
2D/3D-QSAR Model Development Based on a Quinoline Pharmacophoric Core for the Inhibition of Plasmodium falciparum: An In Silico Approach with Experimental Validation

 , , ,
, , ,  , and
, and
Abstract

1. Introduction
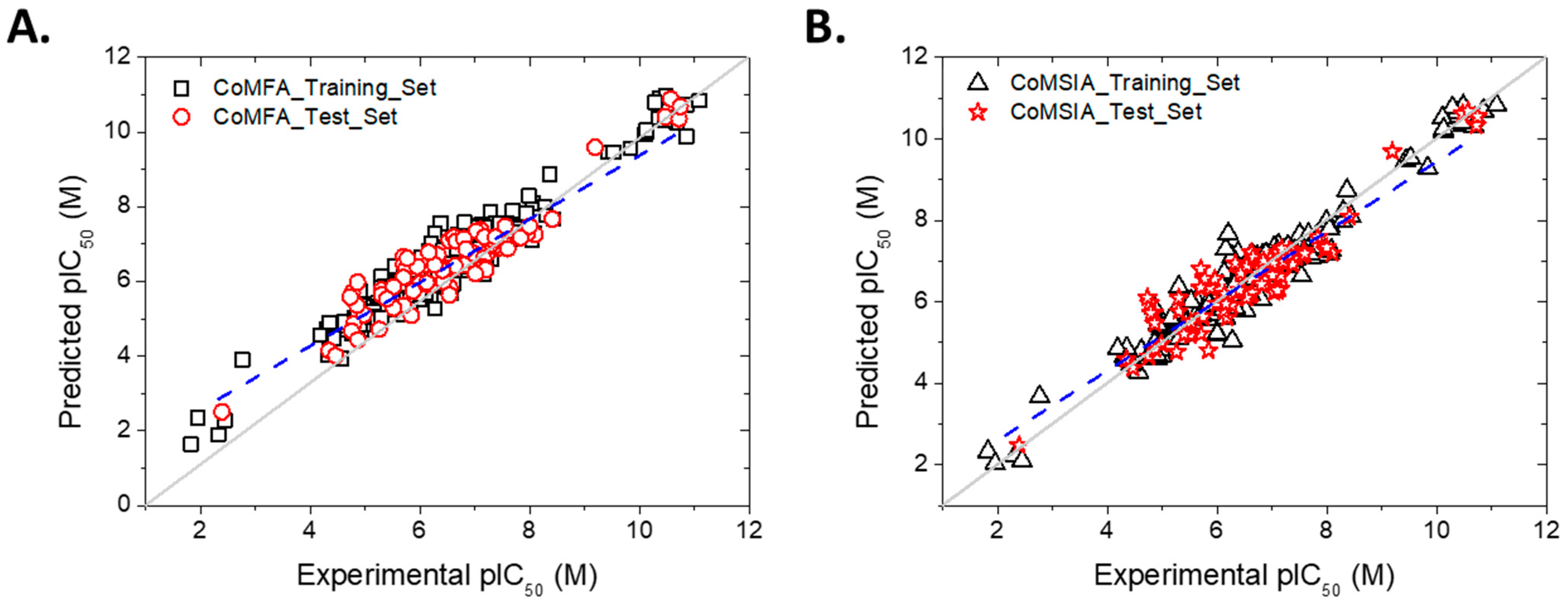
2. Results and Discussion
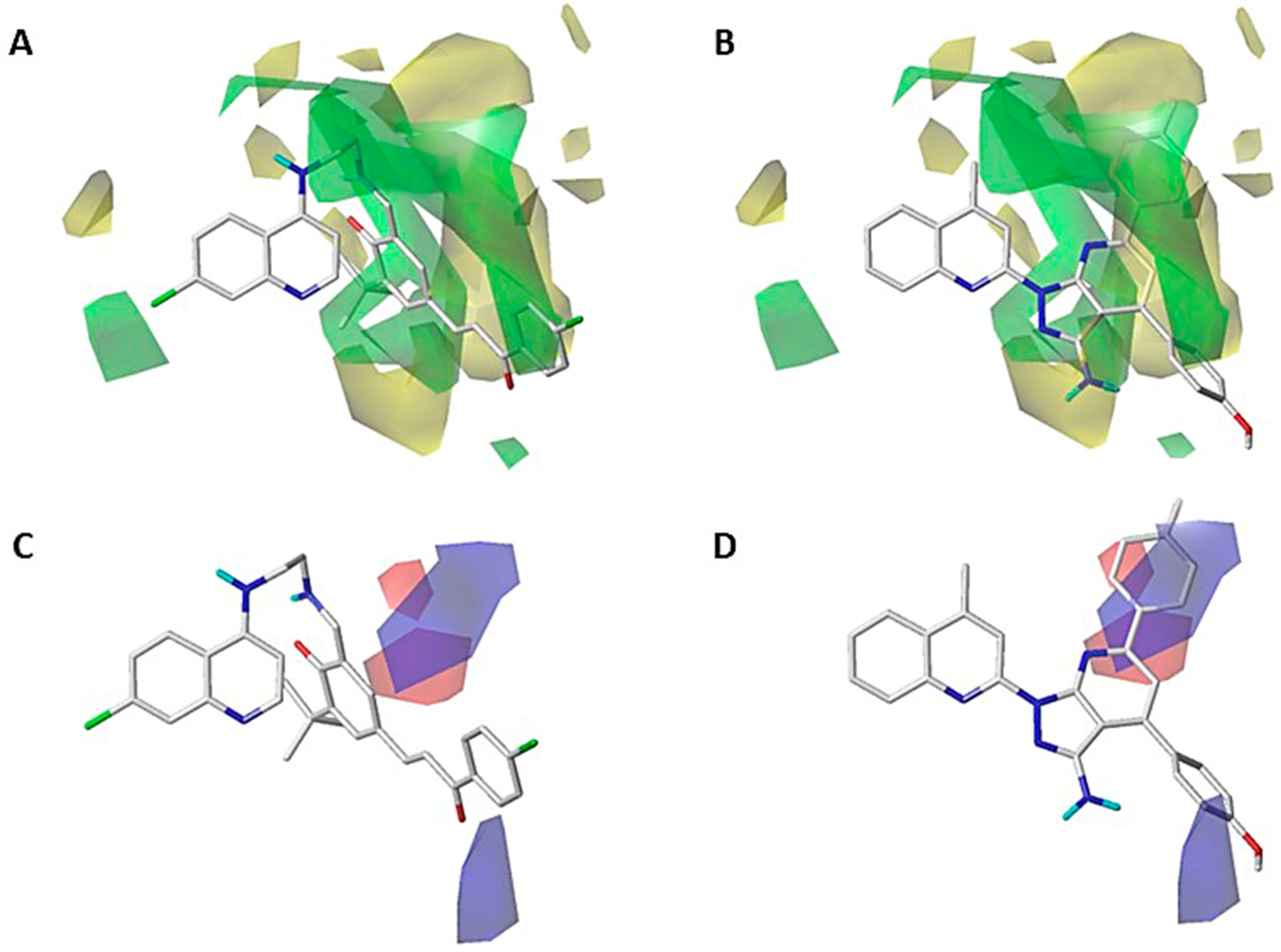
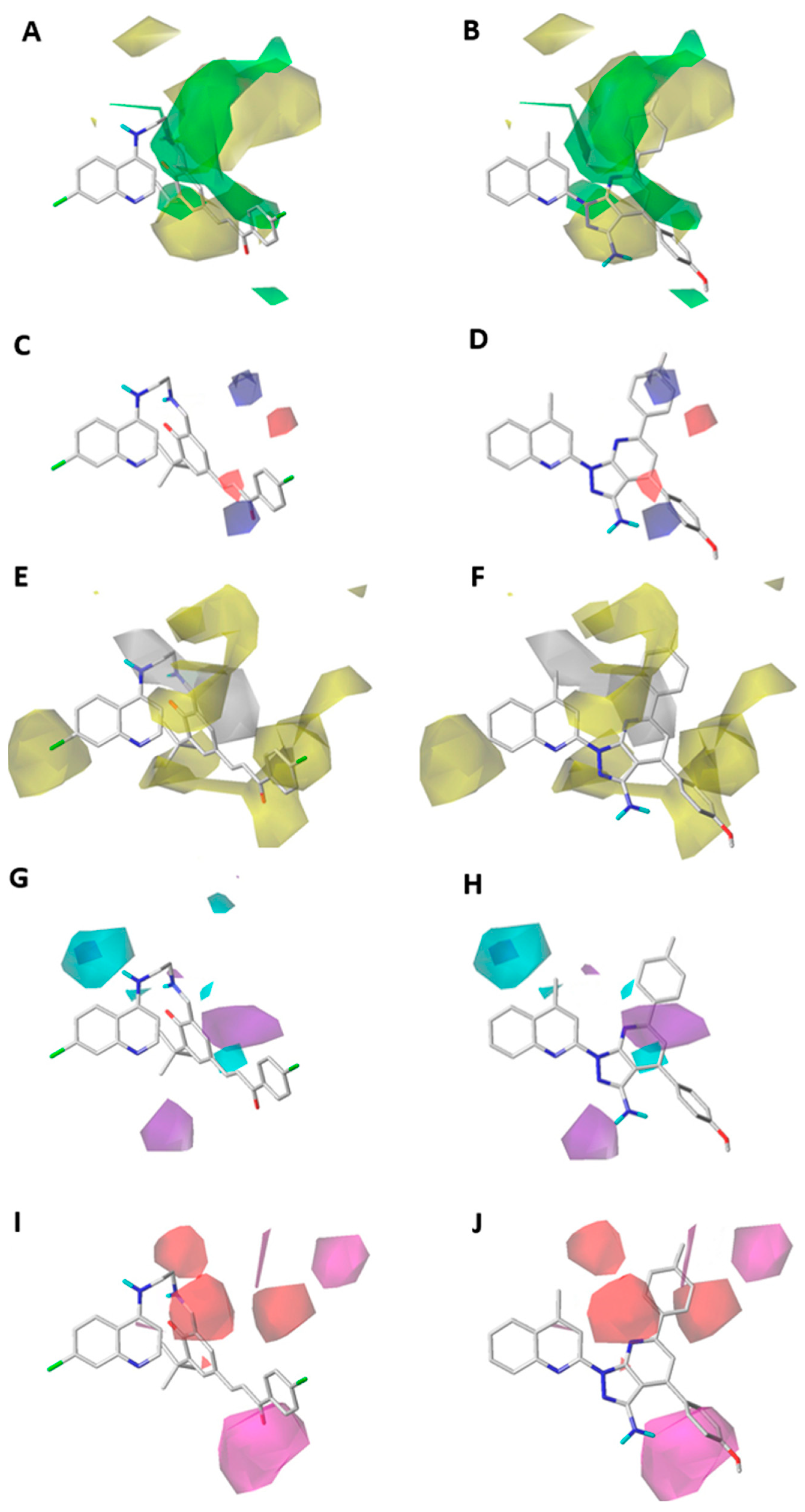
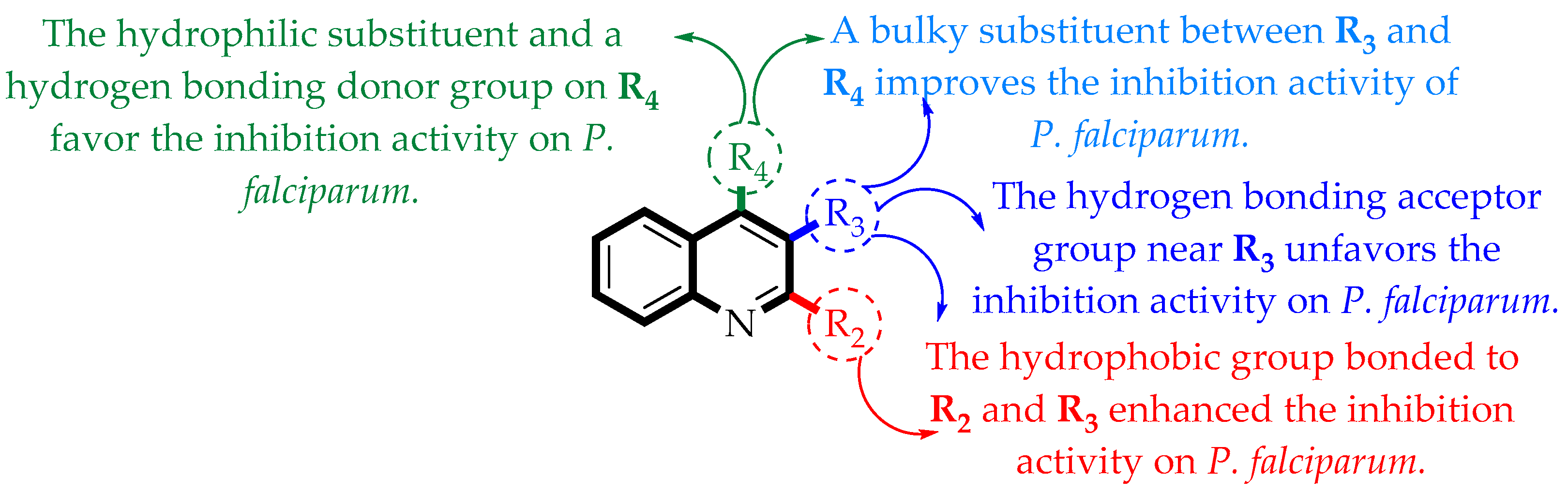
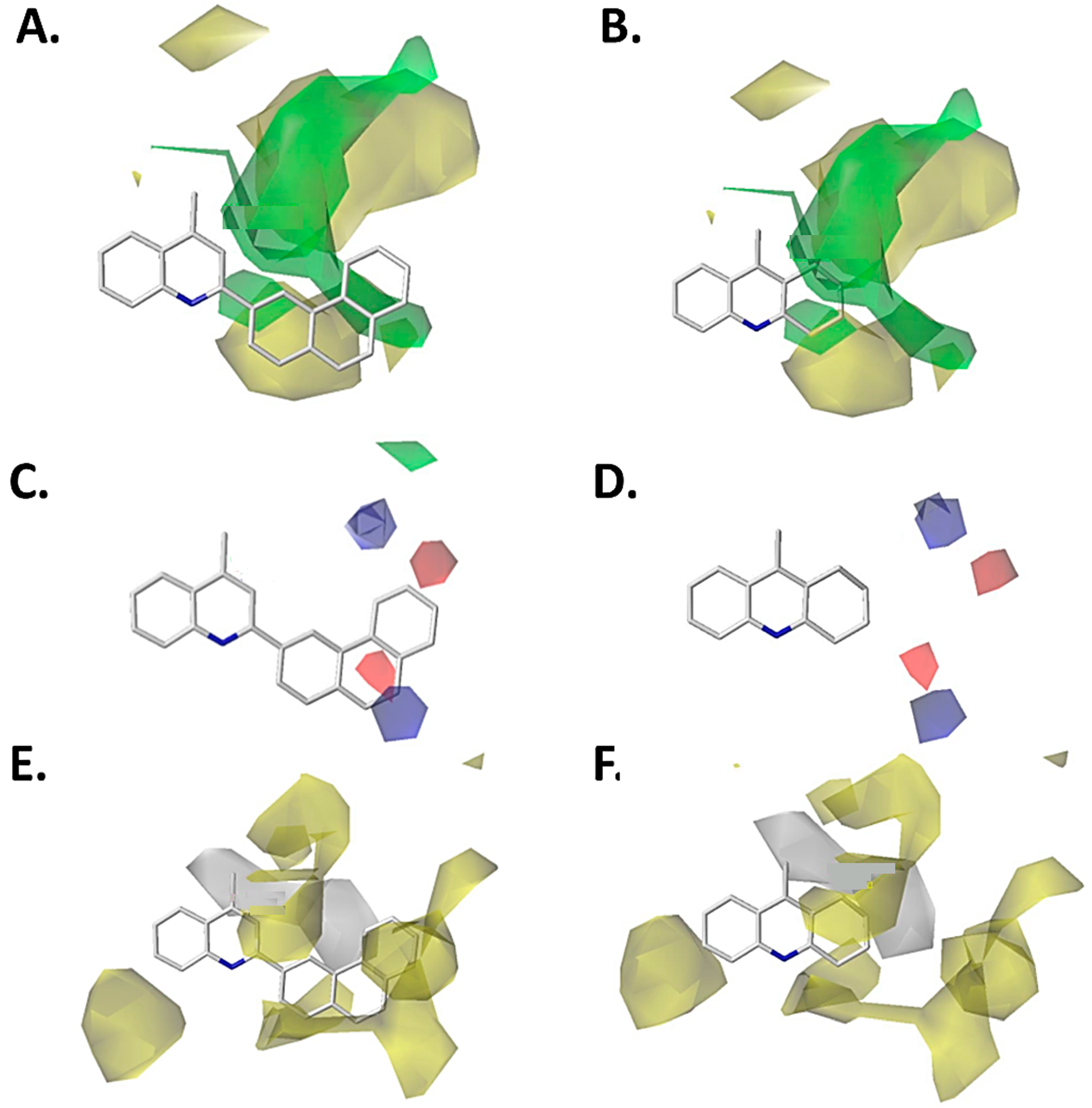
2.1. Contour Map Analysis
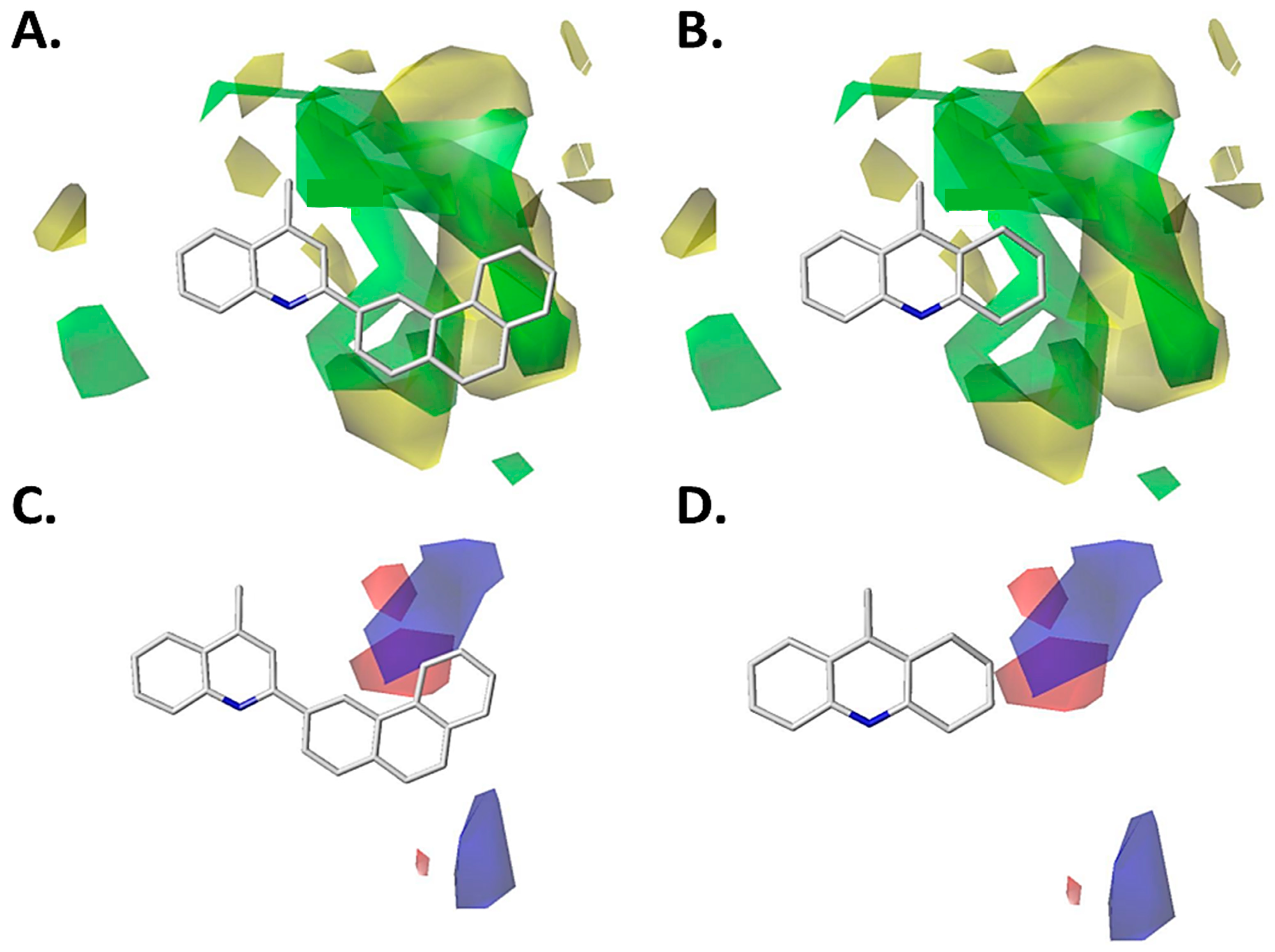
2.1.1. Steric Contour Map
2.1.2. Electrostatic Contour Map
2.1.3. Hydrophobic Contour Map
2.1.4. Hydrogen-Bonding Donor Contour Map
2.1.5. Hydrogen-Bonding Acceptor Contour Map
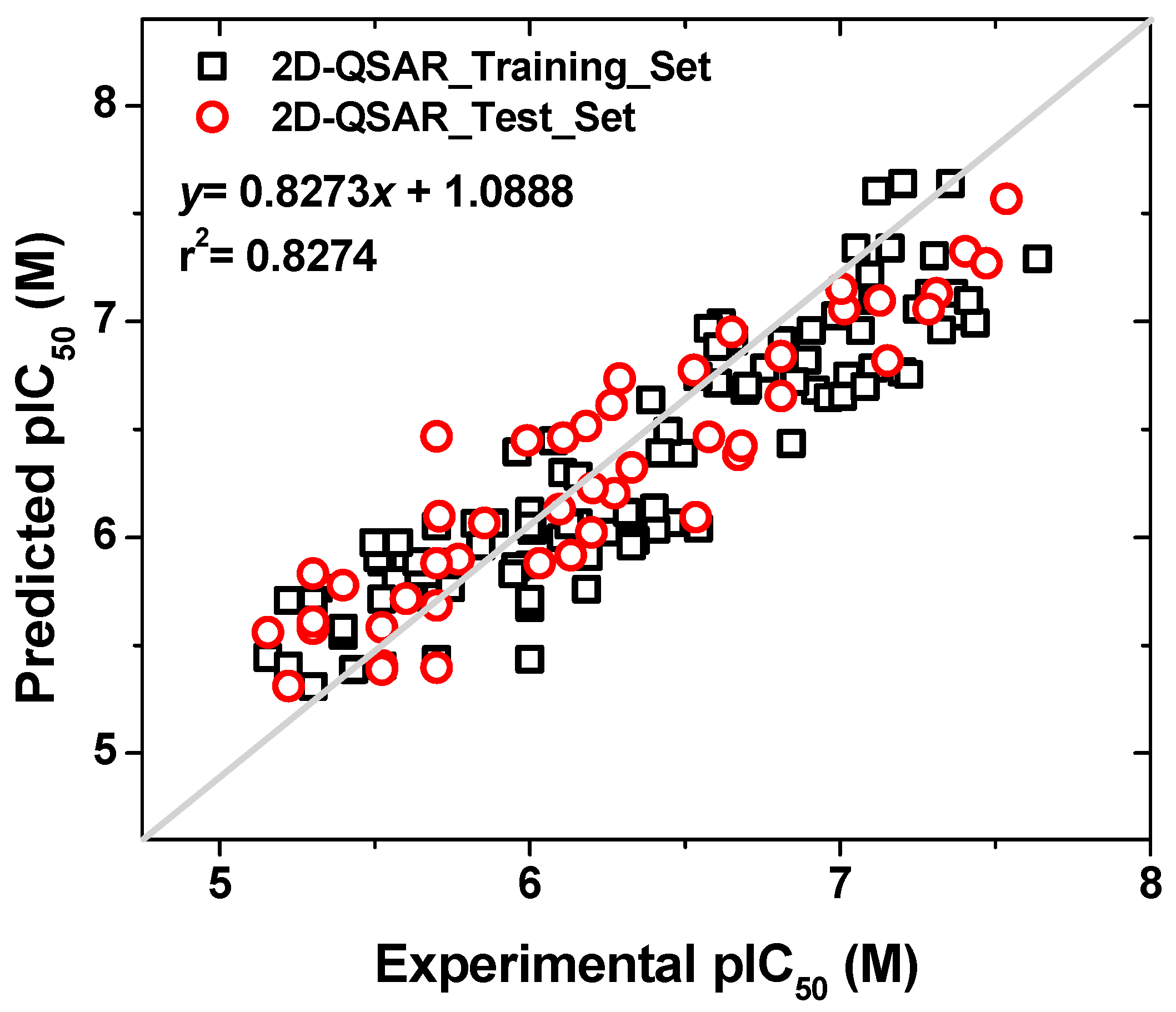
2.1.6. 2D-QSAR Model
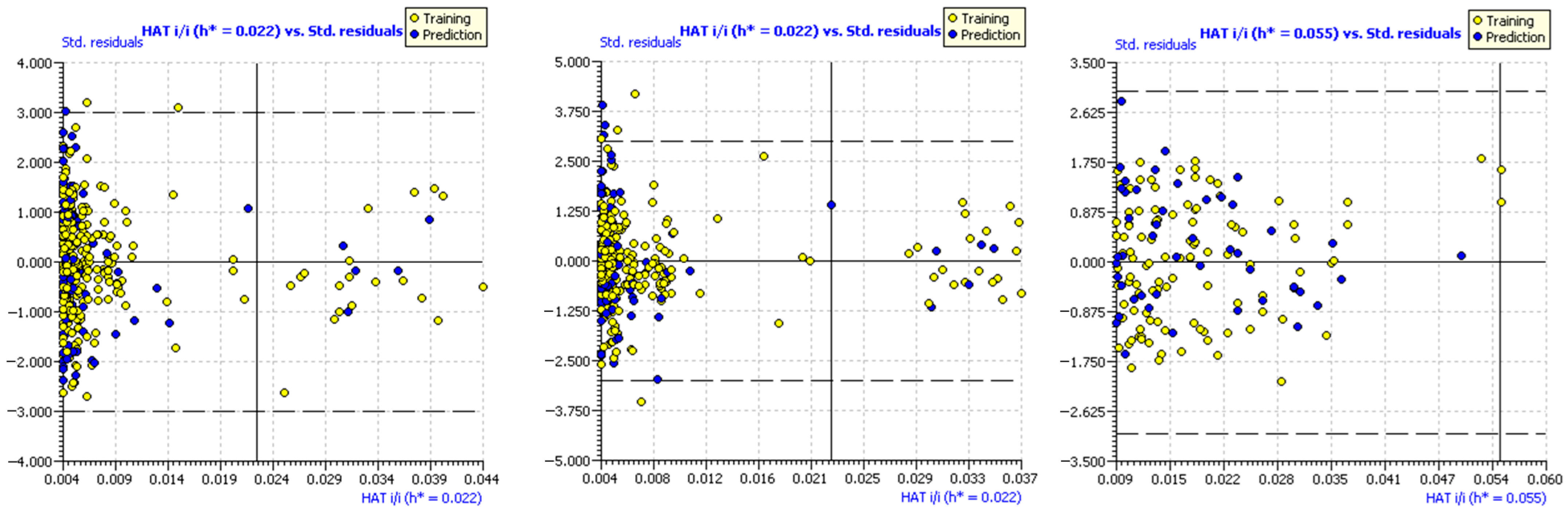
2.1.7. Applicability Domain of the Models
2.1.8. Summary of the Principal Results Based on the Theoretical Models
2.1.9. Limitations of the Models
2.2. Experimental Validation of the Theoretical Models
2.3. Evaluation of Physicochemical and Pharmacokinetic Properties
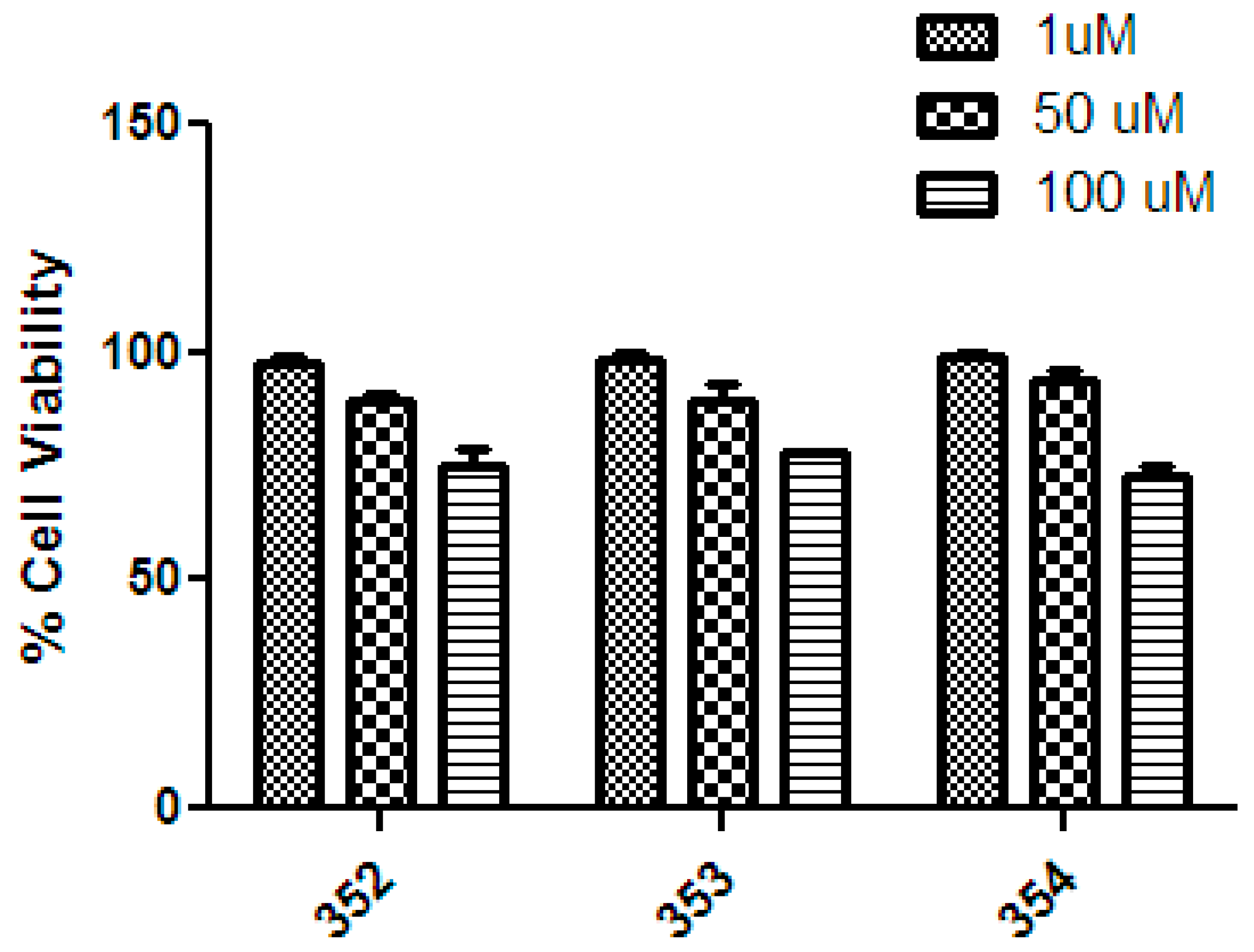
2.4. Cytotoxic Properties
3. Materials and Methods
3.1. Theoretical Models
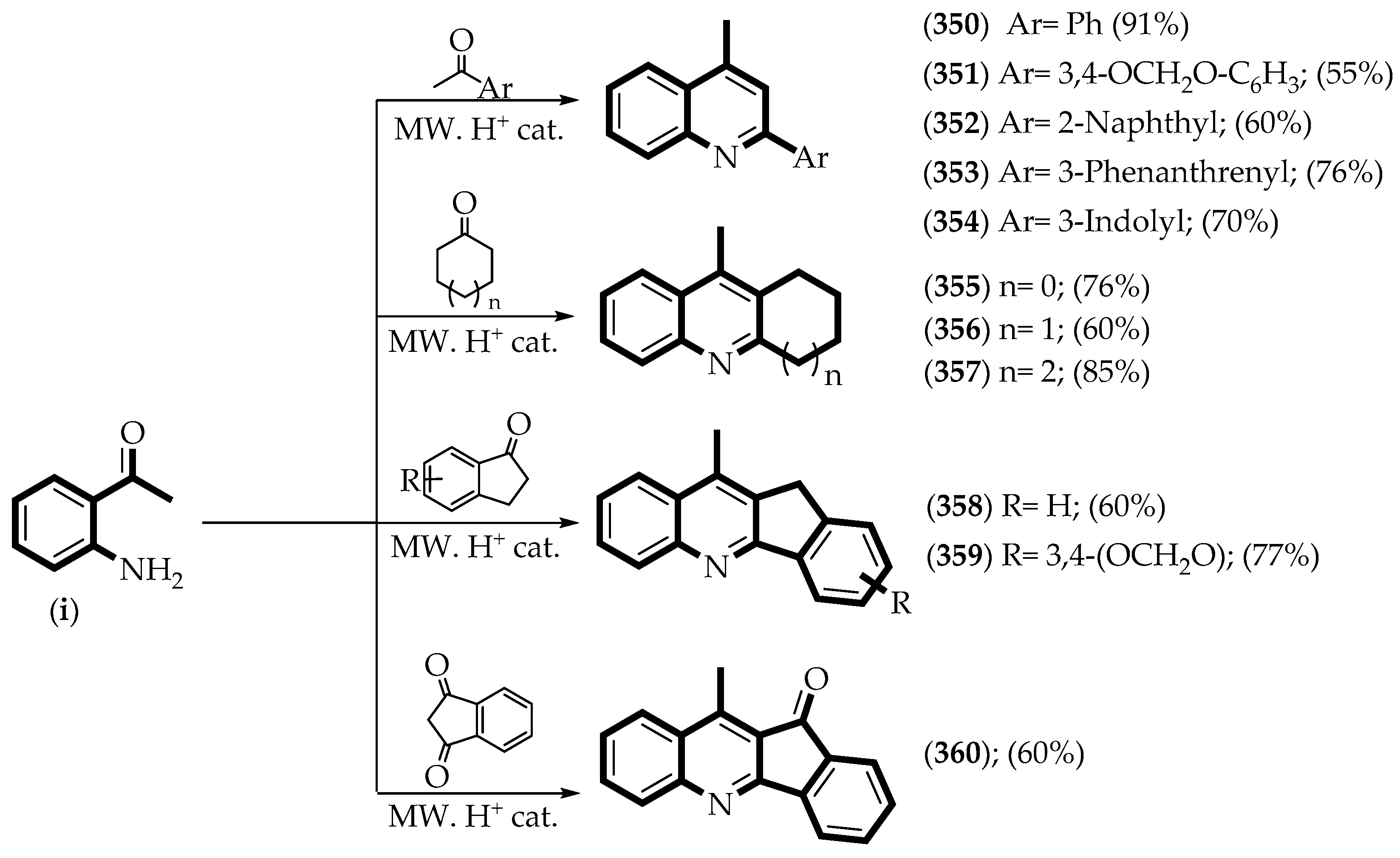
3.2. Synthesis of Compounds
3.2.1. Instrument and Chemicals
3.2.2. Synthetic Procedures
3.3. Biology
3.3.1. Maintenance of In Vitro Culture
3.3.2. SYBR Green I Inhibition Assay for the Asexual Stages of P. falciparum
3.3.3. Hepatocarcinoma Cell Cultures and Cytotoxicity Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fact Sheet about Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 27 October 2023).
- van der Pluijm, R.W.; Amaratunga, C.; Dhorda, M.; Dondorp, A.M. Triple Artemisinin-Based Combination Therapies for Malaria—A New Paradigm? Trends Parasitol. 2021, 37, 15–24. [Google Scholar] [CrossRef] [PubMed]
- El Rhabori, S.; El Aissouq, A.; Chtita, S.; Khalil, F. QSAR, Molecular Docking and ADMET Studies of Quinoline, Isoquinoline and Quinazoline Derivatives against Plasmodium Falciparum Malaria. Struct. Chem. 2023, 34, 585–603. [Google Scholar] [CrossRef]
- Mahmud, A.W.; Shallangwa, G.A.; Uzairu, A. QSAR and Molecular Docking Studies of 1,3-Dioxoisoindoline-4-Aminoquinolines as Potent Antiplasmodium Hybrid Compounds. Heliyon 2020, 6, e03449. [Google Scholar] [CrossRef] [PubMed]
- Shallangwa, G.A.; Mahmud, A.W.; Uzairu, A.; Ibrahim, M.T. 2,4-Disubstituted 6-Fluoroquinolines as Potent Antiplasmodial Agents: QSAR, Homology Modeling, Molecular Docking and ADMET Studies. J. Taibah Univ. Med. Sci. 2024, 19, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Bhati, S.; Sharma, M.; Gehlot, P.; Patel, N.; Dalai, S. 3D-QSAR-Based Design, Synthesis and Biological Evaluation of 2,4-Disubstituted Quinoline Derivatives as Antimalarial Agents. SAR QSAR Environ. Res. 2023, 34, 639–659. [Google Scholar] [CrossRef]
- Mellado, M.; Espinoza, L.; Madrid, A.; Mella, J.; Chavez-Weisser, E.; Diaz, K.; Cuellar, M. Design, Synthesis, Antifungal Activity, and Structure-Activity Relationship Studies of Chalcones and Hybrid Dihydrochromane-Chalcones. Mol. Divers. 2020, 24, 603–615. [Google Scholar] [CrossRef]
- Mellado, M.; Mella, J.; Gonzalez, C.; Vina, D.; Uriarte, E.; Matos, M.J. 3-Arylcoumarins as Highly Potent and Selective Monoamine Oxidase B Inhibitors: Which Chemical Features Matter? Bioorgan. Chem. 2020, 101, 103964. [Google Scholar] [CrossRef]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some Case Studies on Application of “r(m)(2)” Metrics for Judging Quality of Quantitative Structure-Activity Relationship Predictions: Emphasis on Scaling of Response Data. J. Comput. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inf. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Q. What Is a Randomization Test? J. Am. Stat. Assoc. 2023, 118, 2928–2942. [Google Scholar] [CrossRef]
- Uddin, R.; Lodhi, M.U.; Ul-Haq, Z. Combined Pharmacophore and 3D-QSAR Study on A Series of Staphylococcus Aureus Sortase A Inhibitors. Chem. Biol. Drug Des. 2012, 80, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Cheng, L.; Wang, Y.; Lyu, C.; Hou, C.; Zhang, Q. Exploration of 2D and 3D-QSAR Analysis and Docking Studies for Novel Dihydropteridone Derivatives as Promising Therapeutic Agents Targeting Glioblastoma. Front. Pharmacol. 2023, 14, 1249041. [Google Scholar] [CrossRef]
- Wong, W.; Bai, X.-C.; Sleebs, B.E.; Triglia, T.; Brown, A.; Thompson, J.K.; Jackson, K.E.; Hanssen, E.; Marapana, D.S.; Fernandez, I.S.; et al. The Antimalarial Mefloquine Targets the Plasmodium Falciparum 80S Ribosome to Inhibit Protein Synthesis. Nat. Microbiol. 2017, 2, 17031. [Google Scholar] [CrossRef] [PubMed]
- Slater, A.F.G. Chloroquine: Mechanism of Drug Action and Resistance in Plasmodium Falciparum. Pharmacol. Ther. 1993, 57, 203–235. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.L.M.; Palczewska, A.; Palczewski, J.; Kidley, N. Comparison of the Predictive Performance and Interpretability of Random Forest and Linear Models on Benchmark Data Sets. J. Chem. Inf. Model. 2017, 57, 1773–1792. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26, Reprinted in Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the General Solubility Equation: In Silico Prediction of Aqueous Solubility Incorporating the Effect of Topographical Polar Surface Area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef]
- Lin, J.; Sahakian, D.C.; de Morais, S.M.F.; Xu, J.H.; Polzer, R.J.; Winter, S.M. The Role of Absorption, Distribution, Metabolism, Excretion and Toxicity in Drug Discovery. Curr. Top. Med. Chem. 2003, 3, 1125–1154. [Google Scholar] [CrossRef] [PubMed]
- Toennesmann, E.; Kandolf, R.; Lewalter, T. Chloroquine Cardiomyopathy—A Review of the Literature. Immunopharmacol. Immunotoxicol. 2013, 35, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Ilamathi, M.; Sivaramakrishnan, V. Artesunate Acts as Fuel to Fire in Sensitizing HepG2 Cells towards TRAIL Mediated Apoptosis via STAT3 Inhibition and DR4 Augmentation. Biomed. Pharmacother. 2017, 88, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, P.; Luo, Z.; Chen, X.; Zhang, J.; Wang, C.; Chen, P.; Dong, Z. Chloroquine Inhibits Hepatocellular Carcinoma Cell Growth in Vitro and in Vivo. Oncol. Rep. 2016, 35, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Reyna-Jeldes, M.; Weinstein-Oppenheimer, C.; Covarrubias, A.A.; Aguilar, L.F.; Coddou, C.; Mella, J.; Cuellar, M.A. QSAR-Driven Synthesis of Antiproliferative Chalcones against SH-SY5Y Cancer Cells: Design, Biological Evaluation, and Redesign. Arch. Pharm. 2022, 355, e2200042. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Gonzalez, C.; Mella, J.; Aguilar, L.F.; Vina, D.; Uriarte, E.; Cuellar, M.; Matos, M.J. Combined 3D-QSAR and Docking Analysis for the Design and Synthesis of Chalcones as Potent and Selective Monoamine Oxidase B Inhibitors. Bioorgan. Chem. 2021, 108, 104689. [Google Scholar] [CrossRef]
- Cabezas, D.; Mellado, G.; Espinoza, N.; Garate, J.A.; Morales, C.; Castro-Alvarez, A.; Matos, M.J.; Mellado, M.; Mella, J. In Silico Approaches to Develop New Phenyl-Pyrimidines as Glycogen Synthase Kinase 3 (GSK-3) Inhibitors with Halogen-Bonding Capabilities: 3D-QSAR CoMFA/CoMSIA, Molecular Docking and Molecular Dynamics Studies. J. Biomol. Struct. Dyn. 2023, 41, 13250–13259. [Google Scholar] [CrossRef] [PubMed]
- Beus, M.; Fontinha, D.; Held, J.; Rajic, Z.; Prudencio, M.; Zorc, B. Synthesis and Antiplasmodial Evaluation of Novel Mefloquine-Based Fumardiamides. Acta Pharm. 2019, 69, 233–248. [Google Scholar] [CrossRef]
- Aggarwal, S.; Paliwa, D.; Kaushik, D.; Gupta, G.K.; Kumar, A. Synthesis, Antimalarial Evaluation and SAR Study of Some 1,3,5-Trisubstituted Pyrazoline Derivatives. Lett. Org. Chem. 2019, 16, 807–817. [Google Scholar] [CrossRef]
- Saini, D.; Jain, S.; Kumar, A.; Jain, N. Synthesis and Antimalarial Potential of Some Novel Quinoline-Pyrazolopyridine Derivatives. EXCLI J. 2016, 15, 730–737. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Avula, S.R.; Palnati, G.R.; Singh, S.V.; Srivastava, K.; Puri, S.K.; Saxena, J.K. Synthesis and in Vitro Evaluation of New Chloroquine-Chalcone Hybrids against Chloroquine-Resistant Strain of Plasmodium Falciparum. Bioorg. Med. Chem. Lett. 2012, 22, 5455–5459. [Google Scholar] [CrossRef]
- Mishra, A.; Batchu, H.; Srivastava, K.; Singh, P.; Shukla, P.K.; Batra, S. Synthesis and Evaluation of New Diaryl Ether and Quinoline Hybrids as Potential Antiplasmodial and Antimicrobial Agents. Bioorg. Med. Chem. Lett. 2014, 24, 1719–1723. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, A.; Saini, D. Novel Arylidene Derivatives of Quinoline Based Thiazolidinones: Synthesis, in Vitro, in Vivo and in Silico Study as Antimalarials. Exp. Parasitol. 2018, 185, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, S.P.; Kaushik, N.K.; Zaveri, M.; Mohanakrishanan, D.; Kawathekar, N.; Sahal, D. Synthesis, Characterization and Antimalarial Evaluation of New Beta-Benzoylstyrene Derivatives of Acridine. Arab. J. Chem. 2017, 10, S274–S280. [Google Scholar] [CrossRef]
- Singh, K.; Kaur, H.; Chibale, K.; Balzarini, J.; Little, S.; Bharatam, P.V. 2-Aminopyrimidine Based 4-Aminoquinoline Anti-Plasmodial Agents. Synthesis, Biological Activity, Structure-Activity Relationship and Mode of Action Studies. Eur. J. Med. Chem. 2012, 52, 82–97. [Google Scholar] [CrossRef]
- Shah, N.M.; Patel, M.P.; Patel, R.G. New N-Arylamino Biquinoline Derivatives: Synthesis, Antimicrobial, Antituberculosis, and Antimalarial Evaluation. Eur. J. Med. Chem. 2012, 54, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Tukulula, M.; Little, S.; Gut, J.; Rosenthal, P.J.; Wan, B.; Franzblau, S.G.; Chibale, K. The Design, Synthesis, in Silico ADME Profiling, Antiplasmodial and Antimycobacterial Evaluation of New Arylamino Quinoline Derivatives. Eur. J. Med. Chem. 2012, 57, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Prada, J.; Robledo, S.M.; Velez, I.D.; del Pilar Crespo, M.; Quiroga, J.; Abonia, R.; Montoya, A.; Svetaz, L.; Zacchino, S.; Insuasty, B. Synthesis of Novel Quinoline Based 4,5 Dihydro-1H Pyrazoles as Potential Anticancer, Antifungal, Antibacterial and Antiprotozoal Agents. Eur. J. Med. Chem. 2017, 131, 237–254. [Google Scholar] [CrossRef]
- Bentzinger, G.; De Souza, W.; Mullie, C.; Agnamey, P.; Dassonville-Klimpt, A.; Sonnet, P. Asymmetric Synthesis of New Antimalarial Aminoquinolines through Sharpless Aminohydroxylation. Tetrahedron-Asymmetry 2016, 27, 1–11. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Kumar, M.; Modukuri, R.K.; Srivastava, R.K.; Soni, A.; Srivastava, K.; Singh, S.V.; Saxena, J.K.; Gauniyal, H.M.; Puri, S.K. Antiplasmodial Activity of Novel Keto-Enamine Chalcone-Chloroquine Based Hybrid Pharmacophores. Bioorg. Med. Chem. 2012, 20, 2971–2981. [Google Scholar] [CrossRef]
- Barteselli, A.; Parapini, S.; Basilico, N.; Mommo, D.; Sparatore, A. Synthesis and Evaluation of the Antiplasmodial Activity of Novel Indeno[2,1-c]Quinoline Derivatives. Bioorg. Med. Chem. 2014, 22, 5757–5765. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kumar, R.; Kumar, R.; Upadhyay, P.; Sahal, D.; Sharma, U. Rh(III)-Catalyzed C(8)-H Functionalization of Quinolines via Simultaneous C-C and C-O Bond Formation: Direct Synthesis of Quinoline Derivatives with Antiplasmodial Potential. J. Org. Chem. 2018, 83, 12702–12710. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, W.; Huang, X.; Lu, X.; Patel, P.R.; Kim, M.; Orr, M.J.; Fisher, R.M.; Tanaka, T.Q.; McKew, J.C.; et al. Efficient Synthesis of 1,9-Substituted Benzo[h][1,6]Naphthyridin-2(1H)-Ones and Evaluation of Their Plasmodium Falciparum Gametocytocidal Activities. ACS Comb. Sci. 2017, 19, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Dana, S.; Keshri, S.K.; Shukla, J.; Vikramdeo, K.S.; Mondal, N.; Mukhopadhyay, P.; Dhar, S.K. Design, Synthesis and Evaluation of Bifunctional Acridinine-Naphthalenediimide Redox-Active Conjugates as Antimalarials. ACS Omega 2016, 1, 318–333. [Google Scholar] [CrossRef]
- Dola, V.R.; Soni, A.; Agarwal, P.; Ahmad, H.; Raju, K.S.R.; Rashid, M.; Wahajuddin, M.; Srivastava, K.; Haq, W.; Dwivedi, A.K.; et al. Synthesis and Evaluation of Chirally Defined Side Chain Variants of 7-Chloro4-Aminoquinoline To Overcome Drug Resistance in Malaria Chemotherapy. Antimicrob. Agents Chemother. 2017, 61, e01152-16. [Google Scholar] [CrossRef]
- Guillon, J.; Cohen, A.; Boudot, C.; Valle, A.; Milano, V.; Das, R.N.; Gudin, A.; Moreau, S.; Ronga, L.; Savrimoutou, S.; et al. Design, Synthesis, and Antiprotozoal Evaluation of New 2,4-Bis[(Substituted-Aminomethyl)Phenyl]Quinoline, 1,3-Bis[(Substituted-Aminomethyl)Phenyl]Isoquinoline and 2,4-Bis[(Substituted-Aminomethyl)Phenyl]Quinazoline Derivatives. J. Enzym. Inhib. Med. Chem. 2020, 35, 432–459. [Google Scholar] [CrossRef]
- Yu, X.-M.; Ramiandrasoa, F.; Guetzoyan, L.; Pradines, B.; Quintino, E.; Gadelle, D.; Forterre, P.; Cresteil, T.; Mahy, J.-P.; Pethe, S. Synthesis and Biological Evaluation of Acridine Derivatives as Antimalarial Agents. ChemMedChem 2012, 7, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Ramazani, A.; Khosravani, B.; Taran, J.; Ramazani, A. Evaluation of Novel Alpha-(Acyloxy)-Alpha-(Quinolin-4-Yl) Acetamides as Antiplasmodial Agents. Iran. J. Pharm. Res. 2017, 16, 924–928. [Google Scholar]
- Beus, M.; Fontinha, D.; Held, J.; Rajic, Z.; Uzelac, L.; Kralj, M.; Prudencio, M.; Zorc, B. Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents. Molecules 2019, 24, 2812. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Madrid, A.; Reyna, M.; Weinstein-Oppenheimer, C.; Mella, J.; Salas, C.O.; Sánchez, E.; Cuellar, M. Synthesis of Chalcones with Antiproliferative Activity on the SH-SY5Y Neuroblastoma Cell Line: Quantitative Structure–Activity Relationship Models. Med. Chem. Res. 2018, 27, 2414–2425. [Google Scholar] [CrossRef]
- Luczywo, A.; Soto, M.; Muscia, G.C.; Romanelli, G.P.; Sathicq, G.; Gonzalez, C.; Mella, J.; Mellado, M.; Asis, S.E. QSAR-Guided Study for the Microwave-Assisted Synthesis of 4-Methylquinoline Derivatives with Antimycobacterial Activity. ChemistrySelect 2023, 8, e202300042. [Google Scholar] [CrossRef]
- Yang, C.H.; Tai, C.C.; Huang, Y.T.; Sun, I.W. Ionic Liquid Promoted Palladium-Catalyzed Suzuki Cross-Couplings of N-Contained Heterocyclic Chlorides with Naphthaleneboronic Acids. Tetrahedron 2005, 61, 4857–4861. [Google Scholar] [CrossRef]
- Palimkar, S.S.; Siddiqui, S.A.; Daniel, T.; Lahoti, R.J.; Srinivasan, K.V. Ionic Liquid-Promoted Regiospecific Friedlander Annulation: Novel Synthesis of Quinolines and Fused Polycyclic Quinolines. J. Org. Chem. 2003, 68, 9371–9378. [Google Scholar] [CrossRef] [PubMed]
- Tufail, F.; Saquib, M.; Singh, S.; Tiwari, J.; Singh, M.; Singh, J.; Singh, J. Bioorganopromoted Green Friedlander Synthesis: A Versatile New Malic Acid Promoted Solvent Free Approach to Multisubstituted Quinolines. New J. Chem. 2017, 41, 1618–1624. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J. Human Malaria Parasites in Continuous Culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J. Synchronization of Plasmodium-Falciparum Erythrocytic Stages in Culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival—Application to Proliferation and Cyto-Toxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- de Madureira, M.D.; Martins, A.P.; Gomes, M.; Paiva, J.; da Cunha, A.P.; do Rosário, V. Antimalarial Activity of Medicinal Plants Used in Traditional Medicine in S. Tome and Principe Islands. J. Ethnopharmacol. 2002, 81, 23–29. [Google Scholar] [CrossRef]
- Mellado, M.; Gonzalez, C.; Mella, J.; Aguilar, L.F.; Celik, I.; Borges, F.; Uriarte, E.; Delogu, G.; Vina, D.; Matos, M.J. Coumarin-Resveratrol-Inspired Hybrids as Monoamine Oxidase B Inhibitors: 3-Phenylcoumarin versus trans-6-Styrylcoumarin. Molecules 2022, 27, 928. [Google Scholar] [CrossRef] [PubMed]
- Lorca, M.; Faundez, M.; Pessoa-Mahana, C.D.; Recabarren-Gajardo, G.; Diethelm-Varela, B.; Millan, D.; Celik, I.; Mellado, M.; Araque, I.; Mella, J.; et al. Design of benzimidazoles, benzoxazoles, benzothiazoles and thiazolopyridines as leukotriene A4 hydrolase inhibitors through 3D-QSAR, docking and molecular dynamics. J. Serb. Chem. Soc. 2023, 88, 25–39. [Google Scholar] [CrossRef]
- Erol, M.; Celik, I.; Saglik, B.N.; Karayel, A.; Mellado, M.; Mella, J. Synthesis, molecular modeling, 3D-QSAR and biological evaluation studies of new benzimidazole derivatives as potential MAO-A and MAO-B inhibitors. J. Mol. Struct. 2022, 1265, 133444. [Google Scholar] [CrossRef]
- Çevik, U.A.; Celik, I.; Mella, J.; Mellado, M.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, and Molecular Modeling Studies of a Novel Benzimidazole as an Aromatase Inhibitor. ACS Omega 2022, 7, 16152–16163. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular Similarity Indexes in a Comparative-Analysis (comsia) of Drug Molecules to Correlate and Predict Their Biological-Activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Parameters | Threshold Value | CoMFA-SE | CoMSIA-SEHDA |
|---|---|---|---|---|
| 1 | >0.5 | 0.765 | 0.796 | |
| 2 | >0.6 | 0.878 | 0.876 | |
| 3a | 0.954 | 0.954 | ||
| 3b | 0.935 | 0.935 | ||
| 4a | < 1.15 | 0.990 | 0.990 | |
| 4b | < 1.15 | 1.005 | 1.005 | |
| 5a | <0.1 | 0.089 | 0.089 | |
| 5b | <0.1 | 0.067 | 0.067 | |
| 6 | <0.3 | 0.019 | 0.019 | |
| 7 | >0.5 | 0.720 | 0.720 |
| Compound | IC50 (µM) | pIC50 | ||||||
|---|---|---|---|---|---|---|---|---|
| Experimental | CoMFA | CoMSIA | 2D-QSAR | |||||
| Prediction | Residual | Prediction | Residual | Prediction | Residual | |||
| 350 | >40.0 | − | − | − | − | − | ||
| 351 | 35.51 | 4.45 | 6.37 | −1.92 | 5.317 | −0.867 | 5.124 | −0.67 |
| 352 | 15.14 | 4.82 | 6.235 | −1.415 | 5.481 | −0.661 | 5.074 | −0.25 |
| 353 | 1.51 | 5.821 | 6.02 | −0.199 | 5.432 | 0.389 | 5.067 | 0.75 |
| 354 | 5.24 | 5.281 | 5.91 | −0.629 | 5.398 | −0.117 | 5.381 | −0.10 |
| 355 | 22.60 | 4.646 | 6.093 | −1.447 | 5.391 | −0.745 | 5.023 | −0.38 |
| 356 | 30.36 | 4.518 | 6.16 | −1.642 | 5.428 | −0.91 | 4.989 | −0.47 |
| 357 | 22.48 | 4.648 | 6.127 | −1.479 | 5.414 | −0.766 | 4.955 | −0.31 |
| 358 | 23.00 | 4.638 | 5.91 | −1.272 | 5.425 | −0.787 | 5.086 | −0.45 |
| 359 | 22.11 | 4.655 | 6.08 | −1.425 | 5.384 | −0.729 | 5.130 | −0.47 |
| 360 | 27.04 | 4.568 | 5.908 | −1.34 | 5.359 | −0.791 | 5.165 | −0.60 |
| CQ | 0.285 | 6.545 | 7.86 | −1.315 | 7.49 | −0.945 | 5.667 | 0.88 |
| RMSEP | 1.3576 | 0.7381 | 0.5315 | |||||
| MAE | 1.2803 | 0.7006 | 0.4849 | |||||
| Comp. | MW a | CLogP b | nOH c | nOHNH d | TPSA e | NV f | HIE(p) g | Caco-2(p) h | BBB(p) i |
|---|---|---|---|---|---|---|---|---|---|
| 353 | 319.40 | 3.53 | 1 | 0 | 12.89 | 1 | +(1.00) | +(0.72) | +(0.98) |
| 354 | 258.32 | 2.50 | 1 | 1 | 24.68 | 0 | +(1.00) | +(0.78) | +(0.97) |
| 356 | 197.28 | 2.61 | 1 | 0 | 12.89 | 0 | +(1.00) | +(0.73) | +(0.98) |
| CQ j | 319.87 | 3.95 | 8 | 2 | 28.16 | 0 | +(0.99) | +(0.66) | +(1.00) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorca, M.; Muscia, G.C.; Pérez-Benavente, S.; Bautista, J.M.; Acosta, A.; González, C.; Sabadini, G.; Mella, J.; Asís, S.E.; Mellado, M. 2D/3D-QSAR Model Development Based on a Quinoline Pharmacophoric Core for the Inhibition of Plasmodium falciparum: An In Silico Approach with Experimental Validation. Pharmaceuticals 2024, 17, 889. https://doi.org/10.3390/ph17070889
Lorca M, Muscia GC, Pérez-Benavente S, Bautista JM, Acosta A, González C, Sabadini G, Mella J, Asís SE, Mellado M. 2D/3D-QSAR Model Development Based on a Quinoline Pharmacophoric Core for the Inhibition of Plasmodium falciparum: An In Silico Approach with Experimental Validation. Pharmaceuticals. 2024; 17(7):889. https://doi.org/10.3390/ph17070889
Chicago/Turabian StyleLorca, Marcos, Gisela C. Muscia, Susana Pérez-Benavente, José M. Bautista, Alison Acosta, Cesar González, Gianfranco Sabadini, Jaime Mella, Silvia E. Asís, and Marco Mellado. 2024. "2D/3D-QSAR Model Development Based on a Quinoline Pharmacophoric Core for the Inhibition of Plasmodium falciparum: An In Silico Approach with Experimental Validation" Pharmaceuticals 17, no. 7: 889. https://doi.org/10.3390/ph17070889
APA StyleLorca, M., Muscia, G. C., Pérez-Benavente, S., Bautista, J. M., Acosta, A., González, C., Sabadini, G., Mella, J., Asís, S. E., & Mellado, M. (2024). 2D/3D-QSAR Model Development Based on a Quinoline Pharmacophoric Core for the Inhibition of Plasmodium falciparum: An In Silico Approach with Experimental Validation. Pharmaceuticals, 17(7), 889. https://doi.org/10.3390/ph17070889