
Unveiling Moroccan Nature’s Arsenal: A Computational Molecular Docking, Density Functional Theory, and Molecular Dynamics Study of Natural Compounds against Drug-Resistant Fungal Infections
 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results and Discussion
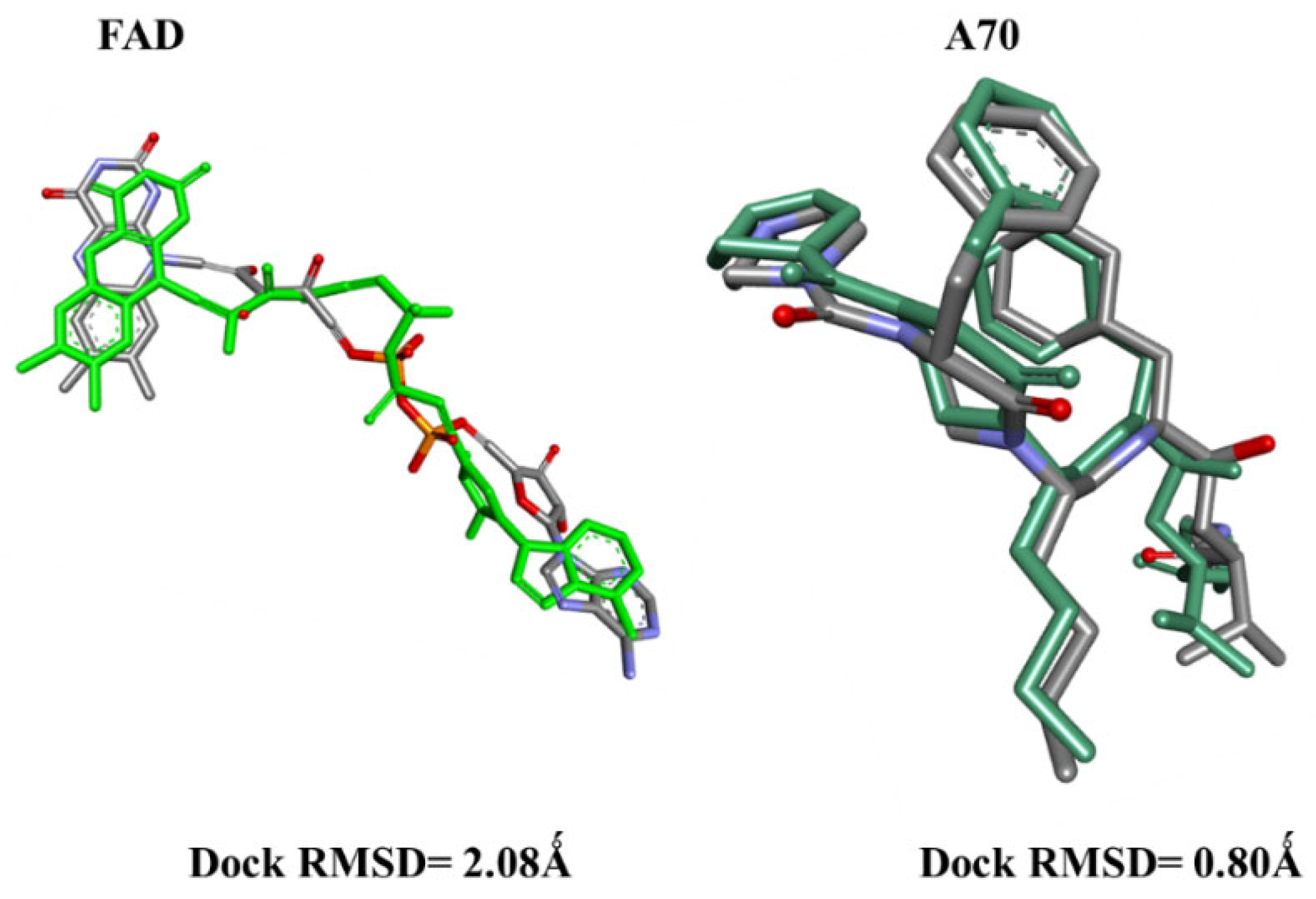
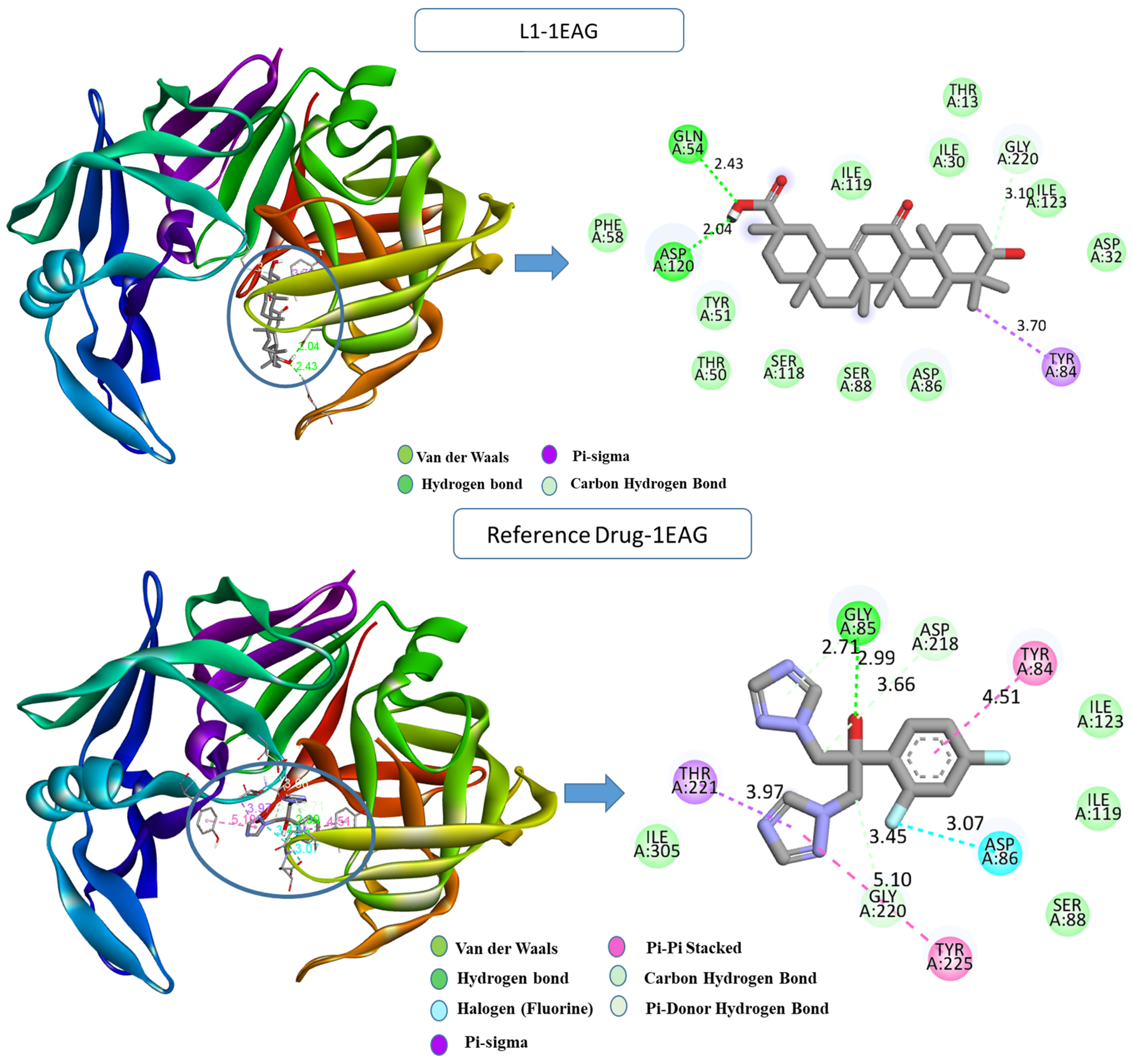
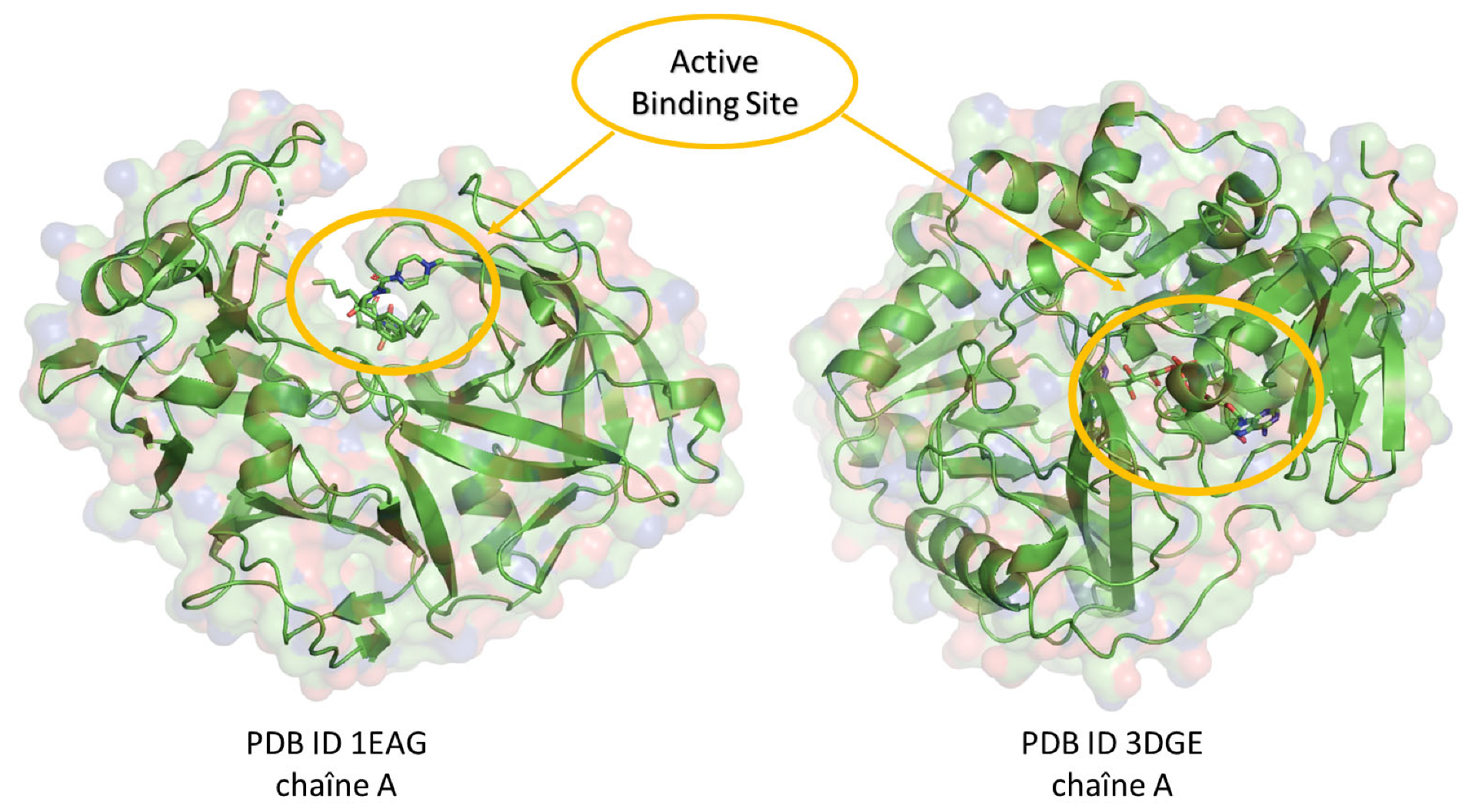
2.1. Molecular Docking
2.2. Computational ADME-Tox and Drug-Likeness
2.3. Molecular Quantum Analysis
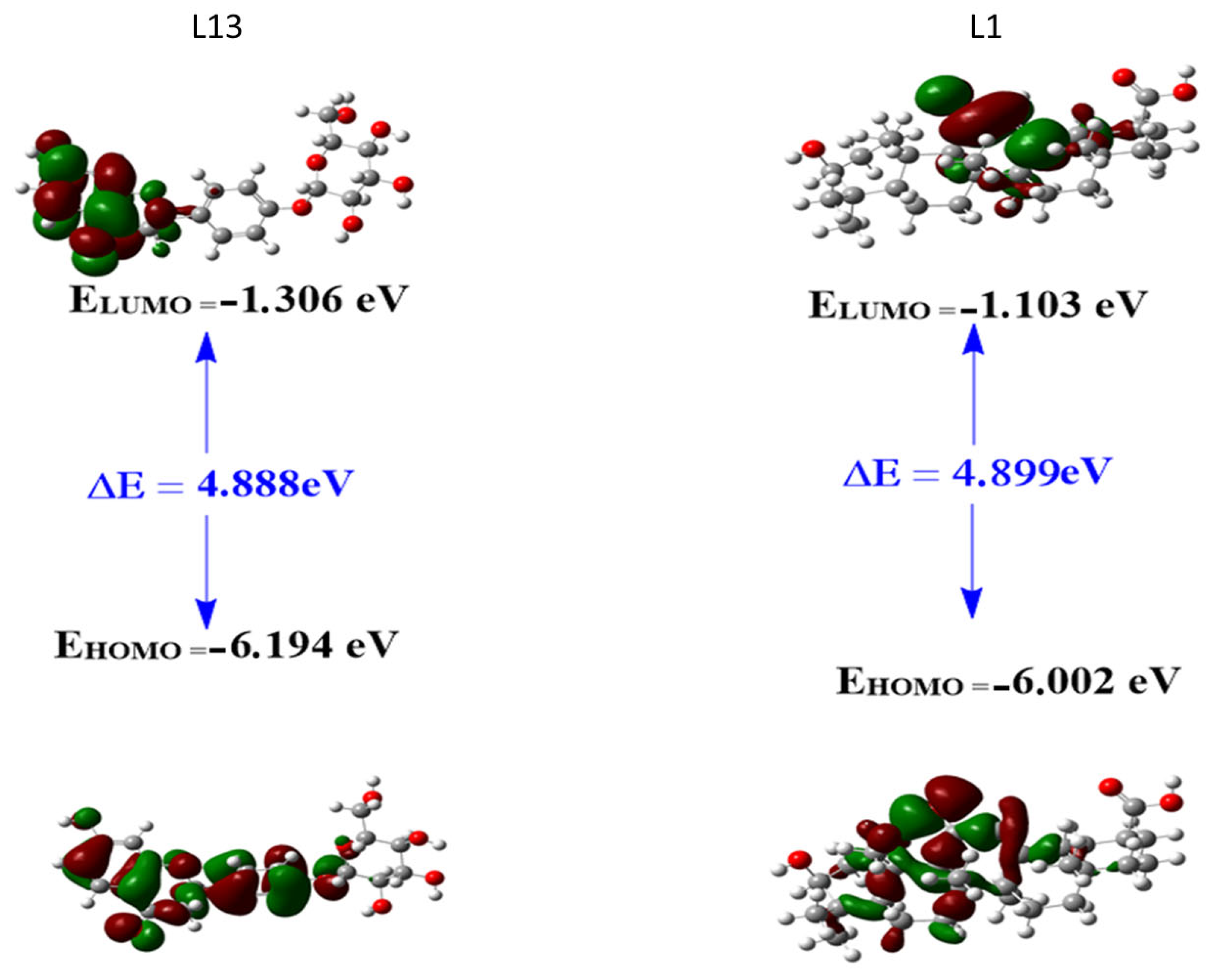
2.3.1. Frontier Molecular Orbitals (FMOs)
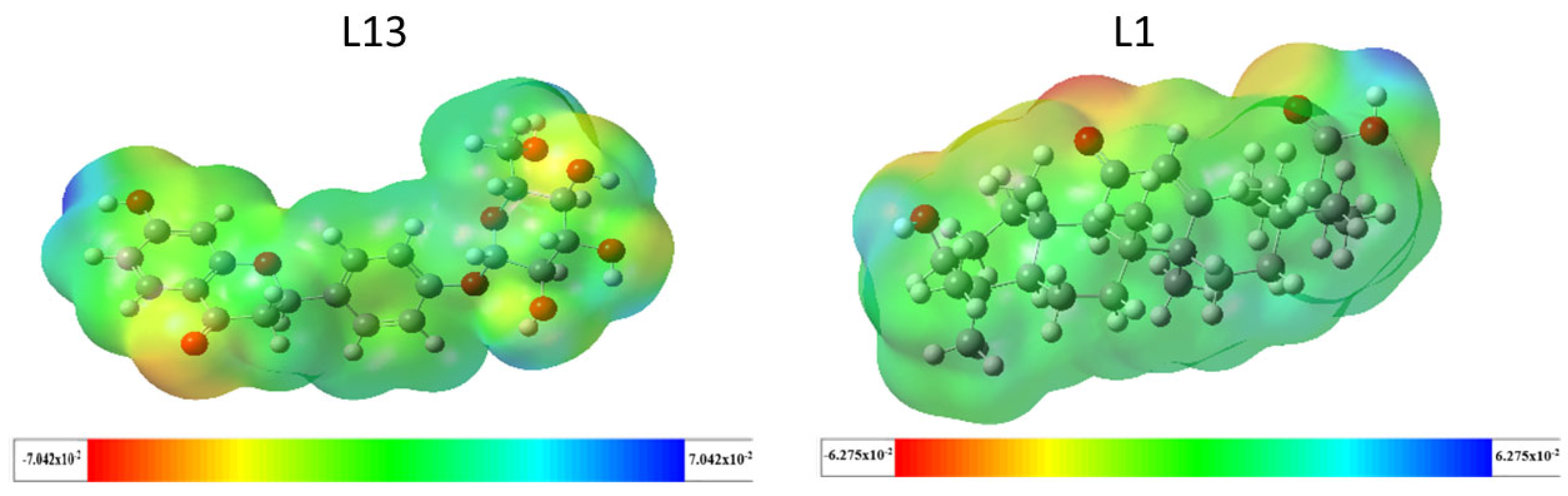
2.3.2. Molecular Electrostatic Potential (MEP)
2.4. Stability of Protein–Ligand Interactions MD Simulation Analysis
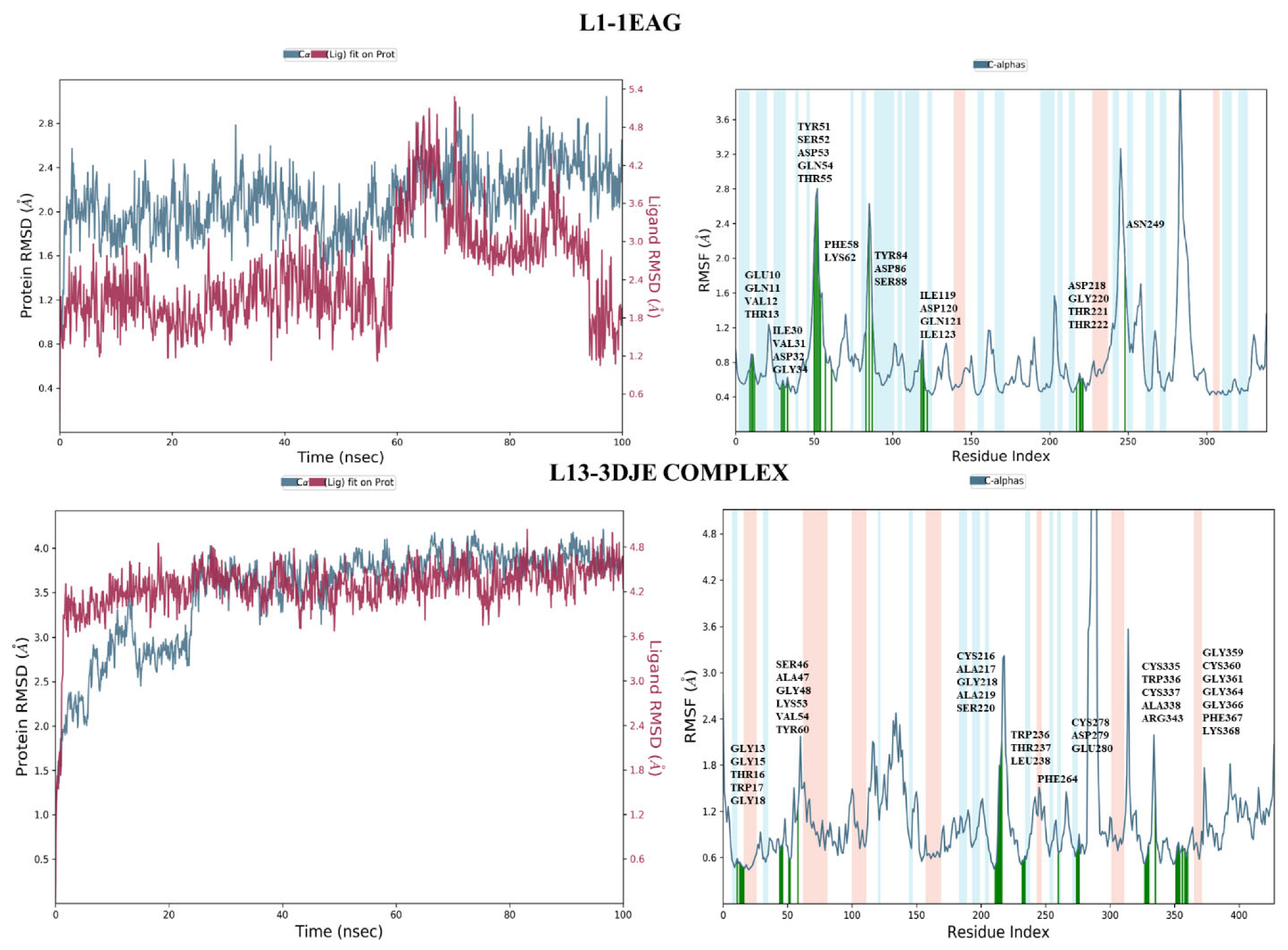
2.4.1. Root-Mean-Square Deviation Analysis and the Root-Mean-Square Fluctuation
2.4.2. Protein–Ligand Contact
3. Materials and Methods
3.1. Database Collection
3.2. Molecular Docking Procedure
3.3. Computational ADME and Drug-Likeness Analysis
3.4. Quantum Chemical Investigation
3.5. Dynamics Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kainz, K.; Bauer, M.A.; Madeo, F.; Carmona-Gutierrez, D. Fungal infections in humans: The silent crisis. Microb. Cell 2020, 7, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Gubbins, P.O.; Anaissie, E.J. Antifungal therapy. In Clinical Mycology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 161–195. [Google Scholar]
- Siscar-Lewin, S.; Hube, B.; Brunke, S. Emergence and evolution of virulence in human pathogenic fungi. Trends Microbiol. 2022, 30, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Priest, E.; Naglik, J.R.; Richardson, J.P. Fungal Toxins and Host Immune Responses. Front. Microbiol. 2021, 12, 643639. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Le Mauff, F.; Sheppard, D.C.; Zhang, S. Filamentous fungal biofilms: Conserved and unique aspects of extracellular matrix composition, mechanisms of drug resistance and regulatory networks in Aspergillus fumigatus. NPJ Biofilm. Microbiomes 2022, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Costa-Orlandi, C.B.; Sardi, J.C.O.; Pitangui, N.S.; de Oliveira, H.C.; Scorzoni, L.; Galeane, M.C.; Medina-Alarcón, K.P.; Melo, W.C.M.; Marcelino, M.Y.; Braz, J.D.; et al. Fungal Biofilms and Polymicrobial Diseases. J. Fungi 2017, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.P.; Lionakis, M.S. Pathogenesis and virulence of Candida albicans. Virulence 2022, 13, 89–121. [Google Scholar] [CrossRef] [PubMed]
- Shinu, P.; Gupta, G.L.; Sharma, M.; Khan, S.; Goyal, M.; Nair, A.B.; Kumar, M.; Soliman, W.E.; Rahman, A.; Attimarad, M.; et al. Pharmacological Features of 18β-Glycyrrhetinic Acid: A Pentacyclic Triterpenoid of Therapeutic Potential. Plants 2023, 12, 1086. [Google Scholar] [CrossRef] [PubMed]
- Arastehfar, A.; Carvalho, A.; Houbraken, J.; Lombardi, L.; Garcia-Rubio, R.; Jenks, J.D.; Rivero-Menendez, O.; Aljohani, R.; Jacobsen, I.D.; Berman, J.; et al. Aspergillus fumigatus and aspergillosis: From basics to clinics. Stud. Mycol. 2021, 100, 100115. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K. The Role of Natural Products as Sources of Therapeutic Agents for Innovative Drug Discovery. In Comprehensive Pharmacology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 408–422. [Google Scholar]
- Yamari, I.; Abchir, O.; Siddique, F.; Zaki, H.; Errougui, A.; Talbi, M.; Bouachrine, M.; ElKouali, M.; Chtita, S. The anticoagulant potential of Lippia alba extract in inhibiting SARS-CoV-2 Mpro: Density functional calculation, molecular docking analysis, and molecular dynamics simulations. Sci. Afr. 2024, 23, e01986. [Google Scholar] [CrossRef]
- Yamari, I.; Mouhib, A.; Es-Sounni, B.; Nejjari, R.; Mazoir, N.; Bakhouch, M.; Mouzdahir, A.; Benharref, A.; El Kouali, M.; Chtita, S. Oxidative functionalization of triterpenes isolated from Euphorbia resinifera latex: Semisynthesis, ADME-Tox, molecular docking, and molecular dynamics simulations. Chem. Phys. Impact 2023, 7, 100372. [Google Scholar] [CrossRef]
- Abchir, O.; Yamari, I.; Nour, H.; Daoui, O.; Elkhattabi, S.; Errougui, A.; Chtita, S. Structure-Based Virtual Screening, ADMET analysis, and Molecular Dynamics Simulation of Moroccan Natural Compounds as Candidates α-Amylase Inhibitors. ChemistrySelect 2023, 8, e202301092. [Google Scholar] [CrossRef]
- Zhang, C.-W.; Zhong, X.-J.; Zhao, Y.-S.; Rajoka, M.S.R.; Hashmi, M.H.; Zhai, P.; Song, X. Antifungal natural products and their derivatives: A review of their activity and mechanism of actions. Pharmacol. Res.-Mod. Chin. Med. 2023, 7, 100262. [Google Scholar] [CrossRef]
- Hsu, H.; Sheth, C.C.; Veses, V. Herbal Extracts with Antifungal Activity against Candida albicans: A Systematic Review. Mini-Rev. Med. Chem. 2021, 21, 90–117. [Google Scholar] [CrossRef] [PubMed]
- Cheraghipour, K.; Ezatpour, B.; Masoori, L.; Marzban, A.; Sepahvand, A.; Rouzbahani, A.K.; Moridnia, A.; Khanizadeh, S.; Mahmoudvand, H. Anti-Candida Activity of Curcumin: A Systematic Review. Curr. Drug Discov. Technol. 2021, 18, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Heard, S.C.; Wu, G.; Winter, J.M. Antifungal natural products. Curr. Opin. Biotechnol. 2021, 69, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A review of its effects on human health. Foods 2017, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Jawad, M.; Schoop, R.; Suter, A.; Klein, P.; Eccles, R. Safety and Efficacy Profile of Echinacea purpurea to Prevent Common Cold Episodes: A Randomized, Double-Blind, Placebo-Controlled Trial. Evid.-Based Complement. Altern. Med. 2012, 2012, 841315. [Google Scholar] [CrossRef] [PubMed]
- Nour, H.; Daoui, O.; Abchir, O.; ElKhattabi, S.; Belaidi, S.; Chtita, S. Combined computational approaches for developing new anti-Alzheimer drug candidates: 3D-QSAR, molecular docking and molecular dynamics studies of liquiritigenin derivatives. Heliyon 2022, 8, e11991. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, T.; Hussein, A.-S.; Beshir, S.; Hamed, A.R.; Ali, E.; El-Tanany, S.S. Antimicrobial Activity of Terpenoids Extracted from Annona muricata Seeds and its Endophytic Aspergillus niger Strain SH3 Either Singly or in Combination. Open Access Maced. J. Med. Sci. 2019, 7, 3127–3131. [Google Scholar] [CrossRef] [PubMed]
- McPhillie, M.J.; Cain, R.M.; Narramore, S.; Fishwick, C.W.G.; Simmons, K.J. Computational Methods to Identify New Antibacterial Targets. Chem. Biol. Drug Des. 2015, 85, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Scrocco, E.; Tomasi, J. Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials. In Advances in Quantum Chemistry; Academic Press: Cambridge, MA, USA, 1978; pp. 115–193. [Google Scholar]
- Harouak, H.; Falaki, K.; Bouiamrine, E.H.; Ibijbijen, J.; Nassiri, L. Diversity of medicinal plants used on oral disease in the city of Meknes, Morocco. J. Med. Plants Stud. 2018, 6, 117–122. [Google Scholar]
- Stanzione, F.; Giangreco, I.; Cole, J.C. Use of molecular docking computational tools in drug discovery. Prog. Med. Chem. 2021, 60, 273–343. [Google Scholar] [PubMed]
- Fluconazol Drug. Available online: https://go.drugbank.com/drugs/DB00196 (accessed on 3 June 2024).
- Helen, M. Berman RCSB protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Guex, N.; Diemand, A.; Peitsch, M.C.; Schwede, T. SwissPdbViewer, v4.1. Available online: https://spdbv.unil.ch/(accessed on 3 June 2024).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systéme. BIOVIA Discovery Studio 2021; Dassault Systèmes: Waltham, MA, USA, 2021. [Google Scholar]
- Palmer, M.E.; Andrews, L.J.; Abbey, T.C.; Dahlquist, A.E.; Wenzler, E. The importance of pharmacokinetics and pharmacodynamics in antimicrobial drug development and their influence on the success of agents developed to combat resistant gram negative pathogens: A review. Front. Pharmacol. 2022, 13, 888079. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo[d]thiazole-2(3H)-imine and its para-substituted derivatives: Solvent and substituent effects. J. Chem. Res. 2021, 45, 147–158. [Google Scholar] [CrossRef]
- Aydogdu, S.; Hatipoglu, A. Electronic Structures and Reactivities of COVID-19 Drugs: A DFT Study. Acta Chim. Slov. 2022, 69, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Harismah, K.; Dhumad, A.M.; Ibraheem, H.S.; Zandi, H.; Majeed, H.J. A DFT approach on tioguanine: Exploring tio-tiol tautomers, frontier molecular orbitals, IR and UV spectra, and quadrupole coupling constants. J. Mol. Liq. 2021, 334, 116018. [Google Scholar] [CrossRef]
- Badar, M.S.; Shamsi, S.; Ahmed, J.; Alam, M.A. Molecular Dynamics Simulations: Concept, Methods, and Applications. In Transdisciplinarity; Springer International Publishing: Cham, Switzerland, 2022; pp. 131–151. [Google Scholar]
- Schrödinger. Maestro, Version 12.5.139; Schrödinger: San Diego, CA, USA, 2021.
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Gong, X.; Liao, S.; Duan, C.; Li, L. Effects of thermostats/barostats on physical properties of liquids by molecular dynamics simulations. J. Mol. Liq. 2022, 365, 120116. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Score (kcal/mol) | Ligand | Score (kcal/mol) | Ligand | Score (kcal/mol) | Ligand | Score (kcal/mol) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1EAG | 3DJE | 1EAG | 3DJE | 1EAG | 3DJE | 1EAG | 3DJE | ||||
| L1 | −9.6 | −8.3 | L11 | −8.3 | −9.2 | L21 | −7.8 | −8.6 | L31 | −7.4 | −8.1 |
| L2 | −9.3 | −9.6 | L12 | −8.3 | −9 | L22 | −7.8 | −9.2 | L32 | −7.4 | −8.3 |
| L3 | −8.9 | −8.5 | L13 | −8.3 | −10.1 | L23 | −7.7 | −8.8 | L33 | −7.4 | −8.9 |
| L4 | −8.8 | −8.4 | L14 | −8.2 | −10.8 | L24 | −7.7 | −9 | L34 | −7.4 | −9.6 |
| L5 | −8.7 | −9 | L15 | −8.2 | −8.9 | L25 | −7.6 | −9 | L35 | −7.3 | −9.3 |
| L6 | −8.6 | −9.9 | L16 | −8.2 | −8.8 | L26 | −7.6 | −8.8 | L36 | −7.3 | −8.9 |
| L7 | −8.5 | −8.4 | L17 | −8.1 | −8.3 | L27 | −7.5 | −8.4 | L37 | −7.2 | −8.4 |
| L8 | −8.5 | −8.6 | L18 | −8 | −7.9 | L28 | −7.5 | −9.4 | L38 | −7.1 | −9.6 |
| L9 | −8.3 | −8.7 | L19 | −7.9 | −9.3 | L29 | −7.5 | −8.5 | L39 | −7 | −7.9 |
| L10 | −7.1 | −8.7 | L20 | −7.8 | −8.3 | L30 | −7.4 | −8.5 | L40 | −7 | −8.5 |
| Scoring for the reference drug (kcal/mol) | |||||||||||
| Candida Albicans/fluconazole | −6.7 | Aspergillus fumigatus/fluconazole | −7.9 | ||||||||
| Molecule | L1 | L13 |
|---|---|---|
| MW (size) | 470.68 | 418.39 |
| GI absorption | High | Low |
| FractionCsp3 (insaturation) | 0.87 | 0.38 |
| #Rotatable bonds (flexibility) | 1 | 4 |
| #H-bond acceptors | 4 | 9 |
| #H-bond donors | 2 | 5 |
| TPSA (polarity) | 74.60 | 145.91 |
| XLOGP3 (lipophilicity) | 5.49 | 0.39 |
| MLOGP | 4.87 | −0.92 |
| ESOL LogS (insolubility) | −6.15 | −2.71 |
| Lipinski #violations | 1 | 0 |
| Bioavailability score | 0.85 | 0.55 |
| PAINS #alerts | 0 | 0 |
| Synthetic accessibility | 6.08 | 4.91 |
| ADMET | Properties | Compounds | |
|---|---|---|---|
| L1 | L13 | ||
| Absorption | Water solubility (log mol/L) | −4.909 | −3.669 |
| Caco2 permeability (log Papp in 10−6 cm/s) | 0.912 | 0.371 | |
| Intestinal absorption (human)% | 97.389 | 56.977 | |
| Skin permeability (log Kpp) | −2.713 | −2.744 | |
| Distribution | VDss (human) (log L/kg) | −0.916 | −0.070 |
| BBB permeability log BB | 0.101 | −1.167 | |
| CNS permeability log PS | −1.320 | −3.853 | |
| Metabolism | CYP2D6 substrate | No | No |
| CYP3A4 substrate | Yes | Yes | |
| CYP1A2 inhibitor | No | No | |
| CYP2C19 inhibitor | No | No | |
| CYP2C9 inhibitor | No | No | |
| CYP2D6 inhibitor | No | No | |
| CYP3A4 inhibitor | No | No | |
| Excretion | Total clearance (log mL/min/kg) | −0.114 | 0.734 |
| Toxicity | AMES toxicity | No | No |
| hERG I inhibitor | No | No | |
| Hepatotoxicity | No | No | |
| Skin sensitization | No | No | |
| Compound | L13 | L1 |
|---|---|---|
| ELUMO (eV) | −1.3056 | −1.1034 |
| EHOMO (eV) | −6.1941 | −6.0022 |
| ΔE | 4.8885 | 4.8989 |
| Χ | 3.7498 | 3.5528 |
| η | 2.4443 | 2.4494 |
| pi | −3.7499 | −3.5528 |
| σ | 0.4091 | 0.4082 |
| ω | 2.876 | 2.577 |
| Dipole moment µ (DEBYE) | 1753.767 | 5735.663 |
| Electronic energy | −40,558.4 | −40,046.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamari, I.; Abchir, O.; Nour, H.; Khedraoui, M.; Rossafi, B.; Errougui, A.; Talbi, M.; Samadi, A.; Kouali, M.E.; Chtita, S. Unveiling Moroccan Nature’s Arsenal: A Computational Molecular Docking, Density Functional Theory, and Molecular Dynamics Study of Natural Compounds against Drug-Resistant Fungal Infections. Pharmaceuticals 2024, 17, 886. https://doi.org/10.3390/ph17070886
Yamari I, Abchir O, Nour H, Khedraoui M, Rossafi B, Errougui A, Talbi M, Samadi A, Kouali ME, Chtita S. Unveiling Moroccan Nature’s Arsenal: A Computational Molecular Docking, Density Functional Theory, and Molecular Dynamics Study of Natural Compounds against Drug-Resistant Fungal Infections. Pharmaceuticals. 2024; 17(7):886. https://doi.org/10.3390/ph17070886
Chicago/Turabian StyleYamari, Imane, Oussama Abchir, Hassan Nour, Meriem Khedraoui, Bouchra Rossafi, Abdelkbir Errougui, Mohammed Talbi, Abdelouahid Samadi, MHammed El Kouali, and Samir Chtita. 2024. "Unveiling Moroccan Nature’s Arsenal: A Computational Molecular Docking, Density Functional Theory, and Molecular Dynamics Study of Natural Compounds against Drug-Resistant Fungal Infections" Pharmaceuticals 17, no. 7: 886. https://doi.org/10.3390/ph17070886