid Phenelzine |
| Cytokine | Effect |
|---|---|
| IFN-α | Fatigue, depression, thought disorders, psychosis and suicidal ideation, stress, anxiety, decreased substance P, myalgia, psychomotor retardation, anorexia, social isolation, irritability, and cognitive disorders (lack of concentration, memory impairment, and bradypsychia) |
| IFN-β | Fatigue, depression, and bradypsychia |
| IFN-γ | Modulates social behavior by regulating the connection of social interaction brain areas |
| TNF-α | Anorexia, fatigue, stress, upregulation of substance P expression, rapid eye movements during sleep, and increased release of excitatory neurotransmitters; noradrenaline and adrenaline stimulate its release |
| IL-1β | Somnolence, confusion, hallucinations, hyperalgesia, fatigue, fever, sleepiness, myalgia, and substance P antinociception (increased GABA and decreased NMDA); noradrenaline and adrenaline stimulate its release |
| IL-2 | Confusion, delusions, depression, psychosis, myalgias, and cognitive dysfunction |
| IL-4 | Regulates higher mental functions such as memory and learning |
| IL-6 | Stress, fatigue, hyperalgesia, depression, and activation of the sympathetic nervous system; noradrenaline, adrenaline, and substance P stimulate their release |
| IL-8 | Mediates sympathetic pain; substance P stimulates its production |
| IL-10 | Blocks pain |
| IL-17A | Modulates anxiety through meningeal γδ T cells |
| Variables | Effect |
|---|---|
| Cortisol | ↑ |
| T helper cells | ns |
| T cytotoxic cells | ↑ |
| NK cells | ↑ |
| B cells | ↓ |
| IL-1β | ↓ |
| TNF-α | ↑ |
| IL-6 | ns |
| IL-2 | ↓ |
| IFN-γ | ↓ |
| IL-4 | ↑ |
| IL-13 | ↑ |
| Variables | Comparison of Patients with MDD vs. Healthy Volunteers | ||
|---|---|---|---|
| W0 vs. HV | W20 vs. HV | W52 vs. HV | |
| Cortisol | ↑ | ns | ↑ |
| IL-1β | ↓ | ↑ | ↑ |
| IL-2 | ↓ | ns | ↓ |
| IFN-γ | ↓ | ns | ↑ |
| IL-10 | ↑ | ↓ | ↓ |
| Variables | Comparison of Patients with MDD vs. Healthy Volunteers (HV) | |||||
|---|---|---|---|---|---|---|
| W0 | W20 | W52 | ||||
| SSRI + hDLE vs. HV | SSRI + hDLE vs. SSRI | SSRI + hDLE vs. HV | SSRI + hDLE vs. SSRI | SSRI + hDLE vs. HV | SSRI + hDLE vs. SSRI | |
| Cortisol | ↑ | ns | ns | ↓ | ns | ↓ |
| IL-1β | ↓ | ns | ↑ | ns | ns | ↓ |
| IL-2 | ↓ | ns | ns | ns | ns | ↑ |
| IFN-γ | ↓ | ns | ns | ↑ | ns | ↑ |
| IL-10 | ↑ | ns | ns | ↑ | ns | ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maldonado-García, J.L.; García-Mena, L.H.; Mendieta-Cabrera, D.; Pérez-Sánchez, G.; Becerril-Villanueva, E.; Alvarez-Herrera, S.; Homberg, T.; Vallejo-Castillo, L.; Pérez-Tapia, S.M.; Moreno-Lafont, M.C.; et al. Use of Extracellular Monomeric Ubiquitin as a Therapeutic Option for Major Depressive Disorder. Pharmaceuticals 2024, 17, 841. https://doi.org/10.3390/ph17070841
Maldonado-García JL, García-Mena LH, Mendieta-Cabrera D, Pérez-Sánchez G, Becerril-Villanueva E, Alvarez-Herrera S, Homberg T, Vallejo-Castillo L, Pérez-Tapia SM, Moreno-Lafont MC, et al. Use of Extracellular Monomeric Ubiquitin as a Therapeutic Option for Major Depressive Disorder. Pharmaceuticals. 2024; 17(7):841. https://doi.org/10.3390/ph17070841
Chicago/Turabian StyleMaldonado-García, José Luis, Lissette Haydee García-Mena, Danelia Mendieta-Cabrera, Gilberto Pérez-Sánchez, Enrique Becerril-Villanueva, Samantha Alvarez-Herrera, Toni Homberg, Luis Vallejo-Castillo, Sonia Mayra Pérez-Tapia, Martha C. Moreno-Lafont, and et al. 2024. "Use of Extracellular Monomeric Ubiquitin as a Therapeutic Option for Major Depressive Disorder" Pharmaceuticals 17, no. 7: 841. https://doi.org/10.3390/ph17070841